

Summary
The precursor cells for metabolically beneficial beige adipocytes can alternatively become fibrogenic and contribute to adipose fibrosis. This adipogenic versus fibrogenic fate decision is poorly understood. Cold exposure or β3-adrenergic agonist treatment of mice decreased the fibrogenic profile of precursor cells and stimulated beige adipocyte differentiation. This fibrogenic-to-adipogenic transition was impaired in aged animals, correlating with reduced adipocyte expression of the transcription factor PRDM16. Genetic loss of Prdm16 mimicked the effect of aging in promoting fibrosis, whereas increasing PRDM16 in aged mice reduced fibrosis and restored beige adipogenesis. PRDM16-expressing adipose cells secreted the metabolite β-hydroxybutyrate (BHB), which blocked precursor fibrogenesis and enhanced beige adipogenesis. BHB catabolism in precursor cells, mediated by BDH1, was required for beige fat differentiation in vivo. Finally, dietary BHB supplementation in aged animals reduced adipose fibrosis and restored beige fat formation. Together, our results demonstrate that adipocytes secrete a metabolite signal that controls beige fat remodeling.
eTOC blurb
Wang et al., show that mature adipocytes secrete beta-hydroxybutyrate (BHB), which acts on adipose precursor cells to suppress fibrosis responses and enhance beige adipocyte differentiation. Raising BHB levels ameliorates adipose fibrosis and restores beige fat development in aged animals.
Graphical Abstract
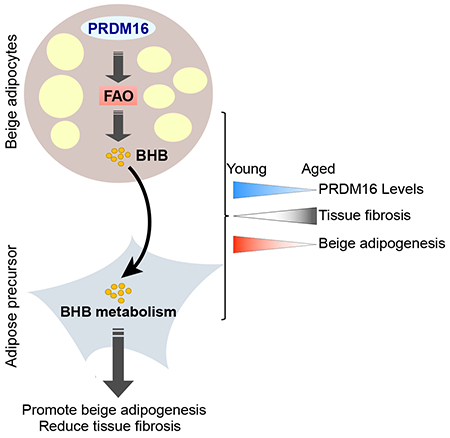
Introduction
Brown and beige adipocytes burn energy for heat production and have garnered much attention because of their capacity to counteract metabolic disease (Harms and Seale, 2013; Kajimura et al., 2015). Brown adipocytes are located in discrete deposits of brown adipose tissue (BAT), whereas beige adipocytes develop in white adipose tissue (WAT) in response to environmental cold and other stimuli (Wang and Seale, 2016). Brown and beige adipocytes have abundant mitochondria that contain Uncoupling protein-1 (UCP1) (Cannon and Nedergaard, 2004). Upon activation by cold/β-adrenergic signaling, UCP1 catalyzes proton leak across the inner mitochondrial membrane, resulting in increased oxygen consumption and heat production (Cannon and Nedergaard, 2004; Fedorenko et al., 2012; Klingenberg and Huang, 1999; Ricquier, 2011).
Increasing the amount and/or activity of beige adipocytes in mice promotes a lean and healthy metabolic phenotype (Wu et al., 2013). Conversely, reductions in beige fat activity predispose animals to the harmful metabolic effects of a high fat diet (Cohen et al., 2014). Beige adipocytes develop from PDGFRα+ precursor cells that are also capable of differentiating into fibrosis-inducing “myofibroblast-like” cells (Berry et al., 2016; Lee et al., 2012; Lin et al., 2018; Long et al., 2014; Marcelin et al., 2017; McDonald et al., 2015; Shao et al., 2016; Wang et al., 2013). Analogous mesenchymal cell populations possessing adipogenic and fibrogenic potential called “fibro-adipogenic progenitor” (FAP) cells are also found in skeletal muscle and heart (Joe et al., 2010; Lemos et al., 2015; Uezumi et al., 2010).
Myofibroblast differentiation in adipose tissue is regulated by various pathways, including hypoxia/HIF1α and TGFβ (Halberg et al., 2009; Marcelin et al., 2017; Sun et al., 2013a; Sun et al., 2013b; Vila et al., 2014). Both of these pathways also inhibit adipogenesis. In adipose tissue, HIF1α-activation is sufficient to cause fibrosis, whereas pharmacological inhibition of HIF1α reduces fibrosis and improves systemic metabolism (Halberg et al., 2009; Jiang et al., 2011; Sun et al., 2013a). TGFβ promotes fibrogenesis through activation of Rho-associated protein kinase (ROCK) and resultant assembly of actin stress fibers (Carthy, 2018). Notably, deletion of Mrtfa, a downstream effector in the TGFβ pathway, blocks the myofibrogenic programming of adipose precursors and enhances beige fat differentiation (Lin et al., 2018; McDonald et al., 2015). However, the mechanisms that regulate the fibrogenic vs. adipogenic activity of precursor cells during beige fat development are currently unknown.
The zinc-finger transcriptional factor PRD1-BF1-RIZ1 homologous domain-containing protein 16 (PRDM16) is a powerful driver of brown and beige adipocyte identity (Cohen et al., 2014; Harms et al., 2015; Ishibashi and Seale, 2015; Kajimura et al., 2008; Ohno et al., 2013; Seale et al., 2011; Seale et al., 2007). PRDM16 co-activates PPARγ and PPARα in adipocytes to activate the expression of thermogenic genes (Hondares et al., 2011; Seale et al., 2008). Interestingly, a recent paper showed that PRDM16 overexpression in adipose tissue reduces high fat diet-induced fibrosis (Hasegawa et al., 2018). However, if and how PRDM16-action in adipocytes affects the fibrogenic vs. beige adipogenic fate of precursor cells was unclear.
In this study, we identify a PRDM16-regulated adipocyte-to-precursor paracrine signal that suppresses fibrogenesis and promotes beige fat development. Specifically, PRDM16 drives a fatty acid oxidation and ketogenesis program in adipose cells, leading to secretion of the metabolite β-hydroxybutyrate (BHB). BHB acts on precursor cells to block HIF1α- or TGFβ-induced myofibroblast differentiation and promote beige adipocyte differentiation. This action of BHB in precursor cells is dependent on the ketyolytic enzyme BDH1, revealing an important role for ketone metabolism in adipose tissue remodeling. Finally, raising PRDM16 or BHB levels in mice reverses aging-induced fibrosis and restores beige fat developmental potential.
Results
Aging impairs beige adipogenesis and promotes fibrogenesis
Human brown/beige fat activity levels decline with aging, correlating with a decrease in metabolic rate and increase in fat mass (Cypess et al., 2009; Ouellet et al., 2011; Pfannenberg et al., 2010; Yoneshiro et al., 2011). Similarly, the development of UCP1+ beige adipocytes in response to cold or the β3-agonist CL316,243 (CL) was drastically reduced in aged (12-month-old) compared to young (2-month-old) mice (Fig.1A,B; Fig. S1A–D), consistent with the results from other studies (Berry et al., 2017; Rogers et al., 2012). Beige fat cells in iWAT develop via the activation of thermogenic genes in mature adipocytes (i.e. beige conversion of mature adipocytes) or through the beige adipogenic differentiation of precursor cells (Berry et al., 2016; Lee et al., 2015; Lee et al., 2012; Shao et al., 2016; Wang et al., 2013). To determine if aging reduces the differentiation of beige adipocytes, we compared the beiging response in young and aged AdipoChaser mice (AdipoqrtTA; TRECre; Rosa26RmT/mG) (Wang et al., 2013). Doxycycline (dox) treatment of these mice induced GFP expression in mature (PLIN1+) adipocytes (“pulse”) (Fig. S1E,F). After CL treatment (“chase”), the iWAT of young mice contained large areas of GFP(−) (i.e. new) multilocular beige adipocytes, with the proportion of GFP(−) adipocytes increasing from ~5% to >35% (Fig. S1F,G). By contrast, CL-treatment did not increase the proportion of unlabeled adipocytes in old mice, indicative of impaired adipogenesis (Fig. S1F,G).
Fig.1. Cold/β3-agonism induces a fibrogenic-to-adipogenic phenotypic shift in adipose stromal cells.

(A-E) Young (2-month-old) and aged (12-month-old) mice were acclimated to thermoneutrality and treated with vehicle or CL for 4 days to induce beiging.
(A) Immunofluorescence staining for UCP1 (green) in iWAT. Nuclei (DAPI, blue); scale bar, 1 mm.
(B) Relative mRNA levels of brown fat marker genes Ucp1 and Cidea. n=4 mice per group.
(C) Heat map of gene expression profiles comparing iWAT from young and aged mice, treated with vehicle or CL.
(D) Gene ontology (GO) and pathway analysis of a gene cluster downregulated by CL selectively in young vs. aged mice (red cluster, panel C). KG, KEGG Pathway; MF, Molecular Function; BP, Biological Process; CC, Cellular Component.
(E) Relative mRNA levels by RT-qPCR of fibrosis marker genes in iWAT. n=4 mice per group.
(F) Relative mRNA levels of fibrosis marker genes in stromal vascular cells (SVC) and mature adipocytes from young mice housed at thermoneutrality or exposed to 4°C for 1 week. n=3 mice per group.
(G) Relative mRNA levels of fibrosis marker genes in SVC from young and aged mice housed at thermoneutrality and treated with vehicle or CL for 4 days. n=3 mice per group.
(H) Picrosirius red staining of collagen fibers in iWAT from young and aged mice treated with vehicle (Ctl) or CL. Scale bar, 50 μM.
All data presented as mean ± s.e.m; *p<0.05, ** p<0.01, ***p<0.001 as analyzed by two-tailed Student’s t-test.
We used RNAseq to profile iWAT gene expression in young and aged mice, treated with either vehicle or CL. As expected, iWAT from aged mice displayed a blunted up-regulation of many thermogenic genes in response to CL-treatment, including genes involved in oxidative phosphorylation and electron transport chain activity (Fig. S1H). CL-treatment also downregulated the expression of extracellular matrix (ECM) and fibrosis marker genes in young iWAT, whereas these genes were unchanged or increased in iWAT from aged animals (Fig. 1C–E, S1I). These fibrosis-signature genes (e.g. Col1a1, Col3a1, Loxl1, Lox, Fbln2) were expressed at higher levels in stromal-vascular cells (SVC; a population enriched for precursor cells) than in mature adipocytes and their expression levels in SVCs were decreased by cold exposure (Fig. 1F, S1J). Finally, CL treatment decreased the expression of fibrosis genes in SVCs from young but not aged mice (Fig.1G). At a histological level, CL-treatment decreased collagen deposition and fibrosis in young mice but augmented WAT fibrosis in old animals (Fig. 1H). Notably, aging did not impair BAT activation or induce fibrotic changes in BAT (Fig. S1A–C). Together, these findings indicate that thermogenic stimuli induce a fibrogenic-to-adipogenic phenotypic shift in iWAT stromal cells and that this process is impaired by aging.
