

Summary
Pancreatic ductal adenocarcinoma (PDA) is characterized by immune-tolerance and immunotherapeutic resistance. We discovered upregulation of receptor-interacting serine/threonine-protein kinase 1 (RIP1) in tumor-associated macrophages (TAMs) in PDA. To study its role in oncogenic progression, we developed a selective small molecule RIP1 inhibitor with high in vivo exposure. Targeting RIP1 reprogrammed TAMs toward an MHCIIhiTNFα+IFNγ+ immunogenic phenotype in a STAT1-dependent manner. RIP1 inhibition in TAMs resulted in cytotoxic T cell activation and T-helper cell differentiation towards a mixed Th1/Th17 phenotype, leading to tumor-immunity in mice and in organotypic models of human PDA. Targeting RIP1 synergized with PD1- and ICOS-based immunotherapies. Tumor-promoting effects of RIP1 were independent of its co-association with RIP3. Collectively, our work describes RIP1 as a checkpoint kinase governing tumor-immunity.
Keywords: Pancreatic cancer, macrophage polarization, inflammation
Graphical Abstract
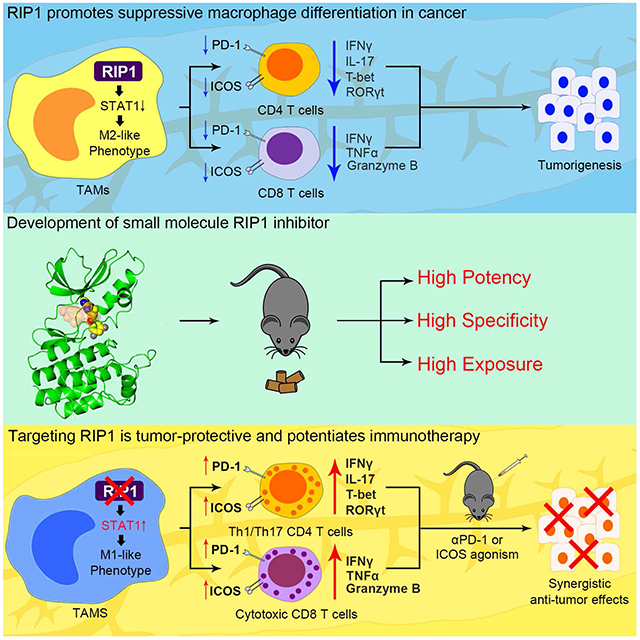
Wang et al. synthesize a selective RIP1 inhibitor that can be used in vivo. Inhibition of RIP1, which is highly expressed in tumor-associated macrophages in pancreatic ductal adenocarcinoma, reverses local immune-suppression and enhances the efficacy of checkpoint and costimulatory receptor-based immunotherapy.
Introduction
Pancreatic ductal adenocarcinoma (PDA) carries a dire prognosis with a mortality rate that approaches its incidence rate (Yadav and Lowenfels, 2013). PDA is characterized by a modest T cell infiltrate whose terminal differentiation status can influence disease outcome by either combating cancer growth via antigen-restricted tumoricidal immune responses or, more commonly, promoting tumor progression via the induction of immune tolerance (Andren-Sandberg et al., 1997; Clark et al., 2007; Zheng et al., 2013). Specifically, CD8+ T cells and Th1-polarized CD4+ T cells mediate tumor-protection in murine models of PDA and are associated with prolonged survival in human disease (Fukunaga et al., 2004). Conversely, Treg and Th2-polarized CD4+ T cells are associated with tumor-permissive anergy (De Monte et al., 2011; Fukunaga et al., 2004; Hiraoka et al., 2006; Ochi et al., 2012). The phenotype of tumor associated macrophages (TAMs) largely regulates immunogenic or immune-suppressive T-cell programming in PDA. Specifically, CD206+IL-10+ M2-like macrophages promote the expansion of Th2 cells and Tregs in PDA whereas MHCIIhiTNFα+ M1-like macrophages recruit Th1 cells and promote anti-tumor cytotoxic T lymphocyte (CTL) activity (Daley et al., 2017a; Seifert et al., 2016a). However, the regulators of macrophage polarization in PDA and the optimal approach to macrophage-based immunotherapy remain uncertain.
We have shown that targeting M-CSF dampens M2-like macrophage recruitment in PDA in the context of ionizing radiation (Seifert et al., 2016b). MCP-1 is also a potent macrophage chemoattractant in PDA (Nywening et al., 2016). CD40 ligand similarly has the potential to reprogram intra-tumoral macrophages and is being tested in the clinic in metastatic PDA (Beatty et al., 2011). We have shown that innate immune receptors specific for microbial-derived molecular motifs or damage-associated molecular patterns (DAMPs) promote immune anergy in PDA by recruiting suppressive macrophages. For example, ligation of NLRP3, Dectin-1, and select toll-like receptors (TLRs) in PDA promote IL-10 expression in macrophages, leading to the differentiation of Th2 cells or Tregs (Daley et al., 2017a; Ochi et al., 2012). Another intriguing potential target in PDA is receptor interacting protein (RIP) kinase 3, which mediates organized cellular necrosis, or necroptosis. We showed that RIP3 induces immune tolerance in PDA via the recruitment of myeloid-derived suppressor cells (MDSC) by driving cellular release of CXCL1 and inducing ligation of Mincle, a death ligand receptor (Seifert et al., 2016a).
RIP1 is a serine/threonine protein kinase closely aligned with RIP3 in that their co-association results in necroptosis (Shutinoski et al., 2016). However, RIP1 additionally drives NF-κB and MAP kinase signaling in response to inflammatory stimuli independently of its association with RIP3 (Meylan et al., 2004; Ofengeim and Yuan, 2013). RIP1 is also a putative master upstream regulator of TLR signaling (Ofengeim and Yuan, 2013). An unbiased CRISPR screen in melanoma identified RIP1 as a top candidate contributing to PD-1 immunotherapy resistance (Manguso et al., 2017). Hence, we postulated that RIP1 may have pleiotropic influences on immune tolerance in cancer.
Results
RIP1 is a promising target in PDA
Analysis of the TCGA database suggested robust RIPK1 expression in human PDA (Figure S1A), and at higher levels in tumors than in surrounding normal pancreas (Figure S1B). Immunohistochemical analysis confirmed high RIP1 expression in human PDA compared to normal pancreas (Figure S1C). Orthotopic PDA derived from Pdx1Cre;KrasG12D;Trp53R172H (KPC) mice similarly exhibited high RIP1 expression in contrast to normal mouse pancreas (Figure S1D). Immune fluorescence microscopy suggested high RIP1 expression in PDA in both transformed epithelial cells and in TAMs (Figure S1E, F). Notably, RIP1 kinase-dead knock-in (KD/KI) mice, which contain a point mutation in the catalytic lysine (K45A) in exon 3 of Ripk1 (Kaiser et al., 2014), were protected against orthotopically implanted PDA tumor, indicating that targeting RIP1 specifically in the extra-tumoral compartment confers protection (Figure S1G). By contrast, shRNA-mediated knockdown of RIP1 in KPC cells did not alter tumor growth, indicating that RIP1 expression in malignant epithelial cells is not critical to PDA progression (Figure S1H).
Development of a RIP1 inhibitor suitable for in vivo testing in PDA
We recently reported GSK’963 as a potent and selective inhibitor of both murine and human RIP1, however, its low oral exposure makes it unsuitable for in vivo administration (Berger et al., 2015; Harris et al., 2016). We endeavored to develop a small molecule that would maintain high potency against both RIP1 orthologues with improved pharmacokinetic features. We identified an analog of GSK’963, (S)-6-(4-(5-(3,5-difluorophenyl)-4,5-dihydro-1H-pyrazole-1-carbonyl)piperidin-1-yl)pyrimidine-4-carbonitrile (GSK’547, RIP1i) (Figure 1A), that is a highly selective and potent inhibitor of RIP1 (Figures S1I–L, and Table S1). RIP1i exhibited a 400-fold improvement in mouse pharmacokinetic oral exposure compared to GSK’963 (Figure S1M). We were able to co-crystallize RIP1i in a kinase domain fragment of RIP1 refined to 3.49 Å, which demonstrated that RIP1i binds in an allosteric pocket between the N-terminal and C-terminal domains at the back of the ATP binding site (Figure 1B, C). This binding mode, indicative of a type III kinase inhibitor, accounts for the observed high level of RIP1 kinase selectivity (Roskoski, 2016). Pharmacodynamic modeling based on mouse oral pharmacokinetic profiles (Figure S1M) and L929 potency (Figure S1K) indicated that RIP1i would maintain blood concentrations sufficient for >90% inhibition of RIP1 activity for sustained periods (Figure S1N). Administration of RIP1i in mouse chow achieved in vivo steady state concentrations above the L929 IC90 over a 24-hour period (Figure S1O). Further, high serum concentrations of RIP1i were sustained over a 6-week treatment course (Figures S1P). By comparison, Nec-1s achieved plasma concentrations ~40-fold below the L929 IC90 inhibition level (Table S2). RIP1i treatment was well-tolerated without evident pathology (Table S3). Hence, RIP1i is a mono-selective small molecule RIP1 inhibitor that is suitable for testing in vivo.
Figure 1. Targeting RIP1 using a selective kinase inhibitor is protective against PDA.
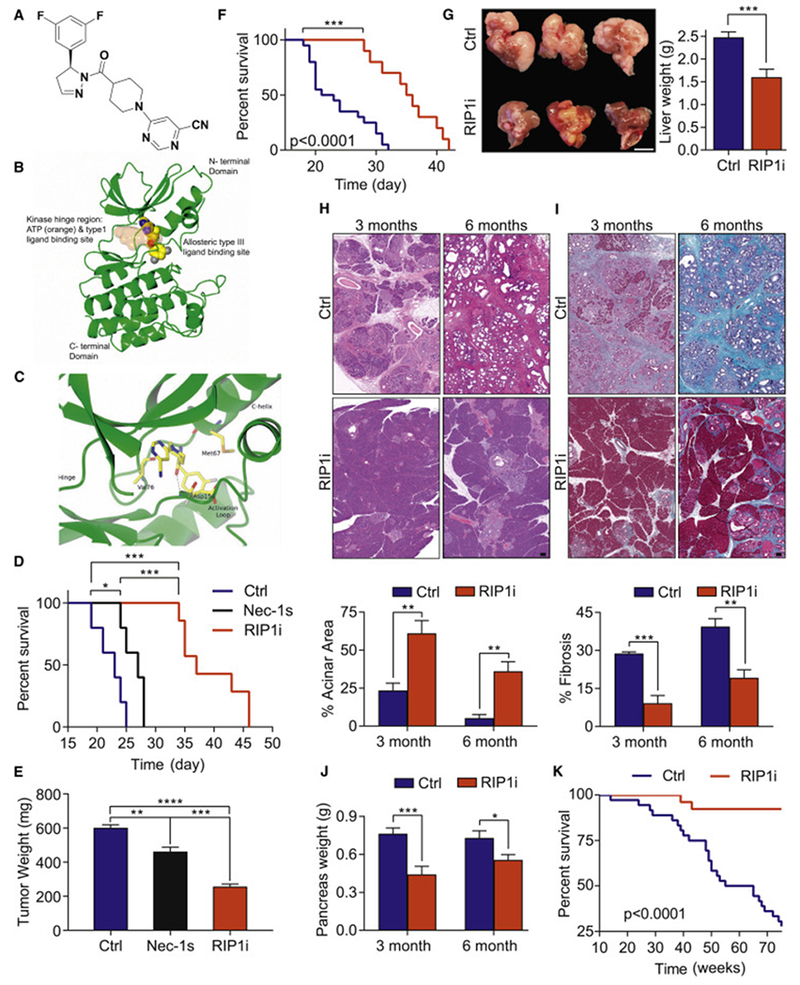
(A) Chemical structure of GSK’547 (RIP1i). (B) The binding orientation of RIP1i in an allosteric pocket of the RIP1 kinase domain produced by single crystal X-ray crystallography. Atoms of RIP1i are represented as spheres with color coding: carbon (yellow), nitrogen (blue), oxygen (red), fluorine (grey). The ATP binding pocket is shaded in orange. The allosteric pocket is located at the back of the hinge region in a deep cleft located between the north and south domains of the kinase. (C) Close-up view of RIP1i bound in the allosteric pocket of the RIP1 kinase domain. The same colour coding is used as for Figure 1B. Note the direct hydrogen-bond interaction between the amide carbonyl of RIP1i and the backbone amide NH of Asp156. (D) Survival of WT mice orthotopically implanted with KPC-derived tumor cells treated with RIP1i, Nec1s, or vehicle beginning on the day of tumor implantation quantified with the Kaplan-Meier estimator (n=5-7/group). (E) Tumor weight from additional cohorts (n=5/group) similar to (D) sacrificed on day 21. One representative of more than five repeats. (F) Kaplan-Meier survival analysis of WT mice administered RIP1i (n=10) or control (n=15) beginning on day 10 after orthotopically implanted with KPC-derived tumor cells. This experiment was repeated twice. (G) Representative images of livers (scale bar = 1 cm) and quantification of liver weights of WT mice injected with KPC-derived tumor cells via the portal vein and treated with RIP1i or control beginning on day 5 after tumor challenge. Mice were sacrificed at 21 days (n=8/group). This experiment was repeated 4 times. (H-J) Representative images of H&E (H) and Gomori Trichrome (I) staining (scale bars = 100 μm) and the percentages of preserved acinar (H) and fibrotic (I) area as well as pancreatic weights (J) of pancreas harvested from KC mice fed with RIP1i-containing or control chow beginning at 6 weeks of life at 3 months and 6 months (n=8). (K) Kaplan-Meier survival analysis in cohorts of RIP1i-treated (n=26) and control (n=36) KC 97 mice as in (H-J). Data are displayed as average ± SEM (*p<0.05; **p<0.01; ***p<0.001; ****p<0.0001). See also Figure S1 and Tables S1–S4.
RIP1 inhibition is protective against PDA in vivo
To determine the efficacy of RIP1i against PDA, we treated mice with RIP1i simultaneously challenged with orthotopic PDA tumor cells derived from KPC mice. RIP1i reduced tumor burden and extended survival compared to mice treated with controls or Nec-1s (Figure 1D, E). RIP1i also protected against established tumors (Figure 1F) and liver metastases (Figure 1G). In a slowly progressive autochthonous model of PDA, P48Cre;KrasG12D (KC) mice treated with RIP1i continuously beginning at 6 weeks old exhibited delayed development of pancreatic dysplasia, diminished peri-tumoral fibrosis, reduced pancreatic tumor weights, and extended survival (Figure 1H–K).
