

Abstract
Rates of production and degradation together specify microRNA (miRNA) abundance and dynamics. Here, we used approach-to-steady-state metabolic labeling to assess these rates for 176 miRNAs in contact-inhibited mouse embryonic fibroblasts (MEFs), 182 miRNAs in dividing MEFs, and 127 miRNAs in mouse embryonic stem cells (mESCs). MicroRNA duplexes, each comprising a mature miRNA and its passenger strand, are produced at rates as fast as 110 ± 50 copies/cell/min, which exceeds rates reported for any mRNAs. These duplexes are rapidly loaded into Argonaute, with <30 min typically required for duplex loading and silencing-complex maturation. Within Argonaute, guide strands have stabilities that vary by 100-fold. Half-lives also vary globally between cell lines, with median values ranging from 11 to 34 h in mESCs and contact-inhibited MEFs, respectively. Moreover, relative half-lives for individual miRNAs vary between cell types, implying the influence of cell-specific factors in dictating turnover rate. The apparent influence of miRNA regions most important for targeting, together with the effect of one target on miR-7 accumulation, suggest that targets fulfill this role. Analysis of the tailing and trimming of miRNA 3′ termini showed that the flux was typically greatest through the isoform tailed with a single uridine, although changes in this flux did not correspond to changes in stability, which suggested that the processes of tailing and trimming might be independent from that of decay. Together, these results establish a framework for describing the dynamics and regulation of miRNAs throughout their life cycle.
MicroRNAs (miRNAs) are small RNAs that mediate posttranscriptional gene regulation by targeting mRNAs for degradation and translational repression. In animals, miRNAs typically recognize their mRNA targets through base-pairing of the miRNA seed region (miRNA nucleotides [nt] 2–8) and sites in mRNA 3′ untranslated regions (UTRs) (Bartel 2018). Each miRNA has many sites throughout the transcriptome, and more than 60% of human protein-coding genes have evolutionarily conserved sites to at least one of the 90 miRNA families that are conserved to fish (Friedman et al. 2009). When knocked out in mice, most miRNA families conserved to fish exhibit abnormal phenotypes, including embryonic or postnatal lethality, infertility, and developmental defects (Bartel 2018). Many miRNAs are regulated both spatially and temporally; the prevalence and conservation of such regulation implies the importance of precise and specific control of miRNA accumulation in each cell type (Houbaviy et al. 2003; Wienholds et al. 2005; Rybak et al. 2008; Thornton and Gregory 2012; Chen and Qin 2015).
The intracellular accumulation of an RNA species is a function of both its production and its destruction, but how the balance between these two processes impinges on miRNA levels is poorly understood. MicroRNA production is a multistep pathway with opportunities for regulation at each step. Canonical miRNAs are initially transcribed as long, hairpin-containing transcripts known as “pri-miRNAs” (Lee et al. 2002). In animal cells, each miRNA hairpin within the pri-miRNA is cropped in the nucleus by the Microprocessor complex, producing a “pre-miRNA” that is then exported to the cytoplasm and cleaved by Dicer to yield the mature miRNA duplex (Ha and Kim 2014). One strand of this duplex is loaded into an Argonaute (AGO) protein to generate a mature complex capable of targeting genes for repression, whereas the other strand is rapidly degraded (Kawamata and Tomari 2010). Strand selection is typically biased, dictated both by the sequence and the thermodynamic stability of the 5′ ends of each duplex strand (Khvorova et al. 2003; Schwarz et al. 2003; Suzuki et al. 2015), and thus, for most miRNA duplexes the strand that is preferentially loaded as the guide strand is readily distinguished from the one that usually acts as the passenger strand. Production of the mature complex can be modulated at each of the steps of biogenesis in a miRNA-specific manner (Han et al. 2007; Lee et al. 2007; Heo et al. 2008; Viswanathan et al. 2008; Trabucchi et al. 2009; Krol et al. 2010; Ma et al. 2010).
In contrast to our understanding of miRNA biogenesis, less is known about miRNA degradation. For example, the extent to which miRNAs degrade through a common pathway characterized by stereotypic changes is unknown. Some mature miRNAs within AGO can have nucleotides added to or removed from their 3′ ends, processes referred to as tailing and trimming, respectively (Ameres and Zamore 2013). Although such modifications can alter stability of some miRNAs, levels of many miRNAs are insensitive to depletion of individual tailing factors (Jones et al. 2009; Katoh et al. 2009; Mansur et al. 2016). Tailing and trimming is also associated with target RNA-directed miRNA degradation (TDMD), a phenomenon in which a highly complementary target site that contains extensive pairing to the 3′ end of a miRNA can trigger degradation of that miRNA (Ameres et al. 2010; Cazalla et al. 2010; Baccarini et al. 2011; Marcinowski et al. 2012; Xie et al. 2012; de la Mata et al. 2015; Bitetti et al. 2018; Ghini et al. 2018; Kleaveland et al. 2018). However, whether tailing and trimming plays a general role in the miRNA decay pathway remains to be determined. Indeed, elimination of tailing by knockout of the TENT2 (GLD2/PAPD4) cytoplasmic poly(A) polymerase has no effect on TDMD-mediated destabilization of miR-7 by the Oip5os1 (also known as Cyrano) long noncoding RNA (lncRNA) in mammalian tissues (Kleaveland et al. 2018).
Questions also remain regarding the rates of miRNA turnover and the extent to which degradation is regulated in a miRNA-specific manner. Most miRNAs that have been examined are highly stable. In rat hearts, miR-208 persists for weeks after its production has ceased, and in several human and mouse cell lines, the levels of most miRNAs do not detectably change after up to 24 h of transcriptional inhibition (van Rooij et al. 2007; Bail et al. 2010; Guo et al. 2015). Furthermore, miRNAs persist long after inducible deletion of Dicer1 in mouse embryonic fibroblasts (MEFs), implying half-lives of over 100 h (Gantier et al. 2011). In contrast, some neuronal-specific miRNAs exhibit more rapid turnover (Krol et al. 2010), and miR-503, a member of the miR-16 extended family proposed to reinforce cell cycle arrest in G0, turns over with a half-life of ∼4 h (Rissland et al. 2011). The correlation between short half-life and functions in regulating rapid cell transitions (i.e., response to neuronal stimulation and exit from cell cycle arrest) implies that miRNA half-lives might be selectively regulated to enable dynamic behavior.
Until recently, a challenge for studying miRNA degradation has been the lack of a high-throughput but nondisruptive method for assessing turnover rates. Inhibition of transcription dramatically disrupts cellular homeostasis, limiting the duration over which decay can be assessed and calling into question the relevance of the half-lives observed (Bensaude 2011; Sun et al. 2012; Lugowski et al. 2018). Metabolic labeling is less invasive and has long been applied to study mRNA half-lives (Ross 1995; Dölken et al. 2008; Rabani et al. 2011; Schwanhäusser et al. 2011; Duffy et al. 2015). In these studies, mRNAs labeled with 4-thiouridine are biotinylated, which enables their isolation and analysis. Two studies use metabolic labeling with 4-thiouridine to assess rates of miRNA turnover in HEK293T cells and 3T9 mouse fibroblasts, respectively (Duffy et al. 2015; Marzi et al. 2016). However, the former study does not provide half-life values but instead classifies miRNAs as having either fast or slow turnover relative to the bulk population, and the latter study reports that only ∼30% of miRNAs have half-lives of >24 h, which seems at odds with results from previous approaches. The pulldown procedures used to obtain these results rely on efficient and selective biotinylation of species that contain a single metabolic label, a challenge that even optimized biotinylation protocols struggle to overcome (Duffy et al. 2015). One way to circumvent this challenge is to couple metabolic labeling with thiol-mediated alkylation, which causes the labeled nucleotides to be read as cytidines during reverse transcription, such that, after sequencing, labeled molecules can be distinguished based on their U-to-C substitutions (Herzog et al. 2017). This method, called SLAM-seq (thiol-linked alkylation for the metabolic sequencing of RNA), has been successfully applied to describe the metabolism of Drosophila miRNAs and siRNAs in S2 cells (Reichholf et al. 2019). However, the need to sequence both the labeled and unlabeled RNA can limit the sensitivity of this method, which might explain why only 42 miRNAs and 18 passenger strands are analyzed.
Metabolic incorporation of 5-ethynyluridine (5EU) into nascent RNA, followed by attachment of a biotin or fluorescent label through click chemistry acting at the ethynyl group of 5EU, provides another method for isolating or detecting newly synthesized RNA (Jao and Salic 2008; Chan et al. 2019; Kwasnieski et al. 2019). The adaptation of this approach to efficiently isolate ∼30-nt fragments of newly synthesized mRNAs (Eisen et al. 2019a) suggested that when examining the production and decay dynamics of RNAs as short as miRNAs, 5EU metabolic labeling might offer a favorable alternative to 4-thiouridine labeling. Here, we applied 5EU-labeling to assess the dynamics of miRNA production and decay in contact-inhibited MEFs, dividing MEFs, and mouse embryonic stem cells (mESCs).
Results
Reproducible high-throughput measurement of miRNA half-lives
Approach-to-steady-state labeling experiments generate information regarding both the production and degradation rates of the species examined (Neymotin et al. 2014). For such experiments, a label is introduced to cells at time zero, and labeled molecules are collected and quantified over subsequent time intervals (Fig. 1A). We used this strategy to measure miRNA production and decay, starting in contact-inhibited MEFs, collecting cells 1, 2, 4, 8, 24, 72, and 168 h (1 wk) after introducing 5EU to the media (refreshing media daily for time points longer than 24 h). From each of these samples, ∼5 µg of total RNA was set aside for small-RNA sequencing, which reported the miRNA profile of the unselected input for each time interval. To the remainder of the total RNA, radiolabeled quantification standards were added (a mixture of three 22- to 25-nt RNAs that each contained a single 5EU and a 30-nt RNA that contained no 5EU), and 5EU-containing small RNAs were enriched and sequenced (Fig. 1A).
Figure 1.

Labeling with 5EU generates reproducible miRNA half-life values. (A) Schematic of the experimental setup. 5EU is added at time zero, and samples are collected at subsequent time intervals. RNA is extracted from each sample, and a small fraction of this extracted RNA is used to generate small-RNA sequencing libraries. The remainder of the RNA is mixed with quantitative standards (stds.), and molecules with one or more 5EU nucleotides are biotinylated and captured using streptavidin beads. RNA eluted from the beads is prepared for small-RNA sequencing. (B) Equations used for fitting approach-to-steady-state data. Equation 1 represents the single-exponential model used to fit a single decay rate for guide strands. Equation 2 represents the biexponential model used to fit two decay rates for both passenger and guide strands when assessing passenger-strand half-lives. Fits returned estimates for α (production rate), βn (degradation rate, with n distinguishing the two rates of Equation 2), f (fraction of molecules exhibiting the corresponding degradation rate), and t0 (time offset). The variable g describes the rate of cell doubling and was set manually based on experimental observations. Fits were carried out for each miRNA guide or passenger strand that satisfied an expression cut-off of 60 reads per million miRNA-mapping reads (i.e., 60 RPM) at each time point. (C) Representative fits to data from contact-inhibited MEFs. Data are plotted with black points, and single-exponential fits with a red line. Time courses representing the 10th through 90th deciles in goodness-of-fit are shown in order of increasing goodness-of-fit (from top left to bottom right). (D) Reproducibility of half-life measurements for guide strands. Shown is the relationship between values determined independently from each of the two biological replicates from contact-inhibited MEFs.
