

Abstract
Hybridization drives rapid speciation by shaping novel genotypic and phenotypic profiles. Genomic incompatibility and transcriptome shock have been observed in hybrids, although this is rarer in animals than in plants. Using the newly sequenced genomes of the blunt snout bream (Megalobrama amblycephala [BSB]) and the topmouth culter (Culter alburnus [TC]), we focused on the sequence variation and gene expression changes in the reciprocal intergeneric hybrid lineages (F1–F3) of BSB × TC. A genome-wide transcriptional analysis identified 145–974 expressed recombinant genes in the successive generations of hybrid fish, suggesting the rapid emergence of allelic variation following hybridization. Some gradual changes of gene expression with additive and dominance effects and various cis and trans regulations were observed from F1 to F3 in the two hybrid lineages. These asymmetric patterns of gene expression represent the alternative strategies for counteracting deleterious effects of the subgenomes and improving adaptability of novel hybrids. Furthermore, we identified positive selection and additive expression patterns in transforming growth factor, beta 1b (tgfb1b), which may account for the morphological variations of the pharyngeal jaw in the two hybrid lineages. Our current findings provide insights into the evolution of vertebrate genomes immediately following hybridization.
Megalobrama amblycephala (blunt snout bream [BSB]; 2n = 2x = 48) and Culter alburnus (topmouth culter [TC]; 2n = 2x = 48), members of the family Cyprinidae, are economically important freshwater fish (Chen 1998; Zhou et al. 2008). The BSB and the TC have distinct feeding habits (the BSB is herbivorous, whereas the TC is carnivorous) and shapes (the BSB has a higher dorsal fin and a shorter body than the TC). Furthermore, the progenies of intergeneric reciprocal crosses between these fishes (BSB [♀] × TC [♂] and TC [♀] × BSB [♂]) show different degrees of phenotypic variation. For example, the hybrid lineages of reciprocal crosses had intermediate shapes between those of their parents (Xiao et al. 2014). Hybrid lineages of BSB and TC also show many physiological advantages over their parents, such as faster growth rates, higher hypoxia tolerance, and greater disease resistance (Xiao et al. 2014; Li et al. 2018), as observed in other hybrid fishes, including tilapia hybrids (Oreochromis mossambicus × O. aureus) (Cnaani et al. 2003), catfish hybrids (Ictalurus punctatus × I. furcatus) (Wolters et al. 1996), and striped bass hybrids (Morone chrysops × M. saxatilis) (Li et al. 2004). Similar to the BSB, the hybrid lineages of the reciprocal crosses were all herbivorous. In fish cultivation, breeding varieties with herbivorous habits helps reduce the raising costs.
Variations in gene expression, including differential homoeolog gene expression and homoeolog silencing (nonexpression of one homoeolog), have been observed in some allopolyploid plants and a few animals (Adams 2007; Liu et al. 2016). These changes derived from hybridization and polyploidization lead to bias in homoeolog expression and expression dominance (Rapp et al. 2009; Yoo et al. 2013). Such asymmetric expression is related to the imbalanced expression of homoeologous genes in polyploids caused by cis-regulatory changes, which affect transcription initiation, transcription rates, and/or transcript stability in homoeologous genes, whereas trans-regulatory changes affect the efficiency of cis-trans interaction by modifying the activity or expression of transcription factors (Wittkopp et al. 2004; Maheshwari and Barbash 2012). The co-evolution always occurred in the interaction of cis-regulatory sequences with trans-regulatory factors, causing divergence of gene expression and changes of biochemical processes after hybridization (McManus et al. 2010). The conservation of cis-regulatory activity is strongly correlated with the ratio of the number of nonsynonymous substitutions per nonsynonymous site to the number of synonymous substitutions per synonymous site (Ka/Ks) (Emerson et al. 2010; McManus et al. 2010). Furthermore, rapid genomic changes related to DNA copy number, expressed sequences, and retrotransposons are always found in intergeneric hybrids and allopolyploids.
Genome-wide allelic expression changes have rarely been explored in successive generations of hybrid vertebrates (Xiao et al. 2016). In this study, we assembled accurate and nearly complete genomes of BSB and TC. Combining genome sequences with RNA-seq data from the reciprocal cross hybrids and their parents, we investigated the expressed recombinant transcripts related to alleles of the two subgenomes and examined their expression patterns, including the asymmetric expression of the alleles, to elucidate the transcriptional regulation in these two successive generations of allodiploid lineages. Finally, the joint effects of allelic recombination and asymmetric expression of the early hybrid generations provided novel insights into the evolution of vertebrate genomes immediately following hybridization.
Results
Procedure for generating hybrid lineages
Blunt snout bream (M. amblycephala [BSB]; 2n = 48) and topmouth culter (C. alburnus [TC]; 2n = 48), which reached sexual maturity in natural waters of the Yangtze River in China, were collected for the hybrid experiments. The procedure for generating the hybrid lineages investigated in this study is shown in Figure 1. BSB and TC were used as the parents in the reciprocal cross hybrids to form two types of hybrid lineages. In the first cross group, a BSB (♀) × TC (♂) cross was performed to produce F1 hybrids (BT, 2n = 48). Then, the intercrossing within F1 females and males produce F2 hybrids, which generated F3, forming the hybrid lineage (F1–F3). In the second cross group, a TC (♀) × BSB (♂) cross was performed to produce F1 hybrids (TB, 2n = 48). Then, the intercrossing within F1 females and males produce F2 hybrids, which generated F3, establishing the hybrid lineage (F1–F3) (Supplemental Methods).
Figure 1.

Procedure for generating the reciprocal cross hybrids of BSB and TC. (A) BSB. (B) TC. (C) BTF1 of BSB (♀) × TC (♂). (D) BTF2 produced by intercrossing within BTF1. (E) BTF3 obtained by intercrossing within BTF2. (F) TBF1 of TC (♀) × BSB (♂). (G) BTF2 produced by intercrossing within TBF1. (H) TBF3 obtained by intercrossing within TBF2. The images of the fish hybrids are only representative of certain progeny. (Scale bar) 3 cm.