RIP1 inhibition in PDA results in T cell activation in situ
Since genetically targeting RIP1 in the extra-tumoral compartment was protective against PDA (Figure S1G), we postulated that RIP1i acts on inflammatory cells. We analyzed the adaptive immune infiltrate in orthotopic KPC tumors in RIP1i-treated vs control mice. RIP1i treatment increased pan-T cell infiltration (Figure 2A, B) and the CD8:CD4 ratio (Figure 2C). In addition to increase in number, PDA-infiltrating T cells were markedly activated in RIP1i-treated hosts. CD4+ and CD8+ T cells upregulated CD44, CD69, PD-1, ICOS, IFNγ, and TNFα (Figure 2D–I). CD4+ T cells in RIP1i-entrained tumors also upregulated IL-17, LFA-1, and CD40, and downregulated CD62L (Figure 2J), while CD8+ T cells upregulated Perforin expression (Figure 2K). Analysis of transcription factor expression in CD4+ T cells suggested that RIP1 inhibition upregulated T-bet and RORγt and reduced FoxP3 (Figure 2L). Collectively, these data suggest that targeting RIP1 in PDA results in enhanced Th1/Th17 differentiation of CD4+ T cells and cytotoxic CD8+ T cell activation in situ. In our liver metastases model, RIP1i similarly induced an activated intratumoral CD4+ and CD8+ T cell phenotype, including Th1/Th17 polarization of CD4+ T cells (Figure S2A–F). Immunogenic intratumoral T cell activation was also observed after RIP1i treatment of established PDA tumors (Figure S2G, H). T cells infiltrating PDA in RIP1 KD/KI mice also exhibited enhanced immunogenicity (Figure S2I–L).
Figure 2. RIP1 inhibition provides adaptive anti-tumor immunity in PDA.
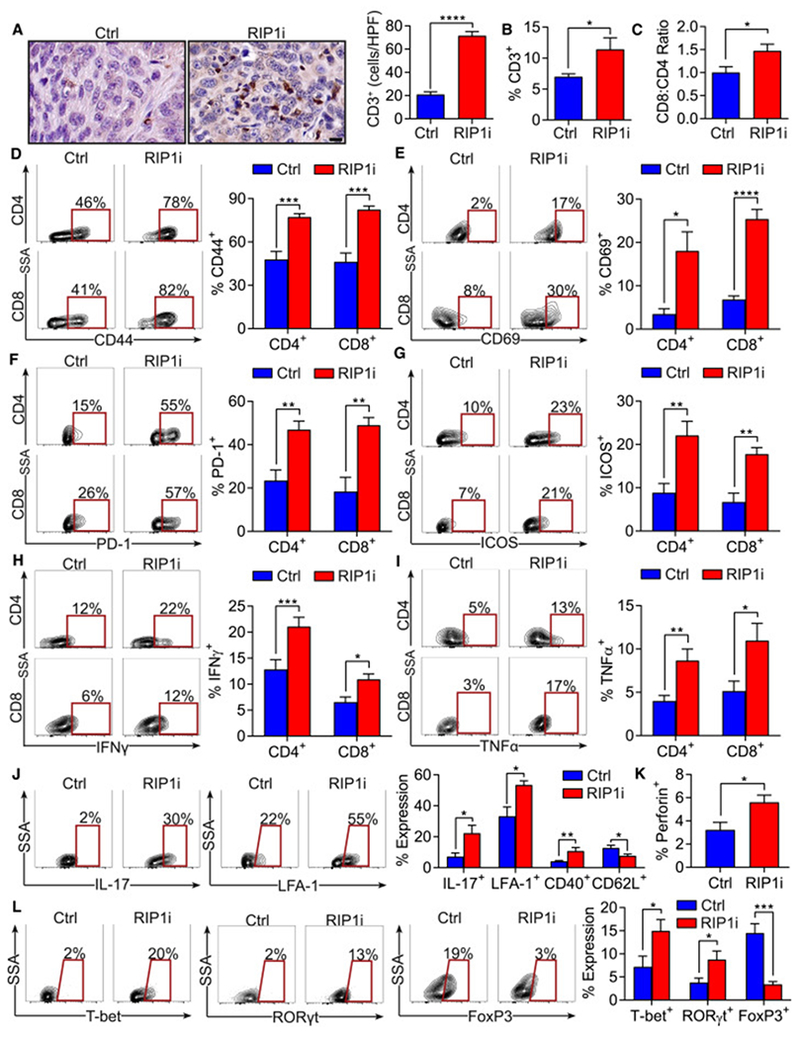
WT mice bearing orthotopic KPC tumors were treated with RIP1i or control and were sacrificed at 21 days (n=5/group). (A) IHC of PDA tumors for CD3 (scale bar = 10 μm). (B, C) The percentage of intra-tumoral CD3+ T cells (B) and the CD8:CD4 T cell ratio (C) determined by flow cytometry. (D-I) CD4+ and CD8+ T cells were gated and tested for expression of CD44 (D), CD69 (E), PD-1 (F), ICOS (G), IFNγ (H), and TNFα (I). (J) Expression of IL-17, LFA-1, CD40, and CD62L in CD4+ T cells. (K) Expression of Perforin in CD8+ T cells . (L) Expression of T-bet, RORγt, and FoxP3 in CD4+ T cells. These experiments were repeated more than 5 times with similar results (*p<0.05; **p<0.01; ***p<0.001; ****p<0.0001). Data are displayed as average ± SEM. See also Figure S2.
Tumor-protection in the context of RIP1 inhibition is T cell dependent and synergizes with PD-1 and ICOS-based immunotherapy
To establish whether the tumor-protective effects of targeting RIP1 are T cell dependent, we depleted CD4+ or CD8+ T cells in vivo in PDA-bearing control and RIP1i-treated mice. Whereas T cell depletion did not affect tumor growth in controls, as reported (Daley et al., 2016; Seifert et al., 2016a), both CD4+ and CD8+ T cell depletion abrogated the protective effects of RIP1 inhibition (Figure 3A). Similarly, RIP1i was not protective in athymic Foxn1nu mice or in Rag1−/− mice (Figure 3B, C). Further, adoptive transfer of tumor-infiltrating T cells from RIP1i-treated mice, but not from control mice, protected against orthotopic KPC tumor growth (Figure 3D). These data confirm that PDA-infiltrating T cells gain tumor-protective capacity in the context of RIP1i. Moreover, consistent with the upregulation of PD-1 in T cells in RIP1i-treated primary and metastatic tumors (Figures 2F, S2B), combined targeting of RIP1 and PD-1 offered synergistic protection against primary PDA and liver metastases, whereas αPD-1 monotherapy did not, as reported (Daley et al., 2016) (Figure 3E, F). The tumor-protection offered by RIP1i was similar to that of RIP1 KD/KI mice. Further, consistent with higher T cell expression of ICOS in tumors of RIP1i-treated mice (Figure 2G), combined RIP1 inhibition plus ICOS agonism conferred additive protection (Figure 3G). Of note, the T cell activation induced by RIP1i were specific to the PDA tumor microenvironment (TME) as we did not observe adaptive immune reprogramming in PBMC or spleen of RIP1i-treated hosts (Figure 3H–M). Splenic T cells in RIP1 KD/KI mice also had a similar baseline phenotype to WT mice (Figure S3A, B). Notably, RIP1i did not have direct effects on T cell phenotype in vitro (Figure S3C, D). Further, consistent with our RIP1 shRNA knockdown data, RIP1i treatment had no direct effect on tumor cell proliferation, colony formation, or migration in three distinct PDA models (Figure S3E–K).
Figure 3. RIP1i-mediated tumor-protection is CD4+ and CD8+ T cell dependent and is synergistic with PD-1 blockade and ICOS agonism.
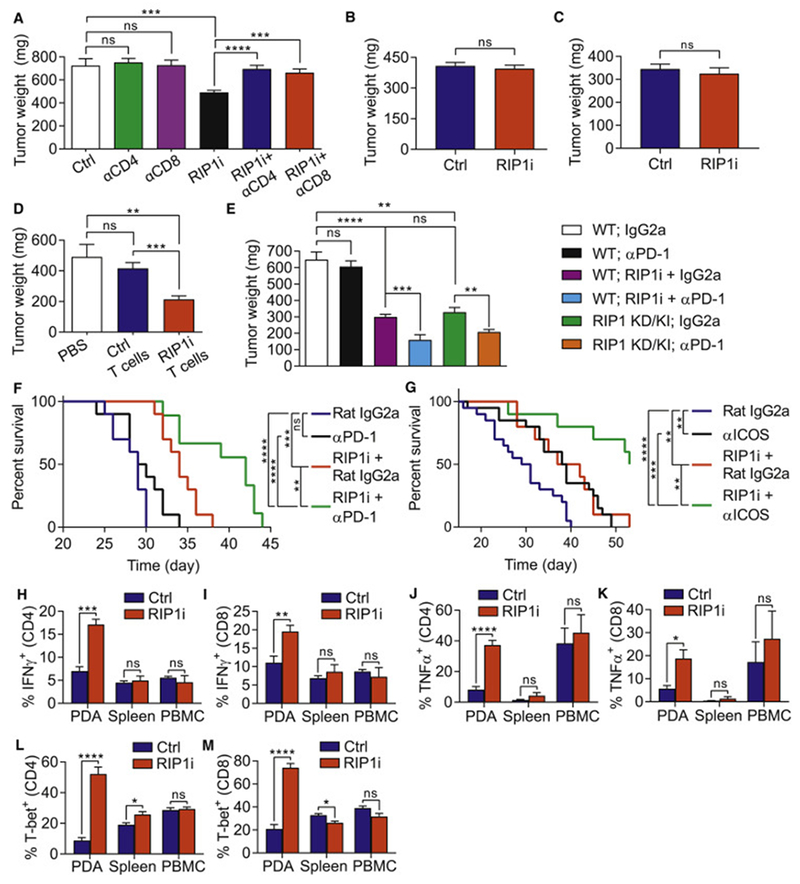
(A) Tumor weight of WT mice bearing KPC-derived tumor treated with neutralizing mAbs against CD4 or CD8 or with isotype control alone or in combination with RIP1i or control and sacrificed at 21 days (n=10/group). This experiment was repeated twice. (B, C) Tumor weights of WT and Foxn1nu mice (B, n=7/group) and of WT and Rag1−/− mice (C, n=5/group) challenged with orthotopic KPC tumor cells and sacrificed at 3 weeks. Each experiment was repeated twice. (D) Tumor weights of WT mice orthotopically implanted with KPC-derived tumor cells alone or admixed with tumor-infiltrating T cells harvested from control PDA or from PDA in RIP1i-treated mice (n=8/group) and were sacrificed on day 21. This experiment was repeated twice. (E) Tumor weight of WT or RIP1 KD/KI mice (n=8/group) orthotopically implanted with KPC-derived tumor cells and treated as indicated. Mice were sacrificed at 21 days. This experiment was repeated 3 times. (F) Kaplan-Meier survival analysis of WT mice administered with a portal venous injection of KPC-derived tumor cells and treated as indicated beginning on day 5 after tumor challenge (n=10/group). This data represents one of two repeat experiments. (G) Kaplan-Meier survival analysis of WT mice orthotopically implanted with KPC-derived tumor cells and treated as indicated (n=10-20/group). This data represents one of two repeat experiments. (H-M) Expression of IFNγ (H, I), TNFα (J, K), and T-bet (L, M) in PBMC, spleen, and PDA-infiltrating CD4+ (H, J, and L) and CD8+ (I, K, and M) T cells from WT mice orthotopically implanted with KPC-derived tumor cells and treated with RIP1i or control and sacrificed at 21 days. These experiments were repeated twice. Data are displayed as average ± SEM (*p<0.05; **p<0.01; ***p<0.001; ****p<0.0001). See also Figure S3.
RIP1 inhibition induces immunogenic reprogramming of intra-tumoral macrophages
Since we observed that RIP1i did not directly activate T cells (Figure S3C, D) but TAMs expressed high RIP1 (Figure S1F), we postulated that RIP1 signaling promotes the expansion and tolerogenic programming of macrophages in PDA, which can secondarily corrupt adaptive tumor immunity. We found that RIP1i-treatment resulted in a ~40% reduction in TAMs infiltrating orthotopic KPC tumors (Figure 4A, B). Moreover, RIP1i reprogrammed TAMs towards an immunogenic M1-like phenotype, including upregulation of MHC-II, CD86, CD80, TNFα, and IFNγ with a concomitant reduction in CD206, IL-10, and TGF-β (Figure 4C–H). Arg1 expression was also reduced in RIP1i tumors (Figure 4I). TAMs infiltrating PDA tumors in RIP1 KD/KI mice similarly exhibited M1-like programming (Figure 4J). Further, in our liver metastases model, RIP1i treatment also led to higher macrophage expression of MHCII and TNFα, and reduced expression of CD206 and IL-10 (Figure 4K).
Figure 4. Inhibition of RIP1 signaling mitigates TAM infiltration and reprograms TAMs in PDA.
