

 Repurposing strategies present an enormous advantage for drug discovery, especially in malaria, where resources are scarce.
Repurposing strategies present an enormous advantage for drug discovery, especially in malaria, where resources are scarce.
Abstract
Malaria is a serious tropical disease that kills thousands of people every year, mainly in Africa, due to Plasmodium falciparum infections. Salirasib is a promising cancer drug candidate that interferes with the post-translational modification of Ras. This S-farnesyl thiosalicylate inhibits isoprenylcysteine carboxyl methyltransferase (ICMT), a validated target for cancer drug development. There is a high homology between the human and the parasite enzyme isoforms, in addition to being a druggable target. Looking to repurpose its structure as an antimalarial drug, a collection of S-substituted derivatives of thiosalicylic acid were prepared by introducing 1,2,3-triazole as a diversity entry point or by direct alkylation of the thiol. We further investigated the in vitro toxicity of FTS analogues to Plasmodium falciparum in the asexual stages and in Vero cells. An antiplasmodial activity assay was performed using a simple, high-sensitivity methodology based on nanoluciferase (NLuc)-transfected P. falciparum parasites. The results showed that some of the analogs were active at low micromolar concentration, including Salirasib. The most potent member of the series has S-farnesyl and the 1,2,3-triazole moiety substituted with phytyl. However, the compound substituted with methyl-naphthyl shows promising physicochemical and activity values. The low cytotoxicity in eukaryotic cells of the most active analogs provided good therapeutic indices, being starting-point candidates for future antimalarial drug development.
Malaria is one of the most serious tropical diseases, and the World Health Organization describes malaria as one of the top ten causes of death worldwide.1 In 2016, malaria infected 216 million and killed more than 400 thousand people; most deaths occurred in Africa and were due to Plasmodium falciparum infections. To date, there are no vaccines for malaria immunization. Therefore, the disease prophylaxis, control and treatment are based on the existing chemotherapy and vector control.
Currently, the WHO suggests the use of combination therapies for the treatment of malaria.2 Unfortunately, its expanding resistance puts at risk the ongoing plans to control the disease.3 For these reasons, new antimalarial compounds effective against Plasmodium's asexual stages in humans are required to treat infected people and for use in collective eradication programs.1,4
To facilitate drug discovery, efforts are being made to develop more sensitive, robust, easier, and less expensive methods for drug screening.5,6 Two well-known research strategies for the discovery of etiological treatments for parasitic protozoan diseases are drug repositioning and improving drug effectiveness through structural modifications.7
Salirasib (farnesyl thiosalicylic acid, FTS) is a promising drug candidate that was first reported more than 20 years ago.8,9 This simple compound is a potent inhibitor of isoprenylcysteine carboxyl methyltransferase (ICMT) in cell-free systems. Surprisingly, its activity did not correlate with whole-cell experiments, being a poor methylation inhibitor. It has been shown that Salirasib acts on the Ras signaling cascade, which is required for cell proliferation and differentiation. This process starts with the removal of all Ras isoforms from their membrane anchoring sites.10 Since its discovery, different clinical trials have been conducted using Salirasib for pancreatic, colon, lung and breast cancers.11
One possible target for the development of potential antimalarial drugs is the isoprenoid biosynthesis.12
Isoprenoids are synthesized in P. falciparum, plant plastids and most bacteria (among other organisms) via the 2-C-methyl-d-erythritol-4-phosphate (MEP) pathway.12 In contrast, most animal cells, several eubacteria, some archaea and fungi synthesize isoprenoid precursors only via the mevalonate pathway. In 2004, Goulart et al.13 showed that for P. falciparum strain 3D7 cultured in vitro, FTS exhibits a dose–response growth inhibition effect with a half maximum inhibitory concentration (IC50) of 14 μM (confidence interval: 13.61–19.18 μM). The greatest lethal effects were observed between the schizont stage and the formation of rings.13 The same authors showed that FTS specifically decreased radioactive uptake in immunoprecipitated Ras isoprenylated proteins radiolabeled with [1-(n)-3H] farnesyl pyrophosphate triammonium salt.13 This effect was only observed in the schizont stage, for which the authors suggested a higher Ras protein expression.13 It was also suggested that, as it occurs in other cell systems, Ras proteins could be involved in cell signaling or proliferation in the parasite.14,15 Based on these precedents, we envisioned FTS as a promising scaffold to develop new antimalarial agents. To explore this hypothesis, a collection of thiosalicylic acid derivatives were prepared and assayed against P. falciparum.