Assembly and annotation of the BSB and TC genomes
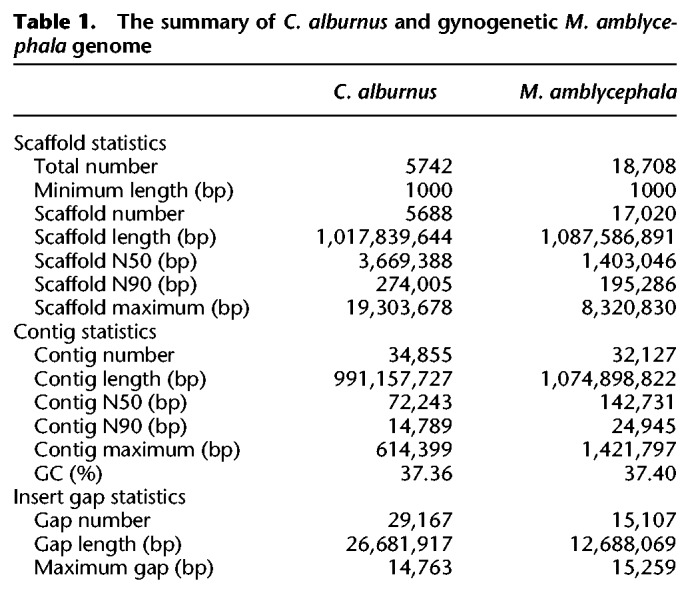
We generated ∼192.54 Gb (180.95 Gb of Illumina plus 11.60 Gb of Pacific Biosciences [PacBio] reads) and 176.33 Gb (171.05 Gb of Illumina plus 5.28 Gb of PacBio reads) of sequencing data for BSB and TC, respectively (Supplemental Tables S1, S2; Supplemental Methods). The resulting assemblies were 1.09 Gb (BSB) and 1.02 Gb (TC) (Table 1). These sizes were very similar to the genome sizes estimated through k-mer analysis and flow cytometry measures (Supplemental Fig. S1; Supplemental Methods). The lengths of contig N50/scaffold N50 were 142.73 kb/1.40 Mb and 72.24 kb/3.67 Mb for BSB and TC, respectively (Table 1). Assessments of genome assembly quality showed that the assemblies covered 99.50%/98.12% unigenes (41,825/41,614 for the BSB; 151,448/148,604 for the TC) derived from RNA-seq across multiple tissues in BSB and TC and had few missing conserved core eukaryotic genes (CEGs) and benchmarking sets of universal single-copy orthologs (BUSCOs) (Supplemental Table S3; Supplemental Methods; Simão et al. 2015). In total, 983.7 Mb (93.86%) of sequences of the BSB assembly were anchored to 24 chromosomal groups by mapping Hi-C contact data (Supplemental Fig. S2; Supplemental Tables S4, S5); for the TC, 760.1 Mb (74.68%) of sequences were anchored to the 24 pseudochromosomes using 6377 single-nucleotide polymorphism (SNP) markers (Supplemental Table S6; Supplemental Methods). Then, based on de novo, homolog-based, and RNA-seq approaches, 29,994 and 30,443 protein-coding genes were identified in BSB and TC assemblies, respectively (Supplemental Tables S7, S8). Based on the 27 Gb of RNA-seq data, 2163 and 1536 noncoding RNA genes were predicted in BSB and TC (Supplemental Table S9). In addition, no obvious difference in coding sequence (CDS) number, CDS length, intron length, or gene length was detected between BSB, TC, zebrafish (Danio rerio), and grass carp (Ctenopharyngodon idellus) (Supplemental Fig. S3; Supplemental Methods).
Table 1.
The summary of C. alburnus and gynogenetic M. amblycephala genome
Genome evolution
A phylogenetic tree was constructed using 796 single-copy genes from 10 species (Fig. 2A). The results indicated that the ancestral lineage of the BSB and the TC diverged from that of the grass carp ∼27.35 million years ago (MYA) (Fig. 2A; Supplemental Table S10; Supplemental Methods). The distribution of Ks statistics for BSB/TC orthologs contains a distinct peak (Ks = 0.0265) that corresponded to a divergence time of 12.74 MYA (Fig. 2A,B; Supplemental Methods). Expansion and contraction analysis of gene clusters showed that 403 and 318 expansion events and 433 and 493 contraction events occurred in the TC and BSB, respectively (Fig. 2A). Annotations of these gene clusters suggested that most of the contraction events were associated with the biological process category of homophilic cell adhesion via plasma membrane adhesion molecules (GO:0007156), whereas most of the expansion events were associated with the G-protein-coupled receptor signaling pathway (GO:0007186). Among the expansion and contraction events, there were also contraction events associated with nine biological process categories related to bone development and skeletal muscle function (Supplemental Table S11), and 69 genes in expansion events were related to the detection of chemical stimuli in olfactory perception (GO:0050911). These factors might contribute to the diversity of bone morphology and feeding habits between BSB and TC. Based on 20,130 orthologous genes found in BSB and TC, 347 genes were identified as positively selected genes (PSGs) (Ka/Ks > 1). Six PSGs were detected among 89 genes associated with the immune response (GO: 0006955), and two PSGs were detected among 11 genes related to regulation of the vascular endothelial growth factor receptor signaling pathway (GO: 0030947), implying that a rapid diversification of growth development and immune response might contribute to speciation. Multiple genome alignments showed that the genomes of the BSB and TC had a high degree of collinearity with that of the zebrafish, despite several types of structural variation existing between them (Fig. 2C; Supplemental Fig. S4).
Figure 2.

Evolution of the BSB and the TC. (A) A phylogenetic tree was constructed from 10 species, including five cyprinids. The time of divergence and the expansion and contraction of gene clusters are described with a maximum-likelihood tree. The number of expansion events in each gene clusters is indicated in red, and contraction events are indicated in blue. The photographs of Xiphophorus maculatus were obtained from Schartl et al. (2013). (B) The distribution of the synonymous substitution rates (Ks) of orthologous genes for interspecies comparisons was identified by 4dTv analysis. The peaks of the Ks distributions of orthologs indicate speciation events. (C) A comparative analysis of the BSB and TC genomes was performed. Maps of 24 BSB chromosomes and of 24 TC chromosomes based on the positions of 20,130 orthologous gene pairs showed highly conserved synteny between them. The A track represents gene density (window size = 100 kb), and the B track represents repeat content within a 500-kb sliding window.
Detection of allelic recombinant genes by Illumina, PacBio, and Sanger sequencing
To detect the allelic recombinant (AR) genes related to alleles of subgenomes in the hybrid lineages, we used a combination of Illumina, PacBio, and Sanger sequencing based on their respective analysis pipelines (Supplemental Methods). After low-quality reads were filtered out, 960.11 million clean RNA-seq reads (273.12 Gb) were obtained from 36 samples (Supplemental Table S12), including the two hybrid lineages (BTF1-BTF3 and TBF1-TBF3), as well as their inbred parents. We detected 348 to 8940 recombinant reads (0.0033%–0.0624% in all uniquely mapped reads) in each of the hybrid transcriptomes (Supplemental Table S13; Supplemental Methods). Although an average of only 0.0166% recombinant reads was detected, a distribution of 145 to 974 AR genes was found in all transcriptomes, in which the average number of recombinant reads per gene was 5.93 (Supplemental Fig. S5). To validate the AR events from Illumina data, 51,351 (76.14%) of 67,446 reads, which were detected as AR reads in the former analyses, were further confirmed by using the other analysis pipeline (Supplemental Table S13; Supplemental Fig. S6A). A gradually increasing trend of number of AR events was found from F1 to F3 in TB lineage. But this trend was not observed in BT lineage (Supplemental Fig. S6B).
To further validate the AR events in 15 genes of F1 and 19 genes of F3 in the two hybrid lineages (Supplemental Table S14), which were identified as AR genes from the Illumina data, we obtained their sequences at the genomic DNA level by using Sanger sequencing. The sequences of AR genes were confirmed in 14 genes in BTF1, 14 genes in TBF1, 14 genes in BTF3, and 13 gens in TBF3 (Supplemental Figs. S7–S22; Supplemental Methods). The long read length of the PacBio sequencing helped us obtain full-length transcripts with no assembly (Supplemental Table S15; Supplemental Methods). Most of the AR genes (48 of 49 in BTF3 and 50 of 52 in TBF3) were consistent with the Illumina data (Supplemental Fig. S7). Combined with the data from F1 and F3 of the two hybrid lineages by Sanger and Illumina sequencing methods, after ruling out five genes, 10 of 15 genes were identified as the AR genes in F1 and F3 of the two hybrid lineages (Supplemental Figs. S7–S22) and were used to further analyze the AR patterns.