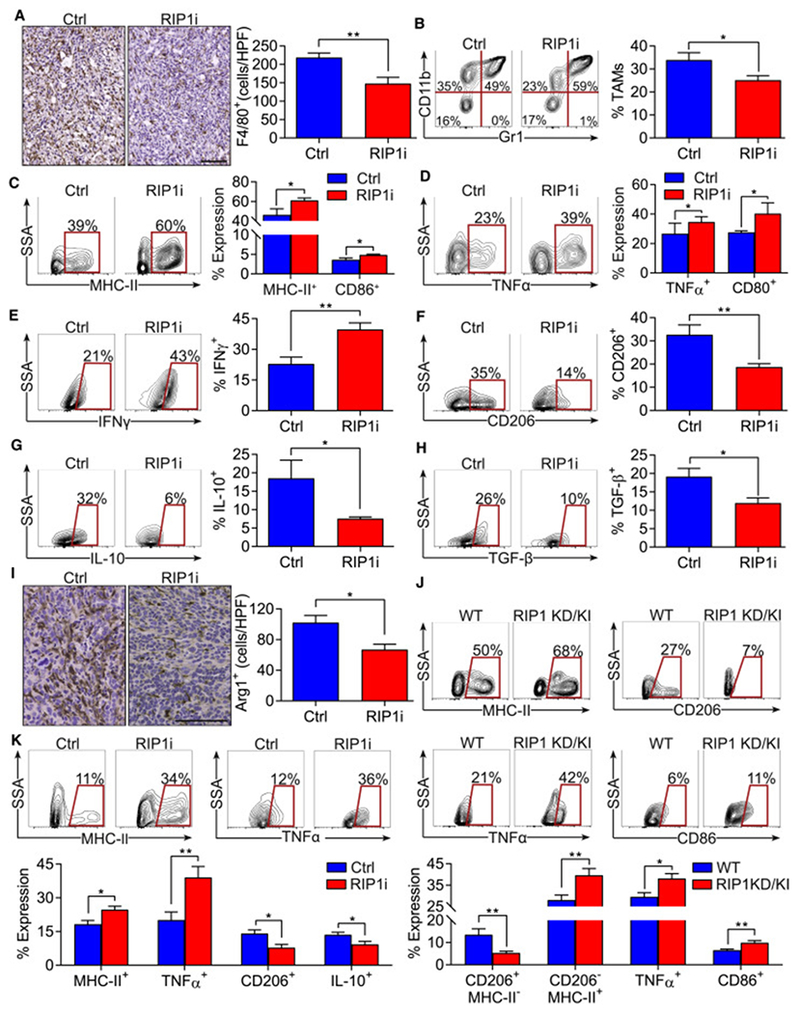
(A-I) WT mice bearing orthotopic KPC tumors were treated with RIP1i or control and were sacrificed at 21 days (n=5/group). IHC of PDA tumors for F4/80 (scale bar = 100 μm) (A). The percentage of Gr1−CD11c−F480+CD11b+ TAM was determined by flow cytometry (B). TAMs were tested for expression of MHC-II, CD86 (C), TNFα, CD80 (D), IFNγ (E), CD206 (F), IL-10 (G), and TGF-β (H). Pancreata were analyzed for expression of Arg1 by IHC (scale bar = 100 μm) (I). (J) WT and RIP1 KD/KI mice bearing orthotopic KPC tumors were sacrificed at 21 days and tumors were analyzed by flow cytometry. TAMs were tested for expression of MHC-II, CD206, TNFα, and CD86 (n=10/group). (K) Expression of MHC-II, TNFα, CD206, and IL-10 in TAMs isolated at day 21 from WT mice injected with KPC-derived tumor cells via portal venous and treated with RIP1i or control beginning on the day of tumor administration (n=5/group). Macrophage phenotyping experiments were repeated more than 5 times (*p<0.05; **p<0.01). Data are displayed as average ± SEM.
RIP1 signaling directly influences macrophage polarization and their capacity to induce T cell differentiation
We postulated that RIP1 signaling governs macrophage differentiation. Consistent with our in vivo data in PDA, RIP1i treatment in vitro directed the programing of bone marrow-derived macrophages (BMDM) towards an immunogenic phenotype, upregulating MHCII, TNFα, and IFNγ, while concomitantly reducing CD206, IL-10 and TGF-β expression (Figure 5A). RIP1i induction of M1-like programming in BMDM was confirmed using qPCR (Figure S4A). Moreover, RIP1i upregulated STAT1 signaling in BMDM, which is associated with M1-programming, but reduced STAT3, STAT5, and STAT6 signaling, which are linked to M2-like macrophage differentiation (Figure S4B–D). BMDM generated from RIP1 KD/KI mice similarly exhibited increased M1 polarization and upregulated STAT1 signaling (Figure S4E). Further, RIP1i-treated macrophages displayed enhanced ability to capture antigen (Figure S4F).
Figure 5. RIP1 regulates macrophage differentiation and immunogenicity.
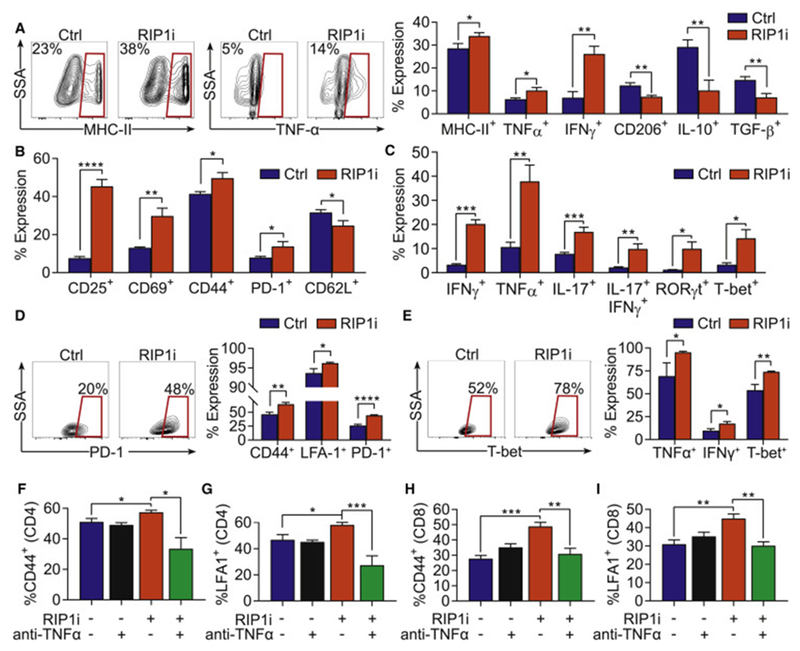
(A) Expression of MHC-II, TNFα, IFNγ CD206, IL-10, and TGF-β in day 7 BMDM treated with RIP1i or vehicle for 18 hr. This experiment was performed more than 5 times in replicates of 5. (B, C) Expression of surface activation markers (B) and cytokines and transcription factors (C) in Ova-restricted CD4+ T cells co-cultured for 96 hr with TAMs harvested from day 21 PDA tumors of mice treated with RIP1i or control chow and pulsed with Ova323-339 peptide. This experiment was performed 4 times in replicates of 5. (D, E) Expression of surface activation markers (D) and cytokines and transcription factors (E) in Ova-restricted CD8+ T cells co-cultured for 96 hr with TAMs harvested from day 21 PDA tumors of mice treated with RIP1i or control chow and pulsed with Ova257-263 peptide. This experiment was performed twice in replicates of 5. (F, G) Expression of CD44 (F) and LFA1 (G) in Ova-restricted CD4+ T cells co-cultured for 96 hr with BMDM treated with RIP1i or vehicle plus a neutralizing α-TNFα mAb or isotype and pulsed with Ova323-339 peptide. This experiment was performed twice in replicates of 5. (H, I) Expression of CD44 (H) and LFA1 (I) in Ova-restricted CD8+ T cells co-cultured for 96 hr with BMDM treated with RIP1i or vehicle plus a neutralizing α-TNFα mAb or isotype and pulsed with Ova257-264 peptide. This experiment was performed twice times in replicates of 5. Data are displayed as average ± SEM (*p<0.05; **p<0.01; ***p<0.001; ****p<0.0001). See also Figure S4.
We postulated that the immunogenic reprogramming of macrophages observed with inhibition of RIP1 directly reverses the characteristic immune-suppressive phenotype of PDA-infiltrating T cells. To model this in vitro, we activated polyclonal splenic T cells using CD3/CD28 co-ligation in co-culture with PDA-infiltrating TAMs harvested from control or RIP1i-treated mice. Whereas control TAMs mitigated T cell activation, RIP1i-treated TAMs exhibited minimal inhibitory effects (Figure S4G). To assess whether RIP1i enhances the capacity of TAMs to induce immunogenic CD4+ T cell differentiation, we pulsed TAMs ex vivo with Ova323-339 peptide before plating with Ova-restricted CD4+ T cells. Compared to TAMs harvested from control PDA tumors, antigen-pulsed TAMs harvested from RIP1i-treated PDA induced higher CD4+ T cell activation, including upregulation of CD25, CD69, CD44, and PD-1, but reduced CD62L (Figure 5B). Further, consistent with our in vivo data in the PDA TME, antigen-pulsed RIP1i-treated TAMs augmented Th1 and Th17 polarization (Figure 5C). Notably, IFNγ and IL-17 were co-expressed in a substantial fraction of CD4+ T cells entrained by RIP1i-treated TAMs (Figure 5C). Ova257-263 peptide-pulsed TAMs from RIP1i-treated mice also induced higher CD8+ T cell activation (CD44, LFA-1, PD-1) and promoted their expression of a cytotoxic phenotype (TNFα, IFNγ, T-bet) (Figure 5D, E). Similarly, RIP1i-treated macrophages were more efficient at activating antigen-restricted CD4+ and CD8+ T cells after loading with Ovalbumin (Figure S4H, I). Since RIP1i-treated macrophages upregulated TNFα, which can drive immunogenic T cell differentiation (Croft, 2009), we postulated that the observed effects were TNFα-dependent. Accordingly, TNFα blockade abrogated the enhanced capacity of RIP1i-treated macrophages to activate CD4+ and CD8+ T cells (Figure 5F–I). Further, akin to T cells, macrophage activation in vivo after RIP1i treatment was seen exclusively in the PDA TME but not in spleen or PBMC (Figure S4J–L).
Targeting RIP1 reprograms macrophages from tumor-promoting to tumor-protective entities
To definitively determine whether TAMs are responsible for RIP1i-induced tumor protection and adaptive immune activation in vivo, we harvested TAMs from control and RIP1i-treated PDA tumors and adoptively transferred the TAMs to WT mice together with KPC tumor cells. Control TAMs did not protect against tumor progression whereas TAMs harvested from RIP1i-treated tumors did (Figure 6A, B) and induced higher CD4+ and CD8+ T cell activation in the TME (Figure 6C–E). Adoptive transfer of RIP1i-treated TAMs also synergized with αPD1 therapy (Figure 6B). Further, similar to our findings using TAMs harvested from RIP1i-treated tumors, adoptive transfer of BMDM from RIP1 KD/KI mice were tumor protective and synergized with αPD1, whereas control BMDM were not efficacious (Figure S4M).
Figure 6. RIP1 inhibition endows TAMs with tumor-protective and immunogenic properties.
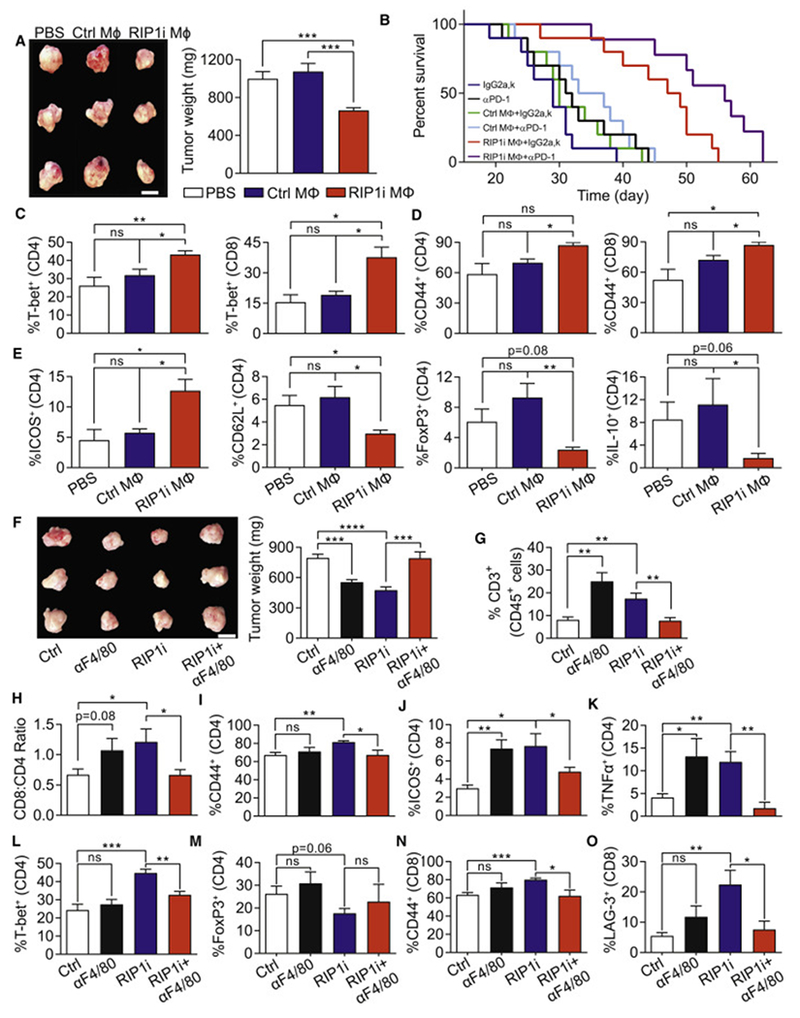
(A) Representative images (scale bar = 1 cm) and weight of tumors from WT mice on day 21 after orthotopic administration of KPC-derived tumor cells alone, tumor cells admixed with TAMs harvested from control PDA, or tumor cells admixed with TAMs harvested from PDA in RIP1i-treated mice (n=5/group). (B) Kaplan-Meier survival analysis of WT mice orthotopically implanted with KPC-derived tumor cells alone, tumor cells admixed with TAMs harvested from control PDA, or tumor cells admixed with TAMs harvested from PDA in RIP1i-treated mice. Cohorts were additionally treated with αPD1 or isotype (n=10/group; p values: Isotype vs. αPD1 0.15, Ctrl TAMs+Isotype vs. Isotype 0.37, Ctrl TAMs+isotype vs. Ctrl TAMs+αPD1 0.29, RIP1i TAMs+Isotype vs. Isotype 0.0001, RIP1i TAMs+Isotype vs. RIP1i TAMs+αPD1 0.01, Ctrl TAMs+Isotype vs. RIP1i TAMs+Isotype 0.0004). Macrophage adoptive transfer experiments were repeated 3 times. (C-E) Expression of T-bet (C) and CD44 (D) in tumor-infiltrating CD4+ and CD8+ T cells and expression of ICOS, CD62L, FoxP3, and IL-10 in tumor-infiltrating CD4+ T cells (E) harvested from WT mice 21 days after orthotopically implanted with KPC-derived tumor cells alone, tumor cells admixed with TAMs harvested from control PDA, or tumor cells admixed with TAMs harvested from PDA in RIP1i-treated mice (n=5/group). This experiment was repeated twice. (F) Representative images (scale bar = 1 cm) and weight of tumors from WT mice bearing KPC-derived tumor treated with an αF4/80 neutralizing or isotype alone or in combination with RIP1i and sacrificed at 21 days (n=7/group). (G-O) Mice were treated as in (F). The percentage of intra-tumoral CD3+ T cells (G) and the CD8:CD4 T cells ratio (H) determined by flow cytometry, the expression of CD44 (I), ICOS (J), TNFα (K), T-bet (L), and FoxP3 (M) in CD4+ T cells, and the expression of CD44 (N) and LAG-3 (O) in CD8+ T cells from (K). This experiment was repeated twice. Data are displayed as average ± SEM (*p<0.05; **p<0.01; ***p<0.001; ****p<0.0001). See also Figure S4.