Bioinformatics studies of the molecular target
In order to position ICMT as a possible molecular target for repositioning drugs, multiple sequence alignments and homology modelling were refined (Fig. S1†).16 The primary human and infectious apicomplexan ICMT sequences (in particular, Toxoplasma and Plasmodium genera) were analyzed. All the sequences correspond to functional proteins belonging to the ICMT superfamily containing a conserved C-terminal substrate-binding site, (particularly, between amino acids 160 and 279 in H. sapiens). There is complete conservation on the amino acids of the active site and those interacting with the S-adenosyl methionine cofactor. Particularly, there is a 36% identity and 54% similarity between Hs-ICMT and Pf-ICMT. These values are considerably higher when the domains involved in the catalytic sites are analyzed. Expanding the sequence analysis towards different species allows us to conclude that the catalytic domain of the enzyme is highly conserved throughout evolution (Fig. S2†). In addition, the TDR Targets database17 indicates that ICMT would be a druggable target and is potentially essential in P. falciparum. Promisingly, the TDR Targets database predicts that Salirasib would be a potential inhibitory drug for Pf-ICMT. Based on these facts, ICMT is an interesting molecular target and Salirasib is an attractive starting point for repositioning drugs toward P. falciparum and possible for use in consequent reengineering.
Design and synthesis
Salirasib activity has been undoubtedly linked to the presence of thiosalicylic acid on the structure.9 The simplicity of its structure only allowed minimal structural modifications to explore a structure–activity relationship and improve the activity. It has been observed that a free carboxyl group is absolutely required for its activity. Introduction of a halogenic substituent into the benzene ring causes a decrease or a loss of activity on the target.18 Therefore, we decided to preserve the 2-mercaptobenzoic acid portion in our design and introduce modifications on the thiol group. The diversity was introduced by direct alkylation of the thiol, preparing a collection of thioethers and additionally, further diversity was added by 1,2,3-triazol formation via click chemistry (Fig. 1).
Fig. 1. Proposed Salirasib analogs.

First, to prepare the thioether collection, selective thiol alkylation was optimized. Propargyl bromide was selected as an alkylating agent for that purpose because it was the key intermediate for the 1,2,3-triazole library preparation (Scheme 1).
Scheme 1. Alkylation of thiosalicylic acid.

Starting from commercial thiosalicylic acid, different reactions were assayed using different bases and solvents.
The attempts using potassium hydroxide and potassium carbonate in water were unsuccessful (Table 1, entries 1 and 2).
Table 1. Optimization of thiosalicylic S-alkylation.
| Entry | Reagents a | Results |
| 1 | KOH (1 eq.), H2O | NR |
| 2 | K2CO3 (1 eq.), H2O | NR |
| 3 | KOH (1 eq.), MeOH | Decomposition |
| 4 | K2CO3 (1 eq.), MeOH | S-Propargyl propargyl thiosalicylate |
| 5 | Guanidinium carbonate (1 eq.) acetone, r.t. | S-Propargyl thiosalicylic acid, 81% yield |
aThiosalicylic acid (1 eq.), propargyl bromide (1 eq.), reflux, 12 h.
When these bases were used, but the solvent was exchanged with MeOH, KOH caused complete product decomposition and with K2CO3, an alkylated ester product was obtained (Table 1, entries 3 and 4). Fortunately, when using guanidinium carbonate as a base in acetone, a complete selectivity was achieved obtaining S-propargyl thiosalicylic acid 1 as the only product (Table 1, entry 5).
With those conditions optimized, we proceeded to prepare a collection of analogs. Alkylating agents were selected aiming to cover a wide spectrum of steric and lipophilic demands. Most of the required alkylating agents were commercial. For those not commercially accessible, bromides were easily prepared from the alcohol precursor by substitution with phosphorous tribromide in Et2O. Isoprenols (geraniol, farnesol, and phytol) were transformed into their respective chlorides under the Corey–Kim modified conditions,19 using N-chlorosuccinimide and DMS in DCM.
The final collection of the alkylated thiosalicylic acid was prepared by S-alkylation (Table 1, entry 5), using the optimized conditions providing twelve analogs with an average yield of 79% after purification by column chromatography (Scheme 2, Table 2, entries 1 to 11).
Scheme 2. Preparation of alkylated thiosalicylic acid library.