It is possible that the AR regions are the same or slightly different in a certain AR gene, depending on different individuals because of the bias of AR read coverage. We classified AR events with the overlapped AR regions as the same AR pattern. Two AR patterns (I and II) were formed based on AR regions in which the 5′ upstream and 3′ downstream sequences were from the sequences derived from one of the parents (Supplemental Fig. S6C). Focusing on global AR patterns in the two hybrid lineages, a gradually increasing trend of number of AR patterns was found from F1 to F3 in the TB lineage, whereas no such trend was observed in the BT lineage (Supplemental Fig. S6D). We focused on the analyses on the 10 AR genes, which were confirmed by Illumina and Sanger sequencing methods in both F1 and F3 of the two hybrid lineages. The distribution of the two AR patterns among the 10 AR genes (Supplemental Fig. S6E) was presented, in which two AR genes (gene order: No. 1–2) showed pattern I in each generation in the two hybrid lineages, and pattern I + II in BTF2-BTF3 and TBF1-TBF3, suggesting the increasing trend of pattern number in BT lineage. One AR gene (gene order: No. 3) showed pattern II in each generation in the two hybrid lineages, and pattern I + II in BTF2 and BTF3, indicating the increasing trend of pattern number in BT lineage. Three AR genes (gene order: No. 4–6) showed pattern I or pattern II in each generation in BT lineage or TB lineage. Three AR genes (gene order: No. 7–9) presented pattern I or pattern II in each generation in BT lineage or TB lineage, and pattern I + II in BTF2, BTF3, and TBF3, suggesting the increasing trend of pattern number in TB lineage (gene order: No. 7) and in BT lineage (gene order: No. 8–9). One AR gene (gene order: No. 10) showed pattern II in each generation in BT lineage, but no regular model in BT lineage, suggesting that the AR events vary depending on different individuals with different mechanisms involved in the allelic recombination.
Expression dominance
It is reasonable to speculate that the merger of different genomes leads to dominance and additive effects in the hybrid offspring (Reif et al. 2007). The mapped reads (68.10 million in BSB and 64.34 million in TC) of the two inbred parents were aligned to the respective genomes (Supplemental Table S16; Supplemental Methods). The expression values in the hybrid were calculated from a combined genome of two inbred parents (Supplemental Methods). Analyses of pairwise alterations of global expression level showed a high degree of expression similarity in comparison to the two inbred parent genomes. This result also revealed that tissue-specific expression samples clustered with each other, yet the hybrid offspring clustered with TC, except for TBF2 (Supplemental Fig. S23). The distribution of a few silent genes in the hybrids was shown in 20,130 orthologous gene pairs between BSB and TC (Supplemental Table S17; Supplemental Methods).
To assess the changes at the global expression level in hybrids, we performed differential expression analysis on the 12,322 expressed orthologous gene pairs in the liver (Supplemental Methods). The results showed 12.14%–23.78% and 6.63%–16.13% differentially expressed genes (DEGs) in BT compared with BSB and TC, respectively (Supplemental Fig. S24). Meanwhile, analyses of TB showed 8.61%–17.17% DEGs compared with the BSB, whereas 7.08%–10.80% were found compared with the TC (Supplemental Fig. S24). The smaller number of DEGs between hybrids and TCs revealed the potential dominance of TC expression in both reciprocal cross hybrids, except for TBF2 (Supplemental Fig. S24). In addition, up-regulated expressed genes were increasingly observed in BTF1-BTF3, TBF1, and BTF3 (Supplemental Fig. S24). To clarify the process of gene expression changes, we determined the average parental expression (APE) based on the expression of the two parents and compared it with expression in the hybrids, revealing increasing trends in up-regulated expressed genes from F1 to F3, whereas an increasing trend in down-regulated expressed genes was observed from BTF1 to BTF3 (Fig. 3A; Supplemental Methods). However, there were more DEGs (742, 6.02%) in TBF3 than in TBF1 (697, 5.66%) and TBF2 (609, 4.94%) (Fig. 3A). Focusing on conservation of epigenetic regulation in successive hybrid generation, the same expression in liver tissue of BTF1-BTF3 was observed in 10,265 (83.31%) genes, including 20 up-regulated genes, 49 down-regulated genes, and 10,196 (82.75%) non-DEGs. Meanwhile, 11 up-regulated genes, 59 down-regulated genes, and 10,710 non-DEGs were detected in the liver of TBF1-TBF3 (Supplemental Fig. S25). In addition, six of 99 DEGs were shared in the liver of BT and TB (Fig. 3B,C).
Figure 3.

Changes in global expression levels between hybrids and their parents. (A) Differential expression analysis was performed between APE and expression in hybrids. The black line indicates that the genes related to APE were ordered by their normalized, standardized expression intensity. Orange scatter points represent up-regulated expression in hybrids, and green scatter points represent down-regulated expression in hybrids. Gradual increases and decreases in the number of DEGs were observed from F1 to F3. (B) The distribution of shared DEGs was observed in the liver of reciprocal cross hybrids in a differential expression analysis between APE and expression in hybrids. (C) Seven shared DEGs were observed between BT and TB; these genes were all down-regulated. (D) Module of 12 expression patterns represents the change in gene expression state between the two inbred parents and their hybrid offspring. (E) Distribution of expression divergence of each parent relative to the hybrid offspring in three generations of BT and TB, with the TC comparison on the x-axis, the BSB comparison on the y-axis, and colors corresponding to pattern I to pattern XII.
To better identify expression dominance, we established 12 patterns as described in Rapp et al. (2009). A greater number of “TC expression dominance” genes (II and XI) than “BSB expression dominance” genes (IV and IX) were observed in three generations of BT (1559 in BTF1, 1651 in BTF2, and 1095 in BTF3) and TB (1248 in F1, 1428 in TBF2, and 1382 in TBF3) (P = 0.0026) (Fig. 3D,E; Supplemental Fig. S26; Supplemental Methods), revealing the potential TC expression dominance in BT and TB. High ratios of “TC expression dominance” in up-regulated genes and “BSB expression dominance” in down-regulated genes were observed based on more genes in patterns II (75.89%) and IX (58.24%) than in XI (24.11%) and IV (41.76%) in BT and TB (Supplemental Fig. S26). Meanwhile, some gradual decreasing trends of “additive” (I and XII) and “expression dominance” (II, XI, IV, and IX) genes and an increasing trend of “Transgressive up-/down-regulation” (III, VII, X, V, VI, and VIII) genes were found from F1 to F3, revealing a gradual weakening of parental effect in hybrids.
Expression divergence and expression bias
To investigate the coregulation of alleles derived from two subgenomes in the hybrids, 9753 orthologous genes were selected through detection of 103,190 species-specific SNPs, and the distribution of species-specific SNPs in each gene was shown (Supplemental Fig. S27; Supplemental Methods). After assessing the BSB and TC allelic expression, the seven genes with TC allelic silencing were shared in the liver of TBF1-TBF3, and eight were shared in BTF1-BTF3 (Supplemental Fig. S28). A cluster analysis of allelic expression data was performed, revealing the close relationship in BSB and TC allelic expression of the hybrids, yet a clear separation was detected in allelic expression of the liver of BTF3 (Supplemental Fig. S29).
Focusing on the direction and magnitude of allelic expression, the log2 (TC/BSB) values in hybrids were calculated based on the sums of the depth of BSB and TC allelic reads (800.12 million clean reads) in all hybrids. The values of BSB and TC allelic expression gradually approached each other from F1 to F3 in both BT and TB (P < 0.01) based on |log2 (TC/BSB)| values (Fig. 4A). Meanwhile, more genes related to overall BSB allelic expression bias [log2 (TC/BSB) < −1] were found than those related to overall TC allelic expression bias [log2 (TC/BSB) > 1] (BT: 304 vs. 165 in F1; 265 vs. 183 in F2; 189 vs. 177 in F3; TB: 454 vs. 185 in F1; 266 vs. 187 in F2), except in TBF3 (154 vs. 157) (Supplemental Fig. S30). The similar phenomenon was detected in the liver tissues based on potential genes of BSB/TC allelic expression bias (Supplemental Table S18). However, the clear BSB allelic expression bias gradually weakened from F1 to F3 in both BT and TB (Supplemental Fig. S31). In addition, a similar number of BSB and TC allelic expression-biased genes (215 vs. 235 in the muscle of BTF3; 162 vs. 168 in the gonad of BTF3; TB: 185 vs. 202 in the muscle of TBF3; 182 vs. 156 in the gonad of TBF3) were observed (Supplemental Fig. S32).