To further test whether the M1-like reprogramming of TAMs is responsible for the tumor-protective T cell phenotype in RIP1i-treated mice, we serially neutralized macrophages in PDA-bearing control and RIP1i-treated mice. Targeting macrophages protected against PDA in WT hosts, as previously reported (Seifert et al., 2016a), but accelerated tumor growth in RIP1i-treated mice (Figure 6F). These data imply that, consistent with our macrophage transfer data, whereas TAMs are tumor-promoting at baseline, they are immunogenically reprogrammed in the absence of RIP1 signaling. Further, whereas macrophage neutralization enhanced T cell expansion and immunogenicity in control PDA, it decreased both pan-T cell infiltration and the CD8:CD4 ratio (Figure 6G, H) and abrogated CD4+ and CD8+ T cell activation in RIP1i-treated tumors (Figure 6I–O).
Targeting RIP1 activates innate and adaptive immunity and induces tumor-protection in organotypic models of human PDA
Consistent with our mouse data, RIP1i treatment directed human PBMC-derived macrophage differentiation towards an immunogenic M1-like phenotype, upregulating HLA-DR, IFNγ, and TNFα, and concomitantly reducing IL-10 expression (Figure 7A). To evaluate the efficacy of RIP1i against human PDA, we treated patient-derived organotypic tumor spheroids (PDOTS) from human PDA specimens with RIP1i or vehicle using a 3-dimensional microfluidic system (Jenkins et al., 2017). Treatment with RIP1i reduced tumor viability and mitigated spheroid growth (Figure 7B, C). Further, RIP1i treatment upregulated a spectrum of immunogenic cytokines, including IL-1α, TNFα, MIP1α, and IL-15, but mitigated expression of suppressive cytokines linked to intra-tumoral immune-suppression and PDA growth, including G-CSF, IL-1β, IL-10, and IL-8 (Figure 7D) (Barber et al., 2000; Daley et al., 2017b; Kuwada et al., 2003; Pickup et al., 2017). Consistent with our murine data, RIP1i induced intra-tumoral macrophage (Figure 7E) and T cell (Figure 7F) activation. Moreover, co-targeting of RIP1 and PD-1 resulted in synergistic efficacy (Figure 7G–I).
Figure 7. Inhibition of RIP1 in organotypic models of human PDA activates innate and adaptive immunity and decreases tumor viability.
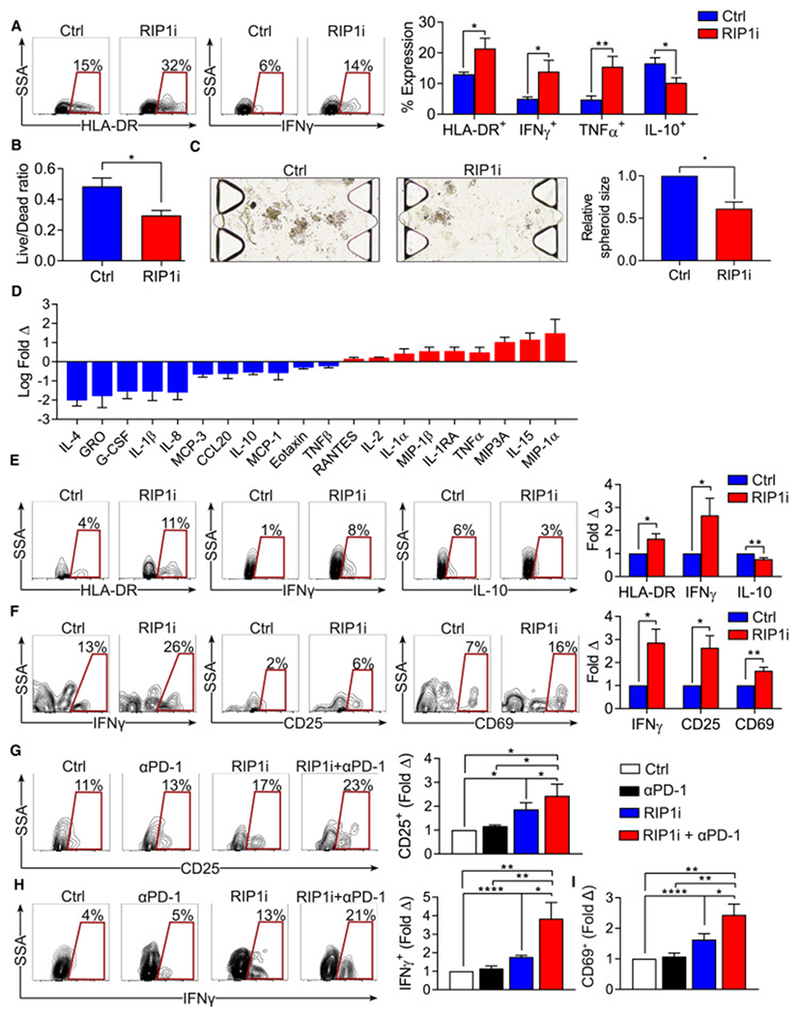
(A) Select contour plots and quantification of the expression of HLA-DR, 1FNγ, TNFα, and IL-10 in PBMC-derived monocytes treated with RIP1i or vehicle (n=9). (B, C) Tumor cell viability (B) and the relative sizes of spheroids (C) of PDOTS (n=5 patient samples) treated with RIP1i or vehicle. (D) The relative levels of indicated inflammatory mediators in supernatant of PDOTS from PDA patients (n=5) treated with RIP1i vs. vehicle. (E, F) Expression of HLA-DR, IFNγ and IL-10 in TAMs (E) and expression of IFNγ, CD25, and CD69 in CD4+ T cells (F) from PDOTS (n=10) treated with RIP1i or vehicle. Representative contour plots and quantitative data are shown as fold change compared to vehicle treatment. (G-I) Expression of CD25 (G), IFNγ (H), and CD69 (I) in CD4+ T cells from PDOTS (n=6 patients) treated with vehicle, RIP1i alone, αPD-1 alone, or RIP1i + αPD-1. Representative contour plots and quantitative data are shown as fold change compared to vehicle treatment. Data are displayed as average ± SEM (*p<0.05; **p<0.01; ***p<0.001; ****p<0.0001).
RIP1i induces STAT1-dependent macrophage activation
Since we observed that targeting RIP1 upregulates STAT1 signaling in macrophages, we postulated that RIP1i-mediated immunogenic reprogramming of macrophages is dependent on STAT1 signaling. Indeed, STAT1 inhibition prevented M1-like macrophage differentiation (Figure 8A) and RIP1i failed to induce immunogenic changes in BMDM derived from STAT1-deficient mice (Figure 8B). To investigate possible additional mechanisms by which RIP1 regulates macrophage phenotype, we analyzed changes in the global transcriptome after RIP1i treatment (Figure 8C). Gene ontology analysis suggested that targeting RIP1 induced changes in diverse innate inflammatory processes (Figure 8D). Ingenuity analysis demonstrated that pathways upregulated by RIP1i included ERK5/MAP kinase and PPAR signaling, whereas select apoptosis pathways were inhibited (Figure 8E). Since Rps6ka2, which encodes the kinase RSK that is activated by ERK5 in MAP kinase signal pathway (Ranganathan et al., 2006), was among the most highly upregulated genes in RIP1i-treated BMDM (Figure 8C), we postulated that RIP1i effects on macrophage polarization are RSK-dependent. Accordingly, inhibition of RSK mitigated RIP1i-induced immunogenic reprogramming of macrophages (Figure 8F). We also confirmed by qPCR that Pparg was upregulated in RIP1i-treated macrophages (Figure 8G). Further, whereas RIP1i mitigated Il1b expression in macrophages, this effect was reversed by PPARγ inhibition (Figure 8H). Since we previously reported that IL-1β signaling is required for suppressive macrophage function in PDA (Daley et al., 2017a), we hypothesized that immunogenic macrophage programming induced by RIP1i is PPARγ-dependent. Accordingly, inhibition of PPARγ reversed the M1-like macrophage phenotype promoted by RIP1i (Figure 8I). Of note, macrophages from RIP1 KD/KI mice also markedly upregulated Pparg and Rps6ka2 (Figure 8J, K).
Figure 8. Signaling changes in macrophages induced by RIP1i.
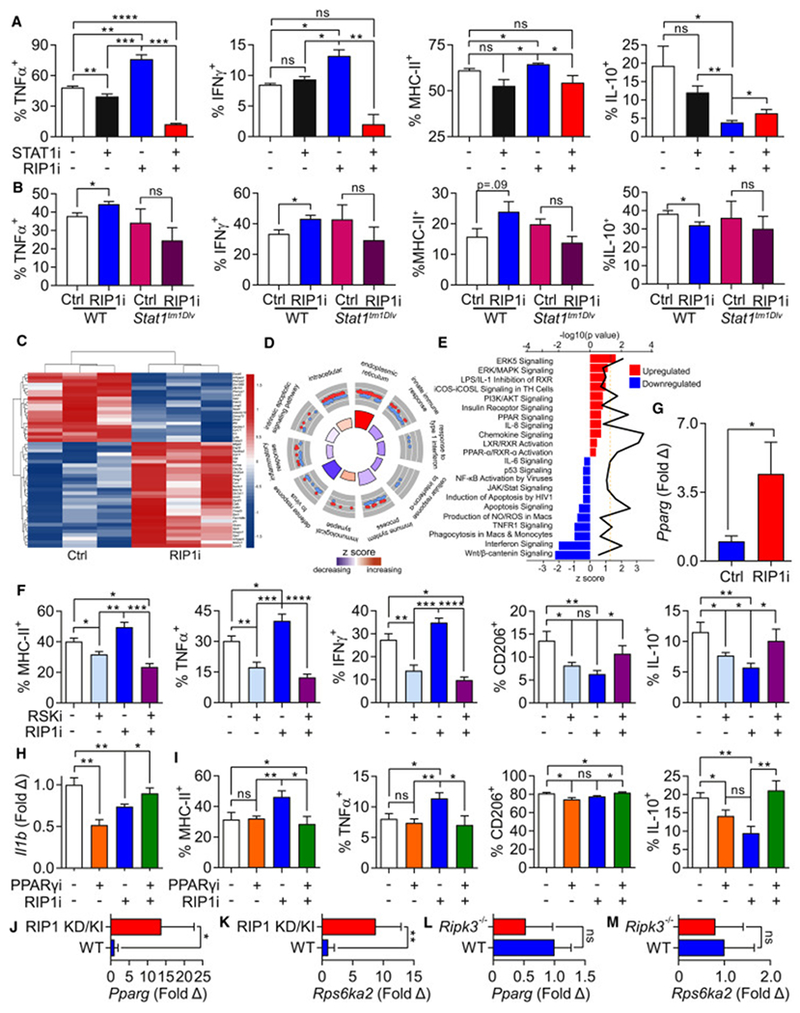
(A) Expression of TNFα, IFNγ, MHC-II, and IL-10 in day 7 BMDM harvested from WT mice treated with RIP1i or vehicle, either alone or in combination with a STAT1 inhibitor (STAT1i) for 18 hr determined by flow cytometry. This experiment was performed 4 times in replicates of 5. (B) Expression of TNFα, IFNγ, MHC-II, and IL-10 in day 7 BMDM harvested from WT or Stat1tm1Dlv mice treated with RIP1i or vehicle for 18 hr determined by flow cytometry. This experiment was performed twice in replicates of 5. (C-E) Heatmap of top 50 differentially expressed genes (C), top scoring Gene Ontology (GO) terms related to innate inflammatory and immune responses (D), and canonical pathway perturbations derived using ingenuity pathway analysis (IPA) (E) from RNA-Seq analysis (in triplicates) of day 7 BMDM treated with RIP1i or vehicle for 18 hr. In (D) red (up-regulated) and blue (down-regulated) dots in outer circle show the log2FC of genes in each GO term. Bar plot colors in the inner circle are based on z-scores, and the height of each bar represents each GO term’s significance. In (E) up-regulated (red) and down-regulated (blue) pathways specific to macrophage and immune response were identified after RIP1i treatment. Z score is shown on the lower x-axis and corresponds to the length of the respective bars. P value for each pathway is indicated by the black line and the corresponding upper x-axis. The dashed line represents the threshold for statistical significance. (F) Expression of MHC-II, TNFα, IFNγ, CD206, and IL-10 in day 7 BMDM treated with RIP1i or vehicle, alone or in combination with RSK inhibitor (RSKi) for 18 hr determined by flow cytometry. This experiment was repeated three times in 5 replicates. (G) The expression of Pparg in day 7 BMDM treated for 18 hr with RIP1i or vehicle measured by qPCR. (H) The expression of Il1b in day 7 BMDM treated for 18 hr with RIP1i alone, PPARγ inhibitor (PPARγi) alone, or RIP1i + PPARγi determined by qPCR. This experiment was performed in replicates of 5 and repeated twice. (I) Expression of MHC-II, TNFα, CD206, and IL-10 in day 7 BMDM treated as in (H). This experiment was repeated twice in 5 replicates. (J, K) Expression of Pparg (J) and Rps6ka2 (K) in day7 BMDM from WT and RIP1 KD/KI mice analyzed by qPCR (n=5/group). (L, M) Expression of Pparg (L) and Rps6ka2 (M) in day 7 WT and Ripk3−/− BMDM were analyzed by qPCR (n=4/group). qPCR experiments were repeated twice. Data are displayed as average ± SEM (*p<0.05; **p<0.01; ***p<0.001; ****p<0.0001). See also Figure S5.