PRDM16 protects against aging- and HFD-induced adipose fibrosis
The transcription factor PRDM16, which regulates the transcription of thermogenic genes in adipocytes, is required for maintaining BAT fate during aging (Harms et al., 2014). Interestingly, aging reduced PRDM16 levels in iWAT, but not in BAT of mice housed at 30°C (Fig. 2A, S2A), correlating with the selective loss of beige fat developmental potential. The reduction of PRDM16 expression in iWAT was more pronounced in adipocytes than in SVC.
Fig.2. PRDM16 promotes a fibrogenic-to-adipogenic cell fate shift during WAT beiging.

(A) Western blot analysis of PRDM16, PPARγ, and α-Actin protein levels in whole iWAT (left) or in isolated adipocytes and stromal-vascular cells (SVC) from iWAT of young and aged mice acclimated to thermoneutrality.
(B,C) Wildtype (Wt) and Prdm16+/− mutant mice were housed at thermoneutrality and treated with vehicle (Control) or CL for 4 days. n=3-4 mice per group.
(B) Relative mRNA levels of Prdm16 and fibrosis marker genes in iWAT.
(C) UCP1 immunofluorescence (top) and Picrosirius red staining of collagen fibers (bottom) in iWAT. Scale bar, 50 μM.
(D,E) Thermoneutral-acclimated wildtype (Wt) and adipocyte-selective Prdm16-knockout (AdipoqCrePrdm16-KO) mice were treated with vehicle control or CL for 4 days. n=3-4 mice per group.
(D) Picrosirius red staining of collagen fibers in iWAT. Scale bar, 50 μM.
(E) Relative mRNA levels of fibrosis marker genes in iWAT.
(F,G) young (2-month-old) and aged (12-month-old) wildtype (Ctl) and Fabp4-Prdm16 mice were acclimated to thermoneutrality and treated with CL for 4 days. n=3-5 mice per group.
(F) Picrosirius red staining of collagen fibers. Scale bar, 50 μM.
(G) Relative mRNA levels of fibrosis marker genes in iWAT.
All data presented as mean ± s.e.m; * p<0.05, ** p<0.01, ***p<0.001 as analyzed by two-tailed Student’s t-test.
To assess if PRDM16 regulates fibrogenic vs. adipogenic precursor fate, we compared the beiging response in littermate wildtype and heterozygous Prdm16+/− mutant mice. Prdm16 expression was reduced by ~50% in the iWAT of Prdm16+/− animals, approximating the PRDM16 reduction observed during aging (Fig. 2A,B,S2B). In wildtype animals, CL induced the formation of UCP1+ beige adipocytes and decreased the number and the thickness of fibrotic/collagen streaks (Fig. 2C, S2C). By contrast, Prdm16+/− iWAT displayed a near-complete loss of CL-induced beige fat development and retained high levels of collagen deposition (Fig. 2C). Fibrosis genes that were downregulated by CL in wildtype iWAT were either unchanged or increased by CL in Prdm16+/− iWAT (Fig. 2B, S2C). The anti-fibrogenic and pro-browning effects of CL-treatment in iWAT were similarly decreased by adipocyte-selective Prdm16 deficiency (AdipoqCre) (Fig. 2D,E; S2D).
We next assessed if elevating PRDM16-expression in adipocytes (of Fabp4-Prdm16 transgenic mice) could prevent aging-induced fibrosis and prevent the loss of beige fat development. CL-treatment decreased collagen deposition and lowered the levels of fibrosis genes (i.e. Collal, Col3a1) in the iWAT of 1-year-old Prdm16-transgenic but not control (wildtype) mice (Fig. 2F,G; S2E,F). Transgenic PRDM16 expression also facilitated the development of UCP1+ beige adipocytes in the iWAT of aged animals (Fig. S2F). Given that the Fabp4 promoter is not entirely restricted to adipocytes (Lee et al., 2013; Mullican et al., 2013; Stine et al., 2015), it is possible that PRDM16 function in another cell type contributes to the phenotype of Fabp4-Prdm16 animals. However, the reciprocal effects of PRDM16 gain- and loss-of-function strongly suggest that PRDM16-action in adipocytes suppresses fibrogenic responses and stimulates beige fat development.
Adipocyte PRDM16 controls precursor cell fate through a paracrine pathway
The HIF1α and TGFβ pathways are major drivers of myofibroblast differentiation. HIF1α activation, in particular, is sufficient to drive adipose fibrosis in the setting of obesity (Halberg et al., 2009). We found that HIF1α levels were elevated in the iWAT of aged versus young animals (Fig. S3A). Activation of HIF1α in primary iWAT precursor cells via treatment with dimethyloxaloylglycine (DMOG) induced a myofibroblast phenotype, including elevated Smooth muscle actin (ACTA2) expression and the formation of actin stress fibers (Fig. S3B,C). HIF1α-activation also blocked adipocyte differentiation, including suppressing lipid accumulation and preventing the induction of adipocyte marker genes (Pparg, Adipoq) (Fig. S3D,E). DMOG did not induce myofibroblast development or inhibit adipogenesis in Hif1α-deficient precursor cells, showing that the effects of DMOG were HIF1α-dependent (Fig. S3B–E).
To determine if PRDM16 interacts with HIF1α to regulate precursor cell fate, we transduced iWAT precursor cells with PRDM16-expressing or control retrovirus and used DMOG to activate HIF1α (Fig. 3A). PRDM16-expression did not affect HIF1α protein levels or vice-versa (Fig. 3B, Fig. S3F). Under vehicle-treated (standard) conditions, PRDM16-expression did not influence adipocyte differentiation efficiency (Fig. 3C,D). Strikingly however, PRDM16 blocked the HIF1α-mediated induction of ACTA2+ stress fibers (Fig. 3C). PRDM16 also restored the adipocyte differentiation capacity of DMOG-treated cells, including promoting lipid accumulation and activating the expression of adipocyte marker genes (Pparg, Fabp4, Ucp1) (Fig. 3C,D).
Fig.3. Adipocyte PRDM16 controls precursor fate through a paracrine pathway.

(A-D) iWAT precursor cells were transduced with control (CTL) or PRDM16-expressing retrovirus and treated with either vehicle control (CTL) or DMOG. After 2 days, cells were immunostained for ACTA2 or induced to differentiate into adipocytes for 5 days.
(A) Experimental schema.
(B) Western blot analysis of HIF1α, PRDM16 and α-Actin protein levels.
(C) ACTA2 immunostaining (green) (top panel) and LipidTOX staining (red) of lipid droplets in differentiated adipocytes (bottom panel). Scale bar, 100 μM (top); 200 μM (bottom).
(D) Relative mRNA levels of adipocyte genes. n=3 per group.
(E-K) iWAT precursor cells were treated with: (1) control medium (DMEM), conditioned medium (CM) from GFP-expressing adipocytes (GFP-CM), or CM from PRDM16-expressing adipocytes (P-CM); and (2) vehicle, DMOG or recombinant TGFβ. After 24 h, cultures were immunostained for ACTA2 or induced to differentiate into adipocytes for 5 days.
(E) Experimental schema.
(F) ACTA2 immunostaining (green). Scale bar, 100 μm.
(G) Violin plots showing relative fluorescent intensity per cell in indicated groups. The white solid boxes represent percentiles (bottom of box = 25th, horizontal line =50th, top of box = 75th). n=50-80 cells per condition.
(H) Oil-Red-O staining (red) of adipocyte lipid droplets.
(I) Relative mRNA levels of adipocyte markers in cultures from (H). n=3-5 per group.
(J) ACTA2 immunostaining (top; green) and LipidTOX staining (bottom; red) of adipocyte lipid droplets in cultures treated with indicated CM and either vehicle or TGFβ. Scale bar, 100 μm. Violin plots showing relative fluorescent intensity per cell (right).
(K) Relative mRNA levels of adipocyte marker genes in cultures from (J). n=3-5 per group.
All data presented as mean ± sem. Analysis by two-way ANOVA with Tukey correction.
Surprisingly, even a low level of PRDM16-transduction efficiency (~20% of cells) prevented myofibrogenic differentiation across the culture (Fig.S3G), suggesting that PRDM16 might act via a cell non-autonomous pathway. Therefore, we tested if conditioned medium (CM) from PRDM16- or GFP-expressing (control) adipocytes could modulate precursor differentiation (Fig. 3E). CM from PRDM16-expressing adipocytes (P-CM) recapitulated the effect of PRDM16-expression in suppressing HIF1α-induced stress-fiber formation whereas CM from GFP-expressing cells (GFP-CM) had a more limited effect (Fig. 3F,G). Moreover, P-CM strongly stimulated beige adipocyte differentiation in HIF1α-expressing cells, including promoting lipid droplet formation and inducing the expression of adipocyte genes (Pparg, Adipoq, Ucp1) (Fig. 3H,I; S3H). P-CM also suppressed the pro-fibrogenic and anti-adipogenic effects of TGFβ-treatment (Fig. 3J,K; S3I). These results indicate that P-CM represses a common downstream step in the myofibrogenic program and enhances adipogenic commitment of precursor cells.
To test if PRDM16 is required for the production of adipogenic paracrine signal(s) by adipocytes, we compared the activity of CM from control (wildtype) and Prdm16-knockout (KO) adipocytes from iWAT (Fig. S3J). For these assays, we used a lower dose of DMOG to achieve modest elevation of HIF1α, which blocked adipocyte differentiation (Fig. S3K). Treatment of these cultures with CM from control adipocytes enhanced adipocyte differentiation, while CM from Prdm16 KO cells had markedly less activity (Fig. S3K,L). Collectively, these data demonstrate that PRDM16 promotes the production of a paracrine factor(s) that suppresses myofibrogenesis and stimulates adipogenic competency in precursor cells.