To assess the efficacy of 5EU labeling and the applicability of approach-to-steady-state labeling for determining rates of miRNA production and degradation, we examined enrichment of both the 5EU-containing standards and cellular miRNAs over nonlabeled species. Monitoring the fate of the internal quantification standards after affinity purification of 5EU-containing species showed that standards with a single 5EU were enriched >150-fold relative to the standard that lacked a 5EU (Supplemental Fig. S1A,B). Levels of cellular miRNAs at each time point were also compared to background levels, which were determined by subjecting samples from unlabeled cells to biotinylation and affinity enrichment (Supplemental Fig. S1C). As the duration of 5EU treatment lengthened, yields of isolated miRNAs progressively increased over background levels, presumably due to higher proportions of 5EU-containing miRNAs following longer labeling intervals (Supplemental Fig. S1C). As expected, miRNAs with fewer uridines tended to be recovered somewhat less efficiently, but only one uridine-depleted miRNA was poorly recovered in the pulldown (Supplemental Fig. S1D). Importantly, treatment for up to 1 wk with 5EU had little effect on cellular miRNA levels (Supplemental Fig. S1E). Together, these observations indicated that metabolic labeling with 5EU could provide an effective method to measure miRNA production and decay, with minimal disruption to miRNA homeostasis.
We fit an exponential function to the results of this approach-to-steady-state experiment, which generated values for the production and degradation rates (α and β, respectively) of each miRNA strand that exceeded our expression threshold (Fig. 1B, Equation 1; Supplemental Table S1). In addition to the sequencing data, the exponential model took as input the rate of cell division (g), which was experimentally determined for each cell line studied. Data for all miRNAs were fit simultaneously to yield, in addition to miRNA-specific production and degradation rates, a single time-offset parameter (t0) for the experiment that accounted for the lag in the signal due to the time required for 5EU incorporation and miRNA processing (Supplemental Fig. S1F,G). The value of 37 min obtained for t0 provided an upper limit on the time required for the typical miRNA to undergo Drosha-catalyzed processing, nucleocytoplasmic export, and Dicer-catalyzed processing. The single-exponential model fit the data well (Fig. 1C), and improvement of fits obtained when using a biphasic, biexponential model was insufficient to justify use of this more complex model.
MicroRNA guide-strand half-lives and production rates determined from two biological replicates correlated well (Pearson R2 = 0.65 and 0.92, respectively) (Fig. 1D; Supplemental Fig. S1H). This reproducibility allowed data from replicates to be pooled and fit simultaneously, which generated higher-confidence values for production and degradation rates (Supplemental Table S1). The half-life measured for miR-503 in contact-inhibited MEFs using these pooled data, at 6.9 h, agreed well with its previously published half-life in 3T3 cells (Rissland et al. 2011). In addition, bootstrap analyses indicated that half-lives and production rates were robust to resampling (Supplemental Fig. S1I,J), which together with analysis of the biological replicates and validation of the miR-503 turnover rate confirmed that rates of production and degradation determined from the single-exponential fits to the metabolic-labeling data were both reproducible and accurate.
Rates of miRNA guide-strand production and degradation
For the guide strands of miRNA duplexes, rates of both production and degradation varied broadly, spanning three and two orders of magnitude, respectively (Fig. 2A,B; Supplemental Table S1). Knowledge of the absolute number of miR-7 molecules in contact-inhibited MEF cells enabled production rates to be converted to absolute values of copies/h/cell (Supplemental Fig. S2A). These analyses revealed that miR-26a-5p, the third most abundant miRNA in these cells, was produced at the fastest rate, with 20 ± 7 (±s.d.) molecules being produced each minute in the average cell. Four additional miRNAs were produced at rates exceeding 10 molecules/min/cell. Production rates of species deriving from genes separated by <10,000 bp were significantly more similar than those of genes separated by >100,000 bp (P = 0.00016, Mann–Whitney U test). This clustering of similar production rates agreed well with previous estimates of pri-miRNA transcript length (Baskerville and Bartel 2005; Chang et al. 2015), which further supported the accuracy of our production-rate measurements. Rates of production and degradation were not correlated, reflecting independence of the miRNA biogenesis and decay pathways (Supplemental Fig. S2B). Although most guide strands (127/176) had half-lives exceeding 24 h (median half-life, 34 h), nine guide strands had half-lives <10 h (Fig. 2B). These relatively short-lived species included miR-503, as previously mentioned, as well as miR-7a, the miRNA with the shortest measured half-life (1.7 h) (Fig. 2B) and a known substrate of TDMD (Kleaveland et al. 2018).
Figure 2.

Dynamics of miRNA guide strands in contact-inhibited MEFs. (A) Distribution of production rates for guide strands in contact-inhibited MEFs. (B) Distribution of half-life values for guide strands in contact-inhibited MEFs. (C) Relationship between steady-state abundance (reads in the 1-wk input sample) and production rate of guide strands in contact-inhibited MEFs. Outliers that passed an expression threshold (production rate >5 copies/h/cell) and differed significantly from the best linear fit (|z-score| > 2) are labeled. (D) Relationship between steady-state abundance (reads in the 1-wk input sample) and half-life of guide strands in contact-inhibited MEFs. The eight most rapidly turned over miRNAs, in order from least to most stable, were miR-7a-5p, miR-154-3p, miR-328-3p, miR-503-5p, miR-450b-5p, miR-329-5p, miR-495-3p, and miR-376b-3p.
Production rate correlated well with miRNA abundance (Pearson R2 = 0.84) (Fig. 2C), suggesting that regulated production is the primary cause of differing miRNA levels. Nevertheless, as exemplified by a handful of miRNAs with unusually short half-lives that included both miR-7a and miR-503, some miRNAs were produced at rates substantially higher than would have been expected for their overall abundances (Fig. 2C). Moreover, although the correlation between half-life and abundance was low (Pearson R2 = 0.10), it was nonetheless significant (P < 10–4), and the eight miRNAs with the fastest turnover each had below-median abundance (Fig. 2D). These results indicated that differential stability can substantially impact miRNA accumulation.
Rates of duplex loading and passenger-strand degradation
Compared to miRNA guide strands, passenger strands have a more ephemeral existence, starting the moment of Dicer-catalyzed duplex production and lasting through the process of duplex loading and silencing-complex maturation, which culminates with either slicing of the passenger strand or its release as a single-stranded species vulnerable to rapid degradation. As such, monitoring the half-life of the passenger strand would be expected to provide a window into the rate of duplex loading and silencing-complex maturation. Complicating this picture, however, strands that most often acted as passenger strands, much more so than guide strands, exhibited a biphasic approach to steady state, shooting up rapidly at early time intervals and then increasing at a slower rate (Fig. 3A). This behavior indicated a mixture of two populations, a major one that was rapidly degraded and a minor one that was more stable. Such behavior would be expected if loading into AGO was not perfectly consistent, and strands that were normally passenger strands were occasionally loaded into AGO as guide strands and thereby stabilized. Of course, this explanation implied that the strand designated as the guide strand of these duplexes occasionally failed to load into AGO and raised the question of why these guide strands also did not exhibit a biphasic approach to steady state. The answer to this question lies with the fact that, for strands that most often acted as guide strands, the minor population was the one that was rapidly degraded, and rapidly degraded minor populations have imperceptible influence on curves describing the approach to steady state as well as the half-life values inferred from these curves, as demonstrated from simulations (Supplemental Fig. S3A). In contrast, for strands that most often acted as passenger strands, the minor population was much more stable than the major one and thus could represent a sizable fraction of the accumulating RNA. The minor population could thereby influence the shape of the curve to inflate the apparent half-life of the passenger strand such that its value did not accurately reflect the rapid rate of turnover of the majority of the passenger species (represented by the initial burst in accumulation) (Supplemental Fig. S3A).
Figure 3.
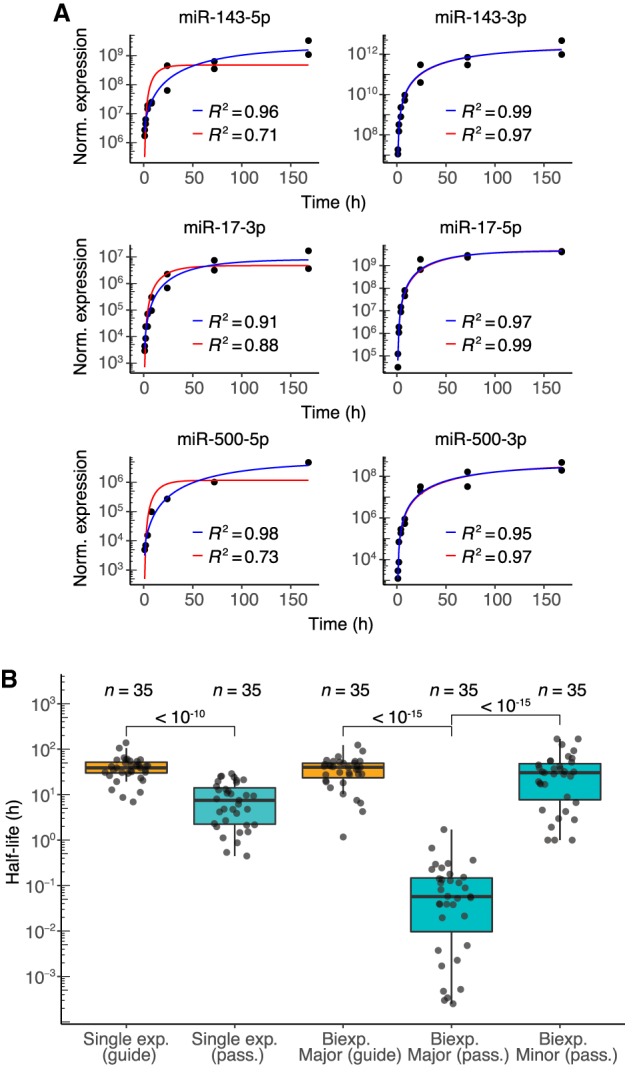
Dynamics of passenger strands in contact-inhibited MEFs. (A) Examples of fits for passenger strands and corresponding guide strands (left and right, respectively). Single- and biexponential fits are indicated (red and blue lines, respectively) with corresponding R2 values. (B) Half-life distributions for guide and passenger strands (orange and teal, respectively) obtained using either the single-exponential (single exp.) or biexponential (biexp.) fitting methods (box, quartiles; whiskers, at most 1.5 times the inter-quartile range). The major half-life was the one that corresponded to the majority of the specified strand (i.e., for the passenger strand, the molecules not incorporated into the mature silencing complex, and for the guide strand, the molecules incorporated into the mature complex), whereas the minor half-life was the one that corresponded to the minority of the specified strand. P values (two-sample t-test) are shown for the indicated comparisons.