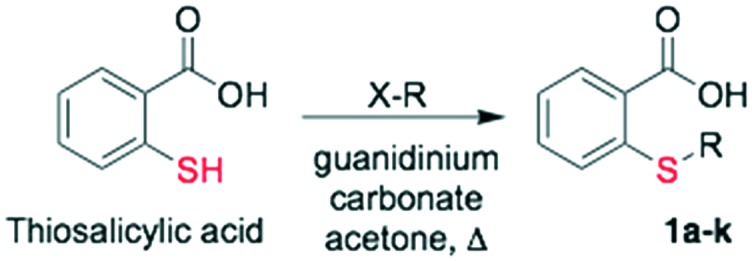
Table 2. In vitro antimalarial activity and yield of alkylated thiosalicylic acid derivatives and of 1,2,3-triazolyl thiosalicylic acid derivatives.
| Entry | Cmpd | R | Yield (%) | P. falciparum a IC50 μM | Z value | R 2 | |
| 1 | 1 | Propargyl | 81 | >200 |
|
||
| 2 | 1a | Cyclohexyl | 87 | >200 | |||
| 3 | 1b | Benzyl | 73 | >200 | |||
| 4 | 1c | 3-Phenyl-propyl | 75 | >200 | |||
| 5 | 1d | Cinnamyl | 68 | >200 | |||
| 6 | 1e | Octyl | 83 | >200 | |||
| 7 | 1f | Decyl | 81 | >200 | |||
| 8 | 1g | CH2CH2CO2CH2CH3 | 78 | >200 | |||
| 9 | 1h | Prenyl | 72 | >200 | |||
| 10 | 1i | Geranyl | 88 | >200 | |||
| 11 | 1j | Farnesyl (Salirasib) | 79 | 20.34 ± 5.52 | 0.93 ± 0.06 | 0.95 ± 0.02 | |
| 12 | 1k | Phytyl | 85 | 13.40 ± 1.53 | 0.93 ± 0.06 | 0.98 ± 0.02 | |
| 13 | 2a | Cyclohexyl | 84 | >200 |
|
||
| 14 | 2b | Benzyl | 80 | >200 | |||
| 15 | 2c | 3-Phenyl-propyl | 88 | >200 | |||
| 16 | 2d | Cinnamyl | 86 | 65.33 ± 10.71 | 0.89 ± 0.09 | 0.87 ± 0.01 | |
| 17 | 2e | Octyl | 78 | >200 | |||
| 18 | 2f | Decyl | 88 | >200 | |||
| 19 | 2g | CH2CH2CO2CH2CH3 | 91 | >200 | |||
| 20 | 2h | Prenyl | 89 | >200 | |||
| 21 | 2i | Geranyl | 84 | >200 | |||
| 22 | 2j | Farnesyl | 90 | 25.97 ± 1.32 | 0.94 ± 0.06 | 0.96 ± 0.02 | |
| 23 | 2k | Phytyl | 83 | 9.75 ± 1.97 | 0.92 ± 0.06 | 0.98 ± 0.01 | |
| 24 | 2l | CH2CO2CH2CH3 | 92 | >200 | |||
| 25 | 2m | Cetyl | 81 | 14.44 ± 5.43 | 0.93 ± 0.06 | 0.97 ± 0.01 | |
| 26 | 2n | Tridecanyl | 55 | 19.83 ± 5.14 | 0.88 ± 0.08 | 0.97 ± 0.02 | |
| 27 | 2o | 2-Phenyl-ethyl | 94 | >200 | |||
| 28 | 2p | 2-Naphthylmethyl | 33 | 20.54 ± 5.45 | 0.92 ± 0.06 | 0.96 ± 0.04 | |
| CQ | 8.15 ± 1.83 nM | 0.75 ± 0.18 | 0.96 ± 0.02 | ||||
| ART | 1.64 ± 0.08 nM | 0.86 ± 0.11 | 0.96 ± 0.03 |
aEvaluated by the NanoLuc luciferase method.