PRDM16-driven FAO is required for the production of paracrine effectors
The adipogenic activity of P-CM was decreased by charcoal treatment but resistant to heating, suggesting that the active component could be a water-soluble metabolite (Fig. S4A). RNA-seq and gene ontology analysis identified “fatty acid triacylglycerol and ketone body metabolism” as enriched pathways in PRDM16-expressing vs. control adipocytes (Fig. 4A). PRDM16-expression increased the levels of many fatty acid oxidation (FAO) and ketone metabolism genes (e.g. Cpt1, Cpt2, Acadl, Acadm, Hmgcs2) in adipocytes (Fig. 4B, S4B). In particular, PRDM16-expressing adipocytes expressed 10-fold higher levels of Cpt1b (Fig. S4B), which encodes the rate-limiting enzyme in FAO. Moreover, transgenic expression of PRDM16 in iWAT adipocytes increased FAO gene levels and CPT1b protein levels (Fig. S4C,D). Reciprocally, aging-induced or genetic loss of PRDM16 in iWAT or isolated adipocyte cultures decreased FAO gene levels and CPT1b protein levels (Fig. S4E–H). At a functional level, PRDM16-expressing cells generated >20-fold higher levels of acetyl-coA from 13C-labeled palmitate than control cells, indicating much higher rates of FAO (Fig. 4C, S4I). Finally, metabolomic analyses revealed increased levels of tricarboxylic acid (TCA) cycle intermediates and reduced levels of glycolytic metabolites in PRDM16-expressing adipocytes (Fig. 4D).
Fig.4. PRDM16-driven FAO is required for the production of paracrine effectors.

(A) Gene Set Enrichment Analysis (GSEA) of genes upregulated in PRDM16-expressing vs. control cells. Enriched pathways were defined by a normalized enrichment score > 1.3 and FDR < 0.05.
(B) Expression heat map of “fatty acid oxidation (FAO) and ketone body metabolism” genes in PRDM16-expressing vs. control cells.
(C) Quantification of FAO. Adipocytes were cultured in medium containing U-13C-palmitate for 6 h prior to metabolite analysis by LC-MS. Relative molar quantity of 13C2-labeled acetyl-CoA in GFP- and PRDM16-expressing adipocytes was calculated by multiplying % molar enrichment of 13C2-labeled acetyl-CoA by relative total acetyl-CoA quantitation. n=3-4 per group.
(D) Heat map showing standardized amounts of indicated metabolites in GFP- and PRDM16-expressing adipocytes, across samples (Z score). n=3 per group
(E-G) PRDM16-expressing adipocytes (donor) were treated with vehicle or Etomoxir (Eto) for 24 hr prior to collecting conditioned medium (CM). HIF1α-expressing (DMOG-treated) precursors were treated with CM and induced to undergo adipocyte differentiation. To control for effects of Eto carried over in CM, a subset of recipient cells were directly exposed to Eto [Eto(recipient)] by mixing Eto with P-CM prior to treatment.
(E) Experimental schema
(F) Oil-Red-O staining of cultures after adipogenic induction.
(G) Relative mRNA levels of adipocyte genes in cultures from (F). n=4-5 per group.
(H,I) AdipoChaser (AdipoqrtTA;TRECre;Rosa26mTmG) mice were pulse labelled by feeding them with doxycyxline (dox) chow (600 mg/kg) for 9 days. During the 3-day washout period, mice were treated with Eto or vehicle for 3 days, followed by treatment with CL and either Eto or vehicle for another 4 days (chase).
(I) Immunofluorescence staining of PLIN1 (red) in adipocytes and GFP reporter (green). Quantification of adipogenesis (% GFP (−)/total PLIN1+ cells) (right).
All data presented as mean ± s.e.m; * p <0.05, **p<0.01, ***p < 0.001 as analyzed by two-tailed Student’s t-test.
To determine if FAO was necessary for the paracrine activity of PRDM16-expressing adipocytes, we treated PRDM16-expressing cells with the CPT1-inhibitor, Etomoxir (Eto) or vehicle (control) for 24 h prior to collecting CM (Fig. 4E). Eto-treatment blocked FAO in adipocytes, as assayed by 3H-palmitate oxidation (Fig. S5A). The CM from Eto-treated adipocytes had a severely diminished capacity to promote adipogenesis and adipocyte gene expression in HIF1α-expressing cells (Fig. 4F,G). Treatment of precursor cultures directly with Eto resulted in a mild impairment of adipocyte differentiation.
We further investigated whether acute inhibition of FAO in mice disrupts CL-induced beige fat remodeling. Mice housed at thermoneutrality were treated with either vehicle or Eto along with CL to induce beiging (Fig. S5B). Eto-treatment blocked the induction of UCP1+ beige adipocytes and decreased the expression levels of thermogenic genes, including a 90% reduction in Ucp1 levels (Fig. S5C,D). We used AdipoChaser reporter mice (Wang et al., 2013) to determine if FAO is required for adipogenesis (Fig. 4H,I). Doxycycline administration in these mice induced the expression of mGFP in adipocytes (pulse). This was followed by a chase period, during which mice were treated with CL to induce beiging along with either vehicle or Eto for 5 days. iWAT from control mice (CL+vehicle) underwent beige adipogenesis, as evidenced by the development of GFP-negative multilocular adipocytes (induced from ~5% to 25%; Fig. 4I, middle-bottom panel; red PLIN+ adipocytes without GFP). By contrast, GFP-negative adipocytes remained at background levels in the iWAT of Eto-treated mice (Fig. 4I, bottom-right panel; yellow = red PLIN+ colocalized with green GFP+). In addition to blocking beige adipogenesis, Eto prevented the beiging of mature adipocytes. Furthermore, Eto-treatment induced the appearance of fibrotic streaks and augmented the expression of fibrosis genes (Fig. S5C–D). These findings suggest that PRDM16-driven FAO is required for the production of a paracrine factor(s) that affects precursor differentiation.
PRDM16 drives the production and export of β-hydroxybutyrate (BHB)
The ketone body β-hydroxybutyrate (BHB) is a small polar metabolite that has signaling functions in a variety of biological contexts (Newman and Verdin, 2014). We noted that PRDM16 regulates the expression of the rate limiting ketogenic enzyme Hmgcs2 (Figs. 4B, S4B,C). LC-MS analysis of metabolites identified a striking ~10-fold increase of BHB levels in P16-CM (~200 μM) compared to that in GFP-CM (~20 μM) (Fig. 5A). The concentration of BHB in CM increased in a time-dependent manner, indicative of an active process (Fig. 5B). The ketogenic precursor 3-hydroxy-3-methylglutaryl-Coenzyme A (HMG-CoA) also accumulated over the same period (Fig. 5C). The secretion rate of BHB in PRDM16-expressing cells was ~12 nmol/mg protein/min, 6-fold higher than that in control adipocytes (Fig. 5D). Metabolic tracing showed elevated incorporation of 13C from 13C-palmitate into BHB in PRDM16-expressing vs. control adipocytes, suggesting that adipocyte FAO-flux fuels BHB production (Fig. 5E). Notably, BHB levels increased in iWAT by 1 day of cold exposure and remained elevated at 7 days (Fig. 5F). This cold-induced increase in iWAT BHB levels was diminished in Prdm16+/− animals (Fig. 5G). Additionally, the cold-stimulated rise in iWAT tissue BHB levels was absent in aged mice, though serum BHB levels rose to a similar degree in young and aged animals (Fig. S5E). Finally, CL-treatment increased BHB levels in BAT and iWAT (Fig. S5F). These data suggest that BHB is produced locally or selectively accumulates in the iWAT of young animals.
Fig.5. PRDM16 drives the production of β-hydroxybutyrate (BHB), which suppresses fibrogenesis.

(A) BHB concentration in control (GFP) and adipocyte conditioned medium in the presence or absence of Eto-treatment, as determined by LC-MS.
(B) Absolute quantity of BHB in GFP-CM and P-CM collected at 1h and 6h time points. n=4 per group.
(C) Relative quantity of HMG-CoA in GFP- and PRDM16-expressing adipocytes at 1h and 6h time points. AUC, area under the curve. AUC for HMG-CoA was normalized to 13C315N1HMG-CoA internal standard (STD) and total protein in each sample. n = 4 per group.
(D) BHB efflux rate in GFP- and PRDM16-expressing adipocytes. n=4 per group.
(E) GFP- and PRDM16-expressing adipocytes were incubated with 13C-palmitate. The absolute amount of 13C-labeled BHB in CM was measured at 1h and 6h time points. n=4 per group
(F) Relative BHB levels in iWAT from mice housed at thermoneutrality or exposed to 5°C cold for indicated times. n=5 per group.
(G) BHB levels in IWAT of wildtype (wt) control and Prdm16+/− mice housed at thermoneutrality or exposed to cold for 8 d. n= 3-6 per group.
(H-K) Adipose precursors were treated with: (1) vehicle or BHB (250 μM); and (2) vehicle, DMOG, or TGFβ. After 2 days, cells were either immunostained for ACTA2 or induced to differentiate into adipocytes for 5 days.
(H) Experimental schema.
(I) Immunofluorescence staining of ACTA2. Scale bar, 100 μM.
(J) Violin plots showing relative fluorescent intensity per cell in indicated groups. The white solid boxes represent percentiles (bottom of box = 25th, horizontal line =50th, top of box = 75th). n=50-80 cells per condition.
(K) LipidTOX staining of adipocyte lipid droplets. Scale bar, 200 μM.
BHB inhibits myofibrogenesis and enhances beige adipogenesis
We next examined whether BHB could modulate the differentiation activity of precursor cells (Fig. 5H). Exposure of cells to BHB mimicked the effect of P-CM in blocking the formation of ACTA2+ stress fibers upon HIF1α-activation or TGFβ-treatment (Fig. 5I,J). BHB also restored the adipocyte differentiation potential of HIF1α-expressing or TGFβ-treated precursors (Fig. 5K). Similar adipogenic effects of BHB were observed in primary human subcutaneous adipose-derived stem cells (hASCs) (Fig S5G,H). These data demonstrate that BHB is sufficient to provoke a “myofibrogenic to adipogenic” shift in precursor cell fate.
BHB is catabolized by cells to generate intermediary metabolites, including acetyl-coA. Additionally, BHB can have a variety of signaling functions, including inhibiting class 1 histone de-acetylases and acting as a ligand for certain G-protein coupled receptors (Newman and Verdin, 2014). To determine if the metabolism of BHB is required for its effects in adipose precursors, we used lentiviral shRNA constructs to reduce the expression of 3-Hydroxybutyrate Dehydrogenase 1 (BDH1), which catalyzes the first step in BHB-catabolism (Fig. 6A). Depletion of Bdh1 modestly decreased the adipogenic conversion of precursor cells under control/basal conditions (Fig. 6B,C). However, the capacity for BHB to restore adipocyte differentiation capacity in HIF1α-expressing cells was ablated by Bdh1-depletion (Fig. 6B,C).