Figure 4.

Allelic expression and cis- and trans-regulation in hybrids. (A) BSB and TC allelic expression values were observed in BT and TB. The black line in the box represents the median value. (B,C) The density distribution of log2(TC/BSB) values was determined in the parents and their hybrid offspring. This graph depicts the extent of allelic expression bias in three tissues: (L) liver; (G) gonad; (M) muscle. (D) Cis- and trans-regulation were observed in the livers of BT and TB. The arrows and the dotted lines represent gradual increasing and decreasing trends in gene number, respectively, from F1 to F3. The solid line represents the absence of an obvious trend.
To investigate allelic expression changes under parental effects, the log2 (TC/BSB) values in parents were calculated and compared with the log2 (TC/BSB) values of the hybrid offspring (Supplemental Methods). The results revealed a gradual decrease of the magnitude of allelic expression across three generations of hybrid progenies (P < 0.01) (Fig. 4B,C; Supplemental Fig. S31). Among the 9753 orthologous gene pairs in the parents, 1373 (14.07%) DEGs were observed between two parents in which only 440 (4.52%) and 586 (6.02%) DEGs between two alleles were found in BTF1 and TBF1, respectively. Then, fewer DEGs were found in the next generations (4.40% in TBF2, 3.48% in TBF3, 4.22% in BTF2, and 3.04% in BTF3) (Supplemental Fig. S31). Furthermore, a gradual decrease in “cis + trans,” “cis only,” and “conserved” genes (31.46%–18.37% in BTF1 to BTF3; 29.37%–17.04% in TBF1 to TBF3) showed the gradual weakening of the parental effect from F1 to F3 (Supplemental Table S19). Meanwhile, novel bias in allelic expression, including “compensatory” and “cis × trans,” also gradually weakened in successive generations (BT: 18.57%–7.38% from F1 to F3; TB: 15.75%–7.03% from F1 to F3) (Supplemental Table S19). In addition, the increase in “trans only” genes, whose differential expression between parents changed to an equal state of allelic expression in hybrids, also reflected the gradual weakening of parental effect from F1 to F3 (Fig. 4D; Supplemental Table S19; Supplemental Methods). In “trans only” genes, the gene ratios related to log2 (TC/BSB) > 1 in parents were more than those related to log2 (TC/BSB) < −1 (up-regulated in TC) in parents (P = 0.023) (average ratio: 21.87% vs. 18.12%) (Supplemental Fig. S33; Supplemental Table S19). The same phenomenon was also found in “cis × trans” genes (P = 0.033) (average: 7.05% vs. 2.27%). However, a higher frequency of “cis + trans” (2.95% vs. 0.32%), “cis only” (1.16% vs. 0.07%), and “compensatory” (0.75% vs. 0.11%) genes was found in the genes with log2 (TC/BSB) < −1 in hybrids (up-regulated in BSB) than in those with log2 (TC/BSB) > 1 in hybrids (all P = 0.023) (Supplemental Fig. S33; Supplemental Table S19). Focusing on the allelic expression changes from F1 to F3, gradually increasing gene numbers were found in “cis + trans” (B < T), “trans only,” and “cis × trans” (B > T), whereas numbers of “conserved,” “cis + trans” (B > T), “cis only” (B > T), “compensatory” (B > T), and “cis × trans” (B < T) genes decreased in both BT and TB (Fig. 4D; Supplemental Table S19).
Evolutionary constraint in expression polymorphism
To study the association of evolutionary constraint with cis- and trans-regulation, we analyzed the correlation between the magnitude of allelic expression (|log2 (TC/BSB)| in parents or/and hybrids, respectively) and Ka/Ks values in parents (Supplemental Methods). The distribution of Ka/Ks showed different patterns for cis- and trans-regulated genes (Supplemental Fig. S34). Specifically, the “trans only” genes were obviously enriched at Ka/Ks of 0.03 (BT) and 0.028 (TB); the Ka/Ks in parents were significantly positively correlated with values of |log2 (TC/BSB)| in parents (R = 0.3052 in BTF1, R = 0.3808 in BTF2, and R = 0.2158 in TBF1) and hybrids (R = 0.2706 in BTF1, R = 0.355 BTF2, and R = 0.2504 in TBF1) as for “cis only” genes (all P-values ≤ 0.0001, Pearson correlation coefficient), respectively (Supplemental Fig. S34); a positive correlation was found between the Ka/Ks in parents and the values of |log2 (TC/BSB)| both in parents (R = 0.1696 in BTF3, R = 0.2164 TBF2) and hybrid (r = 0.1868 in BTF3), respectively (all P-values < 0.005) (Supplemental Fig. S34); in addition, the stronger positive correlation for “cis only” genes detected in shared BT genes (Fig. 5A–C). Furthermore, in successive hybrid generations of the reciprocal cross hybrids, the decreasing correlations of Ka/Ks in the parents and the value of |log2 (TC/BSB)| in hybrids were detected in “cis only” genes of BT (Supplemental Table S20); however, no significant correlation in “cis only” genes was detected in TB.
Figure 5.

Correlation analyses between evolutionary constraint and expression polymorphism. (A–C) Correlation analyses were performed between Ka/Ks values in parents and relevant log2 |TC/BSB| of parents (red) and hybrids (blue) in cis-regulatory genes (17 shared genes in BT). The correlation coefficient between Ka/Ks values and relevant log2 |TC/BSB| in parents was 0.5635 (P-values < 0.05, Pearson correlation coefficient). The correlation coefficients between Ka/Ks values in parents and relevant log2 |TC/BSB| in hybrids are listed in the graph. (D) The log2 |TC/BSB| values of the parents were determined in conserved genes (1055 in BTF1, 912 in BTF2, 966 in BTF3, 1194 in TBF1, 1045 in TBF2, and 803 in TBF3). (****) P < 0.0001 for Spearman's rank correlation coefficient; (**) P < 0.001. (E) The log2 |TC/BSB| values of the hybrids were determined in “cis + trans” (from 565 to 1488 genes in all samples) and “cis × trans” genes (from 643 to 1504 genes in all samples) (all P < 0.0001, Spearman's rank correlation coefficient).
The evolutionary constraint was also presented by variations in exon numbers and coding sequence length, which showed a strong positive correlation with each other in both BSB (R = 0.7435) and TC (R = 0.7768) (all P-values < 0.0001, Pearson correlation coefficient) (Supplemental Fig. S35; Supplemental Methods). The values of |log2 (TC/BSB)| in parents showed a significant correlation with the variation of the coding sequence length in conserved genes of both BT and TB (all P-values < 0.01 for Spearman's rank correlation coefficient) (Fig. 5D). A gradual increase in the correlation coefficient was found in successive TB hybrid generations. The value of |log2 (TC/BSB)| in hybrids also had a significant correlation with the variation of the coding sequence length in “cis + trans” and “cis × trans” genes of both BT and TB (all P-values < 0.0001 for Spearman's rank correlation coefficient) (Fig. 5E). However, no significant correlation was found between Ka/Ks in parents and the variation of CDS length across all samples.