These insights indicated that a biexponential model for the approach to steady state, which included rates for both the minor and major populations (Fig. 1B, Equation 2), was more appropriate for fitting the data of strands that usually acted as passenger strands. However, simulations revealed that fits to the biexponential model were insufficiently constrained when applied to only passenger strands; accurate determination of parameters required passenger-and-guide pairs instead to be fit simultaneously. This simultaneous fitting of passenger–guide pairs reduced the number of fitted variables by specifying a single production rate for both strands, which was a reasonable assumption when considering that both strands were produced simultaneously from the same molecule. The simultaneous fitting also constrained the fractions of passenger and guide species loaded into AGO to add up to 1.0, a reasonable assumption from the perspective that only one strand of a duplex can be loaded as the guide (although this constraint did not allow for the possibility that duplex molecules might be degraded before either strand is loaded; see Discussion). Simultaneous fitting of parameters for passenger–guide pairs enabled accurate determination of major and minor half-lives of simulated passenger species as well as major half-lives of simulated guide species (Supplemental Fig. S3B,C).
This model was then applied to 35 passenger–guide pairs in the contact-inhibited MEF data sets for which both strands surpassed our expression threshold but exhibited greater than fivefold differences in steady-state accumulation (Supplemental Table S2). For guide species deriving from each of these 35 pairs, the major half-lives resembled half-lives obtained from the single-exponential fits (Supplemental Fig. S3D). For the exceptions in which half-lives fit by the two methods deviated, the biexponential model typically predicted shorter half-lives, which was attributable to the difficulty of accurately fitting the minor half-life of these guide strands, which in turn impacted the guide strand major half-life. Thus, for guide strands, half-lives from the single-exponential fits were considered more accurate and used for subsequent analyses. The passenger-strand major half-lives, however, were significantly lower than those obtained from the single-exponential fits (P < 10−15, t-test) and almost three orders of magnitude below guide-strand major half-lives (Fig. 3B; Supplemental Table S2). Moreover, the distribution of passenger-strand minor half-lives did not significantly differ from that of the guide-strand major half-lives (Fig. 3B; P = 0.16, t-test), which implied that strands that normally act as passengers, when occasionally loaded into AGO, generally behave no differently than do strands that normally act as guides.
The major half-lives of the passenger strands, interpreted as the half-lives of these strands when they failed to load into AGO, provided insight into the rates of duplex loading and silencing-complex maturation in contact-inhibited MEF cells. The median major half-life of passenger strands was 0.057 h (Fig. 3B), which suggested that, once produced, about 5 min is typically required for the duplex to be loaded into AGO and the passenger strand to be either expelled (and subsequently degraded) or sliced. Moreover, the broad distribution of passenger-strand major half-lives, which ranged from <0.01 to 1 h, implied that the rates of duplex loading and complex maturation vary substantially for different miRNA duplexes.
Sequence differences and other features that mediate differences in guide half-lives
To address what might specify differences in guide half-lives, we assessed whether species with similar sequences had similar half-lives. MicroRNAs with the same seed region target largely overlapping sets of mRNAs and are classified as members of the same family (Bartel 2009). To assess whether these seed nucleotides also help to specify half-life, we compared differences in half-life for pairs of family members to those for random, nonfamily miRNA pairs. Half-lives were not significantly more similar for family members (Fig. 4A), which indicated that the seed sequence alone does not specify half-life. Because family members target essentially the same mRNAs, these results do not support a proposal that the sheer abundance of target mRNAs can influence miRNA stability (Chatterjee et al. 2011).
Figure 4.

Features associated with rapid miRNA turnover. (A) Differences in half-life for pairs of family members or randomly paired guides. For each pair-wise comparison of all members within each miRNA family, half-life percent differences were computed as the absolute value of the difference between half-lives, divided by the mean of the two half-lives, with this fraction then multiplied by 100. The log2-transformed percent-difference values of all such comparisons in contact-inhibited MEFs are plotted (box, quartiles; whiskers, at most 1.5 times the inter-quartile range). Twenty-five equally sized cohorts of randomly paired guides were analyzed in the same way, with the cohort shown for comparison being the one with the median value of significance (P value, two-sample t-test). (B) Relationship between half-life difference and the number of nonidentical nucleotides (Hamming distance) when comparing pairs of family members. Only nucleotides 3′ of the seed region were considered for the Hamming-distance measurement. Greater Hamming distances represent more dissimilar sequences. Significance of the correlation was determined by a t-test. (C) The effect of Cyrano on steady-state miR-7 levels in contact-inhibited MEFs. Shown is a scan of a northern blot analyzing RNA from wild-type and Cyrano-knockout contact-inhibited MEFs. For each cell line, four biological replicates were analyzed, sequentially probing for miR-17, which served as a loading control, and then for miR-7. The fold-difference in miR-7 relative to the average miR-7 level in wild-type MEFs is shown below each lane.
The failure of the seed region alone to specify half-life indicated that the remainder of the miRNA plays a role in dictating turnover. Indeed, when comparing pairs of family members, sequence similarity outside of the seed region, as measured by Hamming distance, significantly correlated with more similar rates of turnover (R2 = 0.13) (Fig. 4B). This observation did not extend to pairs of non-family-member miRNAs (R2 < 0.01) (Supplemental Fig. S4A), which indicated that, although the seed region alone is not sufficient to specify half-life, it does play a role in half-life specification. To identify portions of the nonseed region that might be especially important in driving half-life similarities, we repeated the correlation analysis for all 4-nt nonseed segments of family-member pairs. Although hamming distance and half-life similarity correlated for most 4-nt segments, the correlation peaked for the segment spanning miRNA nucleotides 13–16 (Supplemental Fig. S4B). This region of the miRNA has the greatest propensity to engage in supplemental pairing to targets, and ∼5% of conserved seed-matched sites exhibit conserved pairing to this segment (Grimson et al. 2007; Friedman et al. 2009). Together, these observations indicate that regions that preferentially engage in target pairing also help to specify half-life.
In an attempt to identify the features that specify half-life, we searched for common motifs or traits among guide strands that rapidly turned over, considering overall nucleotide content, overall dinucleotide content, position-specific k-mers 1–4 nt in length, and predicted pairing stabilities of the seed and supplemental regions. However, no consistent or striking enrichments or relationships were observed (Supplemental Fig. S4C–F). Moreover, no correlation was observed between half-life and either the abundance of all seed-matched, high-confidence predicted targets or the abundance of all such targets with extended supplemental pairing (Supplemental Fig. S4G). Our inability to identify a general feature specifying rapid miRNA turnover implied either that the causative factor had not been properly probed or that unique factors specify turnover of the different rapidly degraded species.
Of the guide strands expressed in contact-inhibited MEF cells, the least stable was miR-7a (hereafter referred to as miR-7, because miR-7b, the other miR-7 isoform, was below the detection threshold in MEFs and mESCs), with a half-life of only 1.7 h measured in our high-throughput analysis. This miRNA pairs extensively to a site within the Cyrano lncRNA, which can promote its efficient destruction through TDMD (Kleaveland et al. 2018). To assess whether Cyrano might be influencing miR-7 stability in contact-inhibited MEFs, we measured miR-7 steady-state levels and half-life in both wild-type and Cyrano-knockout cells. In the absence of Cyrano, miR-7 accumulation and half-life both increased >10-fold (Fig. 4C; Supplemental Fig. S4H), suggesting that the presence of one highly complementary target can dramatically alter the stability of a miRNA. This strong influence of a single target might explain why no correlation was observed between half-life and the overall abundance of predicted targets with supplemental pairing. When, instead, target abundance of the top n species with the most supplemental pairing to each miRNA was assessed for n from 1 to 50, however, no comparison had a Pearson R2 > 0.03. This result suggested either that we have not yet identified the Cyrano-like targets for the other rapidly turned-over guide strands or that miR-7 is an outlier in having its half-life largely shaped by its interaction with a single highly complementary target.
Different miRNA dynamics in dividing MEFs and mESCs
To get a sense of the cell-specific nature of miRNA dynamics, we extended our high-throughput, metabolic-labeling approach to both dividing MEFs and mESCs (Supplemental Table S1; Supplemental Fig. S5A–D). As observed for contact-inhibited MEFs, production rates varied broadly for individual miRNAs within each cell line—spanning three or more orders of magnitude (Supplemental Fig. S5E,F). Comparing rates of production across the different experiments reflected the degree of similarity between the cell lines compared, with contact-inhibited MEFs correlating well with dividing MEFs but hardly at all with mESCs (R2 = 0.78 and 0.078, respectively) (Supplemental Fig. S5G,H). To compare absolute rates of production in the two MEF lines, the number of miR-7 molecules in each dividing MEF cell was determined and used to calculate absolute production rates in dividing MEFs (Supplemental Fig. S2C). These analyses revealed that miR-21a-5p, the most abundant miRNA in these cells, was produced at a rate of 110 ± 50 molecules/min/cell and that seven other miRNAs were produced at rates >10 molecules/min/cell. Although this maximum production rate was approximately sixfold greater than what was observed in contact-inhibited MEFs, similar numbers of miRNAs had production rates exceeding 10 molecules/min/cell in the two cell states.
Half-lives in dividing MEFs tended to be slightly shorter than in contact-inhibited MEFs (median half-lives of 25 h and 34 h, respectively; distributions significantly different, P = 9 × 10−5, Kolmogorov–Smirnov test), and half-lives in mESCs tended to be much shorter (median, 11 h; P < 10−15) (Fig. 5A,B). In addition to an overall shift in miRNA turnover dynamics, half-lives for individual miRNA guide strands in different cell states or lines exhibited low correlation (Fig. 5C,D). For example, the correlation observed when comparing half-lives in the two MEF states was substantially lower than that observed between replicates for either cell state (Figs. 5C, 1D; Supplemental Fig. S5I), which indicated that different cell states can have significant differences in individual miRNA turnover over and above differences observed in bulk turnover. The correlation observed between cell types was even lower (Fig. 5D). A notable exception to this cell-type–specific behavior was miR-7, which had a half-life <5 h in all three data sets. Nonetheless, the observation that individual miRNAs exhibited cell-state– and cell-type–specific posttranscriptional regulation implied differential expression of miRNA-extrinsic factors that influence miRNA degradation.
Figure 5.

Dynamics of miRNA guide strands in dividing MEFs and mESCs. (A,B) Half-life distributions for guide strands in dividing MEFs (A) and mESCs (B). Half-lives were capped at the duration of the longest time interval (72 h and 24 h in dividing MEFs and mESCs, respectively), as modeling indicated that longer half-lives would be fit less accurately. (C,D) Guide-strand half-lives in dividing MEFs (C) and mESCs (D) as a function of those half-lives in contact-inhibited MEFs. (E,F) Relationship between steady-state abundance and half-life in dividing MEFs (E) and mESCs (F). Steady-state abundance is measured as reads in the 72- and 24-h input sample, respectively.