Immune-suppressive effects of RIP1 on innate and adaptive immunity are independent of RIP3
We previously reported that inhibiting necroptosis by deleting RIP3 protects against pancreatic oncogenesis (Seifert et al., 2016a). To determine whether the oncogenic effects of RIP1 depend on its association with RIP3 in necroptosis, cohorts of WT and Ripk3−− mice were treated with RIP1i and then challenged with an orthotopic injection of KPC tumor cells. RIP1i-treatment offered addititive protection in Ripk3−/− mice, suggesting that targeting RIP1 in PDA is independent of RIP3 (Figure S5A, B). Accordingly, PDA-infiltrating CD4+ T cells exhibited synergistic Th1/Th17 differentiation (Figure S5C) and CD8+ T cells displayed synergistic activation (Figure S5D) in the Ripk3−/− cohort treated with RIP1i. Similarly, RIP1i-treatment induced immunogenic polarization of Ripk3−/− BMDM in vitro, including upregulation of MHC-II, TNFα, and IFNγ (Figure S5E). RIP1i also upregulated pSTAT1, but downregulated pSTAT5 in Ripk3−/− BMDM confirming that the effects of RIP1i on M1-like macrophage differentiation are RIP3-independent (Figure S5F). Moreover, akin to our findings in WT BMDM, STAT1 inhibition prevented RIP1i-induced immunogenic macrophage differentiation in Ripk3−/− BMDM; however, STAT1 inhibition did not have suppressive effects in Ripk3−/− BMDM in the absence of RIP1i (Figure S5G–I). Further, unlike genetic or pharmacologic targeting of RIP1, deletion of Ripk3 in macrophages did not upregulate Pparg and Rps6ka2 expression (Figure 8L, M). Collectively, these observations suggest that RIP1 and RIP3 regulate distinct pathways in innate immune programming.
Discussion
T cell checkpoint-based immunotherapy has become standard of care in several cancers. However, PDA has proven refractory to checkpoint-based immunotherapy (Chen et al., 2017). T cell scarcity, chemokine-mediated exclusion of T cells from the tumor milieu, reduced T cell expression of checkpoint receptors, and inability of therapeutic mAbs to physically access PDA secondary to peri-tumoral fibrosis have each been proposed as potential mechanisms of therapeutic resistance in PDA (Daley et al., 2017a; Feig et al., 2013; Provenzano et al., 2012). Moreover, previous work has shown that suppression of cytotoxic T cell responses in PDA is largely the result of T cell corruption by a diversity of innate inflammatory cells. For example, myeloid-derived suppressor cells are recruited to PDA by tumor expression of GM-CSF and this serves to inhibit CD8+ T cell expansion in the tumor microenvironment (Bayne et al., 2012; Pylayeva-Gupta et al., 2012). Select subsets of dendritic cells (DC) in PDA recruit regulatory T cells, which in turn maintain a suppressive DC phenotype (Jang et al., 2017). We have shown that δ1+Vγ9− γδT cells are highly enriched in human PDA, upregulate PD-L1, and suppress adaptive anti-tumor immunity via PD-1 ligation (Daley et al., 2016). However, perhaps the dominant drivers of immune-tolerance in PDA are CD206+MHC-IIlow TAMs, which promote the expansion of Th2 cells and Tregs (Cui et al., 2016).
A number of approaches have been advanced to reprogram TAMs to an immunogenic phenotype in PDA. The CCL2/CCR2 axis appears critical for TAM mobilization and recruitment in PDA, and interruption of this axis has shown promise in early clinical trials (Nywening et al., 2016). CD40 agonists have also shown efficacy in combination with gemcitabine-based chemotherapy in small cohorts of advanced PDA patients, and murine modeling suggested that CD40-mediated protection was macrophage-dependent (Beatty et al., 2011). Targeting focal adhesion kinase is another strategy that can enhance macrophage immunogenicity and immunotherapeutic responsiveness in PDA and was recently advanced to clinical trial (Jiang et al., 2016). We have shown that radiation therapy (RT) upregulates M-CSF expression in PDA cells leading to recruitment of TAMs and accentuation of their immune-suppressive properties, including Treg induction (Seifert et al., 2016b). This has served as the rationale for the design of a clinical trial targeting M-CSF in combination with RT in patients with locally advanced PDA. We also reported that ligation of select pattern recognition receptors in TAMs by pathogen- or inflammation-derived molecular patterns drives suppressive macrophage differentiation (Daley et al., 2017a; Daley et al., 2017b; Seifert et al., 2016a). However, the cell autonomous signaling mechanisms that govern the tolerogenic macrophage program in PDA are not completely understood.
RIP1 is a serine-threonine kinase with dichotomous functions depending on its interacting partner. RIP1 association with caspase 8 in response to noxious stimuli induces apoptosis, whereas RIP1’s association with RIP3 promotes necroptosis (Ofengeim and Yuan, 2013). However, RIP1 also has an emerging role in immune homeostasis. RIP1 deficiency results in bone marrow failure spurred by cell death (Roderick et al., 2014). RIP1 has also been shown to regulate the survival of innate immune cells during mammalian parturition (Kaiser et al., 2014). Moreover, entirely unrelated to its protein interactions that regulate programmed cell death, RIP1 also interacts with various innate immune-based adaptor proteins. For example, RIP1 associates with TRIF and DAI, influencing downstream signaling following pattern recognition receptor ligation (Kaiser et al., 2008; Meylan et al., 2004). We have previously reported that K45A mutations in RIP1 can affect cytokine signaling in antigen presenting cells outside the context of cell death (Shutinoski et al., 2016).
Emerging evidence suggests that RIP1 kinase activity may modulate tumorigenesis. Contrary to our findings, reports in other cancers suggest RIP1 signaling has protective effects by promoting tumor cell death (Wegner et al., 2017). Most studies that have examined the role of RIP1 in cancer have studied its role as a binding partner for RIP3. Induction of RIP1/RIP3-dependent necroptosis has been shown to be an avenue to eliminate cancer cells that have acquired resistance to apoptosis (Han et al., 2007). Inhibition of cIAP1/2 and XIAP in cancer cells can also drive RIPK1/RIPK3-dependent cell death mechanisms (Feoktistova et al., 2011; Tenev et al., 2011; Wong et al., 2014). RIP1 signaling may also be protective against cancer in a non-cell autonomous manner. For example, Yatim et al. showed that necroptotic cells produce signals that promote cross-priming of CD8+ T cells by dendritic cells in colon cancer (Yatim et al., 2015). This required both release of DAMPs and active RIPK1–NF-κB-dependent pro-inflammatory transcription in dying cells. Similarly, another study showed that RIP3-dependent signaling is necessary for promoting IL-12 production by dendritic cells, which is critical for their induction of antitumor immunity in cervical cancer (Schmidt et al., 2015). However, the latter effects were dependent on RIP3-mediated programmed necrosis and not contingent on RIP1 signaling, suggesting possible dichotomous effects of RIP1 and RIP3 on the modulation of tumor immunity.
In contrast to the above reports, emerging data suggest that RIP1/RIP3 signaling may also be pro-tumorigenic in a non-cell intrinsic manner. We have previously shown that RIP3-induced necroptosis drives MDSC accumulation in PDA by upregulating CXCL1 secretion from dying cells and promoting ligation of Mincle, a receptor for the SAP130 protein that is a known byproduct of cell death (Seifert et al., 2016a). Another study showed that genetic deletion of mediators of necroptosis in murine or human breast cancer cell lines reduced tumor xenograft growth in vivo (Liu et al., 2016). This effect was linked to deficient levels of a number of tumor-promoting cytokines and chemokines induced by necroptosis, including IL-6, IL-8, lipocalin-2 (LCN2), CCL2/MCP-1, and CCL5. Collectively, these reports reveal complex and context-specific roles for RIP1/RIP3 in cancer progression. Moreover, the effects of RIP1 kinase activity alone on tumor immunity, independent of its function in the necrosome, have not been well-studied. Accordingly, our results suggest that both the tumor protective effects of targeting RIP1 and its effects on macrophage programming are independent of RIP3. Further, our data suggest that RIP1 modulates antitumor immunity via its effect on macrophage programming, as opposed to direct influences on tumor cell death.
Notably, the TH phenotype associated with RIP1i-induced macrophage programming in PDA is a mixed Th1/Th17 picture. Indeed, we found high co-expression of both IFNγ and IL-17 as well as T-bet and RORγt in CD4+ T cells in PDA upon inhibiting RIP1. IL-17 expression alone has been reported to have tumor-promoting effects in PDA, as transformed pancreatic epithelial cells express high IL-17R and proliferate upon its ligation (McAllister et al., 2014). However, the combined Th1/Th17 phenotype appears to be distinctly immunogenic and is concomitantly associated with downregulation of FoxP3. “Mixed” Th1/Th17 cells have not been well-described in cancer; however, in Crohn’s disease, CD4+ T cells exhibiting the Th1/Th17 phenotype have been reported (Riaz et al., 2016). Similarly, emerging data in acute lung injury suggest that IFNγ+IL-17+ cells play an important pathogenic role (Poole et al., 2012). Quantitative proteomics of gut-derived Th1 and Th1/Th17 clones in Crohn’s disease suggested higher cytotoxic features of Th1 cells compared to Th1/Th17 clones (Riaz et al., 2016). These findings were validated by staining with anti-granzyme B and anti-perforin, as well as by degranulation assays. However, the tumor-protective capacity of Th1 cells vs Th1/Th17 cells in cancer, and in PDA in particular, requires further detailed study.
In summary, our work shows that RIP1 signaling in macrophages is a master regulator of immune tolerance in PDA and confers resistance to immunotherapy. Inhibition of RIP1 enabled efficacy for checkpoint receptor (PD-1) and costimulatory ligand (ICOS) based immunotherapies suggesting that RIP1 can be regarded as an immune ‘checkpoint kinase’. Consistent with our results, an unbiased CRISPR screen in melanoma recently identified RIP1 as a top candidate contributing to αPD-1 immunotherapy resistance (Manguso et al., 2017). Our data thus support inhibition of RIP1 as a potential new therapeutic avenue in oncology.
STAR Methods text
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, George Miller (george.miller@nyumc.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals and In Vivo Procedures
C57BL/6, OT-I, OT-II, Stat1tm1Dlv, Rag1tm1Mom, and Foxn1nu mice were purchased from Jackson Labs (Bar Harbor, ME) and bred in-house. Ripk3−/− mice were obtained from Genentech (San Francisco, CA). RIP1 KD/KI mice were generated by homologous recombination using a targeting construct that mutated the catalytic lysine residue to alanine (K45A) to eliminate all kinase activity, as we previously described (Kaiser et al., 2014). C57BL/6 mice for use specifically in pharmacokinetic experiments were purchased from Taconic Biosciences (Rensselaer, NY). All mice were housed under pathogen-free conditions. KC mice develop slowly progressive pancreatic neoplasia endogenously by expressing mutant Kras in the progenitor cells of the pancreas (Hingorani et al., 2003). We previously detailed tumor progression and survival in control KC mice (Daley et al., 2016). Pancreatic ductal epithelial cells were harvested from KC mice and cultured in vitro as we previously described (Seifert et al., 2016a). Both male and female mice were used, but animals were age-matched within each experiment. Mice were fed either control chow or RIP1i (~100 mg/kg/day) via food-based dosing (Research Diets, Inc, New Brunswick, NJ). For orthotopic pancreatic tumor challenge, 8-10 week old mice were administered intra-pancreatic injections of FC1242 PDA cells derived from KPC mice, as previously described (Zambirinis et al., 2015). Cells were suspended in PBS with 50% Matrigel (BD Biosciences, Franklin Lakes, NJ) and 1x105 tumor cells were injected into the body of the pancreas via laparotomy. Mice were sacrificed 3 weeks later and tumors harvested for analyses. Alternatively, mice were analyzed in survival experiments. In our liver metastases model, KPC-derived tumor cells were suspended in PBS and 1x106 tumor cells were injected into the portal venous system, as we described (Miller et al., 2003). Mice were sacrificed 3 weeks later and liver weights were recorded. Alternatively, mice were analyzed in survival experiments. Special diets were begun on the day of tumor implantation unless otherwise specified. In select experiments, mice were adoptively transferred with FACS-purified TAMs (2x104) or CD3+ T cells (3x104). In some experiments, CD4+ T cells (GK1.5), CD8+ T cells (53-6.7), or macrophages (CI:A3-1; all BioXcell, West Lebanon, NH) were depleted with neutralizing mAbs using regimens we have previously described10. In other experiments animals were treated i.p. with Nec1s (2.5 mg/kg, daily; BioVision, Milpitas, CA), a neutralizing αPD-1 mAb (29F.1A12, 200 μg) or an agonizing ICOS mAb (7E.17G9, 100 μg, Days 4, 7 and 10 post-tumor challenge; both BioXcell) or respective isotype controls. For TNF+zVAD-induced shock experiments, WT mice were pretreated with RIP1i in a dose response manner (0.01, 0.1, 1, and 10 mg/kg, PO) 15 min prior to i.v. injection of TNFα (1.25 mg/kg, Cell Sciences, Canton, MA) and zVAD-FMK (16.7 mg/kg, Bachem, King of Prussia, PA). Temperature was serially monitored by rectal probe. Data are expressed as % inhibition of temperature loss compared to assay controls. In select experiments, GSK’963 (20 mg/kg) were administered by oral dosing. For pharmacokinetic studies to detect steady state drug levels, blood samples were serially collected following the start of food-based dosing and diluted 1:1 with water prior to storage at −80 °C. Prior to bioanalysis, samples were thawed and analytes were isolated by protein precipitation. The resulting supernatant was injected into an LC/MS/MS system optimized for detection of RIP1i. Data are reported as quantitative drug concentrations as determined by standard calibration curve analysis. Using these optimized conditions, the typical lower limit of quantitation achieved was 1 ng/ml. All studies, including pharmacokinetic experiments, were reviewed by the Institutional Animal Care and Use Committee (IACUC) at either NYU School of Medicine or GSK. Experiments were conducted in accordance with the respective NYU School of Medicine and GSK policies on the care, welfare, and treatment of laboratory animals. All experiments met or exceeded the standards of the Association for the Assessment and Accreditation of Laboratory Animal Care, International (AAALAC), the United States Department of Health and Human Services, and all local and federal animal welfare laws.
Human Samples
Human sample collection were approved by the Institutional Review Board (IRB) of the NYU School of Medicine. Informed consent was obtained from all subjects. Human biological samples, consisting of blood or tumor tissue, were sourced ethically, and their research use was in accord with the terms of the informed consents under the IRB approved protocol.