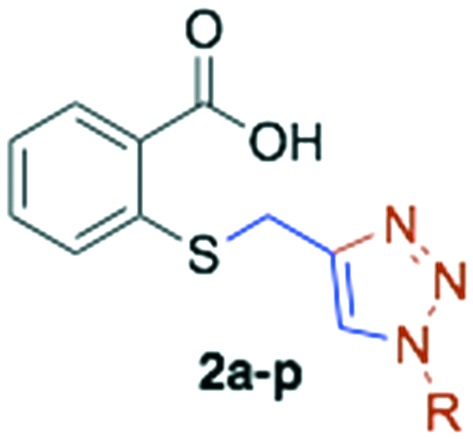
Once the S-alkylated thiosalicylic acid collection was finished, we proceeded to synthesize the 1,2,3-triazole collection. A pool of azides were selected, following the same chemotype used for the alkylated products, and applied to prepare the new derivatives. Azides were mostly prepared by direct substitution of the bromide with sodium azide in DMF. For the isoprenyl azides, isoprenol was transformed in a one-pot reaction into azides, whose regioisomers are in equilibrium.20 The triazoles were synthesized by the reaction of S-propargyl thiosalicylate 1 with the azide under conventional conditions using copper sulphate and sodium ascorbate in tBuOH : H2O (1 : 1).21 The collection of sixteen new products was obtained with an average yield of 81% after purification (Scheme 3, Table 2 entries 12 to 28).
Scheme 3. 1,2,3-Triazolyl thiosalicylate derivatives.

Biological activity
In vitro activity against P. falciparum
The NanoLuc luciferase (NLuc) method for drug screening was applied and validated with results previously obtained6 by the malaria SYBR Green I fluorescence assay (MSF). While the fluorescence-based method relied on quantifying fluorescence on parasite cultures previously incubated with the DNA intercalating dye, the most sensitive and affordable NLuc method involves quantification of bioluminescence in NLuc expressing transgenic parasites.22 In order to confirm that the IC50 values determined by both methods are similar and that the transgenic NLuc expressing parasites are not more resistant to known antimalarials, the wild type 3D7 strain and parasites transfected with the pEF-NLuc expressing vector22 were cultured in serially diluted concentrations of artesunate for 48 h and the growth was quantified by the MSF and/or the NLuc assay. Non-linear regression of growth values generated by both methods produced similar IC50 values suggesting that the results are comparable (Table S1†).
Once NLuc assay was validated, the prepared compounds were screened to select the most effective compounds. For this purpose, the parasites were cultured with FTS or its analogues at 200 μM concentration. The growth was quantified daily by the NLuc assay and compared to that of parasites cultured with the solvent control ethanol. Because most compounds are soluble in ethanol, 200 μM is approximately the highest assayable drug concentration that does not produce significant effects due to ethanol toxicity (0.5–1% at maximum). The inhibition percentage caused by FTS and its analogues at the 200 μM concentration (as compared to an untreated control) was calculated three times. We also performed quality controls with antimalarial drugs, including 200 μM clindamycin (which is a delayed-effect antimalarial compound), both artesunate and chloroquine at 200 nM (as supralethal controls) and 3.125 nM artesunate (as a sublethal control). Compounds 2k and 2m showed haemolysis at 200 μM but not at 100 μM; for this reason, the inhibition percentage study was performed at 100 μM. The results showed that clindamycin, as well as FTS and most of its derivatives, had a higher inhibitory effect at 72 h than that after one or two days (Fig. S3†). On the other hand, inhibition by either sub- or supralethal artesunate concentrations was similar from 1–3 days. Considering the assays initiated with the ring stage parasites, the compounds likely act on late stage parasites as previously reported for FTS.
The IC50 was determined for the FTS analogues which inhibited the parasite growth >90% after 72 h, an effect similar to the observed inhibition of antimalarial control drugs at supralethal concentrations (Table 2, Fig. S4†). The IC50 values of chloroquine and artesunate were determined as positive and quality controls. The IC50 was calculated at 72 h because this condition produced the best antimalarial effect and to be comparable with the previous FTS. We used an initial lethal maximum concentration of 200 μM for FTS (100 μM for compounds 2k and 2m) and its analogues, or 200 nM for chloroquine and artesunate, in addition to several smaller concentrations prepared by serial dilutions.
We also checked all the compounds for which the calculated IC50 values indicated that they may have directly interfered with the NLuc luminescence emission (Fig. S3†). For this purpose, we added a 200 μM concentration of each prepared analog to 100 μL of parasite lysate prepared as previously described from unsynchronized cultures (parasitaemia 1% and haematocrit 2%); we then counted the emitted luminescence with respect to a control without added experimental compounds. The results showed few luminescence modifications which is comparable to those results obtained by other author homologous assays for antimalarial drug screenings.4,5 Again, in this assay, all the samples had Z > 0.5.
The in vitro antimalarial activity of the collection of S-alkylated thiosalicylic acid derivatives (compounds 1a–k) is shown in Table 2 (entries 1–11). Most of the derivatives in this collection were inactive at the maximum concentration tested of 200 μM, including the key intermediate S-propargyl thiosalicylic acid 1.