Fig. 6. BHB-catabolism regulates adipose precursor differentiation.

(A-C) iWAT precursor cells were transduced with scramble (shScr) or Bdh1 shRNA lentivirus and treated with vehicle control (CTL), DMOG alone or DMOG plus BHB. After 2 days, cells were induced to differentiate into adipocytes for 5 days.
(A) Relative mRNA levels of Bdh1 in iWAT precursor cells. n=4 per group
(B) Oil-Red-O staining (red) of lipid droplets in differentiated adipocytes.
(C) Relative mRNA levels of adipogenic genes in differentiated adipocytes. n=4 per group.
(D-G) Bdh1 KO (PdgfraCreERT2; Bdh1flox/flox; R26RtdTomato) and sibling/age-matched control (ctrl; PdgfraCreERT2; Bdh1+/+; R26RtdTomato or PdgfraCreERT2; Bdh1flox/+; R26RtdTomato) mice were treated with tamoxifen for 4 days. iWAT was analyzed from mice at day 0 (pulse) and following 2-week cold exposure to induce beiging (day 23).
(D) Experimental schema.
(E) Flow cytometry analysis showing proportion of PDGFRα+ cells expressing tdTomato in ctrl and Bdh1 KO iWAT (left). Relative mRNA levels of Bdh1 in tdTomato+; PDGFRα+ cells in ctrl and Bdh1 KO iWAT of (right). n=3 mice per group.
(F) Immunofluorescence staining of tdTomato (red), and PLIN1 (green) in iWAT. Scale bar, 200 μM.
(G) Quantification of adipogenesis (% tdTomato+; PLIN1+/total tdTomato+ cells). n=30 randomly chosen 20X magnification fields from 3 mice per group. All data presented as mean ± s.e.m; * p <0.05, **p<0.01, ***p < 0.001 as analyzed by two-tailed Student’s t-test.
We next sought to determine if BDH1 activity in precursor cells is required for beige adipocyte development in vivo. To do this, we generated a mouse model for inducible deletion of Bdh1 in Pdgfrα+ precursor cells. Incorporation of a tdTomato reporter gene allowed us to track the fates of wildtype and Bdh1-null precursors (Bdh1P-KO). We treated thermoneutral-housed adult mice with tamoxifen to induce tdTomato expression in Pdgfrα+ cells and concurrently delete Bdh1 (in Bdh1P-KO animals) (Fig. 6D). Flow cytometry analysis immediately after tamoxifen treatment (pulse) showed that a similar proportion of PDGFRα+ cells were marked by tdTomato-expression in control (wildtype) and Bdh1P-KO animals and that Bdh1 levels were decreased in PDGFRα+ cells from the mutant mice (Fig. 6E, S6A). Immunofluorescence staining shows that tdTomato expression was specifically activated in PDGFRα+ cells (Fig. S6B). Mice were also exposed to 4°C to stimulate beige fat development. In control mice, nearly 40% of tdTomato+ cells contributed to the development of new PLIN1+ adipocytes (Fig. 6F,G). By contrast, there was minimal conversion of Bdh1-deleted cells into mature adipocytes, with most tdTomato+ cells remaining PLIN1-negative (Fig. 6F,G). At this time point, the expression of several fibrosis genes all trended towards higher expression in mutant IWAT. These results reveal an essential role for BHB-metabolism in facilitating the differentiation of adipose precursor cells.
Lastly, we investigated whether raising BHB levels in aged animals could induce beige fat development and ameliorate fibrosis. To do this, we fed 1-year-old mice a diet containing the ketone ester (R)-3-hydroxybutyl (R)-3-hydroxybutyrate (KE). This KE is hydrolyzed in the small intestine to generate BHB and 1,3-butanediol, which is further metabolized by the liver into BHB and acetoacetate (Fig. 7A). Animals were pair-fed KE diet or a calorie-matched control diet for 30 days, concluding with 4 days of cold exposure to induce beiging (Fig. 7B). KE-feeding raised plasma BHB levels by 10-fold compared to that in control fed mice (3-4 mM compared to 0.4 mM; Fig. 7C). Body weights were the same between mice on the KE and control diets (Fig. 7D) while the KE-fed mice had significantly lower blood glucose levels (Fig. 7E).
Fig. 7. Raising BHB levels in aged mice alleviates adipose fibrosis and restores beige adipogenic potential.

(A) Metabolism of the ketone ester (R-1,3-hydroxybutyl R-3-hydroxybutyrate) (KE) in the gut and liver to form BHB.
(B) Composition of control (CTL) and 6% KE diet (left) and schema of experiment (right). Aged (12-month-old) mice were fed with CTL or KE diet for 1 month at room temperature and then switched to 4°C cold for 4 days.
(C-E) Plasma BHB levels (C), body weight (D), and plasma glucose levels (E) of experimental mice. n=10 for control diet group; n=8 for KE diet group.
(F) Hematoxylin & Eosin (H&E) staining (top) and Picrosirius red staining (bottom)of iWAT from mice described above (B). Scale bar, 50 μM.
(G) Relative mRNA levels of thermogenic and fibrosis marker genes in iWAT.
All data presented as mean ± sem; *p< 0.05, **p<0.01, ***p<0.001 as analyzed by two-tailed Student’s t-test.
Histological assessments show that iWAT of control mice contained mostly large unilocular white-like adipocytes, whereas the iWAT of KE-mice developed multilocular beige-like adipocytes in (Fig. 7F). There was also a prominent decrease in the number and size of collagen/fibrotic streaks in the iWAT of KE-fed mice (Fig. 7F). At the molecular level, the iWAT from KE-mice expressed elevated levels of brown/beige fat-marker genes, including a 7-fold increase in Ucp1 and lower levels of fibrosis marker genes (e.g. Col1a1, Col3a1, Acta2 and Lox) (Fig. 7G). KE feeding was not sufficient to reduce fibrosis or promote beige fat development in the absence of cold exposure (Fig. S7A,B). These results demonstrate that BHB-treatment can restore beige adipocyte differentiation competency in old animals.
Discussion
Aging reduces beige fat activity in humans and mice, thereby pre-disposing to metabolic disease and hindering the potential of beige fat-targeted therapeutics (Berry et al., 2017; Cypess et al., 2009; Pfannenberg et al., 2010; Rogers et al., 2012; Saito et al., 2009; Yoneshiro et al., 2011). Our studies reveal that a decrease in adipocyte PRDM16 expression is an important determinant of the aged beige fat phenotype. Loss of PRDM16 in young animals mimics the effect of aging in promoting fibrosis and inhibiting beige fat formation. Reciprocally, raising PRDM16 levels prevents aging-induced adipose fibrosis and preserves beige adipocyte developmental potential.
These results are in line with a recent study showing that PRDM16 activity decreases fibrosis in the context of high fat diet (Hasegawa et al., 2018). In their study, the authors show that PRDM16 interacts with the transcriptional regulator GTF2IRD1 to repress the expression of fibrosis genes in adipocytes. We discovered that PRDM16-action in adipocytes also reduces the fibrogenic activity of precursor cells through a cell non-autonomous mechanism involving the secretion of BHB. Specifically, BHB treatment of precursor cells inhibits HIF1α- and TGFβ-induced myofibroblast development and enhances beige adipogenesis. These effects of BHB depend on BDH1, revealing a requirement for BHB catabolism in stimulating adipogenesis.
BHB metabolism generates ATP and other intermediary metabolites (acetyl CoA, succinyl CoA, NAD), which could affect gene expression in precursor cells through a variety of mechanisms. Interestingly, elevated glycolysis can promote myofibroblast differentiation (Bernard et al., 2015; Ding et al., 2017; Xie et al., 2015) and BHB decreases glycolytic activity in certain cell types (Lund et al., 2011; Lund et al., 2015). Related to this idea, a recent paper shows that downregulation of FAO and increased glycolysis in the skin promotes fibrosis (Zhao et al., 2019). Moreover, a new study from Peter Crawford’s lab shows that acetoacetate produced by hepatocytes reduces liver fibrosis through modulating macrophage metabolism (Puchalska et al., 2019). These studies together with our results highlight an unanticipated relationship between cellular metabolism and extracellular matrix production/structure. In adipose tissue, future studies are needed to determine how BHB modulates the metabolism and function of precursor cells.
The differentiation of precursor cells into myofibroblast-like cells or adipocytes has highly divergent effects on cytoskeletal organization. Myofibroblast differentiation is characterized by cell spreading and the formation of a contractile apparatus, whereas adipocyte differentiation requires a rounding-up of cell morphology, associated with a shift from filamentous to cortical actin. The TGFβ pathway is a potent and well-studied inducer of cytoskeletal remodeling and myofibroblast differentiation in many cell types (Carthy, 2018). Farmer and colleagues elegantly demonstrated that TGFβ controls beige adipogenic vs. smooth muscle-like differentiation in mesenchymal cells (McDonald et al., 2015). Our results show that the fibrogenic profile of adipose precursor cells is downregulated in response to adipogenic stimuli like cold exposure. We postulate that various pathways, including HIF1α and TGFβ, are engaged in vivo under basal conditions to maintain precursor cells in a “fibrogenic” state, thereby preventing adipocyte differentiation. We propose that BHB functions as a key signal to “unlock” the adipogenic potential of precursor cells by suppressing fibrogenic fate (Fig. 7H).
The capacity for adipocytes to produce BHB is intriguing, given that liver is the primary systemic source of BHB. However, several extrahepatic tissues have been shown to produce BHB, including the retinal pigment epithelium (RPE), astrocytes, gut, and skeletal muscle (reviewed by Puchalska and Crawford, 2017). The RPE oxidizes fatty acids to produce BHB, which is then metabolized by neighboring photoreceptor cells (Adijanto et al., 2014). Astrocytes undergo high rates of FAO and produce BHB for use by neurons (Auestad et al., 1991; Blazquez et al., 1998). In the gut, BHB is produced from bacterial-derived butyrate and functions to regulate intestinal cell differentiation (Vanhoutvin et al., 2009; Wang et al., 2017) (Vanhoutvin et al., 2009; Wang et al., 2017). Of particular interest, mammary adipocytes produce BHB, which has been suggested to promote breast cancer growth (Huang et al., 2017). Altogether, these studies provide evidence that certain tissues regulate local BHB levels for use as an energetic substrate and/or signal.