Contribution of evolutionary constraint and expression polymorphism to bone development
Regarding the traits that differed between the two inbred parents, the hybrid offspring showed intermediate traits in bone morphology, number of upper/lower lateral line scales, and dorsal/anal/abdominal fin rays (Fig. 6A,B; Supplemental Tables S21, S22). These phenotypes led us to focus on the 57 expanded and contracted gene clusters related to growth regulation in BSB and TC (Supplemental Table S23). Five relevant genes were positively selected (Ka/Ks > 1) (Supplemental Table S24). Nineteen growth-regulated genes had the same expression pattern in all generations of BT, whereas 14 others showed the same expression pattern in all TB generations (Supplemental Tables S25, S26). Among genes relevant to growth regulation, the ppp2ca, kl, and adcy6a genes belonged to the expanding/contracting gene clusters, and three genes showed the same expression pattern in BT and TB, including transgressive up-regulation (VI) of hsp90b1 and egr1, and no change in expression of hspb1.
Figure 6.

Differences in genomic DNA and allelic expression of the tgfb1b gene contribute to bone morphology. (A) An intermediate trait in bone morphology was observed in BTF1 and TBF1. (Scale bar) 2.5 cm. (B) Intermediate traits were observed on different sides of the pharyngeal jaw. (Scale bar) 1 cm. (C) Alignment of tgfb1b gene sequences in the TC, BSB, and CDS regions in grass carp and zebrafish. The gray represents the same base pair among four species. The black represents the different base pair among four species. (UTR) untranslated region. (D) Ratios of BSB/TC allelic expression were determined in three tissues of parents and reciprocal cross hybrids: (L) liver; (G) gonad; (M) muscle. The spot represents the expression values of the two parents and their hybrid progenies. The line represents the change trend of gene expression.
With respect to cis- and trans-regulatory expression, especially its effects on bone morphology, we detected an increasing trend in “trans only” genes and a decreasing trend in “cis + trans” genes and “cis × trans” genes in successive hybrid generations (Supplemental Table S27). A greater number of genes were identified as having BSB allelic expression bias [log2 (TC/BSB) < −1 in the hybrid] than TC allelic expression bias [log2 (TC/BSB) > 1 in the hybrid] in BT and TB (241 vs. 58 in total) (Supplemental Table S27). We also found an obvious bias toward BSB expression in the growth-regulated traits of the hybrids. However, this difference gradually weakened in successive generations. Concerning the 50 genes with the same cis- and trans-regulatory expression of the reciprocal cross hybrid, 76% (38 genes) had a “trans only” expression pattern, reflecting the convergence effect in allelic expression. For the tgfb1b gene (Ka/Ks = 1.51) between BSB and TC, seven and nine amino acid variations were found in exons 1 and 2 in carnivorous TC in comparison to herbivorous BSB and grass carp and omnivorous zebrafish (Fig. 6C; Supplemental Fig. S36). Meanwhile, coexpression of BSB and TC alleles was detected in the reciprocal cross hybrids, although the expression pattern varied in different generations (Fig. 6D). The expression of the gdf2 gene was “conserved” in BTF1-BTF3 and “trans only” in TBF1-TBF3, suggesting stable coexpression of the BSB and TC alleles. The allelic expression in tgfb1b and gdf2 showed an additive effect.
Discussion
Three polyploidization events occurred in a common ancestor of BSB, TC, zebrafish (D. rerio), grass carp (C. idellus), and common carp (C. carpio) belonging to the family Cyprinidae (Postlethwait et al. 1998). The adaptive radiation of teleost fishes, the most species-rich group of vertebrates, was affected by the joint effect of polyploidization and hybridization, including interspecific hybridization (Lamatsch and Stöck 2009). Through DNA fingerprinting, some fishes, including Xiphophorus clemenciae (Meyer et al. 2006) and Gila seminuda (Demarais et al. 1992), have been found to originate from hybridization. The very recent divergence of the ancestral lineage (12.74 MYA) and high similarity in karyotypes, genome size, and gene characteristics indicate a close genetic relationship between the BSB and the TC in the family Cyprinidae (Fig. 1A–H; Supplemental Fig. S3). These results laid good foundations for exploring the interspecific compatibility in their reciprocal cross hybrids. Different degrees of trait divergence in the two BSB/TC hybrid lineages provide enough samples to investigate potential mechanisms underlying these phenomena (Fig. 6A,B; Supplemental Tables S21, S22; Xiao et al. 2014, 2016).
The results of this study provide new perspectives on the rapid emergence of AR transcripts and the asymmetric expression of alleles in vertebrates immediately following hybridization. The distribution of 145 to 974 AR genes in BTF1-BTF3 and TBF1-TBF3 was identified from RNA-seq data (Supplemental Table S13). The main mechanisms regarding the AR genes include the allelic exchanges of the two subgenomes in genomic DNA and the trans-splicing of pre-mRNAs. Based on the AR genes found in F1 hybrids of the two hybrid lineages, which were validated by Illumina and Sanger sequencing methods (Supplemental Fig. S6E), it is possible that the allelic exchanges of the two subgenomes occur in somatic cells. The allelic recombination in somatic cells was also reported in Nicotiana tabacum (Lebel et al. 1993), in F1 hybrids of Carassius auratus cuvieri (♀) × C. auratus red var. (♂) (Liu et al. 2018), and in F1 hybrids of C. auratus red var. (♀) × Cyprinus carpio L. (♂) (Liu et al. 2016). On the other hand, the trans-splicing of pre-mRNAs leads to the joining of different exons derived from two or more allelic transcripts (Mitchell 2000; Mayer and Floeter-Winter 2005), by which some fractions of the transcript root from one inbred parent, and the remainder roots from the other inbred parent. These AR genes also may result from large-scale DNA repair, nonallelic end-joining, or even transposon activity (Wang et al. 2006; Levin and Moran 2011; Bao and Yan 2012; Fedoroff 2012; Liu et al. 2016). The aforementioned mechanisms may lead to allelic recombination in somatic cells and/or germ cells in F1 and the subsequent generations. In the future, orthogonal methods to investigate AR genes in the two hybrid lineages will be necessary to rule out any ancestral effect (i.e., error in reference genome), sequencing artifacts, and inadequate bioinformatic analyses, which may result from the presented methodology.
Regarding the numbers of AR events and patterns, their increasing trends were found in the successive generations of TB hybrid lineage (Supplemental Fig. S6B–D), but such trends were not observed in BT lineage because of abnormal values with much higher numbers of AR events and AR patterns in sample 1 of BTF1 and BTF2 than the values of other samples in BT lineage (Supplemental Fig. S6B–D). On the other hand, the other values of AR events and AR patterns in sample 2 and sample 3 of BTF1 and BTF2 lineage presented the increasing trends in the successive generations (Supplemental Fig. S6B–D). Based on the distribution of the AR pattern in the 10 AR genes (Supplemental Fig. S6E), pattern I and/or pattern II were found in each generation in BT lineage or TB lineage, suggesting that they were inherited from one generation to another, respectively. The former generation with the single pattern (pattern I or pattern II) and the later generations with the pattern I + II (gene order: No. 1–3 and No. 7–9) indicated the increasing trend of pattern number in the two hybrid lineages (Supplemental Fig. S6E). The unexpected numbers of AR events and AR patterns and unexpected distribution of the AR patterns in the two hybrid lineages (Supplemental Fig. S6; Supplemental Table S13) are probably related to the trans-splicing mechanism (Mitchell 2000; Mayer and Floeter-Winter 2005) that occurred in somatic cells, which will lead to the random AR events and AR patterns, depending on different individuals with different mechanisms involved in the allelic recombination. The AR events effectively eliminate deleterious alleles and improve the adaptability of hybrid fish (Schumer et al. 2018). The high levels of homoeologous recombination have also been observed in some natural allotetraploids, such as Brassica (Mason et al. 2011) and Triticum aestivum L. (Nelson et al. 1995), and a low frequency was detected in Ambystoma for pairing of duplicated premeiotic chromosomes instead of homologs (Neaves and Baumann 2011).