Interestingly, the only active compounds of the series were the prenylated analogs 1j and 1k. Compounds 1j (R = E,E-farnesyl, Salirasib) and 1k (R = phytyl) have IC50 values of 20.34 and 13.40 μM, respectively. The derivatives with smaller prenyl chains (1h R = prenyl and 1i R = geranyl) were inactive at 200 μM, linking the activity with the number of isoprene units. This is not surprising having in mind that the early studies of thiosalicylic as prenylated protein methyl transferase inhibitors have shown it to be very sensitive to the nature of the prenyl chain, being inactive for the geranylated analogs.18,23
On the other hand, it is interesting to note that only the most voluminous molecules of this family that contain a fatty acid chain (1j and 1k) are active in P. falciparum. Members with shorter chains are inactive (1e and 1f). Finally, the remaining inactive analogs that have aromatic, carbocyclic or alkyl esters put some limits to the possible stereo-electronic variations on the structure.
The in vitro antimalarial activity of the collection of the 1,2,3-triazolyl thiosalicylic acid derivatives (compounds 2a–p) is presented in Table 2 (entries 13–28). The behavior of this collection is similar to that of the S-alkylated analogs with long fatty substituents providing a better activity. A detailed look on the activities shows that compounds 2a, 2b, 2c, 2e, 2f, 2g, 2h, 2i, 2l and 2o did not display activity on P. falciparum at the maximum concentration tested (200 μM).
The remaining six analogs displayed IC50 values in the range of 65.33 μM, for the cinnamyl derivative 2d, to 9.75 μM for the phytyl analog 2k, the most active compounds of both collections (twice as active as Salirasib). A detailed look on the nature of the substituents that provide better activities revealed again that farnesyl (2j), phytyl (2k), and long alkyl linear chains (2m = cetyl and 2n = tridecanyl) are among the most active. But in this case, the aromatic substituted derivatives 2d (cinnamyl) and 2p (methyl naphthyl) also appeared among the most active, while compounds 2b (benzyl), 2c (3-phenyl propyl) and 2o (2-phenyl ethyl) are not active at concentrations below 200 μM.
In vitro cytotoxicity assay in Vero cells
The collection prepared was assayed against Vero cells (ATCC CCL-81, already available in our laboratory) to determine the selectivity against P. falciparum. The initial screening showed that all the compounds were non-cytotoxic at the maximum concentration tested of 4.75 μg mL–1. Then, a selection of the most active analogs was subjected to a full analysis to determine the IC50.
Compounds 1j, 1k, 2d, 2j, 2k, 2m, 2n and 2p were assayed showing different degrees of cytotoxicity as was expected because they have an anticancer origin (Table 3). The most toxic compound, analog 2l, has an IC50 of 34 μM while the less toxic analogs, compounds 1j, 2d and 2j, were inactive at the maximum concentration tested of 200 μM. A detailed analysis on the activities shows that compounds with long aliphatic or prenyl chains are the most cytotoxic derivatives. That is the case for analogs 1k, 2k, 2m, 2n and 2p that have IC50 values below 60 μM, but there is no case where their selectivity index is lower than 1.8. Taking these values into consideration, the best selected analogs of the series were compounds 1j, 2j and 2p that have therapeutic indices of 10.3, >7.70 and 6.95, respectively. According to these values, the analogues can be considered as promising starting point candidates24 to move to further in vivo studies.
Table 3. Cytotoxicity of the selected analogs.
| Entry | Cmpd | Family/R | Vero cells IC50 μM | SI |
| 1 | 1j | S-Alk/farnesyl | 210 ± 5 | 10.3 |
| 2 | 1k | S-Alk/phytyl | 52 ± 4 | 3.87 |
| 3 | 2d | 123T/cinnamyl | >200 | >3.06 |
| 4 | 2j | 123T/farnesyl | >200 | >7.70 |
| 5 | 2k | 123T/phytyl | 34 ± 1 | 3.67 |
| 6 | 2m | 123T/cetyl | 27 ± 4 | 1.86 |
| 7 | 2n | 123T/tridecanyl | 59 ± 4 | 2.99 |
| 8 | 2p | 123T/naphthyl | 143 ± 6 | 6.95 |
ADME-Tox calculations
An oral drug requires good bioavailability that can be achieved by balancing its partitioning and solubility properties. These properties are difficult to achieve on any drug development pipeline. Indeed, one third of new chemical entities have poor pharmacokinetic properties and have not been able to reach the clinical trial phase. To avoid these problems, an early estimation of ADME-Tox properties seems mandatory in any drug discovery program. The in silico ADME-Tox gave substantial information for the feasible and pharmacotherapeutic use of the chemical library.