Reasoning that global differences in miRNA turnover dynamics might reflect differences in AGO turnover rates in the different cell states and cell lines, we used pulsed SILAC (Schwanhäusser et al. 2011) to measure the half-life of AGO2 in these different cellular contexts. We focused on AGO2 for two reasons: As the most highly expressed AGO family member in each of these contexts, it was presumably the one that was associated with most miRNA molecules of these cells. In addition, AGO2 was the AGO family member that could be reliably queried in all three cellular contexts using our approach. In contact-inhibited MEFs, the AGO2 half-life was 57.3 ± 1.6 h (±s.d.), whereas in dividing MEFs, the AGO2 half-life decreased to 40.9 ± 2.0 h (Supplemental Fig. S5J). For comparison, we computed an aggregate half-life for all miRNA guide strands in each cell type of interest. In both contact-inhibited MEFs and dividing MEFs, these aggregate half-lives, of 42.5 h and 19.8 h, respectively, were substantially lower than the measured AGO2 half-lives. In mESCs, the AGO2 half-life was difficult to determine because the signal from protein degradation could not be distinguished above the signal due to dilution caused by cell division (Supplemental Fig. S5J). Nonetheless, a lower limit for AGO2 half-life was 100 h (estimated based on the ability to determine half-lives up to this value with this methodology for other proteins in mESCs), which was much higher than the aggregate half-life of 8.4 h calculated for miRNA guide strands in mESCs. Thus, decreased bulk AGO2 stability failed to explain the general changes in miRNA turnover dynamics observed between cell states and cell types, which indicated that differences in other factors must be contributing to these changes. Furthermore, these comparisons showing that, on average, AGO2 is more stable than the miRNA guide strands indicated that most miRNAs are removed from AGO2 in a manner that allows the protein to be reloaded.
To assess the generality of the principles of miRNA dynamics determined from analyses of the contact-inhibited MEFs data set, we applied the same sets of analyses to the dividing MEF and mESC data sets. Abundance of miRNAs was again highly correlated with miRNA production rate and lowly correlated with miRNA degradation rate, with miR-7 in dividing MEFs being the most notable example of a miRNA for which accumulation was substantially affected by degradation rate (Fig. 5E,F; Supplemental Fig. S6A,B). Use of the biexponential model to fit passenger-strand half-lives in dividing MEFs and mESCs corroborated the stability change of over two orders of magnitude attributable to loading into AGO (Supplemental Fig. S6C,D). The median duration of duplex loading followed by passenger-strand decay in dividing MEFs and mESCs was estimated to be 29 and 21 min, respectively—somewhat greater than the 5-min value estimated in contact-inhibited MEFs (Supplemental Fig. S6C,D). Analysis of miRNA family members in dividing MEFs supported the conclusion that regions preferentially involved in target recognition play a greater role in specifying half-life (Supplemental Fig. S6E,F). The analogous analyses for guide RNAs in mESCs were less conclusive, perhaps due to lower numbers of family members and therefore less analytical power (Supplemental Fig. S6G,H). More in-depth analyses of the features that specify miRNA turnover echoed those carried out in contact-inhibited MEFs, with no motifs or commonalities discernible for miRNAs in either dividing MEFs or mESCs.
Acquisition of half-life measurements for miRNAs in dividing MEFs provided the most suitable data set for comparing our results to those that have been previously reported in mammalian cells, which were for miRNAs in dividing mouse 3T9 fibroblasts (Marzi et al. 2016). Half-lives for guide strands determined in both data sets had no correlation (R2 < 0.01) (Supplemental Fig. S7A). This result, which was unexpected given the highly correlated mRNA expression observed in the two cell lines (R2 = 0.87) (Supplemental Fig. S7B), could stem from the different experimental protocols used to determine half-lives. Approach-to-steady-state labeling with 5EU coupled to time-course normalization with quantitative 5EU-containing standards should, in theory, improve the sensitivity and accuracy of metabolic labeling as applied to miRNAs, as biotinylation of 4-thiouridine with HPDP-biotin is known to be inefficient (Duffy et al. 2015) and sample normalization with quantitative standards is more robust than normalization against a selected miRNA.
Rates of tailing and trimming, and their relationship with miRNA stability
After loading into AGO, the 3′ ends of mature miRNAs can change through the action of tailing or trimming. These alternative 3′-end isoforms accumulate over time in a manner that reflects their rates of production, their rates of conversion to other isoforms, and any changes in stability that the modified 3′ end might impart. Our analyses showed that, for most miRNAs, the initial mature species remained the most abundant throughout the time course, but for a few, the level of a tailed or trimmed isoform either approached or surpassed that of the initial isoform, as exemplified by miR-23a-3p and miR-674-5p (Fig. 6A). Classes of isoforms exhibited different behaviors for different miRNAs; although all miR-674-5p derivative isoforms accumulated to greater abundance than did the initial isoform, only the trimmed isoform did so for miR-23a-3p.
Figure 6.
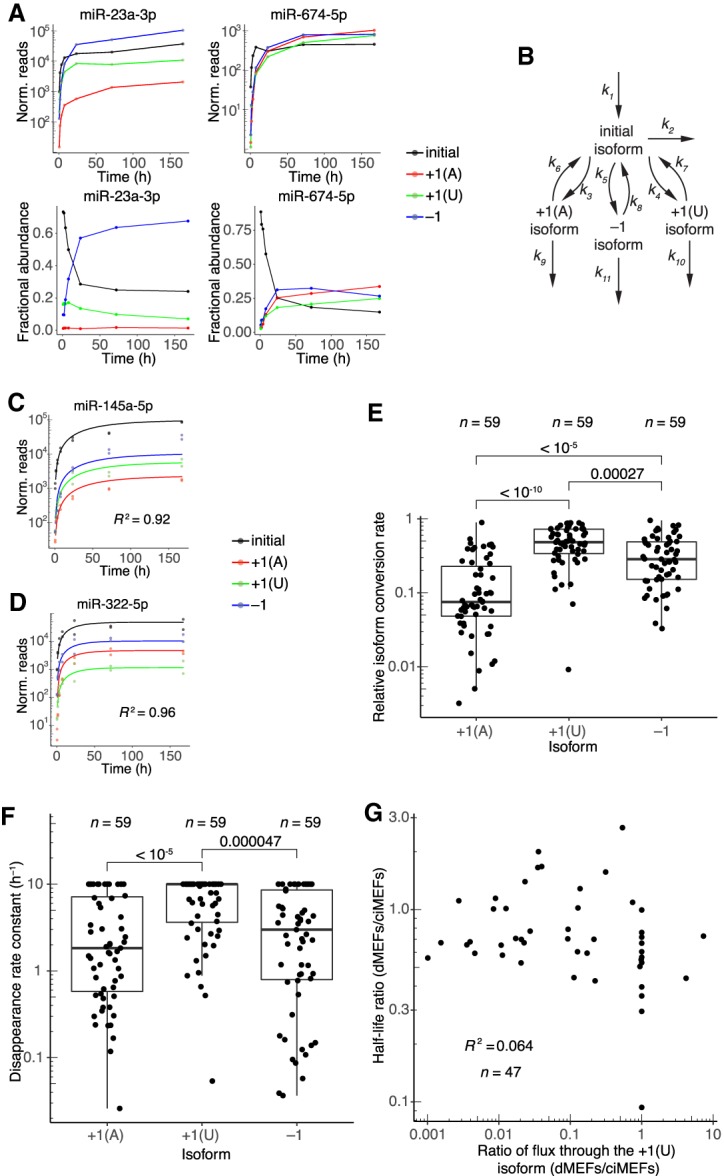
Dynamics of 3′-end isoforms. (A) Isoform accumulation for miR-23a-3p and miR-674-5p, two guide RNAs for which accumulation of derivatives of the initial isoform overtook that of the initial isoform. (Top) Absolute accumulation of the initial isoform (black) and its major derivatives, in which it is either trimmed by 1 nt (−1, blue) or tailed with either a single A (+1[A], red) or a single U (+1[U], green). Lines connect the mean values for each time point and do not represent fits to the data. (Bottom) Fractional abundance of the initial isoform and its major derivatives. (B) Schematic depicting the isoform-conversion model that was fit to the data and used for simulations. Arrows represent processes catalyzed by the cellular milieu of nucleotidyl transferases, phosphodiesterases, and other enzymes; the model is agnostic to the particular enzyme that carries out each reaction. Rate constants extracted from the fit are labeled (kn). Isoform nomenclature is as in A. (C,D) Representative examples of fits to isoform dynamics for miRNAs in contact-inhibited MEFs. Shown is the plot from the middle of the lowest quartile (miR-145a-5p, R2 = 0.92) and the plot from the middle of the highest quartile (miR-322-5p, R2 = 0.96) of goodness-of-fit (R2), when fitting data from both replicates. (E) Relative rates of conversion to the +1(A), +1(U), and −1 isoforms in contact-inhibited MEFs. Rate constants of conversion to each isoform (k3, k4, and k5 for +1(A), +1(U), and −1, respectively) were normalized to the summed conversion rate constant (k3 + k4 + k5) to generate the relative rates of conversion. Simulations indicated that conversion rate constants >1 h−1 could not be accurately fit, and thus conversion rate constants were capped at this value. Significance was evaluated with a Mann–Whitney U test, and P values are shown. (F) Rate constants for disappearance (k6 + k9, k7 + k10, or k8 + k11) of the +1(A), +1(U), and −1 isoforms, respectively, in contact-inhibited MEFs. Simulations indicated that disappearance rate constants >10 h−1 could not be accurately fit, and thus disappearance rate constants were capped at this value. Significance was evaluated with a Mann–Whitney U test, and all significant comparisons are indicated together with their P values. (G) Correlation between half-life ratios (dividing values from dividing MEFs by those from contact-inhibited MEFs) and ratios of the flux through the +1(U) isoform (again dividing values from dividing MEFs by those from contact-inhibited MEFs). Dividing MEFs are denoted as dMEFs; contact-inhibited MEFs are denoted as ciMEFs.