With respect to asymmetric gene expression in the two hybrid lineages, TC expression dominance at the global expression level and slight BSB bias at the allelic expression level were observed (Figs. 3, 4; Supplemental Fig. S30). The very useful classification of cis- and/or trans-regulatory patterns can effectively describe divergence of gene expression and help us investigate their potential mechanisms (McManus et al. 2010). Genes classified as “trans only,” “compensatory,” and “cis × trans” showed novel divergent expression after hybridization (Supplemental Table S19). These genes were more likely to have divergent expression than the genes in “cis only,” “conserved,” and “cis + trans,” implying that the adjustments of allelic expression might be necessary to the improvement of hybrid incompatibilities and contribute to speciation. Meanwhile, genes classified as “cis only,” “conserved,” and “cis + trans” showed that these stable expression in progenies were mainly affected by the parental-specific inheritance (Supplemental Table S19; Emerson et al. 2010; McManus et al. 2010). A similar phenomenon was observed in natural allopolyploid wheat (Feldman et al. 2012) and cotton (Yoo et al. 2013). In addition, decreased and increased trends of gene expression at global and allelic levels were observed from F1 to F3 of the two hybrid lineages (Figs. 3, 4A; Supplemental Fig. S31). The direction and magnitude of gene expression changes in hybrids showed asymmetric patterns, which were often regulated by DNA methylation (Wang et al. 2016; Choi et al. 2018), histone modification (Schotanus et al. 2015), and microRNAs (Ambros 2004).
The obvious trends of gene expression changes raised the question of why these expression patterns could appear in hybrids (Fig. 4D; Supplemental Table S19). To address this issue, a strong correlation was detected between the Ka/Ks in the two parents and the values of |log2 (TC/BSB)| in hybrids for the genes of the “cis only” category (Fig. 5A–C). A similar phenomenon has also been described in F1 hybrid yeast (Emerson et al. 2010). Moreover, the correlation coefficients obviously decreased from F1 to F3 (Fig. 5A–C). This gradual weakening of cis-regulation may result from a series of cumulative genetic changes, including the allelic exchanges of conserved noncoding sequences (CNSs) in genomic DNA (Bird et al. 2018). The shared recombinant events of coding sequences supported the cumulative effect of genomic recombination in CNSs (Supplemental Figs. S6–S22; Supplemental Table S13). The interaction between cis-regulatory sequences and trans-regulatory elements of two alleles in hybrids is enhanced by the exchange of CNSs deriving from the merger of two subgenomes (Wittkopp et al. 2004). Genomic DNA exchanges showed a close relationship with gene expression divergence in animals, contributing to survival and adaptation to environments (Marquès-Bonet et al. 2004). However, no significant correlation between genomic DNA exchanges and presumed cis- and trans-regulatory effects was observed in our study.
Identification of mRNA expression patterns with the corresponding phenotypes was challenging because of the complex gene regulatory network and unknown interactions between RNAs and proteins (Emerson et al. 2010). In this study, the distinctness of traits between two parents (BSB and TC), including appearance and feeding habit, was manifested by the wide diversity of genetic sequences and gene expression patterns. The tgfb1b gene (Ka/Ks = 1.51) was regarded as one of the major genes for pharyngeal jaw development based on the analyses between intermediate traits of BSB and TC and the additive effect of allelic expression (Fig. 6C,D; Supplemental Fig. S36). However, further experiments should be conducted to verify the molecular function of the tgfb1b gene.
In summary, this study indicates the rapid emergence of expressed recombinant genes and asymmetric gene expression patterns in early hybrid generations, providing insights into the evolution of vertebrate genomes immediately following hybridization.
Methods
Samples
A wild, water-captured male adult of C. alburnus (TC) and a gynogenetic M. amblycephala (BSB) that was stimulated by UV-inactivated sperm of C. carpio haematopterus were obtained for whole-genome sequencing (Gong et al. 2019). Then, 24 fish (age 2 yr), including three male TCs with mature testes, three BSBs with mature ovaries, three female individuals each from BTF1-BTF3, and three female individuals each from TBF1-TBF3, were used in the subsequent analyses (Fig. 1A–H). The detailed descriptions about the sample preparation for genome and transcriptome sequencing were presented in the Supplemental Methods.
Genome-wide evolutionary analysis
Genome assembly was performed with a series of analyses. Single-copy genes from all 10 species were used to construct a phylogenetic tree and estimate their divergence time. Orthologous gene pairs between TC and BSB were determined by all-against-all reciprocal BLASTP (v. 2.2.26) comparisons based on the protein sequences. Homologous blocks between species were detected. and the numbers of transversions at fourfold degenerate sites (i.e., 4dTv values) of the blocks were calculated on the CDS alignments using the HKY model. The detailed descriptions related to the sequencing and assembly, assessment of assembly quality, gene prediction and annotation, noncoding RNA prediction, evolutionary analysis, diversifying selection analysis, genetic map construction and scaffold anchoring of TC genome, and Hi-C assembly of the BSB genome are provided in the Supplemental Methods.
Recombinant sequences detected by Illumina, PacBio, and Sanger sequencing
RNA-seq of all samples with three biological replicates were sequenced using paired-end Illumina technology according to the manufacturer's instructions. A custom computational script was used to remove the raw reads containing adapters, poly(N) tails, and those of low quality (see filtering raw reads, Supplemental Script 1). After screening, the hybrid high-quality Illumina reads in the preceding analysis were mapped to a mixed genome of the two parents using STAR (v. 2.4.0) (Dobin et al. 2013). To check for recombinant events, DNA fragments were obtained in three biological replicates of BSB, TC, TBF1, BTF1, TBF3, and BTF3 using Sanger sequencing. After screening out low-quality data in PacBio sequencing, we aligned reads to the reference genome (a mixed genome of the two parents). The detailed descriptions related to the ortholog identification and AR sequence detection by Illumina sequencing, PCR validation of AR genes, library construction and PacBio sequencing, and AR gene detection by genome-wide long-read alignments could be found in the Supplemental Methods.
Global expression patterns
In-house Perl scripts were used to calculate the number of mapped reads in each gene (see calculating mapped reads, Supplemental Scripts 2). The APE values were determined based on the average expression level of the orthologous gene pairs in two parents [HAPE = (B + T)/2], and the other values were obtained based on the average of the two expression values aligned with both parent genomes [Hreal = (HB + HT)/2]. For comparison of HAPE and Hreal, clear changes in expression after hybridization were described in one comparison. Another way to investigate the expression levels in a comparison of the hybrid with both inbred parents was in terms of “additive,” “BSB/TC expression dominance,” and “Transgressive down-/up-regulation” inheritance, according to the magnitude of the expression difference, as described by Rapp et al. (2009). The detailed descriptions regarding the mapping of RNA-seq data, differential expression analysis, and analysis of expression dominance are provided in the Supplemental Methods.