BHB production in adipose cells was tightly linked to FAO, which is the dominant metabolic pathway used by thermogenic brown and beige adipocytes. Our results highlight the important role of PRDM16 as a driver of FAO in beige adipocytes. PRDM16 binds and regulates the expression of many genes involved in FAO, likely via co-activating the transcriptional function of PPARs, including PPARα, a key driver of FAO in many cell types (Harms et al., 2015; Hondares et al., 2011; Seale et al., 2008). Overall, we propose that BHB functions as a metabolic sensor, indicative of high levels of FAO in beige fat and providing a permissive signal to promote beige fat cell differentiation.
Elevating blood ketone levels using a ketogenic diet or ketone ester treatment was previously shown to increase UCP1 expression and mitochondrial content in BAT (Douris et al., 2017; Kennedy et al., 2007; Srivastava et al., 2013; Srivastava et al., 2012). Remarkably, raising circulating BHB levels in aged mice above that normally observed with cold exposure through feeding a ketone ester diet reversed adipose fibrosis and restored beige fat developmental potential. This suggests that even a short exposure to BHB is sufficient to restore the adipogenic differentiation competency of “aged” adipose precursors.
In summary, our findings establish the existence of a metabolite-based paracrine signaling axis in beige adipose tissue, with beige adipocytes providing metabolite cues to regulate adipose precursor cell differentiation. This raises the possibility that increasing ketone metabolism in adipose tissue could ameliorate fibrosis and encourage beige fat development for combating metabolic disease.
Limitations of Study
Emerging studies indicate that adipose precursor cells, marked by their expression of PDGFRα, represent a heterogeneous cell compartment, comprising different cell types, including several putative fibrogenic populations (Burl et al., 2018; Hepler et al., 2018; Marcelin et al., 2017; Merrick et al., 2019; Schwalie et al., 2018). Future studies will determine if BHB acts preferentially on specific subpopulations of stromal cells in adipose. Additionally, we focused on the subcutaneous inguinal adipose tissue of mice, which has a high potential for beige fat development. However, BHB appears to act primarily via facilitating the general program of adipogenesis, common to all adipocytes. This raises the possibility that BHB plays additional roles in regulating white adipocyte differentiation, such as during high fat diet feeding. It also remains uncertain whether acetoacetate, acetate or certain short chain fatty acids could play similar roles to BHB in regulating adipose tissue remodeling responses. Finally, it is unknown whether ketone ester feeding or other ways to increase adipose BHB levels would have anti-fibrotic and/or pro-adipogenic effects in human adipose tissue.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Patrick Seale (sealep@pennmedicine.upenn.edu).
Experimental Model and Subject Details
Mice
All animal experiments were approved by the University of Pennsylvania’s Institutional Animal Care and Use Committee. Mice were housed in temperature-controlled, pathogen-free barrier facility on a 12 hr/12 hr light/dark cycle. 12-month-old C57BL/6 male mice were obtained from the National Institute of Aging. AdipoqCre (strain name: B6; FVB-Tg(Adipoq-cre)1Evdr/J, stock no. 010803), Rosa26tdTomato (strain name: B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J, stock no. 007914), Rosa26CreER (strain name: B6;129-Gt(ROSA)26Sortm1(cre/ERT)Nat/J, stock no. 004847) mouse strains were obtained from the Jackson Laboratory. Prdm16flox/flox, Fabp4-Prdm16 and Prdm16-deficient mice were generated as described previously (Cohen et al., 2014; Harms et al., 2014; Seale et al., 2008; Seale et al., 2011). The AdipoqrtTA; TRECre; Rosa26mT/mG AdipoChaser mice were described before (Shao et al., 2016; Wang et al., 2013). The PdgfrαcreERT2 mouse strain was obtained from Dr. Brigid Hogan (Duke University). Bdh1flox mice were obtained from Dr. Dan Kelly (University of Pennsylvania). All mouse strains were maintained on a C57BL/6J background. Sibling or age matched male mice were used for all the experiments. To induce Cre activity, tamoxifen (Sigma, T5648) was dissolved at 20 mg/ml in corn oil (Sigma, C8267) and 5-7 weeks old mice were intraperitoneally injected with tamoxifen at a dose of 100 mg/kg for 5 consecutive days. Mice were then scarified at the specific days following the last tamoxifen injection. For CL 316,243 (Sigma, C5976) treatment, mice were intraperitoneally injected with 1 mg/kg CL 316,243 for 5 consecutive days. For in vivo etomoxir (Sigma, E1905) treatment, mice were intraperitoneally injected with 45 mg/kg etomoxir for 5 consecutive days. For cold exposure, mice were singly housed at 5 C for 7 days.
Cell Culture
Primary inguinal progenitors were isolated from 7-12 weeks old male mice as previously described (Wang et al., 2014). Briefly, inguinal adipose tissues were dissected, minced into small pieces (approximate 1 cm3 in size) with scissors. Minced samples were then transferred to the DMEM/F12 media containing 1.5 U/ ml collagenase D (Sigma, 11088858001) and 2.4 U/ml Dispase (Sigma, 4942078001) and rotated for 45 min at 37 C. Digested samples were then passed over a 100-μM filter to remove undigested clump, and stromal cells were gently pelleted. Stromal cells were resuspended in DMEM/F12 media containing 100 μg/ml primocin (Invivogen, ant-pm-2), and 10% fetal bovine serum (FBS). Cells were induced to undergo adipocyte differentiation using a standard cocktail (DMEM/F12 containing 10% FBS, 0.5 μM isobutylmethylxanthine, 125 nM indomethacin, 1 μM dexamethasone, 20 nM insulin, 1 nM T3, and 1 μM rosiglitazone). After 48 hr, cells were switched to maintenance medium (DMEM/F12 containing 10% FBS, 20 nM insulin, 1 nM T3, and 1 μM rosiglitazone). To delete Prdm16 in adipocytes, primary inguinal preadipocytes were isolated from Rosa26CreER; Prdm16flox/flox mice, and then treated with 1 μM 4-hydroxy-tamoxifen (Sigma, H6278) for 3 days in growth phase. For CRISPR/Cas9-mediated gene editing, guide RNA sequences against Hif1α or the Rosa26 gene (negative control) were cloned into LentiCRISPR (R, Addgene, 49535). The following retroviral cDNA constructs were used for gain-of-function studies: MSCV-puro, MSCV-GFP, MSCV-PRDM16, MSCV-HIF1αP577A/P402A (generously provided by Dr. Celeste Simon). To activate HIF1α, cells were treated with 250 μM (high dose) or 100 μM (low dose) of Dimethyloxaloylglycine (DMOG, Cayman Chemical, 71210). For Oil-Red-O staining, cells were fixed with 4% paraformaldehyde (PFA), washed with water twice, followed by 60% isopropanol for 5 min at room temperature (RT). Then, cells were stained with oil red o (Sigma, O0625) at RT for 10 min. For LipidTOX staining, cells were fixed with 4% PFA, and stained with lipidTOX (Thermo Scientific, H34476) at RT for half an hour.
To prepare conditioned medium (CM), cells were incubated with 4 mL of DMEM/F12 (with 10% dialyzed FBS or 200 μM palmitic acid) for 24 hrs. CM was then collected, centrifuged at 1000xg for 10 min at 4°C, and the supernatant stored at −80°C. For charcoal purification, CM was incubated with 1% (w/v) activated charcoal (Sigma, 05105) at room temperature for 10 min, and then centrifuged at 1000xg for 15 min to remove charcoal. To study the effects of the FAO inhibitor, etomoxir (Sigma, E1905), on CM activity, “secretor” cells were treated with 25 μM etomoxir for 24 hr.
Method Details
ACTA2/SMA staining and morphological analysis in cultured cells
Cells were trypsinized, counted and plated (3 × 104 cells per well) on chamber slides in the presence or absence of CM or BHB. After 2 h, cells were treated with 500 μM DMOG for another 24 h. Cells were then fixed in 4% paraformaldehyde (PFA) in PBS for 15 min, and permeabilized with 0.4% Triton X-100 in PBS for 15 min. Cells were then stained with SMA antibody (Sigma, A2547). Cell images were captured at low magnification (20 × objective) with a Leica TCS SP8 Confocal. Fluorescence intensity was quantified by using the morphology analysis tools available in the Image J software.
Mass Spectrometry-based SCFA measurements
SCFAs were measured in conditioned media and tissue extracts by LC-MS after derivitization with 2-hydrazinoquinoline (Lu et al., 2013). 13C4-D-α-hydroxybutyrate and 13C22H3-acetate (both from Cambridge Isotope laboratories) internal standards were added to a media samples to a final concentration of 1 mM (for 13C tracing experiments, only 13C22H3-acetate was used as an internal standard). Standard curves for were generated by serial dilution of pure standards (β-hydroxybutyrate and 13C4-β-hydroxybutyrate (Sigma)) and internal standard was added to standard curve samples in the same manner as to the media samples. For derivitization, 5 μl of sample was combined with 50 μl of acetonitrile containing 2 mM triphenylphosphine (TPP) and 2 mM 2,2-dipyridyl disulfide (DPDS), followed by the addition of 50 μl of acetonitrile containing 2 mM 2-hydrazinoquinoline in a screw cap glass tube. The reaction was incubated at 60’C for 15 min after which, samples were placed on ice and the reaction was stopped by the addition of 100 μL of HPLC (Optima) grade water. Samples were centrifuged at 2000 x g at 4°C for 10 min and the upper 100 μL of cleared supernatant was taken for LC-MS analysis. Samples were separated on a UHPLC BEH C18 column (2.1 × 50 mm) attached to an Acquity LC system (Waters). Solvents were - A: 0.05% aqueous acetic acid containing 2 mM ammonium acetate, and B: 95% aqueous acetonitrile containing 0.05% acetic acid and 2 mM ammonium acetate. Samples were run over a 5 minute gradient with starting conditions 1% solvent B ramping to 100% B over 1 min and held at 100% B for 2.5 min before re-equilibration at 1% B for 1.5 min. LC eluate was analyzed with a TSQ triple-quadrupole mass spectrometer (Thermo) with positive electrospray ionization. Data were acquired by selected reaction monitoring of the mass transitions indicted in the table below.
| Parent | ion | |
|---|---|---|
| Metabolite | [M+H] | Product ions |
| Acetate | 202 | 143, 160 |
| 13C2-Acetate | 204 | 143, 160 |
| 13C2,2H3-Acetate | 207 | 143, 160 |
| β-hydroxybutyrate | 246 | 143, 160 |
| 13C4-β-hydroxybutyrate | 250 | 143, 160 |
SCFA extraction from frozen tissues:
Frozen tissue samples were cut to ~20 mg on a super chilled ceramic tile on dry ice and the weight of each piece was recorded. Tissues were extracted in 200 μL 80:20 methanol:water chilled to −80°C in a 1.5 mL plastic tube. Ball bearings (2 per sample) were added and tissues were homogenized by mechanical disruption in a bullet blender (Next Advance) operated at intensity 11 for 5 mins at 4°C. Samples were centrifuged at 17,000 x g for 10 min at 4°C and the cleared supernatant was used for SCFA determination.
in vitro isotopic labeling
Adipocyte cultures were washed with PBS twice, and then incubated with 4 mL DMEM/F12 (no FBS) containing 100 μM U-13C16-Palmitate or unlabeled Palmitate. After 1hr or 6hr incubation, cell culture dishes were placed on ice. 1 mL fresh medium was added to an empty well without any cells as a blank control. Medium was collected, and centrifuged at 1000 x g for 10 min at 4°C. The supernatant was subjected to MS analysis. Cells were washed with ice cold PBS three times, and then harvested in 1 mL 10% (w/v) ice cold trichloroacetic acid for acyl-CoA extraction. Total protein levels were used for normalization.