METHOD DETAILS
Cellular Preparation and Flow Cytometry
Single cell suspensions of PDA tumors were prepared for flow cytometry as described previously with slight modifications (Daley et al., 2017a). Briefly, pancreata were placed in cold 2 % FACS (PBS with 2 % FBS) with Collagenase IV (1 mg/mL; Worthington Biochemical, Lakewood, NJ), Trypsin inhibitor (1 mg/mL; EMD Millipore, Billerica, MA) and DNase I (2 U/mL; Promega, Madison, WI), and minced with scissors to sub-millimeter pieces. Tissues were then incubated at 37 °C for 20 min with gentle shaking every 5 min. Specimens were passed through a 70 μm mesh and centrifuged at 350 g for 5 min. Cell pellets were re-suspended and cell labeling was performed after blocking FcγRIII/II with an anti-CD16/CD32 mAb (eBiosciences, San Diego, CA) by incubating 1x106 cells with 1 μg of fluorescently conjugated mAbs directed against mouse CD44 (IM7), CD206 (C068C2), PD-1 (29F.1A12), CD3 (17A2), CD4 (RM4-5), CD8 (53-6.7), CD45 (30-F11), CD11b (M1/70), CD11c (N418), CD86 (GL-1), Gr1 (RB6-8C5), MHC II (M5/114.15.2), IL-10 (JES5-16E3), IFNγ (XMG1.2), TNFα (MP6-XT22), F4/80 (BM8), ICOS (15F9), CD62L (MEL-14), CD25 (3C7), CD40 (3/23), CD69 (H1.2F3), IL-17A (TC11-18H10.1), TGFβ (TW7-16B4), LFA-1 (H155-78), LAG-3 (C9B7W), pSTAT1 (A15158B), pSTAT3 (13A3-1), pSTAT6 (A15137E; all BioLegend, San Diego, CA), pSTAT5 (47/Stat5(pY694); BD Biosciences), T-bet (eBio4B10), RORγt (AFKJS-9), Granzyme B (NGZB), Perforin (eBioOMAK-D), and FoxP3 (FJK-16s; all eBiosciences). Human flow cytometry antibodies included CD45 (HI30), CD3 (UCHT1), CD4 (A161A1), CD25 (BC96), CD69 (FN50), CD11b (ICRF44 or M1/70), TNFα (MAb11), IFNγ (4S.B3), IL-10 (JES3-19F1) and HLA-DR (L243; all Biolegend). Dead cells were excluded from analysis using zombie yellow (BioLegend). Intracellular staining for cytokines, transcription factors, and Granzyme B was performed using the Fixation/Permeabilization Solution Kit (eBiosciences). Flow cytometry was carried out on the LSR-II flow cytometer (BD Biosciences). Data were analyzed using FlowJo v.10.1 (Treestar, Ashland, OR). BMDM were prepared and cultured as previously described (Greco et al., 2016). In select experiments, LPS (1 ug/mL, InvivoGen, San Diego, CA) or RIP1i (50 nM) were added to day 7 BMDM cultures. In other experiments, cells were treated with inhibitors of STAT1 (Fludarabine, 50 μM; Selleckchem, Houston, TX), RSK (LJI308, 10 μM; Sigma-Aldrich, Saint Louis, MO), or PPARγ (GW9662, 20 μM; Sigma-Aldrich). Antigen capture was performed using FITC-Albumin (1 mg/ml; Sigma-Aldrich) as we described (Miller et al., 2002). Mouse PBMC were prepared for flow cytometry using a Ficoll gradient. Human PBMC were isolated from normal blood using a Ficoll gradient and plated with recombinant human M-CSF (50 ng/mL; R&D, Minneapolis, MN) for 7 days.
PDOTS preparation, treatment, and analysis
PDOTS were prepared as previously described with slight modifications (Jenkins et al., 2017). Briefly, human surgically resected pancreatic tumor specimens were received fresh in DMEM media on ice and minced in 10cm dishes. Minced tumors were resuspended in DMEM +10 % FBS with 100 U/mL collagenase type IV to obtain spheroids. Partially digested samples were pelleted and then re-suspended in fresh DMEM +10 % FBS and strained over both 100 mm and 40 mm filters to generate S1 (>100 mm), S2 (40-100 mm), and S3 (<40 mm) spheroid fractions, which were subsequently maintained in ultra-low-attachment tissue culture plates. An aliquot of the S2 fraction was pelleted and resuspended in type I rat tail collagen at a concentration of 2.5 mg/mL following addition of 10x PBS with phenol red with pH adjusted using NaOH. An aliquot of the S2 fraction was pelleted and re-suspended in type I rat tail collagen and the spheroid-collagen mixture was then injected into the center gel region of the DAX-1 3D microfluidic cell culture chip (Aim Biotech, Singapore). After 30 min at 37 °C, collagen hydrogels containing PDOTS were hydrated with media with indicated treatments. To determine cellular viability, dual labeling was performed by loading the microfluidic device with Nexcelom ViaStainTM AO/PI Staining Solution (Nexcelom, Lawrence, Massachusetts). Following incubation with the dyes, images were captured on a Nikon Eclipse 80i fluorescence microscope equipped with Z-stack (Prior) and CoolSNAP CCD camera (Roper Scientific, Trenton, NJ). Image capture and analysis were performed using NIS-Elements AR software package (Nikon, Tokyo, Japan). Levels of inflammatory mediators were determined using the MILLIPLEX Human Cytokine 41-Plex kit, according to the manufacturer’s protocol (MilliporeSigma).
Histology, Immunohistochemistry, and Microscopy
For histological analysis, pancreatic specimens were fixed with 10 % buffered formalin, dehydrated in ethanol, embedded with paraffin, and stained with H&E or Gomori Trichrome. The percentage of preserved acinar area and fibrosis were calculated, as previously described (Zambirinis et al., 2015). Immunohistochemistry on paraffin-embedded or frozen mouse tissues was performed using antibodies directed against F4/80 (CI:A3-1), Arginase1 (Polyclonal), CD3 (SP7; all Abcam, Cambridge, MA), RIP1 (#5389; ProSci, Poway, CA), CK19 (Troma-III; University of Iowa), and DAPI (#H-1200; Vector Labs, Burlingame, CA). For paraffin-embedded slides, samples were dewaxed in ethanol followed by antigen retrieval with 0.01 M Sodium Citrate with 0.05 % Tween. For frozen slides, samples underwent antigen retrieval (10% SDS) prior to incubation with the primary antibody. Immunofluorescent images were acquired using a Zeiss LSM700 confocal microscope with ZEN 2010 software (Carl Zeiss, Thornwood, New York).
Protein Biochemistry
Recombinant FLAG-tagged RIPK1 protein (residues 1-294) containing C34A, C127A, C233A and C240A substitutions was generated from a recombinant baculovirus (pFastBac1 FLAG-RIPK1 (1-294) C34A, C127A C233A, C240A) expressed in 10 L of Spodoptera frugiperda Super 9 insect cells with a moi of 0.1. After 3 days cells were harvested by centrifugation. 100 g of cell pellet was resuspended in 500ml Buffer A (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 10% (w/v) glycerol, pH 7.5 containing Complete Protease Inhibitor cocktail minus EDTA (Roche, Nutley, NJ) and cells broken using a dounce homogenizer followed by sonication. Clarified supernatant was batch bound to 40 ml anti-FLAG M2 agarose (Sigma-Aldrich), incubated overnight at 4 °C and the resin then packed into a 5 cm column. The resin was washed with Buffer A and FLAG-tagged RIPK1 protein eluted with Buffer A containing 200 μg/ml FLAG peptide. The eluate was concentrated to 10 ml and loaded onto a 140 ml Superdex 200 pg at 1 ml/min. 1.5 ml fractions were collected and those containing RIPK1 were pooled and concentrated to 0.85 ml (final yield 12.6 mg protein at 14.8 mg/ml). Protein was stored in 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM DTT, 10 % (w/v) glycerol, pH 7.5.
X-ray Crystallography
Crystals of the RIP1 complex with RIP1i were obtained by co-crystallization using sitting drop vapor diffusion with all sample manipulation carried out at 4 °C or over ice. 100nL drops of the protein at 14.8 mg/ml in the storage buffer (25 mM Tris-HCl, 150 mM NaCl, 1 mM DTT, 10 % glycerol, pH 7.5) with 4 mM RIP1i (dissolved in DMSO) were dispensed using a Mosquito dispensing robot into a 96-well plate and incubated against the Qiagen PEGS I crystallization screening kit from which 150 nL was dispensed on top of the protein drop. Crystals spontaneously nucleated in many of the drops and grow to full size over approximately one week. Crystals were quickly transferred through a 10 μL drop containing precipitant solution from the Qiagen screen, 10 % ethylene glycol and 4 mM RIP1i before plunge freezing into liquid nitrogen. Data was collected at the Diamond Light Source (Oxford, UK) on beamline I02 using a Pilatus 6M detector where 180° of data was collected using a oscillation increment of 0.2°. Data processing was carried out using the CCP4 (Winn et al., 2011) program imosflm in spacegroup P1, converted to orthorhombic spacegroup P222 by Pointless prior to scaling with Aimless (Table S4). Data was subsequently reindexed to P212121 using Pointless and the operator [k,l,h] to match the details of related structures. Refinement was carried out with the Global Phasing program Buster with a previously solved but undisclosed higher resolution target structure utilized for structural restraints (Smart et al., 2012). The chemical library for RIP1i was generated with phenix.elbow modified by the Cambridge Crystallographic Data Centre program Mogul (Adams et al., 2010). All model building was carried out with COOT (Emsley et al., 2010) and figures generated utilizing the CCP4 program CCP4MG. The Protein Data Bank as accession code will be generated upon publication acceptance.
Kinase selectivity and cell-based viability assays
Kinase selectivity assays were performed as we previously described (Berger et al., 2015). RIP1i (10 μM) was tested against 371 kinases using a P33-radiolabeled assay according to the manufacturer’s protocol (Reaction Biology, Devault, Pennsylvania). Reactions were performed in the presence of 10 μM ATP. Data are reported as % enzyme activity relative to DMSO controls. The efficacy of RIP1 inhibitors was tested in vitro using L929 cells (ATCC, Manassas, VA). Cell death was induced with recombinant TNFα (100 ng/ml; R&D Systems) in the presence of caspase inhibitor QVD-Oph (25 μM; Millipore Sigma). To evaluate the effect of RIP1 inhibition, cells were pretreated with RIP1i at various doses for 30 min. Induced cell death was evaluated 24 hr later by measuring cellular ATP levels using CellTiter-Glo Luminescent Cell Viability Assay according to the manufacturer’s protocol (Promega, Fitchburg, WI).
Fluorescent polarization (FP) binding assay
An FP-based binding assay was used to quantify the interaction between RIP1i and the ATP-binding pocket of RIP1 by competition with a fluorescently labeled ATP-competitive ligand as we previously described (Berger et al., 2015). In brief, purified GST-tagged RIP1 (1-375) was used at a final assay concentration of 200 nM. A fluorescently-labeled ligand (14-(2-{[3-({2-{[4-(cyanomethyl)phenyl]amino}-6-[(5-cyclopropyl-1H-pyrazol-3-yl)amino]-4-pyrimidinyl}amino) propyl]amino}-2-oxoethyl)-16,16,18,18-tetramethyl-6,7,7a,8a,9,10,16,18-octahydrobenzo [2’,3’]indolizino[8’,7’:5′,6′]pyrano [3′,2′:3,4]pyrido[1,2-a]indol-5-ium-2-sulfonate) was used at a final assay concentration of 5 nM. Samples were read on an Analyst multimode reader (Molecular Devices, Sunnyvale, CA) and the inhibition was expressed as percent inhibition of internal assay controls.
Western Blotting and qPCR
For protein extraction, tissues were homogenized in ice-cold RIPA buffer. Total protein was quantified using the DC Protein Assay according to the manufacturer’s instructions (BioRad, Hercules, CA). Western blotting was performed as previously described with minor modifications (Zambirinis et al., 2015). Briefly, 10 % Bis-Tris polyacrylamide gels (NuPage, Invitrogen, Carlsbad, CA) were equiloaded with 10-30 μg of protein, electrophoresed at 200V, and electrotransferred to PVDF membranes. After blocking with 5 % BSA, membranes were probed with primary antibodies to STAT1 (#9172), pSTAT1 (#9167), β-actin (8H10D10, all Cell Signaling, Danvers, MA). Blots were developed by ECL (Thermo Scientific, Asheville, NC). For qPCR, total RNA was extracted using an RNeasy mini kit (Qiagen, Valencia, CA) and cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). Real-time qPCR was performed in duplicate for each sample using the BioRad Real-Time PCR System (BioRad). Each reaction mixture contained 10 μl of SYBR Green Master Mix (Applied Biosystems), 0.5 μl of forward and reverse primers (Invitrogen) and 3 μl of cDNA (corresponding to 50 ng of RNA). The qPCR conditions were: 50 °C for 2 min, 95 °C for 10 min, followed by 40 cycles at 95°C for 15 s, and 60 °C for 1 min. Amplification of specific transcripts was confirmed by melting curve profiles generated at the end of the PCR program. Expression levels of target genes were normalized to the expression of 18S (internal control) and calculated based on the comparative cycle threshold (CT) method (2−ΔΔCt). See primer sequence details in Table S5. Data on gene expression in human tissues was derived from the Human Protein Atlas (https://www.proteinatlas.org/).
Ripk1 knockdown
Lentiviral transfer plasmids against RIP1 SHCLNG-NM_009068 (TRCN0000022466; CCGGCGTGACTTTCACATTAAGATACTCGAGTATCTTAATGTGAAAGTCACGTTTTT) and (TRCN0000278133; CCGGGCATTGTCCTTTGGGCAATATCTCGAGATATTGCCCAAAGGACAATGCTTTTT) were transformed into Stbl3 bacteria. Plasmids were purified using MaxiPrep Kit (Qiagen) and DNA concentration was evaluated by Nanodrop (Thermo Fisher Scientific). The transfer plasmids were co-transfected into HEK293FT cells with packaging plasmids PLP1, PLP2, and VSVG. To evaluate lentivirus concentration, titration the ability of virus to induce puromycin resistant colonies was performed in HEK293FT cell line. Next, KPC tumor cells were transduced for 48 hr followed by selection with puromycin (2 μg/ul) for 10 days. The efficacy of Ripk1 knockdown was measured by qPCR.