Detection of cis- and trans-regulation and correlation with Ka/Ks
According to the comparison of SNPs and other loci in orthologous gene pairs between BSB and TC, completely different loci, including heterozygous and homozygous loci, were considered species-specific SNPs, as in Schaefke et al. (2013) and McManus et al. (2010). In-house Perl scripts were used to calculate the BSB/TC allelic reads in the hybrids based on corresponding BSB/TC species-specific SNPs (see calculating allelic reads with SNPs, Supplemental Scripts 3). The cis- and/or trans-regulatory patterns were established based on significant differences between TC and BSB in parents and hybrids as described in McManus et al. (2010). The cis- and/or trans-regulatory results were sorted by positive/negative values of log2 (TC/BSB) in parents or hybrids. The details regarding the species-specific SNP identification, detection of allelic expression levels in hybrids, allelic expression silencing and bias, cis- and trans-regulatory differences underlying expression divergence between BSB and TC, correlation analysis of cis- and trans-regulatory expression, Ka/Ks, and AR genes are described in the Supplemental Methods.
Data access
The genome assembly from this study was submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA269572 (Zhou et al. 2015). All raw RNA-seq data were submitted to the NCBI Sequence Read Archive SRA; https://www.ncbi.nlm.nih.gov/sra under accession number SRP050891. Sanger sequencing data were submitted to the NCBI Nucleotide database (https://www.ncbi.nlm.nih.gov/nuccore/) under accession numbers MK166917–MK166955, MK202974–MK202987, MK212159–MK212183, MK226325–MK226332, MK093841–MK093849, MN083058–MN083086, MN097778–MN097796, MN517127–MN517145, and MN514403–MN514427. Custom computational scripts used in this study are included as Supplemental Scripts.
Supplementary Material
Acknowledgments
This research was supported by the National Natural Science Foundation of China (Grant No. 31730098, 31430088 and 31702334), the earmarked fund for China Agriculture Research System (Grant No. CARS-45), Hunan Provincial Natural Science and Technology Major Project (Grant No. 2017NK1031), the Key Research and Development Project of Hunan Province (Grant No. 2016NK2128), the Key Research and Development Program of Hunan Province (Grant No. 2018NK2072), High-Level Talent Agglomeration Program of Hunan, China (2019RS1044), and the Cooperative Innovation Center of Engineering and New Products for Developmental Biology of Hunan Province (Grant No. 20134486).
Author contributions: S.L. and L.R. contributed to the conception and design of the study. S.L., L.R., Q.Q., F. Han, and J. Li wrote and modified the manuscript. S.L., K.L., Q.Q., J.X., C.Z., and M.T. made main contribution to the establishment of the hybrid lineages. L.R., S.L., W.L., G.W., G.L., J. Liu, Z.W., X.L., M.L., H.Z., and C.Y. carried out bioinformatics analyses. J.X., X.G., J.C., C.W., C.Z., M.T., Q.Q., J.W., K.L., S.W., F. Hu, R. Zhao, R. Zhu, Y.S., Y.W., Q.L., X.Y., and C.T. assisted in extracting the raw material. J.X., W.L., and C.W. assisted in collecting the photographs. All authors read and approved the final manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.249805.119.
Freely available online through the Genome Research Open Access option.
References
- Adams KL. 2007. Evolution of duplicate gene expression in polyploid and hybrid plants. J Hered 98: 136–141. 10.1093/jhered/esl061 [DOI] [PubMed] [Google Scholar]
- Ambros V. 2004. The functions of animal microRNAs. Nature 431: 350–355. 10.1038/nature02871 [DOI] [PubMed] [Google Scholar]
- Bao J, Yan W. 2012. Male germline control of transposable elements. Biol Reprod 86: 162 10.1095/biolreprod.111.095463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird KA, VanBuren R, Puzey JR, Edger PP. 2018. The causes and consequences of subgenome dominance in hybrids and recent polyploids. New Phytol 220: 87–93. 10.1111/nph.15256 [DOI] [PubMed] [Google Scholar]
- Chen Y. 1998. Fauna sinica: Osteichthyes cypriniformes (II). Science Press, Beijing. [Google Scholar]
- Choi K, Zhao X, Tock AJ, Lambing C, Underwood CJ, Hardcastle TJ, Serra H, Kim J, Cho HS, Kim J, et al. 2018. Nucleosomes and DNA methylation shape meiotic DSB frequency in Arabidopsis thaliana transposons and gene regulatory regions. Genome Res 28: 532–546. 10.1101/gr.225599.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cnaani A, Hallerman EM, Ron M, Weller JI, Indelman M, Kashi Y, Gall GA, Hulata G. 2003. Detection of a chromosomal region with two quantitative trait loci, affecting cold tolerance and fish size, in an F2 tilapia hybrid. Aquaculture 223: 117–128. 10.1016/S0044-8486(03)00163-7 [DOI] [Google Scholar]
- DeMarais BD, Dowling TE, Douglas ME, Minckley W, Marsh PC. 1992. Origin of Gila seminuda (Teleostei: Cyprinidae) through introgressive hybridization: implications for evolution and conservation. Proc Natl Acad Sci 89: 2747–2751. 10.1073/pnas.89.7.2747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson JJ, Hsieh LC, Sung HM, Wang TY, Huang CJ, Lu HH, Lu MY, Wu SH, Li WH. 2010. Natural selection on cis and trans regulation in yeasts. Genome Res 20: 826–836. 10.1101/gr.101576.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedoroff NV. 2012. Presidential address. Transposable elements, epigenetics, and genome evolution. Science 338: 758–767. 10.1126/science.338.6108.758 [DOI] [PubMed] [Google Scholar]
- Feldman M, Levy AA, Fahima T, Korol A. 2012. Genomic asymmetry in allopolyploid plants: wheat as a model. J Exp Bot 63: 5045–5059. 10.1093/jxb/ers192 [DOI] [PubMed] [Google Scholar]
- Gong DB, Xu LH, Wu C, Wang S, Liu QF, Cao L, Mao ZW, Wang YD, Hu FZ, Zhou R, et al. 2019. Two types of gynogenetic blunt snout bream derived from different sperm. Aquaculture 511: 734250 10.1016/j.aquaculture.2019.734250 [DOI] [Google Scholar]
- Lamatsch DK, Stöck M. 2009. Sperm-dependent parthenogenesis and hybridogenesis in teleost fishes. In Lost sex, pp. 399–432. Springer, Dordrecht, Netherlands. [Google Scholar]
- Lebel EG, Masson J, Bogucki A, Paszkowski J. 1993. Stress-induced intrachromosomal recombination in plant somatic cells. Proc Natl Acad Sci 90: 422–426. 10.1073/pnas.90.2.422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin HL, Moran JV. 2011. Dynamic interactions between transposable elements and their hosts. Nat Rev Genet 12: 615–627. 10.1038/nrg3030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Lewis DH, Gatlin DM. 2004. Dietary oligonucleotides from yeast RNA influence immune responses and resistance of hybrid striped bass (Morone chrysops × Morone saxatilis) to Streptococcus iniae infection. Fish Shellfish Immunol 16: 561–569. 10.1016/j.fsi.2003.09.005 [DOI] [PubMed] [Google Scholar]