To assess the effect of tailing and trimming in a more global manner and to tease apart the contributions of production and degradation rates to isoform accumulation, we generated a model to describe mature miRNA production and isoform conversion (Fig. 6B). The model fit the rate constant of production of the initial isoform (k1), the rate constants of conversion to selected trimmed and tailed species (k3, k4, and k5), the rate constants of conversion of these selected species back to the initial isoform (k6, k7, and k8), and the rate constants of disappearance for each of the selected species (k2, k9, k10, and k11). The model was agnostic to what proteins carry out each of the specified steps but instead summarized the actions of potentially many nucleotidyl transferases, nucleases, and other such enzymes in one scheme. The model encompassed all major species that accumulated over the time course but excluded minor species that were detected but never reached a level to be accurately modeled. Thus, each disappearance rate constant accounted for not only decay but also conversion to any isoform not specified by the model. The model was found to accurately fit simulated data sets that spanned a wide array of production, conversion, and degradation rate constants (Supplemental Fig. S8A). These simulations showed that, although not all rate constants modeled could be fit accurately with the limited amount of data provided to the model, the model successfully determined four individual rate constants (k1, k3, k4, and k5) and the sums of three pairs of rate constants (k6 + k9, k7 + k10, k8 + k11). Application of the model to the contact-inhibited MEF data set required annotation of the mature species initially produced for each miRNA (Supplemental Table S1). These initial isoforms were annotated using the 1-h time point; any species that represented more than 20% of the reads from an arm of a miRNA hairpin were considered Dicer products. This threshold seemed reasonable given the assumption that modified species would not be able to accumulate to such levels after such a short labeling time, especially when accounting for the observed lag time of 37 min (Fig. 1B, t0 of Equation 1). Initial isoforms from 3p arms annotated in this manner were compared with published pre-miRNA sequences of Drosha products in human cells lines (Kim et al. 2017). For 77% of the miRNAs present in both data sets, the 3′ terminus of our inferred Dicer product matched the 3′ terminus of the previously reported Drosha products. The annotations that differed presumably resulted from either modification of the pre-miRNA 3′ terminus prior to Dicer cleavage or species-specific differences in Drosha processing. For the 47 miRNAs that passed the read-threshold requirement for isoform analyses in both contact-inhibited MEFs and dividing MEFs, the initial isoforms were identical in both cell states. Initial isoforms were also identical for the five miRNAs satisfying this criterion in both contact-inhibited MEFs and mESCs.
We used these annotations and the model to fit rate constants for initial-isoform production, conversion to other isoforms, and isoform disappearance (which was a combined rate constant describing isoform degradation, isoform conversion to further-modified isoforms, and isoform conversion back to the initial isoform) for all miRNAs in the contact-inhibited MEF data set that passed a threshold requirement of at least 1 read at all time points for all isoforms. The model fit the data well, and as expected, it predicted rates of initial-isoform production that were highly correlated with those determined by the single-exponential fits (Fig. 6C,D; Supplemental Fig. S8B,C). Relative rates of conversion to each isoform for individual miRNAs spanned over two orders of magnitude, indicating a high degree of variation in the preferred modification trajectory for different miRNAs (Fig. 6E). As a general trend, however, trimming was faster than A tailing but slower than U tailing (Fig. 6E). Rate constants of disappearance predicted by the model also spanned up to two orders of magnitude for some isoforms, indicating differential responses for individual miRNAs to modifications (Fig. 6F). Disappearance rate constants were largest for U-tailed species, indicating that species containing this modification were destabilized relative to species containing other modifications in contact-inhibited MEFs (Fig. 6F). Rate constants of isoform conversion and isoform disappearance for a given isoform were correlated; miRNAs with relatively high rate constants for conversion to the A-tailed species also had relatively high rate constants of disappearance for this species (Supplemental Fig. S8D). Correlation of these parameters was not observed in fits to simulated data, and thus these correlations did not appear to be an artifact of the model but rather could represent a real biological phenomenon, such as a coupling or processivity between modification events.
To assess the generalizability of these observations across multiple cell states, we fit the isoform model to data from the dividing MEFs. The parameters fit to these data revealed that global trends in isoform dynamics resembled those observed for contact-inhibited MEFs; in particular, conversion to the U-tailed isoform generally proceeded with the fastest rates, and these U-tailed species were generally less stable than were the other isoforms (Supplemental Fig. S8E,F). At the level of individual miRNAs, rate constants of isoform disappearance were correlated between the two data sets, but rate constants of isoform conversion were less so, as exemplified for the −1 isoform (R2 = 0.44 and 0.17, respectively) (Supplemental Fig. S8G,H). Both the conversion and disappearance rate constants tended to be somewhat decreased in dividing MEFs as compared to contact-inhibited MEFs (Supplemental Fig. S8G,H). Correlations between changes in isoform dynamics and changes in half-lives for miRNAs in these two cell states were examined to assess the relationship between flux through isoforms and miRNA turnover. The strongest correlation was observed between the change in flux through the + 1(U) isoform and the change in half-life (R2 = 0.064) (Fig. 6G). Reasoning that a stronger correlation might have been masked by the inability to accurately determine rate constants above certain values (as determined from the isoform dynamics simulations), we repeated these analyses after removing all species with capped rate constants but still observed only very low R2 values (≤0.072). These low coefficients of determination, in addition to the observations that general flux through isoforms was decreased whereas miRNA turnover rate was increased in dividing MEFs, implied substantial independence between the miRNA modification and degradation pathways.
Discussion
Our global analyses of miRNA metabolism with 5EU provided half-life measurements for the guide strands of 201 miRNAs in unperturbed MEFs and 127 miRNAs in unperturbed mESCs. These measurements showed that in these cells most miRNAs are long-lived, which confirmed the prevailing view of miRNA stability previously drawn from low-throughput analyses of a few miRNAs as well as broader analyses of cells perturbed with transcriptional inhibitors. The median half-life observed for miRNAs in dividing MEFs was 25 h—a value substantially greater than the 2.2-h median observed for mRNA half-lives in a similar cell type (dividing NIH3T3 cells) (Eisen et al. 2019b). This >10-fold difference implies that levels of mRNAs can change more rapidly than those of miRNAs, making mRNAs much more adept at responding quickly to rapid environmental changes or other signaling cues and relegating miRNAs to a more supporting role in lowering the half-lives of many mRNAs, thereby helping mRNAs to achieve this more rapid response. The long half-lives generally observed for miRNAs are nonetheless suitable for changes over longer time frames, such as those typically operating over the course of mammalian development, and they provide regulatory stability to the cell. Perhaps most importantly, they enable miRNAs to reach the high intracellular levels needed to impart consequential regulation, which are typically >1000 molecules per cell—levels much higher than the median mRNA level of ∼17 molecules per cell for mRNAs with detectable protein products (Brown et al. 2007; Schwanhäusser et al. 2011; Denzler et al. 2016).
Although most miRNAs were long-lived, our analysis identified a few with half-lives resembling those of intermediate- to long-lived mRNAs (1.7–10 h), including miR-503, a miRNA whose shorter half-life is proposed to facilitate its role in facilitating exit from cell-cycle arrest (Rissland et al. 2011). Among these relatively short-lived miRNAs, the mechanism of destabilization is known for only one, miR-7, a TDMD substrate (Kleaveland et al. 2018). Indeed, in contact-inhibited MEFs, removal of the Cyrano lncRNA, which triggers miR-7 TDMD, substantially increased miR-7 half-life and steady-state accumulation. Although the mechanism of destabilization of the other short-lived miRNAs is unknown, the observation that the miRNA nucleotides most important for target recognition also appeared to be most important for dictating half-life suggested a role for target pairing, as observed for miR-7 destabilization. A broader role for miRNA–target interactions, and more specifically TDMD, in mediating miRNA stability is supported by other known instances of endogenous TDMD (Bitetti et al. 2018; Ghini et al. 2018) as well as early observations in HEK293T cells, in which the presence of a highly complementary target decreases miR-223 half-life from 46 to 19 h (Baccarini et al. 2011).
Combining metabolic labeling with approach-to-steady-state kinetics enabled production rate constants to be determined alongside of decay rate constants, which provided a more complete understanding of miRNA dynamics than typically achieved using pulse-chase kinetics. These rate constants revealed that miRNAs can be produced at impressively rapid rates, both in proliferative as well as nonproliferative cells. The most abundant miRNA in dividing MEFs, miR-21a-5p, was produced at a rate of 110 ± 50 copies/cell/min. Even at a production rate of 10 copies/cell/min, the rate of miR-21a production would be faster than that of the most rapidly produced mRNA in NIH3T3 fibroblasts and comparable to that of pre-rRNA production from a single pre-rRNA locus in HeLa cells (Schwanhäusser et al. 2011; Turowski and Tollervey 2015). When considering that miR-21a is transcribed from two alleles of a single locus and that RNA polymerase II (RNAPII) elongates with a rate constant of ∼4.3 kb/min and has a footprint of ∼40–50 nt (Darst et al. 1991; Rice et al. 1993; Darzacq et al. 2007; Krebs et al. 2017), the rate of 110 copies/cell/min implies that the Mir21a locus is coated in elongating RNAPII at ∼50% of its maximum density. Such efficient RNAPII recruitment is presumably challenging, and greater than twofold more efficient recruitment would be impossible, which helps explain why some highly expressed miRNAs are transcribed from multiple genes—the extreme being miR-430, which makes up 99% of the miRNA in the early zebrafish embryo and is transcribed from an array of >90 genes (Giraldez et al. 2005; Wei et al. 2012).
Our experiments also provided insight into previously inaccessible portions of the mammalian miRNA life cycle. Investigation of the dynamics of passenger-strand turnover, using the biexponential fit designed to distinguish between molecules acting as passenger strands and those loaded as guides into AGO, revealed that the combined processes of duplex loading and silencing-complex maturation, with slicing or expulsion of the passenger strand, can occur in <1 min and typically within 30 min. These results indicate that the much longer lag time observed for the action of synthetic siRNA duplexes arises from other steps of that pathway, such as entry into cells and endosome release (Wittrup et al. 2015). Despite the ability of AGO to stabilize miRNA guide strands (Winter and Diederichs 2011), we observed that AGO2 often outlived the miRNA, especially in mESCs, indicating that most miRNAs can be degraded in a way that allows AGO2 to be recycled for use with another miRNA.
The biexponential model applied to assess passenger-strand dynamics assumed that one strand from every duplex succeeded in being loaded into AGO, with no degradation of miRNA duplexes prior to loading into AGO. However, in the context of miRNA overexpression, increasing AGO expression leads to increased miRNA abundance (Diederichs and Haber 2007), suggesting that in conditions of artificially increased miRNA levels surplus duplex exists and is degraded before either strand can be loaded as the guide. Although the prevalence of duplex degradation in the context of endogenous miRNA levels is unknown, the concentration of free AGO might be limiting for at least some miRNA duplexes in the cell. The observation that guide and passenger strands have such divergent half-lives implies that such duplexes, if they exist, degrade rapidly compared to the degradation rate of loaded guide strand, in which case degradation of unloaded duplex would not be expected to distort the guide-strand half-life values. With respect to passenger-strand values, if the unloaded duplexes degrade more rapidly than the time required for duplex loading and silencing-complex maturation, then the premature duplex degradation would reduce our passenger-strand half-life values but not substantially (unless a large fraction of the duplex was degraded before loading). Conversely, if the unloaded duplexes degrade less rapidly than the time required for duplex loading and silencing-complex maturation, then the duplex degradation would increase our passenger-strand half-life values, and the values that we report would represent upper limits on the time required for duplex loading and silencing-complex maturation.
The recent report of miRNA production rates and half-lives in S2 cells provides the opportunity to compare the results obtained in Drosophila cells with those that we obtained in mammalian cells. As with mammalian miRNAs, Drosophila miRNAs can be produced very rapidly, as fast as 228 ± 48 molecules/min/cell, whereas incorporation into AGO to form the mature silencing complex appears to take longer, typically more than 1 h in S2 cells (Reichholf et al. 2019). Half-lives vary between miRNAs in Drosophila cells, as they do in mammalian cells, with the median half-live of 11.4 h observed in S2 cells resembling that which we observed in mESCs (Reichholf et al. 2019).