To rationalize the profile of our analogs, computational studies of all the synthesized compounds were performed to predict their absorption, distribution, metabolism and excretion (ADME) properties, Lipinski's rule of five, toxicity liabilities and drug likeness. The calculations were performed using web-based software Molinspiration,25 Osiris,26 ChemAxon,27 and SwissADME.28 The Molinspiration platform uses a sum of fragment-based contributions and correction factors being able to calculate the parameters of most of the organic and even organometallic molecules. The analysis of the collection revealed that only two compounds have a MW >500. Almost half of them (13 analogs) violated the rule with log P > 5. However, our library contains ionizable groups that are charged at physiological pH, which make log D a better descriptor of the lipophilicity of these molecules. The calculated log D at physiological pH (7.4) of the collection shows that 93% of the library have values ≤5 (–1.62 to 6.31). Finally, all the compounds fulfill the restrictions on the number of hydrogen donors and acceptors. The physicochemical profile and distribution of the prepared compounds based on their molecular weight, polar surface area, log D and octanol/water partition coefficient are shown in Fig. 2.
Fig. 2. Distribution of physicochemical parameters of the chemical library.

The OSIRIS Property Explorer platform was used to perform a toxicology analysis while also providing the drug likeness and a drug score. The results revealed that none of the products prepared are potentially mutagenic, irritant, teratogenic or toxic for sexual reproduction. The solubility is a critical property which aids in the circulation of a drug after the administration and into the bloodstream. The calculated drug likeness shows a variable profile with thirteen derivatives having a poor profile (<–3), but the remaining derivatives fall between –3 and 0. The combination of solubility, log P, MW and toxicity risk is used to calculate the drug scores, which were from moderate to good compared with that of a standard drug (0.13–0.61).
Even though there are diverse routes of drug administration, oral dosing is highly preferred, especially for tropical diseases. Early estimation of oral bioavailability (for example, the fraction of the dose that reaches the bloodstream after oral administration) is very useful in the drug development pipeline. Bioavailability is highly multifactorial but is primarily driven by gastrointestinal absorption. SwissADME use the BOILED-Egg method to predict the gastrointestinal absorption and brain penetration of small molecules. At this point, among all the active compounds, the one that stands out the most is compound 2p.
A good proportion of the analogs did not meet the log P requirement. However, the analogs contain ionizable groups, so the log D values calculated at physiological pH are more adequate. We wanted to find a correlation between the compound activity and its partition coefficient. A chart correlating the P. falciparum IC50vs. log D presented in Fig. 3 (on top) showed a clear pattern. An interesting outcome of this chart is the location of the most active compounds. On the one hand, a region of “high activity” can be defined with all the compounds having log D >3. On the other hand, the remaining analogs were inactive except for the methyl-naphthyl derivative 2p. The tendency shows that the highest activity for the library is concentrated for the analogs with log D between 4.7 and 6.5, with a maximum value of 6 (Fig. 3, bottom). The analog 2p seems to conjugate good activity, excellent ADME-Tox values and appropriate physicochemical parameters. Likewise, the difference in the physicochemical properties with respect to the other active molecules of the family suggests that the compound 2p would exert a different mechanism of action.
Fig. 3. Top: Correlation between the in vitro antimalarial activity and log D. Bottom: Expansion of the “high activity” area and tendency.