- Li W, Liu J, Tan H, Yang C, Ren L, Liu Q, Wang S, Hu F, Xiao J, Zhao R, et al. 2018. Genetic effects on the gut microbiota assemblages of hybrid fish from parents with different feeding habits. Front Microbiol 9: 2972 10.3389/fmicb.2018.02972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Luo J, Chai J, Ren L, Zhou Y, Huang F, Liu X, Chen Y, Zhang C, Tao M, et al. 2016. Genomic incompatibilities in the diploid and tetraploid offspring of the goldfish × common carp cross. Proc Natl Acad Sci 113: 1327–1332. 10.1073/pnas.1512955113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Qi Y, Liang Q, Xu X, Hu F, Wang J, Xiao J, Wang S, Li W, Tao M, et al. 2018. The chimeric genes in the hybrid lineage of Carassius auratus cuvieri (♀)×Carassius auratus red var. (♂). Sci China Life Sci 61: 1079–1089. 10.1007/s11427-017-9306-7. [DOI] [PubMed] [Google Scholar]
- Maheshwari S, Barbash DA. 2012. Cis-by-trans regulatory divergence causes the asymmetric lethal effects of an ancestral hybrid incompatibility gene. PLoS Genet 8: e1002597 10.1371/journal.pgen.1002597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquès-Bonet T, Cáceres M, Bertranpetit J, Preuss TM, Thomas JW, Navarro A. 2004. Chromosomal rearrangements and the genomic distribution of gene-expression divergence in humans and chimpanzees. Trends Genet 20: 524–529. 10.1016/j.tig.2004.08.009 [DOI] [PubMed] [Google Scholar]
- Mason AS, Nelson MN, Castello MC, Yan G, Cowling WA. 2011. Genotypic effects on the frequency of homoeologous and homologous recombination in Brassica napus × B. carinata hybrids. Theor Appl Genet 122: 543–553. 10.1007/s00122-010-1468-5 [DOI] [PubMed] [Google Scholar]
- Mayer MG, Floeter-Winter LM. 2005. Pre-mRNA trans-splicing: from kinetoplastids to mammals, an easy language for life diversity. Mem Inst Oswaldo Cruz 100: 501–513. 10.1590/S0074-02762005000500010 [DOI] [PubMed] [Google Scholar]
- McManus CJ, Coolon JD, Duff MO, Eipper-Mains J, Graveley BR, Wittkopp PJ. 2010. Regulatory divergence in Drosophila revealed by mRNA-seq. Genome Res 20: 816–825. 10.1101/gr.102491.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer A, Salzburger W, Schartl M. 2006. Hybrid origin of a swordtail species (Teleostei: Xiphophorus clemenciae) driven by sexual selection. Mol Ecol 15: 721–730. 10.1111/j.1365-294X.2006.02810.x [DOI] [PubMed] [Google Scholar]
- Mitchell LG. 2000. Chimeric RNA molecules generated by trans-splicing. Google Patents. U.S. patent no. 6,013,487.
- Neaves WB, Baumann P. 2011. Unisexual reproduction among vertebrates. Trends Genet 27: 81–88. 10.1016/j.tig.2010.12.002 [DOI] [PubMed] [Google Scholar]
- Nelson JC, Sorrells ME, Van Deynze A, Lu YH, Atkinson M, Bernard M, Leroy P, Faris JD, Anderson JA. 1995. Molecular mapping of wheat: major genes and rearrangements in homoeologous groups 4, 5, and 7. Genetics 141: 721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postlethwait JH, Yan YL, Gates MA, Horne S, Amores A, Brownlie A, Donovan A, Egan ES, Force A, Gong Z, et al. 1998. Vertebrate genome evolution and the zebrafish gene map. Nat Genet 18: 345–349. 10.1038/ng0498-345 [DOI] [PubMed] [Google Scholar]
- Rapp RA, Udall JA, Wendel JF. 2009. Genomic expression dominance in allopolyploids. BMC Biol 7: 18 10.1186/1741-7007-7-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reif JC, Gumpert FM, Fischer S, Melchinger AE. 2007. Impact of interpopulation divergence on additive and dominance variance in hybrid populations. Genetics 176: 1931–1934. 10.1534/genetics.107.074146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefke B, Emerson JJ, Wang TY, Lu MY, Hsieh LC, Li WH. 2013. Inheritance of gene expression level and selective constraints on trans- and cis-regulatory changes in yeast. Mol Biol Evol 30: 2121–2133. 10.1093/molbev/mst114 [DOI] [PubMed] [Google Scholar]
- Schartl M, Walter RB, Shen Y, Garcia T, Catchen J, Amores A, Braasch I, Chalopin D, Volff JN, Lesch KP, et al. 2013. The genome of the platyfish, Xiphophorus maculatus, provides insights into evolutionary adaptation and several complex traits. Nat Genet 45: 567–572. 10.1038/ng.2604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotanus K, Soyer JL, Connolly LR, Grandaubert J, Happel P, Smith KM, Freitag M, Stukenbrock EH. 2015. Histone modifications rather than the novel regional centromeres of Zymoseptoria tritici distinguish core and accessory chromosomes. Epigenetics Chromatin 8: 41 10.1186/s13072-015-0033-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumer M, Xu C, Powell DL, Durvasula A, Skov L, Holland C, Blazier JC, Sankararaman S, Andolfatto P, Rosenthal GG, et al. 2018. Natural selection interacts with recombination to shape the evolution of hybrid genomes. Science 360: 656–660. 10.1126/science.aar3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31: 3210–3212. 10.1093/bioinformatics/btv351 [DOI] [PubMed] [Google Scholar]
- Wang HC, Chou WC, Shieh SY, Shen CY. 2006. Ataxia telangiectasia mutated and checkpoint kinase 2 regulate BRCA1 to promote the fidelity of DNA end-joining. Cancer Res 66: 1391–1400. 10.1158/0008-5472.CAN-05-3270 [DOI] [PubMed] [Google Scholar]
- Wang X, Werren JH, Clark AG. 2016. Allele-specific transcriptome and methylome analysis reveals stable inheritance and cis-regulation of DNA methylation in Nasonia. PLoS Biol 14: e1002500 10.1371/journal.pbio.1002500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittkopp PJ, Haerum BK, Clark AG. 2004. Evolutionary changes in cis and trans gene regulation. Nature 430: 85–88. 10.1038/nature02698 [DOI] [PubMed] [Google Scholar]
- Wolters WR, Wise DJ, Klesius PH. 1996. Survival and antibody response of channel catfish, blue catfish, and channel catfish female × blue catfish male hybrids after exposure to Edwardsiella ictaluri. J Aquat Anim Health 8: 249–254. 10.1577/1548-8667(1996)008<0249:SAAROC>2.3.CO;2 [DOI] [Google Scholar]
- Xiao J, Kang X, Xie L, Qin Q, He Z, Hu F, Zhang C, Zhao R, Wang J, Luo K, et al. 2014. The fertility of the hybrid lineage derived from female Megalobrama amblycephala × male Culter alburnus. Anim Reprod Sci 151: 61–70. 10.1016/j.anireprosci.2014.09.012 [DOI] [PubMed] [Google Scholar]
- Xiao J, Hu F, Luo K, Li W, Liu S. 2016. Unique nucleolar dominance patterns in distant hybrid lineage derived from Megalobrama amblycephala × Culter alburnus. BMC Genet 17: 150 10.1186/s12863-016-0457-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo MJ, Szadkowski E, Wendel JF. 2013. Homoeolog expression bias and expression level dominance in allopolyploid cotton. Heredity (Edinb) 110: 171–180. 10.1038/hdy.2012.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Ren Z, Zeng H, Yao B. 2008. Apparent digestibility of various feedstuffs for bluntnose black bream Megalobrama amblycephala Yih. Aquac Nutr 14: 153–165. 10.1111/j.1365-2095.2007.00515.x [DOI] [Google Scholar]
- Zhou Y, Ren L, Xiao J, Zhong H, Wang J, Hu J, Yu F, Tao M, Zhang C, Liu Y, et al. 2015. Global transcriptional and miRNA insights into bases of heterosis in hybridization of Cyprinidae. Sci Rep 5: 13847 10.1038/srep13847 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.