Some studies have reported effects of certain terminal modifications on individual miRNAs (Jones et al. 2009; Katoh et al. 2009; Mansur et al. 2016), and in many systems, guide-RNA degradation observed upon loss of terminal 2′-O methylation is associated with tailing or trimming (Li et al. 2005; Ameres et al. 2010; Kamminga et al. 2010, 2012; Lim et al. 2015). However, the overall role of tailing and trimming in the degradation pathway of mature metazoan miRNAs had not been investigated. Here, we applied a model of isoform dynamics to extract rate parameters for mature miRNA production, mature miRNA conversion to tailed and trimmed species, as well as disappearance of those tailed and trimmed species. The broad spread of rate constants for each of these processes reflected the miRNA-specific nature of isoform dynamics, with different miRNAs differentially acquiring terminal modifications and then differentially responding to these modifications. Despite this variation, the addition of a single U both occurred at the fastest rate and was associated with the greatest degree of destabilization. Nonetheless, changes in isoform dynamics observed between MEF cell states poorly reflected the changes in miRNA half-lives, which implied substantial independence of the two pathways.
This implied independence contrasts with findings that U-tailing is on-pathway for degradation of the let-7 family of pre-miRNAs in mammals, as well as unmethylated miRNAs in plants and unmethylated piRNAs in zebrafish (Yu et al. 2005; Ibrahim et al. 2010; Kamminga et al. 2010; Heo et al. 2012; Ren et al. 2012; Zhao et al. 2012). As our isoform analyses were carried out only on MEF data sets, we cannot eliminate the possibility that changes in tailing and trimming might play a role more globally in altering stabilities in different cell types. Furthermore, as we could not fit the rate constants for isoform disappearance and back-conversion independently, it is also possible that more significant correlations between differences in miRNA stability and either of these parameters could have been obscured. Nonetheless, the proposed independence of the two pathways agrees with observations that loss of the noncanonical poly(A) polymerase TENT2 (GLD2/PAPD4) does not systematically change miRNA levels in the mouse hippocampus or in THP-1 cells (Burroughs et al. 2010; Mansur et al. 2016). Additionally, although TENT2 loss substantially diminishes tailing of miR-7, it does not impede Cyrano-mediated TDMD (Kleaveland et al. 2018). Taken together, our results and the TENT2 observations suggest that tailed and trimmed species do not represent intermediates on the miRNA decay pathway for all miRNAs but do not exclude the possibility that, for select miRNAs, tailing or trimming might accelerate degradation.
Comparing rates of miRNA turnover in contact-inhibited MEFs, dividing MEFs, and mESCs revealed both specific changes in rates of turnover of individual miRNAs as well as general changes in miRNA turnover rates, as illustrated by the decreases in the median half-life from 34 h in contact-inhibited MEFs to 25 h in dividing MEFs and 11 h in mESCs. Both the miRNA-specific and the more general differences implied roles for miRNA-extrinsic factors in regulating half-life. With respect to miRNA-specific differences, the observation that miRNA nucleotides most important for target interaction were also most associated with shorter half-lives suggested a role for targets in this specification, although with the exception of miR-7 and a few other established TDMD substrates, further studies will be required to identify targets responsible for these differences. With respect to the general differences observed between the cell states and cell types, the differential activities of more broad-spectrum but as-yet-unidentified decay factors might mediate the differences. The faster dynamics exhibited by miRNAs in mESCs as compared to those in either MEF state presumably poises mESCs for differentiation and the more rapid changes in miRNA levels that this entails. The discovery of both miRNA-specific and more general differences in miRNA dynamics will facilitate identification of the miRNA-extrinsic factors that mediate these differences. It also lays the foundation for further exploration of regulated miRNA turnover and how it interfaces with regulation of miRNA production to help drive or reinforce biological transitions.
Methods
5EU labeling and cell collection
For experiments examining contact-inhibited MEFs, cells were plated in 15-cm dishes and allowed to reach confluency. Cells were then left confluent for 4 d, with media changes every 2–3 d. On the fifth day of confluency, a media change was performed, which was timed such that all plates that were to be collected ≤ 24 h after 5EU addition were last fed 24 h prior to collection. The following day, 5EU (Jena Bioscience) was added to the culture media to a final concentration of 400 µM, and cells were collected 0, 1, 2, 4, 8, 24, 72, and 168 h later. For time intervals longer than 24 h, the media and 5EU were refreshed every 24 h, timed such that the last feeding was 24 h prior to collection. A total of 14 dishes were plated for each experiment, with one plate each for the 0-, 24-, 72-, and 168-h time intervals, two plates each for the 4- and 8-h time intervals, and three plates each for the 1- and 2-h time intervals.
For the experiments examining dividing MEFs, cells for each time point were plated 24 h in advance of collection at a density of either 1.25 million (replicate 1) or 1 million (replicate 2) cells per 15-cm plate. 5EU was added to a final concentration of 400 µM, and cells were collected at 0, 1, 2, 4, 8, 24, and 72 h. During the 72-h time interval, cells were split and replated daily with fresh media and 5EU. The same numbers of plates were used for each time interval as were used for the contact-inhibited MEFs.
For the first mESC replicate, cells for each time point were plated 48 h in advance of collection at a density of 200,000 cells per 15-cm dish. The next day, media was changed for the 0-, 1-, 2-, 4-, and 8-h time intervals 24 h prior to planned collection. Media was also changed 24 h in advance for the 16- and 24-h time intervals, and 5EU was added to a final concentration of 125 µM. The following day, 5EU was added at the same concentration for the remaining time intervals, and cells were collected. The second mESC replicate was performed in the same way, except cells were plated 26 h in advance of collection at a density of 750,000 cells per 15-cm dish.
For all experiments, cell collection proceeded by adding TRI Reagent (Life Technologies, 3 mL per 15-cm dish), scraping the cells off the plate, and then transferring this mixture to a 15-mL conical tube which was then snap-frozen and stored at –80°C. For the dividing MEFs, cells were washed with PBS prior to the addition of TRI Reagent. For the mESCs, cells were similarly washed with –CaCl2, –MgCl2 PBS (Gibco).
Biotinylation and pulldown
Biotin was attached to metabolically labeled RNAs in a 10- to 20-µL reaction with 4 mM biotin disulfide azide (Click Chemistry Tools), 5 mM CuSO4 (Sigma-Aldrich), 5 mM THPTA (Click Chemistry Tools), 20 mM sodium L-ascorbate (Sigma-Aldrich), and 50 mM HEPES, pH 7.5. After incubating for 1 h at room temperature with protection from light, the reaction was then quenched with 5 mM EDTA, and RNA was extracted using phenol chloroform (Sigma-Aldrich) and precipitated. For the pulldown, 100 µL MyOne Streptavidin C1 bead slurry (Life Technologies) were used for every 25 µg RNA in the click reaction. The beads were washed twice with B&W buffer (10 mM Tris, pH 7.5, 1 mM EDTA, 2 M NaCl, and 0.01% TWEEN 20), twice with solution A (0.1 M NaOH, 0.05 M NaCl, 0.01% TWEEN 20), twice with solution B (0.1M NaCl, 0.01% TWEEN 20), and twice with water. Beads were then blocked for 30 min at room temperature on an end-over-end rotator with 0.5 µg/µL of yeast total RNA diluted in high-salt wash buffer (HSWB) (10 mM Tris, pH 7.5, 1 mM EDTA, 100 mM NaCl, and 0.01% TWEEN 20). Blocked beads were then washed three times with HSWB. The RNA pellet was dissolved in HSWB (100 µL per 100 µL of beads used), and this solution was used to resuspend the blocked and washed beads. After incubation on an end-over-end rotator for 30 min at room temperature, beads were washed twice with 50°C water, and then twice with 50°C 10× HSWB. During the final wash, beads were transferred to a new tube, the final wash was removed, Tris(2-carboxyethyl)phosphine hydrochloride (TCEP, Sigma-Aldrich) was added (200 µL per 100 µL beads), and the beads were incubated on an end-over-end rotator at 50°C. After 20 min, the TCEP eluate was moved to a new tube, the beads were washed once with 150 µL of water, the TCEP eluate and wash were pooled, NaCl was added to a concentration of 0.3 M, and RNA was precipitated with 2.5 volumes of 100% ethanol. Small RNA libraries were then prepared from these precipitated samples and submitted for sequencing as described in the Supplemental Material (Supplemental Table S3).
Fitting models to data
MicroRNAs were filtered for expression, requiring at least 60 reads per million total reads aligned to miRNAs for every time point. Data for miRNAs passing this threshold were normalized to the sum of the three 5EU-containing quantitative standards. The data were additionally normalized by miRNA levels observed in the input libraries for each time interval, which accounted for fluctuations in steady-state levels of miRNAs over the course of the experiment. When combining data from replicates, data were batch normalized to account for differences in the preparation of the quantitative standards used for different replicates, and then the points for each replicate were plotted on the same graph and fit with a single curve. For MEFs, if only one replicate passed the expression threshold, it was carried forward in isolation, whereas for mESCs, due to the more variable nature of these libraries, analysis focused on miRNAs passing the expression filter in both replicates. Both these combined data sets as well as data from individual replicates were then fit to the single-exponential models. Pairs of guide and passenger strands were selected as those for which (1) both strands mapped to a unique genomic locus, (2) both strands passed the expression filter of 60 reads per million miRNA reads at each time point, and (3) the steady-state level of the guide strand was at least fivefold greater than that of the passenger strand. For all analyses, half-lives were calculated as ln(2) divided by the degradation rate constant. For computational details concerning all fitting, see the Supplemental Methods.
The single-exponential model with a time offset treated the bioavailability of 5EU as a step function that jumped from zero to steady-state 5EU levels at a specified time after 5EU addition. Modeling this bioavailability instead as an increasing exponential function in which 5EU reached half its steady-state availability after 1 h yielded nearly identical production rates and half-lives (Supplemental Fig. S1F,G). Lacking justification for this more complex model of bioavailability, we went forward with the single-exponential model with the simple time offset.
For isoform analyses, miRNA behavior across time was decomposed into time courses for individual isoforms. Due to sequencing depth and the low abundance of miRNA isoforms, for most miRNAs only the initial biogenesis products and their +1(A), +1(U), and −1 isoforms had substantial signal across the time course. All species with more than one raw read for these isoforms at each time interval were fit with the isoform dynamics model, using combined data from replicates with batch normalization.
Calculating aggregate miRNA half-life
For comparison to AGO2 half-lives, a single aggregate half-life was calculated for the combination of all miRNAs in a given cell state or type. To generate the appropriate combination of all guide strands, we had to account for the slight U bias attributable to the pulldown procedure. This correction was accomplished with a U-bias scaling factor determined from the ratio of the measured steady-state value (determined from abundance in the input sample) to the fit steady-state value for each miRNA. The alpha parameters for each guide strand were scaled by this factor and then, in combination with the beta parameters, were used to determine the ratio of 5EU-labeled to unlabeled miRNAs at each time point, which represented the pool of miRNAs synthesized in the time following 5EU addition relative to that synthesized prior to 5EU addition. These ratios were then fit in a manner identical to the SILAC heavy-to-light ratios to extract the representative miRNA half-life (see Supplemental Methods).