RNA-Seq and Analyses
RNA-Seq libraries were prepared using the Illumina TruSeq Stranded Total RNA library prep, after ribodepletion with Ribozero Gold kit (cat# 20020597, Illumina, San Diego, CA) starting from 500 ng of DNase I treated total RNA, following the manufacturer’s protocol, with the exception that 9 cycles of PCR were performed to amplify the libraries. The amplified libraries were purified using AMPure beads, quantified by Qubit and qPCR, and visualized in an Agilent Bioanalyzer (Agilent, Santa Clara, CA). The libraries were pooled equimolarly and sequenced on one lane of an Illumina HiSeq 2500 flow cell, v4 chemistry as paired end 50. The raw fastq reads were aligned to mm10 mouse reference genome using STAR aligner (Dobin et al., 2013). Fastq Screen was used to check for any contaminations in the samples and Picard RnaSeqMetrics was used to obtain the metrics of all aligned RNA-Seq reads. featureCounts was used to quantify the gene expression levels (Liao et al., 2014). The raw gene counts data were used for further differential expression analysis. To identify the differentially expressed genes, DESeq2 R package was used (Love et al., 2014). The resulting genes with adjusted p<0.05 were considered significant. Heatmaps were generated using pheatmap R package. To determine the functional annotation of the significantly expressed genes, Gene Ontology (GO) analysis was performed and the expression levels of genes in each term were represented in a GO circle plot using the R package GOplot. To identify the signaling pathways in which the genes are enriched, Ingenuity Pathway Analysis (IPA) was carried out for genes that were considered significant. The canonical pathways from IPA analysis were represented as a barplot and the regulatory network of genes associated with phagocytosis and cell death were represented using Cytoscape (Shannon et al., 2003).
Human Tisue Expression Profiling
PDAC tumor and Adjacent Normal mRNA raw expression profiles were downloaded from GEO [accession #: GSE16515 and GSE15471] and normalized in one batch using a GC-content background corrected Robust Multi-array Average (RMA) algorithm (GC-RMA) in R: A language and environment for statistical computing, as previously described (Gadaleta et al., 2011). Hierarchical clustering was performed in GENE-E (https://software.broadinstitute.org/GENE-E/) and adjacent normal tissues clustering with PDAC tumors and PDAC tumors clustering with adjacent normals were removed. Patients with expression profiles containing the gene of interest in both PDAC and Adjacent Normal groups were utilized for differential expression analysis.
Tumor Cell Proliferation, Migration, and Clonogenic Assays
For cellular proliferation assays, 2x103 KPC-derived, KC-derived (Pylayeva-Gupta et al., 2012), or Pan02 (ATCC) tumor cells were seeded in 96-well plates and incubated with 100 μL culture medium. Tumor cell proliferation was measured using the XTT assay kit according to the manufacturer’s protocol (Sigma-Aldrich). For cell migration assays, 5x103 tumor cells were seeded in the upper compartment of a Transwell chambers (24-well format; 8 μm pore size; Corning, Teterboro, NJ) and were cultured with FBS-free RPMI, while the lower compartment was filled with complete medium containing DMSO or RIP1i (50 nM). After 48 hr, cells were fixed with 4 % paraformaldehyde (VWR, Philadelphia, PA). The non-migrated cells in the upper chamber were wiped off with cotton swaps and the filter was stained for 10 min with 0.5 % crystal violet (Fisher Scientific, Fair Lawn, NJ) in 20 % methanol and rinsed with PBS. ImageJ (National Institutes of Health, Bethesda, MD) was used to quantify the cells that had migrated across the filters. For cell clonogenic assays, 2x103 tumor cells were seeded in 6-well plates and treated in 2 mL complete RPMI medium with DMSO or RIP1i (50 nM). After 2 weeks, colonies were stained using 0.5 % crystal violet and counted.
T cell Proliferation Assays
For antibody-based T cell proliferation assays, splenic CD3+ T cells were activated using CD3/CD28 co-ligation in 96-well plates, as we previously described (Daley et al., 2016). In selected wells, TAMs were added in a 1:5 macrophage: T cell ratio. For antigen-restricted T cell stimulation assays, splenic OT-I or OT-II T cells were cultured with macrophages pulsed, respectively, with Ova257-264 or Ova323-339 peptide in a 5:1 ratio. Alternatively, macrophages were loaded with Ovalbumin (1 mg/ml, 60 min). In select wells a neutralizing anti-TNFα mAb (10 μg/ml, MP6-XT22, BioLegend) or isotype control was added. T cell activation was determined at 72 hr by flow cytometry.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
Data is presented as mean +/− standard error. Survival was measured according to the Kaplan-Meier method and analyzed by log rank. The sample size for each experiment, n, is included in the results section or the associated figure legends. Statistical significance was determined by the Student’s t test and the Wilcoxon test using GraphPad Prism 7 (GraphPad Software, La Jolla, CA). P-values <0.05 were considered significant.
DATA AND SOFTWARE AVAILABILITY
RNAseq data has been submitted to the GEO repository, and the accession number is GSE111626. The PDB report has been deposited in RCSB Protein Data Bank under ID code 6HHO.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| InVivoMab anti-mouse CD4 | BioXCell | Clone: GK1.5 Cat#: BE0003-1 |
| InVivoMab anti-mouse CD8α | BioXCell | Clone: 53-6.7 Cat#: BE0004-1 |
| InVivoMab anti-mouse F4/80 | BioXCell | Clone: CI:A3-1 Cat#: BE0206 |
| InVivoMab anti-mouse PD-1 | BioXCell | Clone: 29F.1A12 Cat#: BE0273 |
| InVivoMab anti-mouse ICOS | BioXCell |
Clone: 7E.17G9 Cat#: BE0059 |
| WB: Rabbit Ab Stat1 | Cell Signalling | Cat#: 9172 |
| WB: Rabbit mAb phosphor-Stat1 (Tyr701) | Cell Signalling | Cat#: 9167 |
| WB: Mouse mAb β-actin (8H10D10) | Cell Signalling | Cat#: 3700 |
| IHC/IF: anti-F4/80 antibody | Abcam | Clone: CI:A3-1 Cat#: ab6640 |
| IHC: anti-Arginase antibody | Abcam | Cat#: ab60176 |
| IHC: anti-CD3 antibody | Abcam | Clone: SP7 Cat#: ab16669 |
| IHC/IF: anti-RIP1 antibody | ProSci | Cat#: 5389 |
| IF: anti-CK19 antibody | DSHB at University of Iowa | Clone: Troma-III |
| IF: DAPI | Vector Labs | Cat#: H-1200 |
| Purified anti-mouse CD3ε | Biolegend | Clone: 145-2C11 |
| Purified anti-mouse CD28 | Biolegend | Clone: 37.51 |
| FC: anti-mouse CD44 | Biolegend | Clone: IM7 |
| FC: anti-mouse CD206 | Biolegend | Clone: C068C2 |
| FC: anti-mouse PD-1 | Biolegend | Clone: 29F.1A12 |
| FC: anti-mouse CD3 | Biolegend | Clone: 17A2 |
| FC: anti-mouse CD4 | Biolegend | Clone: RM4-5 |
| FC: anti-mouse CD8 | Biolegend | Clone: 53-6.7 |
| FC: anti-mouse CD45 | Biolegend | Clone: 30-F11 |
| FC: anti-mouse/human CD11b | Biolegend | Clone: M1/70 |
| FC: anti-mouse CD11c | Biolegend | Clone: N418 |
| FC: anti-mouse CD86 | Biolegend | Clone: GL-1 |
| FC: anti-mouse Gr1 | Biolegend | Clone: RB6-8C5 |
| FC: anti-mouse MHC II | Biolegend | Clone: M5/114.15.2 |
| FC: anti-mouse IL-10 | Biolegend | Clone: JES5-16E3 |
| FC: anti-mouse IFNγ | Biolegend | Clone: XMG1.2 |
| FC: anti-mouse TNFα | Biolegend | Clone: MP6-XT22 |
| FC: anti-mouse F4/80 | Biolegend | Clone: BM8 |
| FC: anti-mouse ICOS | Biolegend | Clone: 15F9 |
| FC: anti-mouse CD62L | Biolegend | Clone: MEL-14 |
| FC: anti-mouse CD25 | Biolegend | Clone: 3C7 |
| FC: anti-mouse CD40 | Biolegend | Clone: 3/23 |
| FC: anti-mouse CD69 | Biolegend | Clone: H1.2F3 |
| FC: anti-mouse IL-17A | Biolegend | Clone: TC11-18H10.1 |
| FC: anti-mouse TGFβ | Biolegend | Clone: TW7-16B4 |
| FC: anti-mouse LFA-1 | Biolegend | Clone: H155-78 |
| FC: anti-mouse LAG-3 | Biolegend | Clone: C9B7W |
| FC: anti-STAT1 Phospho (Ser727) | Biolegend | Clone: A15158B |
| FC: anti-STAT3 Phospho (Tyr705) | Biolegend | Clone: 13A3-1 |
| FC: anti-STAT6 Phospho (Tyr641) | Biolegend | Clone: A15137E |
| FC: anti-STAT5 Phospho (pY694) | BD Biosciences | Clone: 47/Stat5(pY694) |
| FC: anti-mouse T-bet | eBioscience | Clone: eBio4B10 |
| FC: anti-mouse RORγt | eBioscience | Clone: AFKJS-9 |
| FC: anti-mouse Granzyme B | eBioscience | Clone: NGZB |
| FC: anti-mouse Perforin | eBioscience | Clone: eBioOMAK-D |
| FC: anti-mouse FoxP3 | eBioscience | Clone: FJK-16s |
| FC: anti-human CD45 | Biolegend | Clone: HI30 |
| FC: anti-human CD3 | Biolegend | Clone: UCHT1 |
| FC: anti-human CD4 | Biolegend | Clone: A161A1 |
| FC: anti-human CD25 | Biolegend | Clone: BC96 |
| FC: anti-human CD69 | Biolegend | Clone: FN50 |
| FC: anti-human CD11b | Biolegend | Clone: ICRF44 |
| FC: anti-human TNFα | Biolegend | Clone: Mab11 |
| FC: anti-human IFNγ | Biolegend | Clone: 4S.B3 |
| FC: anti-human IL-10 | Biolegend | Clone: JES3-19F1 |
| FC: anti-human HLA-DR | Biolegend | Clone: L243 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Patient-derived organotypic tumor spheroids | NYU Langone Medical Center | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Mouse TNF-alpha | Cell Sciences | Cat#: CRT192C |
| Recombinant Human M-CSF Protein | R&D | Cat#: 216-MC |
| zVAD-FMK | Bachem | Cat#: N-1510 |
| Nec-1s | BioVision | Cat#: 2263 |
| GSK’963 | GlaxoSmithKline | N/A |
| RIP1i (GSK’547) | GlaxoSmithKline | N/A |
| Fludarabine | Selleckchem | S1491; CAS: 21679-14-1 |
| LJI308 | Sigma-Aldrich | SML1788; CAS: 1627709-94-7 |
| GW9662 | Sigma-Aldrich | M6191; CAS: 22978-25-2 |
| Albumin-fluorescein isothiocyanate conjugate | Sigma-Aldrich | A9771; MDL: MFCD00282182 |
| Rodent Chow (Purina chow 5001) formulated with inhibitor | Research Diets | Cat#: C16102601i |
| QVD-Oph | Millipore Sigma | 551476 |
| Recombinant human TNFα | R&D systems | 210-TA-100 |
| FLAG-RIPK1 1-294 (C34A C127A C233A C240A) | Genscript | N/A |
| fluorescently-labeled ligand (14-(2-{[3-({2-{[4-(cyanomethyl)phenyl]amino}-6-[(5-cyclopropyl-1H-pyrazol-3-yl)amino]-4-pyrimidinyl}amino) propyl]amino}-2-oxoethyl)-16,16,18,18-tetramethyl-6,7,7a,8a,9,10,16,18-octahydrobenzo [2’,3’]indolizino[8’,7’:5′,6′]pyrano [3′,2′:3,4]pyrido[1,2-a]indol-5-ium-2-sulfonate) | GSK | N/A |
| Critical Commercial Assays | ||
| MILLIPLEX Human Cytokine 41-Plex kit | MilliporeSigma | Cat#: HCYTMAG-60K-PX41 |
| Reaction Biology Kinase Assay | Reaction Biology Corp | Wild type kinase panel |
| Cell Titre-Glo Luminescent Cell Viability Assay | Promega | G7573 |
| Deposited Data | ||
| X-ray crystalllography data, maps and structure coordinates: PDB | This paper | 6HHO https://www.rcsb.org/ |
| Raw and analyzed RNAseq data | This paper | GEO: GSE111626 |
| Experimental Models: Cell Lines | ||
| Mouse: KPC Pancreatic Cancer Cell line | Laboratory of Dave Tuveson | Cell line: FC1242 |
| L929 (Mouse L-cells NCTC 929) | ATCC | ATCC CCL-1 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 (H-2Kb) | The Jackson Laboratory | JAX: 000664 |
| Mouse: C57BL/6-Tg(TcraTcrb)1100Mjb/J (OT-I) | The Jackson Laboratory | JAX: 003831 |
| Mouse: B6.Cg-Tg(TcraTcrb)425Cbn/J (OT-II) | The Jackson Laboratory | JAX: 004194 |
| Mouse: B6.129S(Cg)-Stat1tm1Dlv/J | The Jackson Laboratory | JAX: 012606 |
| Mouse: B6.129S7-Rag1tm1Mom/J | The Jackson Laboratory | JAX: 002216 |
| Mouse: B6.Cg-Foxn1nu/J (B6 nude) | The Jackson Laboratory | JAX: 000819 |
| Mouse: Ripk3−/− | Genentech | N/A |
| Mouse: RIP1 KD/KI | GlaxoSmithKline | N/A |
| Mouse: B6. LSL-KrasG12D and p48Cre (KC ) | Laboratory of Dafna Bar-Sagi | N/A |
| Oligonucleotides | ||
| Primers for qPCR, see Table S5 | This paper | N/A |
| Recombinant DNA | ||
| RIP1 shRNA Plasmid: SHCLNG-NM_009068 | Sigma-Aldrich | TRCN0000022466 |
| RIP1 shRNA Plasmid: SHCLNG-NM_009068 | Sigma-Aldrich | TRCN0000278133 |
| Software and Algorithms | ||
| GraphPad Prism 7 | GraphPad Software | |
| Analyst multimode reader | Molecular Devices | 0200 |
| iMOSFLM | CCP4 | N/A |
| Aimless | CCP4 | 0.512 |
| ctruncate | CCP4 | 1.16.9 |
| unique | CCP4 | 6.5.013 |
| freerflag | CCP4 | 1.2 |
| pointless | CCP4 | 1.9.16 |
| Other | ||
RIP1 promotes tolerogenic macrophage differentiation via suppression of STAT1 in cancer.