Conclusions
Drug repositioning has emerged as a valuable strategy for the development of new treatments against tropical diseases including malaria. Drug repurposing and reengineering have been shown as efficient strategies to reduce the cost and time of drug development. Almost 40% of drugs registered by the US FDA found new uses in the treatment of several human diseases.29 Based on that premise, we selected Salirasib as a starting point to reengineer the anticancer drug as a new antimalarial agent, preparing a library of new 28 thiosalicylic acid derivatives. The whole collection was tested against P. falciparum, the etiological agent of malaria. The selectivity towards P. falciparum in comparison with Vero cells could point Salirasib as an example of drug repositioning. Moreover, an island of high activity was found for the derivatives substituted with long alkyl chains or the farnesyl terpene derivative. Compound 2k is the most active structure against P. falciparum (IC50 of 9.75 μM, SI of 3.67). Some compounds have high log P values breaking Lipinski's rules, but log D calculated at physiological pH seems to be a better descriptor, based on the ionizable groups on the prepared analogs. The naphthyl derivative 2p has an antimalarial activity profile similar to Salirasib, good ADME-Tox parameters and an excellent drug score being the best candidate as an antiplasmodial agent. The lack of toxicity of 2p (predicted in silico and validated on Vero cells) together with its structural simplicity prompted us to point out this new analog as a starting point candidate for future antimalarial drug development. The most active members of the collection have activity values that are comparable to other examples of drug repositioning towards malaria, such as verteporfin, amlodipine besylate, triamterene, rocuronium bromide, or apoptozole.30,31 The same conclusion can be reached if the examples of repositioning towards other parasitic diseases are analyzed.32,33 However, in some cases, the selectivity index is low, or they break more than one of Lipinski's rules. Finally, further optimization would be required for the series to become a serious candidate for future drug development.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was supported in part by grants from the National Research Council of Argentina, CONICET (PIP 2009-11/0796 and 2012-14/0448 awarded to G. R. L.); Agencia Nacional de Promoción Científica y Tecnológica, ANPCyT-Argentina (PICT 2011/0589 awarded to G. R. L.); Universidad Nacional de Rosario and Fundación Josefina Prats; Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP 2017/22452-1 awarded to A. M. K. and 2015/19316-3 awarded to M. F. A.) Brasil; and Conselho Desenvolvimento Científico e Tecnológico (CNPq) Brasil. GRL and CB are members of the scientific staff of CONICET-Argentina. E. O. J. P. thanks CONICET for the award of a fellowship. I. B. V. is a fellow from the FAPESP.
Footnotes
†Electronic supplementary information (ESI) available: Detailed experimental procedures. Copies of the 1H and 13C NMR spectra of all new products. See DOI: 10.1039/c9md00298g
References
- WHO, World malaria report 2017.
- Burrows J. N., Duparc S., Gutteridge W. E., Hooft van Huijsduijnen R., Kaszubska W., Macintyre F., Mazzuri S., Möhrle J. J., Wells T. N. C. Malar. J. 2017;16:26. doi: 10.1186/s12936-016-1675-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondorp A. M., Smithuis F. M., Woodrow C., von Seidlein L. Trends Parasitol. 2017;33:353–363. doi: 10.1016/j.pt.2017.01.004. [DOI] [PubMed] [Google Scholar]
- Griffin J. T., Hollingsworth T. D., Okell L. C., Churcher T. S., White M., Hinsley W., Bousema T., Drakeley C. J., Ferguson N. M., Basáñez M. G., Ghani A. C. PLoS Med. 2010;7:e1000324. doi: 10.1371/journal.pmed.1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidock D. A., Rosenthal P. J., Croft S. L., Brun R., Nwaka S. Nat. Rev. Drug Discovery. 2004;3:509–520. doi: 10.1038/nrd1416. [DOI] [PubMed] [Google Scholar]
- Smilkstein M., Sriwilaijaroen N., Kelly J. X., Wilairat P., Riscoe M. Antimicrob. Agents Chemother. 2004;48:1803–1806. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews K. T., Fisher G., Skinner-Adams T. S. Int. J. Parasitol.: Drugs Drug Resist. 2014;4:95–111. doi: 10.1016/j.ijpddr.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marom M., Haklai R., Ben-Baruch G., Marciano D., Egozi Y., Kloog Y. Cancer Res. 1995;270:22263–22270. doi: 10.1074/jbc.270.38.22263. [DOI] [PubMed] [Google Scholar]