Water-soluble metabolite extraction
Adipocyte cultures were washed with ice cold PBS twice, and then harvested in ice cold methanol (HPLC) on dry ice. After 20 minutes of incubation at −80 °C, the resulting mixture was collected and centrifuged at 16000 x g for 5 min. The pellets were re-extracted with 1 mL ice cold methanol on dry ice. The supernatants from two rounds of extraction were combined, dried under nitrogen, and subjected to MS analysis. Total protein levels were used for normalization.
Acyl-CoA extraction and measurement
Acyl-CoAs were extracted from ~20 mg frozen tissue pieces into 1 mL 10%(w/v) ice cold trichloroacetic acid with 13C315N1-labeled acyl-CoA internal standard and purified by solid phase extraction as described previously(Frey et al., 2016). Acyl-CoAs were detected on a Q-Exactive (Thermo) mass spectrometer as described previously(Frey et al., 2016). For isotopic tracer analysis, isotopic enrichment was calculated to compensate for the non-linearity of isotopic enrichment using the FluxFix calculator(Trefely et al., 2016).
Flow cytometry
Isolated cells were filtered through 100 μm and 40 μm filters (Corning), and then incubated with the RBC lysis buffer at RT for 5 min, and then washed with PBS. Cells were stained with antibodies at 1:200 dilution in FACS buffer (HBSS with 3% FBS) for 30 min at 4°C and sorted using a BD FACS Aria. Debris and dead cells were excluded by forward scatter, side scatter, and DAPI gating.
Ketone Ester (KE) Feeding
Animals were provided with a complete liquid diet once per day. Control diet was prepared at 226 g/L Control Liquid Diet powder (Bio-Serv, F1259SP) and supplemented with 2.0 g/L acesulfame K (Sigma, 04054) artificial sweetener and 0.1 mL/L food-grad pure mint extract (McCormick, MD). An isocaloric 6% ketone ester (KE) diet was prepared with 38.3 g/L control diet powder, 187.7 g/L ethanol diet powder, 240 mL/L of 25% ketone ester, plus acesulfame K and mint extract. Animals were housed singly to monitor food consumption. All mice were initially ad-lib fed with control diet to adapt them to liquid diet. The mass of diet consumed was measured each day, and the amount provided to the control group was matched to the amount eaten by the “KE” group during the previous day (pair-feeding). Body masses and food consumption were monitored daily in the morning (~10-12 AM), and fresh food provided in the afternoon (~4-6 PM). The mice were maintained on KE and CTL diet for 30 days, followed by a 4-day cold exposure to elicit beiging. Blood samples were collected, mice were euthanized, and tissues were collected and frozen in liquid nitrogen.
Fatty acid oxidation assay
Cells were incubated in DMEM/F12 supplemented with [9,10-3H(N)]palmitate conjugated on BSA and carnitine for 2 hr. The resultant 3H2O in the incubation solution was separated using ion-exchange columns (Bio Rad, #7316211), and then measured by scintillation counter. Total protein levels were used for normalization.
RNA-seq and analysis
RNA-seq libraries were generated with the illumina Truseq RNA Sample Preparation kit (Version 2, Illumina, RS-122-2001). High-throughput sequencing were performed on the HiSeq2000 at the Next Generation Sequencing Core at the University of Pennsylvania. RNA-seq reads were aligned to mm9 using STAR pipeline (R). Genes with fold-changes >2, and FDR < 0.01 were selected for gene ontology analysis. Gene Set Enrichment Analysis (GSEA) was performed using the Molecular Signatures Database (MSigDB) C2 collection of cellular processes. For GSEA, RNA-seq data was first ranked according to the FDR to generate an ordered gene list for Pre-ranked GSEA. Raw data have been deposited into NCBI’s GEO databank, with the following accession numbers. GSE129083 for datasets comparing young and. aged inguinal WAT with our without CL316,243 treatment. GSE129084 for datasets comparing vehicle control vs. PRDM16-expressing adipose cells.
Histology
For immunohistochemistry, isolated tissues were fixed in 4% PFA overnight, dehydrated, and embedded in paraffin for sectioning. Following de-paraffinization, slides were stained with hematoxylin and eosin, Masson’s Trichrome stain or picro-sirius red (abcam, ab150681). For immunostaining, heat antigen retrieval was performed in a pressure cooker using Bulls Eye Decloaking buffer (Biocare). Slides were incubated in primary antibody overnight followed by multiple washes with PBS. Sections were then incubated with secondary antibody conjugated to peroxidase and developed using Tyramide Signal Amplification (TSA, Perkin Elmer). The following primary antibodies were used: UCP1 (generously provided by AstraZeneca), PLIN1 (Cell Signaling, 3470S), RFP (VWR Scientific, RL600-401-379). Images were captured on a Leica TCS SP8 confocal microscope or a Keyence BZ-X700 fluorescent light microscope.
QPCR and Western blot
Total RNA was extracted by TRIzol (Invitrogen) followed by purification using PureLink RNA columns (Invitrogen). Isolated mRNA was reverse transcribed using the High-Capacity cDNA Synthesis kit (Applied Biosystems) and used in real-time PCR reactions with SYBR Green master mix (Applied Biosystems) on a 7900 HT (Applied Biosystems). Tata-binding protein (Tbp) was used an internal normalization control. Primer sequences are in Table S1. Protein extracts were prepared as previously described (Rajakumari et al., 2013). Cells or tissues were lysed in radioimmunoprecipitation assay (RIPA) lysis buffer containing 0.5% NP-40, 0.1% sodium deoxycholate, 150 mM NaCl, 50 mM Tris-Cl, pH 7.5, protease inhibitor cocktail (Complete; Roche) and 1 mM phenylmethylsulfonyl fluoride (PMSF). The protein content was measured using a detergent-compatible (DC) protein assay kit (Bio-Rad). Lysates or nuclear fractions were resolved onto bis-Tris NuPAGE gels (Invitrogen), transferred to PVDF membrane (Millipore). For HIF1α western blot, 500 μM DMOG was freshly added to lysis buffer to prevent HIF1α degradation. Primary antibodies were: anti-PRDM16 (Seale et al., 2007), anti-PPARγ (Cell Signaling Technology, 81b8), anti-UCP1 (R&D Systems, MAB6158), anti-HIF1α (Cayman Chemical, 10006421), anti-Actin (Millipore, MAB1501), anti-TGFβ (Novus Biologicals, MAB1835), anti-P-Smad3 (AbCam, ab52903), anti-Smad3 (Santa Cruz, sc-101154)
Quantification and Statistical Analysis
Unless specified in the main test or figure legends, all sample numbers (n) represent biological replicates. For analysis of the statistical significance of difference between two or three groups, two-sided unpaired Student’s t-test was used. For analysis of the statistical significance of difference between four or more groups, two-way ANOVA was used All data are shown as the mean with sem. No samples or animals were excluded from analysis. Sample size estimation was not used. Studies were not conducted blinded.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rb anti PRDM16 | P. Seale | Generated in house |
| Rb anti PPARg (81B8) | Cell Signaling | 2443S |
| Ms anti Tubulin (DM1A) | Sigma | T6199 |
| Ms anti Actin, clone C4 | Millipore | MAB1501 |
| Ms anti Actin, α-Smooth Muscle | Sigma | A2547 |
| Ms anti TGFβ 1, 2, 3 | Novus Biologicals | MAB1835-SP |
| Ms anti SMAD3 | Santa Cruz | Sc-101154 |
| Rb anti P-SMAD3 | AbCam | ab52903 |
| Rb anti HIF1α | Cayman Chemical | 10006421 |
| Rb anti Perilipin | Cell Signaling | 3470S |
| Rb anti RFP | VWR Scientific | RL600-401-379 |
| APC anti-mouse PDGFRα | BioLegend | 136007 |
| Goat anti-P-SMAD2/3 | Santa-Cruz | Sc-11769 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tamoxifen (Free Base) | Sigma | T5648 |
| 4-hydroxy-tamoxifen | Sigma | H6278 |
| Etomoxir | Sigma | E1905 |
| Corn Oil | Sigma | C8267 |
| Palmitic Acid | Sigma | 76119 |
| Methanol (HPLC) | VWR | MX0475-1 |
| Water (HPLC) | Fisher Chemical | W5-1 |
| Palmitic Acid [9,10-3H(N)], 1mCi | Perkin Elmer | NET043001MC |
| DMOG | Cayman Chemical | 71210 |
| Recombinant Mouse TGFβ | R & D | 7345-B2 |
| ion-exchange columns | Bio-Rad | 7316211 |
| Palmitic Acid (1-13C, 99%) | Cambridge Isotopes | CLM-150 |
| Sodium-3-hydroxybutyrate | Sigma | 54965 |
| TruSeq RNA Sample Prep Kit v2 set A | Illumina | RS-122-2001 |
| PCR Master Mix, Power SYBR Green | Applied Biosystems | 4367659 |
| CL 316, 243 | Sigma | C5976 |
| Deposited Data | ||
| RNA sequencing data -Young vs. Aged mice (+/− CL316,243) -Control (Puro) vs. PRDM16-expressing adipose cells |
GEO repository |
GSE129083 GSE129084 |
| Experimental Models: Organisms/Strains | ||
| R26CreER | The Jackson Laboratory | 004847 |
| Prdm16flox | The Jackson Laboratory | 024992 |
| Bdh1flox | Dr. Daniel Kelly | NA |
| AdipoqCre | The Jackson Laboratory | 010803 |
| Rosa26tdTomato | The Jackson Laboratory | 007914 |
| PdgfrαcreERT2 | Dr. Brigid Hogan | NA |
| AdipoqrtTA | Dr. Philipp Scherer | NA |
| TreCre | The Jackson Laboratory | 006234 |
| Rosa26mTmG | The Jackson Laboratory | 007676 |
| Prdm16-deficient | Dr. Patrick Seale | NA |
| Fabp4-Prdm16 | Dr. Bruce Spiegelman | NA |
| Oligonucleotides | ||
| See Table S1 for qPCR Primers | Integrated DNA Technologies (IDT) | NA |
| Recombinant DNA | ||
| PLKO.1-shRNA-Scramble | Addgene, Sabatini, PMID15718470 | 1864 |
| PLKO.1-shRNA-Bdh1 | The RNAi Consortium (TRC)- Broad Institute via UPenn High-Throughput Screening Core | TRCN0000041898 |
| pMD2.G | Addgene (Gift from Didier Trono) | 12259 |
| psPAX2 | Addgene (Gift from Didier Trono) | 12260 |
| LentiCRISPR v2 | Addgene (Gift from Feng Zhang) | 52961 |
| MSCV-GFP | Dr. Patrick Seale | NA |
| MSCV-Prdm16 | Dr. Patrick Seale | NA |
| Software and Algorithms | ||
| GraphPad Prism 7 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Image J | NIH | https://imagej.nih.gov/ij/ |
| R | Open source | https://www.r-project.org/ |
| GSEA | Broad Institute | http://software.broadinstitute.org/gsea/index.jsp |
| Reactome (Pathway Browser) | PMID: 29145629 | https://reactome.org/PathwayBrowser/ |
Highlights.