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE129431.
Supplementary Material
Acknowledgments
We thank members of the Bartel lab for helpful discussions; T. Eisen, J. Kwasnieski, S. McGeary, and K. Lin for computational advice; B. Kleaveland for sharing of reagents and samples; and the Whitehead Institute Genome Technology Core for sequencing. This work was supported by National Institutes of Health grant GM118135 (D.P.B.) and a National Science Foundation predoctoral GRFP fellowship (E.R.K.). D.P.B. is an investigator of the Howard Hughes Medical Institute.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.251421.119.
References
- Ameres SL, Zamore PD. 2013. Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol 14: 475–488. 10.1038/nrm3611 [DOI] [PubMed] [Google Scholar]
- Ameres SL, Horwich MD, Hung J-H, Xu J, Ghildiyal M, Weng Z, Zamore PD. 2010. Target RNA–directed trimming and tailing of small silencing RNAs. Science 328: 1534–1539. 10.1126/science.1187058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccarini A, Chauhan H, Gardner TJ, Jayaprakash AD, Sachidanandam R, Brown BD. 2011. Kinetic analysis reveals the fate of a microRNA following target regulation in mammalian cells. Curr Biol 21: 369–376. 10.1016/j.cub.2011.01.067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bail S, Swerdel M, Liu H, Jiao X, Goff LA, Hart RP, Kiledjian M. 2010. Differential regulation of microRNA stability. RNA 16: 1032–1039. 10.1261/rna.1851510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233. 10.1016/j.cell.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. 2018. Metazoan microRNAs. Cell 173: 20–51. 10.1016/j.cell.2018.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskerville S, Bartel DP. 2005. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA 11: 241–247. 10.1261/rna.7240905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaude O. 2011. Inhibiting eukaryotic transcription. Which compound to choose? How to evaluate its activity? Transcription 2: 103–108. 10.4161/trns.2.3.16172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitetti A, Mallory AC, Golini E, Carrieri C, Gutiérrez HC, Perlas E, Pérez-Rico YA, Tocchini-Valentini GP, Enright AJ, Norton WHJ, et al. 2018. MicroRNA degradation by a conserved target RNA regulates animal behavior. Nat Struct Mol Biol 25: 244–251. 10.1038/s41594-018-0032-x [DOI] [PubMed] [Google Scholar]
- Brown BD, Gentner B, Cantore A, Colleoni S, Amendola M, Zingale A, Baccarini A, Lazzari G, Galli C, Naldini L. 2007. Endogenous microRNA can be broadly exploited to regulate transgene expression according to tissue, lineage and differentiation state. Nat Biotechnol 25: 1457–1467. 10.1038/nbt1372 [DOI] [PubMed] [Google Scholar]
- Burroughs AM, Ando Y, de Hoon MJ, Tomaru Y, Nishibu T, Ukekawa R, Funakoshi T, Kurokawa T, Suzuki H, Hayashizaki Y, et al. 2010. A comprehensive survey of 3′ animal miRNA modification events and a possible role for 3′ adenylation in modulating miRNA targeting effectiveness. Genome Res 20: 1398–1410. 10.1101/gr.106054.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazalla D, Yario T, Steitz JA. 2010. Down-regulation of a host microRNA by a Herpesvirus saimiri noncoding RNA. Science 328: 1563–1566. 10.1126/science.1187197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SH, Tang Y, Miao L, Darwich-Codore H, Vejnar CE, Beaudoin J-D, Musaev D, Fernandez JP, Benitez MDJ, Bazzini AA, et al. 2019. Brd4 and P300 confer transcriptional competency during zygotic genome activation. Dev Cell 49: 867–881.e8. 10.1016/j.devcel.2019.05.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang T-C, Pertea M, Lee S, Salzberg SL, Mendell JT. 2015. Genome-wide annotation of microRNA primary transcript structures reveals novel regulatory mechanisms. Genome Res 25: 1401–1409. 10.1101/gr.193607.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Fasler M, Büssing I, Großhans H. 2011. Target-mediated protection of endogenous microRNAs in C. elegans. Dev Cell 20: 388–396. 10.1016/j.devcel.2011.02.008 [DOI] [PubMed] [Google Scholar]
- Chen W, Qin C. 2015. General hallmarks of microRNAs in brain evolution and development. RNA Biol 12: 701–708. 10.1080/15476286.2015.1048954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darst SA, Edwards AM, Kubalek EW, Kornberg RD. 1991. Three-dimensional structure of yeast RNA polymerase II at 16 Å resolution. Cell 66: 121–128. 10.1016/0092-8674(91)90144-N [DOI] [PubMed] [Google Scholar]
- Darzacq X, Shav-Tal Y, De Turris V, Brody Y, Shenoy SM, Phair RD, Singer RH. 2007. In vivo dynamics of RNA polymerase II transcription. Nat Struct Mol Biol 14: 796–806. 10.1038/nsmb1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Mata M, Gaidatzis D, Vitanescu M, Stadler MB, Wentzel C, Scheiffele P, Filipowicz W, Großhans H. 2015. Potent degradation of neuronal miRNAs induced by highly complementary targets. EMBO Rep 16: 500–511. 10.15252/embr.201540078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denzler R, McGeary SE, Title AC, Agarwal V, Bartel DP, Stoffel M. 2016. Impact of microRNA levels, target-site complementarity, and cooperativity on competing endogenous RNA-regulated gene expression. Mol Cell 64: 565–579. 10.1016/j.molcel.2016.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diederichs S, Haber DA. 2007. Dual role for argonautes in microRNA processing and posttranscriptional regulation of microRNA expression. Cell 131: 1097–1108. 10.1016/j.cell.2007.10.032 [DOI] [PubMed] [Google Scholar]
- Dölken L, Ruzsics Z, Rädle B, Friedel CC, Zimmer R, Mages J, Hoffmann R, Dickinson P, Forster T, Ghazal P, et al. 2008. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA 14: 1959–1972. 10.1261/rna.1136108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy EE, Rutenberg-Schoenberg M, Stark CD, Kitchen RR, Gerstein MB, Simon MD. 2015. Tracking distinct RNA populations using efficient and reversible covalent chemistry. Mol Cell 59: 858–866. 10.1016/j.molcel.2015.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen TJ, Eichhorn SW, Subtelny AO, Bartel DP. 2019a. MicroRNAs cause accelerated decay of short-tailed target mRNAs. bioRxiv 10.1101/763367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen TJ, Eichhorn SW, Subtelny AO, Lin KS, McGeary SE, Gupta S, Bartel DP. 2019b. The dynamics of cytoplasmic mRNA metabolism. bioRxiv 10.1101/763599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman RC, Farh KK-H, Burge CB, Bartel DP. 2009. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19: 92–105. 10.1101/gr.082701.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantier MP, McCoy CE, Rusinova I, Saulep D, Wang D, Xu D, Irving AT, Behlke MA, Hertzog PJ, Mackay F, et al. 2011. Analysis of microRNA turnover in mammalian cells following Dicer1 ablation. Nucleic Acids Res 39: 5692–5703. 10.1093/nar/gkr148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghini F, Rubolino C, Climent M, Simeone I, Marzi MJ, Nicassio F. 2018. Endogenous transcripts control miRNA levels and activity in mammalian cells by target-directed miRNA degradation. Nat Commun 9: 3119 10.1038/s41467-018-05182-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldez AJ, Cinalli RM, Glasner ME, Enright AJ, Thomson JM, Baskerville S, Hammond SM, Bartel DP, Schier AF. 2005. MicroRNAs regulate brain morphogenesis in zebrafish. Science 308: 833–838. 10.1126/science.1109020 [DOI] [PubMed] [Google Scholar]
- Grimson A, Farh KK-H, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. 2007. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell 27: 91–105. 10.1016/j.molcel.2007.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Liu J, Elfenbein SJ, Ma Y, Zhong M, Qiu C, Ding Y, Lu J. 2015. Characterization of the mammalian miRNA turnover landscape. Nucleic Acids Res 43: 2326–2341. 10.1093/nar/gkv057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha M, Kim VN. 2014. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 15: 509–524. 10.1038/nrm3838 [DOI] [PubMed] [Google Scholar]
- Han L, Witmer PDW, Casey E, Valle D, Sukumar S. 2007. DNA methylation regulates microRNA expression. Cancer Biol Ther 6: 1290–1294. 10.4161/cbt.6.8.4486 [DOI] [PubMed] [Google Scholar]
- Heo I, Joo C, Cho J, Ha M, Han J, Kim VN. 2008. Lin28 mediates the terminal uridylation of let-7 precursor microRNA. Mol Cell 32: 276–284. 10.1016/j.molcel.2008.09.014 [DOI] [PubMed] [Google Scholar]
- Heo I, Ha M, Lim J, Yoon M-J, Park J-E, Kwon SC, Chang H, Kim VN. 2012. Mono-uridylation of pre-microRNA as a key step in the biogenesis of group II let-7 microRNAs. Cell 151: 521–532. 10.1016/j.cell.2012.09.022 [DOI] [PubMed] [Google Scholar]
- Herzog VA, Reichholf B, Neumann T, Rescheneder P, Bhat P, Burkard TR, Wlotzka W, von Haeseler A, Zuber J, Ameres SL. 2017. Thiol-linked alkylation of RNA to assess expression dynamics. Nat Methods 14: 1198–1204. 10.1038/nmeth.4435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houbaviy HB, Murray MF, Sharp PA. 2003. Embryonic stem cell-specific microRNAs. Dev Cell 5: 351–358. 10.1016/S1534-5807(03)00227-2 [DOI] [PubMed] [Google Scholar]
- Ibrahim F, Rymarquis LA, Kim E-J, Becker J, Balassa E, Green PJ, Cerutti H. 2010. Uridylation of mature miRNAs and siRNAs by the MUT68 nucleotidyltransferase promotes their degradation in Chlamydomonas. Proc Natl Acad Sci 107: 3906–3911. 10.1073/pnas.0912632107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jao CY, Salic A. 2008. Exploring RNA transcription and turnover in vivo by using click chemistry. Proc Natl Acad Sci 105: 15779–15784. 10.1073/pnas.0808480105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MR, Quinton LJ, Blahna MT, Neilson JR, Fu S, Ivanov AR, Wolf DA, Mizgerd JP. 2009. Zcchc11-dependent uridylation of microRNA directs cytokine expression. Nat Cell Biol 11: 1157–1163. 10.1038/ncb1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamminga LM, Luteijn MJ, Den Broeder MJ, Redl S, Kaaij LJ, Roovers EF, Ladurner P, Berezikov E, Ketting RF. 2010. Hen1 is required for oocyte development and piRNA stability in zebrafish. EMBO J 29: 3688–3700. 10.1038/emboj.2010.233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamminga LM, Van Wolfswinkel JC, Luteijn MJ, Kaaij LJ, Bagijn MP, Sapetschnig A, Miska EA, Berezikov E, Ketting RF, Kim SK. 2012. Differential impact of the HEN1 homolog HENN-1 on 21U and 26G RNAs in the germline of Caenorhabditis elegans. PLoS Genet 8: e1002702 10.1371/journal.pgen.1002702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh T, Sakaguchi Y, Miyauchi K, Suzuki T, Kashiwabara S-i, Baba T, Suzuki T. 2009. Selective stabilization of mammalian microRNAs by 3′ adenylation mediated by the cytoplasmic poly(A) polymerase GLD-2. Genes Dev 23: 433–438. 10.1101/gad.1761509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamata T, Tomari Y. 2010. Making RISC. Trends Biochem Sci 35: 368–376. 10.1016/j.tibs.2010.03.009 [DOI] [PubMed] [Google Scholar]