- Marciano D., Ben-Baruch G., Marom M., Egozi Y., Haklai R., Kloog Y. J. Med. Chem. 1995;38:1267–1272. doi: 10.1021/jm00008a004. [DOI] [PubMed] [Google Scholar]
- Bustinza-Linares E., Kurzrock R., Tsimberidou A. M. Future Oncol. 2010;6:885–891. doi: 10.2217/fon.10.71. [DOI] [PubMed] [Google Scholar]
- Tsimberidou A. M., Rudek M. A., Hong D., Ng C. S., Blair J., Goldsweig H., Kurzrock R. Cancer Chemother. Pharmacol. 2010;65:235–241. doi: 10.1007/s00280-009-1027-4. [DOI] [PubMed] [Google Scholar]
- Jordão F. M., Kimura E. A., Katzin A. M. Mem. Inst. Oswaldo Cruz. 2011;106:134–141. doi: 10.1590/s0074-02762011000900018. [DOI] [PubMed] [Google Scholar]
- Goulart H. R., Kimura E. A., Peres V. J., Couto A. S., Duarte F. A. A., Katzin A. M. Antimicrob. Agents Chemother. 2004;48:2502–2509. doi: 10.1128/AAC.48.7.2502-2509.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thélu J., Bracchi V., Burnod J., Ambroise-Thomas P. Cell. Signalling. 1994;6:777–782. doi: 10.1016/0898-6568(94)00038-7. [DOI] [PubMed] [Google Scholar]
- Gisselberg J. E., Zhang L., Elias J. E., Yeh E. Mol. Cell. Proteomics. 2017;16:S54–S64. doi: 10.1074/mcp.M116.064550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame C., Higgins D. G., Heringa J. J. Mol. Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- Agüero F., Al-Lazikani B., Aslett M., Berriman M., Buckner F. S., Campbell R. K., Carmona S., Carruthers I. M., Chan A. W., Chen F., Crowther G. J., Doyle M. A., Hertz-Fowler C., Hopkins A. L., McAllister G., Nwaka S., Overington J. P., Pain A., Paolini G. V., Pieper U., Ralph S. A., Riechers A., Roos D. S., Sali A., Shanmugam D., Suzuki T., Van Voorhis W. C., Verlinde C. L. Nat. Rev. Drug Discovery. 2008;7:900–907. doi: 10.1038/nrd2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciano D., Aharonson Z., Varsano T., Haklai R., Kloog Y. Bioorg. Med. Chem. Lett. 1997;7:1709–1714. [Google Scholar]
- Labadie G. R., Viswanathan R., Poulter C. D. J. Org. Chem. 2007;72:9291–9297. doi: 10.1021/jo7017747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porta E. O. J., Vallejos M. M., Bracca A. B. J., Labadie G. R. RSC Adv. 2017;7:47527–47538. [Google Scholar]
- Porta E. O. J., Jäger S. N., Nocito I., Lepesheva G. I., Serra E. C., Tekwani B. L., Labadie G. R. MedChemComm. 2017;8:1015–1021. doi: 10.1039/c7md00008a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo M. F., Nie C. Q., Elsworth B., Charnaud S. C., Sanders P. R., Crabb B. S., Gilson P. R. PLoS One. 2014;9:e112571. doi: 10.1371/journal.pone.0112571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aharonson Z., Gana-Weisz M., Varsano T., Haklai R., Marciano D., Kloog Y. Biochim. Biophys. Acta, Mol. Basis Dis. 1998;1406:40–50. doi: 10.1016/s0925-4439(97)00077-x. [DOI] [PubMed] [Google Scholar]
- Muller P. Y., Milton M. N. Nat. Rev. Drug Discovery. 2012;11:751–761. doi: 10.1038/nrd3801. [DOI] [PubMed] [Google Scholar]
- Molinspiration v2014.11, 2014, http://www.molinspiration.com (accessed May 11, 2018).
- Osiris version 2, https://www.organic-chemistry.org/prog/peo/ (accessed May 11, 2018).
- ChemAxon Calculator and Predictors, https://chemaxon.com/products/calculators-and-predictors (accessed October 24, 2018).
- Daina A., Michielin O., Zoete V. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburn T. T., Thor K. B. Nat. Rev. Drug Discovery. 2004;3:673–683. doi: 10.1038/nrd1468. [DOI] [PubMed] [Google Scholar]
- Madassary Pazhayam N., Chhibber-Goel J., Sharma A. Drug Discovery Today. 2019;24:263–271. doi: 10.1016/j.drudis.2018.08.006. [DOI] [PubMed] [Google Scholar]
- Chen Y., Murillo-Solano C., Kirkpatrick M. G., Antoshchenko T., Park H. W., Pizarro J. C. Sci. Rep. 2018;8:10333. doi: 10.1038/s41598-018-28608-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simões-Silva M. R., De Araújo J. S., Oliveira G. M., Demarque K. C., Peres R. B., D'Almeida-Melo I., Batista D. G. J., Da Silva C. F., Cardoso-Santos C., Da Silva P. B., Batista M. M., Bahia M. T., Soeiro M. N. C. Biochem. Pharmacol. 2017;145:46–53. doi: 10.1016/j.bcp.2017.08.025. [DOI] [PubMed] [Google Scholar]
- Dichiara M., Marrazzo A., Prezzavento O., Collina S., Rescifina A., Amata E. ChemMedChem. 2017;12:1235–1253. doi: 10.1002/cmdc.201700259. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.