Beiging stimuli decrease the fibrotic phenotype of adipose precursor cells.
Aging reduces adipocyte PRDM16 expression, which impairs beige fat remodeling.
PRDM16 induces beta-hydroxybutyrate (BHB) secretion from adipocytes.
Metabolism of BHB in precursor cells blocks fibrosis and enhances beige adipogenesis.
Context and Significance.
Beige fat cells burn energy and can thereby reduce obesity and metabolic disease. The precursor cells that develop into beige fat also give rise to fibrosis-generating cells that contribute to metabolic dysfunction. This study shows that mature fat cells produce a metabolic signal, β hydroxybutyrate (BHB), which acts on precursor cells to repress fibrosis and stimulate beige fat development. The production of BHB by fat cells is controlled by the regulatory protein PRDM16. PRDM16 and BHB levels decline in fat tissue during aging, resulting in fibrosis and diminished beige fat. Dietary supplementation of BHB in aged mice reduces fibrosis and restores beige fat formation, raising the possibility that this pathway could be targeted therapeutically to reduce obesity.
Acknowledgements
We thank Lan Cheng and the Histology and Gene Expression Core of the Penn Cardiovascular Institute for immunohistochemistry, the Functional Genomics Core and Metabolomics Core of the Penn Diabetes and Endocrinology Center (DK19525) for high-throughput sequencing and metabolomic analyses. We are grateful to Shibiao Wan for help with bioinformatic analysis as well as Danielle Sanchez and Celeste Simon for help with HIF1α experiments. This work was supported by: American Heart Association (AHA) fellowship grant 15POST25700059 to W.W.; American Diabetes Association (ADA) fellowship grant 1-18-PDF-144 to S.T.; NIH/NICHD R03HD092630 to N.W.S.; NIH grants DK098656 and AG043483 to J.A.B; and ADA grant 1-16-IBS-269 and NIH/NIDDK grant DK103008 to P.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References:
- Adijanto J, Du J, Moffat C, Seifert EL, Hurle JB, and Philp NJ (2014). The retinal pigment epithelium utilizes fatty acids for ketogenesis. J Biol Chem 289, 20570–20582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auestad N, Korsak RA, Morrow JW, and Edmond J (1991). Fatty acid oxidation and ketogenesis by astrocytes in primary culture. Journal of neurochemistry 56, 1376–1386. [DOI] [PubMed] [Google Scholar]
- Berry DC, Jiang Y, Arpke RW, Close EL, Uchida A, Reading D, Berglund ED, Kyba M, and Graff JM (2017). Cellular Aging Contributes to Failure of Cold-Induced Beige Adipocyte Formation in Old Mice and Humans. Cell Metab 25, 166–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry DC, Jiang Y, and Graff JM (2016). Mouse strains to study cold-inducible beige progenitors and beige adipocyte formation and function. Nat Commun 7, 10184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazquez C, Sanchez C, Velasco G, and Guzman M (1998). Role of carnitine palmitoyltransferase I in the control of ketogenesis in primary cultures of rat astrocytes. Journal of neurochemistry 71, 1597–1606. [DOI] [PubMed] [Google Scholar]
- Burl RB, Ramseyer VD, Rondini EA, Pique-Regi R, Lee YH, and Granneman JG (2018). Deconstructing Adipogenesis Induced by beta3-Adrenergic Receptor Activation with Single-Cell Expression Profiling. Cell Metab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon B, and Nedergaard J (2004). Brown adipose tissue: function and physiological significance. Physiological reviews 84, 277–359. [DOI] [PubMed] [Google Scholar]
- Carthy JM (2018). TGFbeta signaling and the control of myofibroblast differentiation: Implications for chronic inflammatory disorders. Journal of cellular physiology 233, 98–106. [DOI] [PubMed] [Google Scholar]
- Cohen P, Levy JD, Zhang Y, Frontini A, Kolodin DP, Svensson KJ, Lo JC, Zeng X, Ye L, Khandekar MJ, et al. (2014). Ablation of PRDM16 and Beige Adipose Causes Metabolic Dysfunction and a Subcutaneous to Visceral Fat Switch. Cell 156, 304–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, et al. (2009). Identification and importance of brown adipose tissue in adult humans. N Engl J Med 360, 1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douris N, Desai BN, Fisher FM, Cisu T, Fowler AJ, Zarebidaki E, Nguyen NLT, Morgan DA, Bartness TJ, Rahmouni K, et al. (2017). Beta-adrenergic receptors are critical for weight loss but not for other metabolic adaptations to the consumption of a ketogenic diet in male mice. Mol Metab 6, 854–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorenko A, Lishko PV, and Kirichok Y (2012). Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 151, 400–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey AJ, Feldman DR, Trefely S, Worth AJ, Basu SS, and Snyder NW (2016). LC-quadrupole/Orbitrap high-resolution mass spectrometry enables stable isotope-resolved simultaneous quantification and (1)(3)C-isotopic labeling of acyl-coenzyme A thioesters. Anal Bioanal Chem 408, 3651–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, Wang ZV, Landskroner-Eiger S, Dineen S, Magalang UJ, et al. (2009). Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol 29, 4467–4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms M, and Seale P (2013). Brown and beige fat: development, function and therapeutic potential. Nat Med 19, 1252–1263. [DOI] [PubMed] [Google Scholar]
- Harms MJ, Ishibashi J, Wang W, Lim HW, Goyama S, Sato T, Kurokawa M, Won KJ, and Seale P (2014). Prdm16 is required for the maintenance of brown adipocyte identity and function in adult mice. Cell Metab 19, 593–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms MJ, Lim HW, Ho Y, Shapira SN, Ishibashi J, Rajakumari S, Steger DJ, Lazar MA, Won KJ, and Seale P (2015). PRDM16 binds MED1 and controls chromatin architecture to determine a brown fat transcriptional program. Genes Dev 29, 298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa Y, Ikeda K, Chen Y, Alba DL, Stifler D, Shinoda K, Hosono T, Maretich P, Yang Y, Ishigaki Y, et al. (2018). Repression of Adipose Tissue Fibrosis through a PRDM16-GTF2IRD1 Complex Improves Systemic Glucose Homeostasis. Cell Metab 27, 180–194 e186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepler C, Shan B, Zhang Q, Henry GH, Shao M, Vishvanath L, Ghaben AL, Mobley AB, Strand D, Hon GC, et al. (2018). Identification of functionally distinct fibro-inflammatory and adipogenic stromal subpopulations in visceral adipose tissue of adult mice. eLife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondares E, Rosell M, Diaz-Delfin J, Olmos Y, Monsalve M, Iglesias R, Villarroya F, and Giralt M (2011). Peroxisome proliferator-activated receptor alpha (PPARalpha) induces PPARgamma coactivator 1alpha (PGC-1alpha) gene expression and contributes to thermogenic activation of brown fat: involvement of PRDM16. J Biol Chem 286, 43112–43122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CK, Chang PH, Kuo WH, Chen CL, Jeng YM, Chang KJ, Shew JY, Hu CM, and Lee WH (2017). Adipocytes promote malignant growth of breast tumours with monocarboxylate transporter 2 expression via beta-hydroxybutyrate. Nat Commun 8, 14706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi J, and Seale P (2015). Functions of Prdm16 in thermogenic fat cells. Temperature (Austin) 2, 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Qu A, Matsubara T, Chanturiya T, Jou W, Gavrilova O, Shah YM, and Gonzalez FJ (2011). Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet-fed mice. Diabetes 60, 2484–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joe AW, Yi L, Natarajan A, Le Grand F, So L, Wang J, Rudnicki MA, and Rossi FM (2010). Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol 12, 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura S, Seale P, Tomaru T, Erdjument-Bromage H, Cooper MP, Ruas JL, Chin S, Tempst P, Lazar MA, and Spiegelman BM (2008). Regulation of the brown and white fat gene programs through a PRDM16/CtBP transcriptional complex Genes Dev 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura S, Spiegelman BM, and Seale P (2015). Brown and Beige Fat: Physiological Roles beyond Heat Generation. Cell Metab 22, 546–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy AR, Pissios P, Otu H, Roberson R, Xue B, Asakura K, Furukawa N, Marino FE, Liu FF, Kahn BB, et al. (2007). A high-fat, ketogenic diet induces a unique metabolic state in mice. American journal of physiology. Endocrinology and metabolism 292, E1724–1739. [DOI] [PubMed] [Google Scholar]
- Klingenberg M, and Huang SG (1999). Structure and function of the uncoupling protein from brown adipose tissue. Biochimica et biophysica acta 1415, 271–296. [DOI] [PubMed] [Google Scholar]
- Lee KY, Russell SJ, Ussar S, Boucher J, Vernochet C, Mori MA, Smyth G, Rourk M, Cederquist C, Rosen ED, et al. (2013). Lessons on conditional gene targeting in mouse adipose tissue. Diabetes 62, 864–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YH, Petkova AP, Konkar AA, and Granneman JG (2015). Cellular origins of cold-induced brown adipocytes in adult mice. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 29, 286–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YH, Petkova AP, Mottillo EP, and Granneman JG (2012). In vivo identification of bipotential adipocyte progenitors recruited by beta3-adrenoceptor activation and high-fat feeding. Cell Metab 15, 480–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos DR, Babaeijandaghi F, Low M, Chang CK, Lee ST, Fiore D, Zhang RH, Natarajan A, Nedospasov SA, and Rossi FM (2015). Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting TNF-mediated apoptosis of fibro/adipogenic progenitors. Nat Med 21, 786–794. [DOI] [PubMed] [Google Scholar]