- Khvorova A, Reynolds A, Jayasena SD. 2003. Functional siRNAs and miRNAs exhibit strand bias. Cell 115: 209–216. 10.1016/S0092-8674(03)00801-8 [DOI] [PubMed] [Google Scholar]
- Kim B, Jeong K, Kim VN. 2017. Genome-wide mapping of DROSHA cleavage sites on primary microRNAs and noncanonical substrates. Mol Cell 66: 258–269.e5. 10.1016/j.molcel.2017.03.013 [DOI] [PubMed] [Google Scholar]
- Kleaveland B, Shi CY, Stefano J, Bartel DP. 2018. A network of noncoding regulatory RNAs acts in the mammalian brain. Cell 174: 350–362.e17. 10.1016/j.cell.2018.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs AR, Imanci D, Hoerner L, Gaidatzis D, Burger L, Schübeler D. 2017. Genome-wide single-molecule footprinting reveals high RNA polymerase II turnover at paused promoters. Mol Cell 67: 411–422.e4. 10.1016/j.molcel.2017.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krol J, Busskamp V, Markiewicz I, Stadler MB, Ribi S, Richter J, Duebel J, Bicker S, Fehling HJ, Schübeler D, et al. 2010. Characterizing light-regulated retinal microRNAs reveals rapid turnover as a common property of neuronal microRNAs. Cell 141: 618–631. 10.1016/j.cell.2010.03.039 [DOI] [PubMed] [Google Scholar]
- Kwasnieski JC, Orr-Weaver TL, Bartel DP. 2019. Early genome activation in Drosophila is extensive with an initial tendency for aborted transcripts and retained introns. Genome Res 29: 1188–1197. 10.1101/gr.242164.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Jeon K, Lee JT, Kim S, Kim VN. 2002. MicroRNA maturation: stepwise processing and subcellular localization. EMBO J 21: 4663–4670. 10.1093/emboj/cdf476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EJ, Gusev Y, Jiang J, Nuovo GJ, Lerner MR, Frankel WL, Morgan DL, Postier RG, Brackett DJ, Schmittgen TD. 2007. Expression profiling identifies microRNA signature in pancreatic cancer. Int J Cancer 120: 1046–1054. 10.1002/ijc.22394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Yang Z, Yu B, Liu J, Chen X. 2005. Methylation protects miRNAs and siRNAs from a 3′-end uridylation activity in Arabidopsis. Curr Biol 15: 1501–1507. 10.1016/j.cub.2005.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SL, Qu ZP, Kortschak RD, Lawrence DM, Geoghegan J, Hempfling A-L, Bergmann M, Goodnow CC, Ormandy CJ, Wong L, et al. 2015. HENMT1 and piRNA stability are required for adult male germ cell transposon repression and to define the spermatogenic program in the mouse. PLoS Genet 11: e1005620 10.1371/journal.pgen.1005620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugowski A, Nicholson B, Rissland OS. 2018. Determining mRNA half-lives on a transcriptome-wide scale. Methods 137: 90–98. 10.1016/j.ymeth.2017.12.006 [DOI] [PubMed] [Google Scholar]
- Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, Teruya-Feldstein J, Reinhardt F, Onder TT, Valastyan S, et al. 2010. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol 12: 247–256. 10.1038/ncb2024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansur F, Ivshina M, Gu W, Schaevitz L, Stackpole E, Gujja S, Edwards YJ, Richter JD. 2016. Gld2-catalyzed 3′ monoadenylation of miRNAs in the hippocampus has no detectable effect on their stability or on animal behavior. RNA 22: 1492–1499. 10.1261/rna.056937.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcinowski L, Tanguy M, Krmpotic A, Rädle B, Lisnić VJ, Tuddenham L, Chane-Woon-Ming B, Ruzsics Z, Erhard F, Benkartek C, et al. 2012. Degradation of cellular mir-27 by a novel, highly abundant viral transcript is important for efficient virus replication in vivo. PLoS Pathog 8: e1002510 10.1371/journal.ppat.1002510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzi MJ, Ghini F, Cerruti B, De Pretis S, Bonetti P, Giacomelli C, Gorski MM, Kress T, Pelizzola M, Muller H, et al. 2016. Degradation dynamics of microRNAs revealed by a novel pulse-chase approach. Genome Res 26: 554–565. 10.1101/gr.198788.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neymotin B, Athanasiadou R, Gresham D. 2014. Determination of in vivo RNA kinetics using RATE-seq. RNA 20: 1645–1652. 10.1261/rna.045104.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabani M, Levin JZ, Fan L, Adiconis X, Raychowdhury R, Garber M, Gnirke A, Nusbaum C, Hacohen N, Friedman N, et al. 2011. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat Biotechnol 29: 436–442. 10.1038/nbt.1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichholf B, Herzog VA, Fasching N, Manzenreither RA, Sowemimo I, Ameres SL. 2019. Time-resolved small RNA sequencing unravels the molecular principles of microRNA homeostasis. Mol Cell 75: 756–768.e7. 10.1016/j.molcel.2019.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren G, Chen X, Yu B. 2012. Uridylation of miRNAs by HEN1 SUPPRESSOR1 in Arabidopsis. Curr Biol 22: 695–700. 10.1016/j.cub.2012.02.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GA, Chamberlin MJ, Kane CM. 1993. Contacts between mammalian RNA polymerase II and the template DNA in a ternary elongation complex. Nucleic Acids Res 21: 113–118. 10.1093/nar/21.1.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissland OS, Hong S-J, Bartel DP. 2011. MicroRNA destabilization enables dynamic regulation of the miR-16 family in response to cell-cycle changes. Mol Cell 43: 993–1004. 10.1016/j.molcel.2011.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross J. 1995. mRNA stability in mammalian cells. Microbiol Mol Biol Rev 59: 423–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybak A, Fuchs H, Smirnova L, Brandt C, Pohl EE, Nitsch R, Wulczyn FG. 2008. A feedback loop comprising lin-28 and let-7 controls pre-let-7 maturation during neural stem-cell commitment. Nat Cell Biol 10: 987–993. 10.1038/ncb1759 [DOI] [PubMed] [Google Scholar]
- Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. 2011. Global quantification of mammalian gene expression control. Nature 473: 337–342. 10.1038/nature10098 [DOI] [PubMed] [Google Scholar]
- Schwarz DS, Hutvágner G, Du T, Xu Z, Aronin N, Zamore PD. 2003. Asymmetry in the assembly of the RNAi enzyme complex. Cell 115: 199–208. 10.1016/S0092-8674(03)00759-1 [DOI] [PubMed] [Google Scholar]
- Sun M, Schwalb B, Schulz D, Pirkl N, Etzold S, Larivière L, Maier KC, Seizl M, Tresch A, Cramer P. 2012. Comparative dynamic transcriptome analysis (cDTA) reveals mutual feedback between mRNA synthesis and degradation. Genome Res 22: 1350–1359. 10.1101/gr.130161.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki HI, Katsura A, Yasuda T, Ueno T, Mano H, Sugimoto K, Miyazono K. 2015. Small-RNA asymmetry is directly driven by mammalian Argonautes. Nat Struct Mol Biol 22: 512–521. 10.1038/nsmb.3050 [DOI] [PubMed] [Google Scholar]
- Thornton JE, Gregory RI. 2012. How does Lin28 let-7 control development and disease? Trends Cell Biol 22: 474–482. 10.1016/j.tcb.2012.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabucchi M, Briata P, Garcia-Mayoral M, Haase AD, Filipowicz W, Ramos A, Gherzi R, Rosenfeld MG. 2009. The RNA-binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature 459: 1010–1014. 10.1038/nature08025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turowski TW, Tollervey D. 2015. Cotranscriptional events in eukaryotic ribosome synthesis. Wiley Interdiscip Rev RNA 6: 129–139. 10.1002/wrna.1263 [DOI] [PubMed] [Google Scholar]
- van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. 2007. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 316: 575–579. 10.1126/science.1139089 [DOI] [PubMed] [Google Scholar]
- Viswanathan SR, Daley GQ, Gregory RI. 2008. Selective blockade of microRNA processing by Lin28. Science 320: 97–100. 10.1126/science.1154040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C, Salichos L, Wittgrove CM, Rokas A, Patton JG. 2012. Transcriptome-wide analysis of small RNA expression in early zebrafish development. RNA 18: 915–929. 10.1261/rna.029090.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienholds E, Kloosterman WP, Miska E, Alvarez-Saavedra E, Berezikov E, de Bruijn E, Horvitz HR, Kauppinen S, Plasterk RH. 2005. MicroRNA expression in zebrafish embryonic development. Science 309: 310–311. 10.1126/science.1114519 [DOI] [PubMed] [Google Scholar]
- Winter J, Diederichs S. 2011. Argonaute proteins regulate microRNA stability: Increased microRNA abundance by Argonaute proteins is due to microRNA stabilization. RNA Biol 8: 1149–1157. 10.4161/rna.8.6.17665 [DOI] [PubMed] [Google Scholar]
- Wittrup A, Ai A, Liu X, Hamar P, Trifonova R, Charisse K, Manoharan M, Kirchhausen T, Lieberman J. 2015. Visualizing lipid-formulated siRNA release from endosomes and target gene knockdown. Nat Biotechnol 33: 870–876. 10.1038/nbt.3298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Ameres SL, Friedline R, Hung J-H, Zhang Y, Xie Q, Zhong L, Su Q, He R, Li M, et al. 2012. Long-term, efficient inhibition of microRNA function in mice using rAAV vectors. Nat Methods 9: 403–409. 10.1038/nmeth.1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B, Yang Z, Li J, Minakhina S, Yang M, Padgett RW, Steward R, Chen X. 2005. Methylation as a crucial step in plant microRNA biogenesis. Science 307: 932–935. 10.1126/science.1107130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Yu Y, Zhai J, Ramachandran V, Dinh TT, Meyers BC, Mo B, Chen X. 2012. The Arabidopsis nucleotidyl transferase HESO1 uridylates unmethylated small RNAs to trigger their degradation. Curr Biol 22: 689–694. 10.1016/j.cub.2012.02.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.