GSK’547 (RIP1i) is a mono-selective kinase inhibitor that robustly targets RIP1 in vivo.
RIP1i protects against pancreas cancer via macrophage-mediated Th1/Th17 and CTL activation.
RIP1i synergizes with checkpoint- and costimulatory receptor-based immunotherapies.
Significance.
The drivers of innate and adaptive immune suppression in pancreatic carcinoma are incompletely understood. We show that RIP1 kinase promotes tolerogenic macrophage differentiation in the pancreatic cancer tumor microenvironment. We developed a small molecule RIP1 inhibitor with high specficicity, potency, and in vivo exposure. RIP1 inhibition resulted in immunogenic macrophage differentiation in pancreatic cancer leading to adaptive immune activation and tumor protection. RIP1 inhibition also enabled efficacy for PD-1-based immunotherapy and enhanced the effects of ICOS agonism. Collectively, our work describes a critical regulator of immune tolerance in pancreatic carcinoma and supports inhibition of RIP1 as a potential oncologic therapy.
Acknowledgements
We thank the NYU Langone Health Genome Technology Center (GTC) for expert library preparation and sequencing; the GTC is partially supported by the Cancer Center Support Grant P30CA016087 at the Laura and Isaac Perlmutter Cancer Center. We thank Sara Brett and Meixia Bi at GSK for providing ICOS agonist antibody. We thank Michael Ouellette and Julie Cox for help with L929 and FP binding assays, Helen Sun for help with bioanalysis for PK studies, and Carol Capriotti, Michelle Schaeffer, Bonnie Hoffman, Sze-Ling Ng and Liang Huang for help with in vivo studies.
Grant Support: This work was supported by NIH grants T32GM007308 (EK and JAKR), CA168611 (GM), CA203105 (GM), CA215471 (GM), CA19311 (GM), and DK106025 (GM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
JMM, AMB, MR, JL, PAH, MM, JHT, JEM, and JB are employees and shareholders of GSK.
JMM and JB are inventors on a patent application owned by GSK.
NYU School of Medicine signed a Research Contract Agreement with GSK to fund research in GM’s lab.
GM and DC received a one-time honorarium payment for giving a presentation at GSK.
GM is a co-founder and on the Scientific Advisory Board of NYBO Therapeutics.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andren-Sandberg A, Dervenis C, and Lowenfels B (1997). Etiologic links between chronic pancreatitis and pancreatic cancer. Scand J Gastroenterol 32, 97–103. [DOI] [PubMed] [Google Scholar]
- Barber MD, Powell JJ, Lynch SF, Fearon KC, and Ross JA (2000). A polymorphism of the interleukin-1 beta gene influences survival in pancreatic cancer. British journal of cancer 83, 1443–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, and Vonderheide RH (2012). Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 21, 822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, et al. (2011). CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 331, 1612–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger SB, Harris P, Nagilla R, Kasparcova V, Hoffman S, Swift B, Dare L, Schaeffer M, Capriotti C, Ouellette M, et al. (2015). Characterization of GSK’963: a structurally distinct, potent and selective inhibitor of RIP1 kinase. Cell death discovery 1, 15009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Xiao-Zhong G, and Qi XS (2017). Clinical Outcomes of Specific Immunotherapy in Advanced Pancreatic Cancer: A Systematic Review and Meta-Analysis. Journal of immunology research 2017, 8282391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, and Vonderheide RH (2007). Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer research 67, 9518–9527. [DOI] [PubMed] [Google Scholar]
- Croft M (2009). The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol 9, 271–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui R, Yue W, Lattime EC, Stein MN, Xu Q, and Tan XL (2016). Targeting tumor-associated macrophages to combat pancreatic cancer. Oncotarget 7, 50735–50754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley D, Mani VR, Mohan N, Akkad N, Ochi A, Heindel DW, Lee KB, Zambirinis CP, Pandian GSB, Savadkar S, et al. (2017a). Dectin 1 activation on macrophages by galectin 9 promotes pancreatic carcinoma and peritumoral immune tolerance. Nat Med 23, 556–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley D, Mani VR, Mohan N, Akkad N, Pandian G, Savadkar S, Lee KB, Torres-Hernandez A, Aykut B, Diskin B, et al. (2017b). NLRP3 signaling drives macrophage-induced adaptive immune suppression in pancreatic carcinoma. J Exp Med 214, 1711–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley D, Zambirinis CP, Seifert L, Akkad N, Mohan N, Werba G, Barilla R, Torres-Hernandez A, Hundeyin M, Mani VR, et al. (2016). gammadelta T Cells Support Pancreatic Oncogenesis by Restraining alphabeta T Cell Activation. Cell 166, 1485–1499 e1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, Braga M, Di Carlo V, Doglioni C, and Protti MP (2011). Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. The Journal of experimental medicine 208, 469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, et al. (2013). Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A 110, 20212–20217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, Cain K, MacFarlane M, Hacker G, and Leverkus M (2011). cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell 43, 449–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukunaga A, Miyamoto M, Cho Y, Murakami S, Kawarada Y, Oshikiri T, Kato K, Kurokawa T, Suzuoki M, Nakakubo Y, et al. (2004). CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas 28, e26–31. [DOI] [PubMed] [Google Scholar]
- Gadaleta E, Cutts RJ, Kelly GP, Crnogorac-Jurcevic T, Kocher HM, Lemoine NR, and Chelala C (2011). A global insight into a cancer transcriptional space using pancreatic data: importance, findings and flaws. Nucleic Acids Res 39, 7900–7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco SH, Mahmood SK, Vahle AK, Ochi A, Batel J, Deutsch M, Barilla R, Seifert L, Pachter HL, Daley D, et al. (2016). Mincle suppresses Toll-like receptor 4 activation. Journal of leukocyte biology 100, 185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Li L, Qiu S, Lu Q, Pan Q, Gu Y, Luo J, and Hu X (2007). Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol Cancer Ther 6, 1641–1649. [DOI] [PubMed] [Google Scholar]
- Harris PA, King BW, Bandyopadhyay D, Berger SB, Campobasso N, Capriotti CA, Cox JA, Dare L, Dong X, Finger JN, et al. (2016). DNA-Encoded Library Screening Identifies Benzo[b][1,4]oxazepin-4-ones as Highly Potent and Monoselective Receptor Interacting Protein 1 Kinase Inhibitors. J Med Chem 59, 2163–2178. [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al. (2003). Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450. [DOI] [PubMed] [Google Scholar]
- Hiraoka N, Onozato K, Kosuge T, and Hirohashi S (2006). Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res 12, 5423–5434. [DOI] [PubMed] [Google Scholar]
- Jang JE, Hajdu CH, Liot C, Miller G, Dustin ML, and Bar-Sagi D (2017). Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer. Cell reports 20, 558–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins RW, Aref AR, Lizotte PH, Ivanova E, Stinson S, Zhou CW, Bowden M, Deng J, Liu H, Miao D, et al. (2017). Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, Nywening TM, Hawkins WG, Shapiro IM, Weaver DT, et al. (2016). Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med 22, 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Daley-Bauer LP, Thapa RJ, Mandal P, Berger SB, Huang C, Sundararajan A, Guo H, Roback L, Speck SH, et al. (2014). RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc Natl Acad Sci U S A 111, 7753–7758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Upton JW, and Mocarski ES (2008). Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J Immunol 181, 6427–6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwada Y, Sasaki T, Morinaka K, Kitadai Y, Mukaida N, and Chayama K (2003). Potential involvement of IL-8 and its receptors in the invasiveness of pancreatic cancer cells. International journal of oncology 22, 765–771. [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liu X, Zhou M, Mei L, Ruan J, Hu Q, Peng J, Su H, Liao H, Liu S, Liu W, et al. (2016). Key roles of necroptotic factors in promoting tumor growth. Oncotarget 7, 22219–22233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, Collins NB, Bi K, LaFleur MW, Juneja VR, et al. (2017). In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547, 413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister F, Bailey JM, Alsina J, Nirschl CJ, Sharma R, Fan H, Rattigan Y, Roeser JC, Lankapalli RH, Zhang H, et al. (2014). Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell 25, 621–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meylan E, Burns K, Hofmann K, Blancheteau V, Martinon F, Kelliher M, and Tschopp J (2004). RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat Immunol 5, 503–507. [DOI] [PubMed] [Google Scholar]
- Miller G, Lahrs S, and Dematteo RP (2003). Overexpression of interleukin-12 enables dendritic cells to activate NK cells and confer systemic antitumor immunity. FASEB J 17, 728–730. [DOI] [PubMed] [Google Scholar]
- Miller G, Lahrs S, Pillarisetty VG, Shah AB, and DeMatteo RP (2002). Adenovirus infection enhances dendritic cell immunostimulatory properties and induces natural killer and T-cell-mediated tumor protection. Cancer research 62, 5260–5266. [PubMed] [Google Scholar]
- Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, Toriola AT, Nieman RK, Worley LA, Yano M, et al. (2016). Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol 17, 651–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochi A, Nguyen AH, Bedrosian AS, Mushlin HM, Zarbakhsh S, Barilla R, Zambirinis CP, Fallon NC, Rehman A, Pylayeva-Gupta Y, et al. (2012). MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. J Exp Med 209, 1671–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofengeim D, and Yuan J (2013). Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol 14, 727–736. [DOI] [PubMed] [Google Scholar]
- Pickup MW, Owens P, Gorska AE, Chytil A, Ye F, Shi C, Weaver VM, Kalluri R, Moses HL, and Novitskiy SV (2017). Development of Aggressive Pancreatic Ductal Adenocarcinomas Depends on Granulocyte Colony Stimulating Factor Secretion in Carcinoma Cells. Cancer immunology research 5, 718–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole JA, Gleason AM, Bauer C, West WW, Alexis N, Reynolds SJ, Romberger DJ, and Kielian T (2012). alphabeta T cells and a mixed Th1/Th17 response are important in organic dust-induced airway disease. Ann Allergy Asthma Immunol 109, 266–273 e262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, and Hingorani SR (2012). Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 21, 418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, and Bar-Sagi D (2012). Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 21, 836–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan A, Pearson GW, Chrestensen CA, Sturgill TW, and Cobb MH (2006). The MAP kinase ERK5 binds to and phosphorylates p90 RSK. Arch Biochem Biophys 449, 8–16. [DOI] [PubMed] [Google Scholar]
- Riaz T, Sollid LM, Olsen I, and de Souza GA (2016). Quantitative Proteomics of Gut-Derived Th1 and Th1/Th17 Clones Reveal the Presence of CD28+ NKG2D-Th1 Cytotoxic CD4+ T cells. Mol Cell Proteomics 15, 1007–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roderick JE, Hermance N, Zelic M, Simmons MJ, Polykratis A, Pasparakis M, and Kelliher MA (2014). Hematopoietic RIPK1 deficiency results in bone marrow failure caused by apoptosis and RIPK3-mediated necroptosis. Proc Natl Acad Sci U S A 111, 14436–14441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskoski R Jr. (2016). Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol Res 103, 26–48. [DOI] [PubMed] [Google Scholar]
- Schmidt SV, Seibert S, Walch-Ruckheim B, Vicinus B, Kamionka EM, Pahne-Zeppenfeld J, Solomayer EF, Kim YJ, Bohle RM, and Smola S (2015). RIPK3 expression in cervical cancer cells is required for PolyIC-induced necroptosis, IL-1alpha release, and efficient paracrine dendritic cell activation. Oncotarget 6, 8635–8647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert L, Werba G, Tiwari S, Giao Ly NN, Alothman S, Alqunaibit D, Avanzi A, Barilla R, Daley D, Greco SH, et al. (2016a). The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature 532, 245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert L, Werba G, Tiwari S, Giao Ly NN, Nguy S, Alothman S, Alqunaibit D, Avanzi A, Daley D, Barilla R, et al. (2016b). Radiation Therapy Induces Macrophages to Suppress T-Cell Responses Against Pancreatic Tumors in Mice. Gastroenterology 150, 1659–1672 e1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shutinoski B, Alturki NA, Rijal D, Bertin J, Gough PJ, Schlossmacher MG, and Sad S (2016). K45A mutation of RIPK1 results in poor necroptosis and cytokine signaling in macrophages, which impacts inflammatory responses in vivo. Cell Death Differ 23, 1628–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart OS, Womack TO, Flensburg C, Keller P, Paciorek W, Sharff A, Vonrhein C, and Bricogne G (2012). Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr D Biol Crystallogr 68, 368–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, Zachariou A, Lopez J, MacFarlane M, Cain K, et al. (2011). The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell 43, 432–448. [DOI] [PubMed] [Google Scholar]
- Wegner KW, Saleh D, and Degterev A (2017). Complex Pathologic Roles of RIPK1 and RIPK3: Moving Beyond Necroptosis. Trends Pharmacol Sci 38, 202–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong WW, Vince JE, Lalaoui N, Lawlor KE, Chau D, Bankovacki A, Anderton H, Metcalf D, O’Reilly L, Jost PJ, et al. (2014). cIAPs and XIAP regulate myelopoiesis through cytokine production in an RIPK1- and RIPK3-dependent manner. Blood 123, 2562–2572. [DOI] [PubMed] [Google Scholar]
- Yadav D, and Lowenfels AB (2013). The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 144, 1252–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatim N, Jusforgues-Saklani H, Orozco S, Schulz O, Barreira da Silva R, Reis e Sousa C, Green DR, Oberst A, and Albert ML (2015). RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8(+) T cells. Science 350, 328–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambirinis CP, Levie E, Nguy S, Avanzi A, Barilla R, Xu Y, Seifert L, Daley D, Greco SH, Deutsch M, et al. (2015). TLR9 ligation in pancreatic stellate cells promotes tumorigenesis. J Exp Med 212, 2077–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Xue J, Jaffee EM, and Habtezion A (2013). Role of immune cells and immune-based therapies in pancreatitis and pancreatic ductal adenocarcinoma. Gastroenterology 144, 1230–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNAseq data has been submitted to the GEO repository, and the accession number is GSE111626. The PDB report has been deposited in RCSB Protein Data Bank under ID code 6HHO.