- Lin JZ, Rabhi N, and Farmer SR (2018). Myocardin-Related Transcription Factor A Promotes Recruitment of ITGA5+ Profibrotic Progenitors during Obesity-Induced Adipose Tissue Fibrosis. Cell reports 23, 1977–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Svensson KJ, Tsai L, Zeng X, Roh HC, Kong X, Rao RR, Lou J, Lokurkar I, Baur W, et al. (2014). A smooth muscle-like origin for beige adipocytes. Cell Metab 19, 810–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Yao D, and Chen C (2013). 2-Hydrazinoquinoline as a Derivatization Agent for LC-MS-Based Metabolomic Investigation of Diabetic Ketoacidosis. Metabolites 3, 993–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund TM, Obel LF, Risa O, and Sonnewald U (2011). beta-Hydroxybutyrate is the preferred substrate for GABA and glutamate synthesis while glucose is indispensable during depolarization in cultured GABAergic neurons. Neurochem Int 59, 309–318. [DOI] [PubMed] [Google Scholar]
- Lund TM, Ploug KB, Iversen A, Jensen AA, and Jansen-Olesen I (2015). The metabolic impact of beta-hydroxybutyrate on neurotransmission: Reduced glycolysis mediates changes in calcium responses and KATP channel receptor sensitivity. Journal of neurochemistry 132, 520–531. [DOI] [PubMed] [Google Scholar]
- Marcelin G, Ferreira A, Liu Y, Atlan M, Aron-Wisnewsky J, Pelloux V, Botbol Y, Ambrosini M, Fradet M, Rouault C, et al. (2017). A A PDGFRalpha-Mediated Switch toward CD9high Adipocyte Progenitors Controls Obesity-Induced Adipose Tissue Fibrosis. Cell Metab 25, 673–685. [DOI] [PubMed] [Google Scholar]
- McDonald ME, Li C, Bian H, Smith BD, Layne MD, and Farmer SR (2015). Myocardin-related transcription factor a regulates conversion of progenitors to beige adipocytes. Cell 160, 105–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrick D, Sakers A, Irgebay Z, Okada C, Calvert C, Morley MP, Percec I, and Seale P (2019). Identification of a mesenchymal progenitor hierarchy in adipose tissue. Science 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullican SE, Tomaru T, Gaddis CA, Peed LC, Sundaram A, and Lazar MA (2013). A novel adipose-specific gene deletion model demonstrates potential pitfalls of existing methods. Mol Endocrinol 27, 127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman JC, and Verdin E (2014). Ketone bodies as signaling metabolites. Trends Endocrinol Metab 25, 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno H, Shinoda K, Ohyama K, Sharp LZ, and Kajimura S (2013). EHMT1 controls brown adipose cell fate and thermogenesis through the PRDM16 complex. Nature 504, 163–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouellet V, Routhier-Labadie A, Bellemare W, Lakhal-Chaieb L, Turcotte E, Carpentier AC, and Richard D (2011). Outdoor temperature, age, sex, body mass index, and diabetic status determine the prevalence, mass, and glucose-uptake activity of 18F-FDG-detected BAT in humans. J Clin Endocrinol Metab 96, 192–199. [DOI] [PubMed] [Google Scholar]
- Pfannenberg C, Werner MK, Ripkens S, Stef I, Deckert A, Schmadl M, Reimold M, Haring HU, Claussen CD, and Stefan N (2010). Impact of age on the relationships of brown adipose tissue with sex and adiposity in humans. Diabetes 59, 1789–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puchalska P, and Crawford PA (2017). Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab 25, 262–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puchalska P, Martin SE, Huang X, Lengfeld JE, Daniel B, Graham MJ, Han X, Nagy L, Patti GJ, and Crawford PA (2019). Hepatocyte-Macrophage Acetoacetate Shuttle Protects against Tissue Fibrosis. Cell Metab 29, 383–398 e387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajakumari S, Wu J, Ishibashi J, Lim HW, Giang AH, Won KJ, Reed RR, and Seale P (2013). EBF2 determines and maintains brown adipocyte identity. Cell Metab 17, 562–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricquier D (2011). Uncoupling protein 1 of brown adipocytes, the only uncoupler: a historical perspective. Frontiers in endocrinology 2, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers NH, Landa A, Park S, and Smith RG (2012). Aging leads to a programmed loss of brown adipocytes in murine subcutaneous white adipose tissue. Aging cell 11, 1074–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M, Okamatsu-Ogura Y, Matsushita M, Watanabe K, Yoneshiro T, Nio-Kobayashi J, Iwanaga T, Miyagawa M, Kameya T, Nakada K, et al. (2009). High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes 58, 1526–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalie PC, Dong H, Zachara M, Russeil J, Alpern D, Akchiche N, Caprara C, Sun W, Schlaudraff KU, Soldati G, et al. (2018). A stromal cell population that inhibits adipogenesis in mammalian fat depots. Nature 559, 103–108. [DOI] [PubMed] [Google Scholar]
- Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, Scime A, Devarakonda S, Conroe HM, Erdjument-Bromage H, et al. (2008). PRDM16 controls a brown fat/skeletal muscle switch. Nature 454, 961–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P, Conroe HM, Estall J, Kajimura S, Frontini A, Ishibashi J, Cohen P, Cinti S, and Spiegelman BM (2011). Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J Clin Invest 121, 96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P, Kajimura S, Yang W, Chin S, Rohas LM, Uldry M, Tavernier G, Langin D, and Spiegelman BM (2007). Transcriptional control of brown fat determination by PRDM16. Cell Metab 6, 38–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao M, Ishibashi J, Wang Q, Kusminski CM, MacPherson KA, Vishvanath L, Spurgin SB, Hepler C, Holland WL, Seale P, et al. (2016). Conversion of White to Beige Fat Cells and the Reversal of Obesity Through Inactivation of Adipocyte Zfp423. Cell Metab In Press . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Baxa U, Niu G, Chen X, and Veech RL (2013). A ketogenic diet increases brown adipose tissue mitochondrial proteins and UCP1 levels in mice. IUBMB Life 65, 58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Kashiwaya Y, King MT, Baxa U, Tam J, Niu G, Chen X, Clarke K, and Veech RL (2012). Mitochondrial biogenesis and increased uncoupling protein 1 in brown adipose tissue of mice fed a ketone ester diet. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 26, 2351–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stine R, Shapira SN, Ishibashi J, Lim HW, Harms M, Won KJ, and Seale P (2015). EBF2 promotes beige fat cell recruitment in white adipose tissue. Mol Metab In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Halberg N, Khan M, Magalang UJ, and Scherer PE (2013a). Selective inhibition of hypoxia-inducible factor 1alpha ameliorates adipose tissue dysfunction. Mol Cell Biol 33, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Tordjman J, Clement K, and Scherer PE (2013b). Fibrosis and adipose tissue dysfunction. Cell Metab 18, 470–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trefely S, Ashwell P, and Snyder NW (2016). FluxFix: automatic isotopologue normalization for metabolic tracer analysis. BMC Bioinformatics 17, 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uezumi A, Fukada S, Yamamoto N, Takeda S, and Tsuchida K (2010). Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat Cell Biol 12, 143–152. [DOI] [PubMed] [Google Scholar]
- Vanhoutvin SA, Troost FJ, Hamer HM, Lindsey PJ, Koek GH, Jonkers DM, Kodde A, Venema K, and Brummer RJ (2009). Butyrate-induced transcriptional changes in human colonic mucosa. PLoS One 4, e6759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila IK, Badin PM, Marques MA, Monbrun L, Lefort C, Mir L, Louche K, Bourlier V, Roussel B, Gui P, et al. (2014). Immune cell Toll-like receptor 4 mediates the development of obesity- and endotoxemia-associated adipose tissue fibrosis. Cell reports 7, 1116–1129. [DOI] [PubMed] [Google Scholar]
- Wang Q, Zhou Y, Rychahou P, Fan TW, Lane AN, Weiss HL, and Evers BM (2017). Ketogenesis contributes to intestinal cell differentiation. Cell Death Differ 24, 458–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QA, Tao C, Gupta RK, and Scherer PE (2013). Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat Med 19, 1338–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Kissig M, Rajakumari S, Huang L, Lim HW, Won KJ, and Seale P (2014). Ebf2 is a selective marker of brown and beige adipogenic precursor cells. Proc Natl Acad Sci U S A 111, 14466–14471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, and Seale P (2016). Control of brown and beige fat development. Nature reviews. Molecular cell biology 17, 691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Cohen P, and Spiegelman BM (2013). Adaptive thermogenesis in adipocytes: is beige the new brown? Genes Dev 27, 234–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneshiro T, Aita S, Matsushita M, Okamatsu-Ogura Y, Kameya T, Kawai Y, Miyagawa M, Tsujisaki M, and Saito M (2011). Age-related decrease in cold-activated brown adipose tissue and accumulation of body fat in healthy humans. Obesity (Silver Spring) 19, 1755–1760. [DOI] [PubMed] [Google Scholar]
- Zhao X, Psarianos P, Soltan Ghoraie L, Yip K, Goldstein D, Gilbert R, Witterick I, Pang H, Hussain A, Lee JH, et al. (2019). Metabolic regulation of dermal fibroblasts contributes to skin extracellular matrix homeostasis and fibrosis. Nat Metabolism 1, 147–157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.