

 A series of benzochalcone derivatives have been synthesized and evaluated for CYP1 inhibitory activity and cytotoxic properties against wild type cell lines (MCF-7 and MDA-MB-231) and drug resistant cell lines (LCC6/P-gp and MCF-7/1B1).
A series of benzochalcone derivatives have been synthesized and evaluated for CYP1 inhibitory activity and cytotoxic properties against wild type cell lines (MCF-7 and MDA-MB-231) and drug resistant cell lines (LCC6/P-gp and MCF-7/1B1).
Abstract
A series of benzochalcone derivatives have been synthesized and evaluated for CYP1 inhibitory activity and cytotoxic properties against wild type cell lines (MCF-7 and MDA-MB-231) and drug resistant cell lines (LCC6/P-gp and MCF-7/1B1). All of these compounds were found to have selective inhibition towards CYP1B1 and the most potent two possessed single-digit nanomolar CYP1B1 potency. In addition, some of them showed promising cytotoxic activities not only against wild type cells, but also against drug resistant cells at low micromolar concentrations. More importantly, these multi-functional compounds may surmount drug–drug interactions that frequently occur during the combination of CYP1B1/P-gp inhibitors and anticancer drugs to overcome drug resistance. This study may provide a good starting point for the further development of more potent multi-functional agents with CYP1B1 inhibitory activity and cytotoxic potency in cancer prevention and treatment.
1. Introduction
Cancer remains one of the most serious threats to human health, since its incidence and mortality are rapidly growing worldwide. Many environmental factors have been proposed to promote carcinogenesis, including ultraviolet radiation, chemical carcinogens and infection. Chemical carcinogens are frequently found in everyday life. For example, benzo[a]pyrene, found in tobacco smoke and charred foods, induces tumorigenesis after being subjected to oxidation by cytochrome P450 family 1 enzymes (CYP1s).1,2 The CYP1 family consists of three members, namely CYP1A1, CYP1A2, and CYP1B1. Among them, CYP1B1 is the most extensively studied one. Beyond its role in metabolizing xenobiotics into carcinogenic products, CYP1B1 can catalyze the 4-hydroxylation of 17β-estradiol (E2) subsequently leading to the formation of E2-3,4-quinone, a mutagenic compound able to bind covalently to DNA.3 In contrast, CYP1A1 and CYP1A2 are prone to producing 2-hydroxyestradiol (2-OHE2), while its further-oxidized product E2-2,3-quinone is found to be non-mutagenic.4
To date, although a variety of drugs have been approved for cancer treatment, most patients will eventually develop drug resistance, leading to therapy failure.5 Thus there is still an urgent need for new agents that are effective against this disease. Various mechanisms have been attributed to drug resistance, such as enhanced drug efflux (overexpression of P-glycoprotein (P-gp), etc.), altered drug metabolism and reduced apoptosis.6 CYP1B1 has also been reported to be associated with drug resistance, due to its role in the metabolism of certain anticancer agents such as docetaxel, paclitaxel, and cisplatin.7 More studies have given evidence that CYP1B1 is overexpressed in various cancers and is specially localized to tumors.8 Therefore, development of selective CYP1B1 inhibitors could be a promising strategy for cancer prevention and overcoming drug resistance.9
Flavonoids have long been recognized for their broad spectrum of pharmacological activities.10,11 α-Naphthoflavone (ANF), a 7,8-benzoflavone derivative, is a potent CYP1 inhibitor with no obvious selectivity for CYP1B1 and CYP1A2.12 In the study of reversing CYP1B1-mediated drug resistance, owing to its low cytotoxicity, ANF only acts as a modulator to increase the anticancer efficacy of cytotoxic drugs. As a result, the safety issues caused by drug–drug interactions should be taken into consideration when combining modulators and anticancer drugs. Notably, chalcones, as an important member of the flavonoid family, also display a wide range of biological activities including anticancer and suppression of CYP1 enzymes as well as P-glycoprotein.13–17 Inspired by the biological properties of chalcones, we herein describe our efforts to identify benzochalcone-based bi-functional compounds with both cancer preventive and therapeutic potential that were designed from lead compound ANF (shown in Fig. 1). In this way, their inhibitory potency on the CYP1B1 enzyme and P-gp not only can block the metabolic activation of chemically diverse procarcinogens, but may also increase their efficacy in killing CYP1B1 or P-gp overexpressed cells. More importantly, these multi-functional agents with CYP1B1 and P-gp inhibitory activity as well as cytotoxic potency may be of advantage, especially in terms of surmounting drug–drug interactions that frequently occur during the combined administration of drugs.
Fig. 1. Design and structural modification of benzochalcones.

2. Results and discussion
2.1. Chemistry
These different benzochalcones with varying substituents on rings A and B could be divided into three different subtypes, B ring modified benzochalcones (1–14), A/D ring modified benzochalcones (15–18) and linker modified benzochalcones (19–20). The synthesis of compounds 1–20 followed the general pathway outlined in Scheme 1. Compounds 1–18 were synthesized by Claisen–Schmidt condensation of different substituted acetophenones and benzaldehydes in the presence of ethanolic KOH. Particularly, synthesis of compounds 14, 17 and 18 required the corresponding intermediates 1,4,5,8-tetramethoxy-2-naphthaldehyde (4e), 1-(1-aminonaphthalen-2-yl)ethan-1-one (5c) and 1-(1-hydroxy-4,5,8-trimethoxynaphthalen-2-yl)ethan-1-one (4h) that were prepared according to our reported procedures.18,19 Compound 19 was obtained by selective reduction of the carbon–carbon double bond of α,β-unsaturated ketone of 1.20 Treatment of compound 15 with guanidine hydrochloride in the presence of KOH and H2O2 generated compound 20 in good yield.21
Scheme 1. Reagents and conditions: (i) a) KOH, EtOH, RT.; (ii) b) Zn; NH4Cl, RT.; (iii) c) guanidine hydrochloride, KOH, H2O2, EtOH, reflux; (iv) d) (CH3)2SO4, NaOH, THF/H2O, RT.; e) NBS, CH3CN, –20 °C; f) CH3ONa, CuI, DMF/MeOH, reflux; g) DMF, POCl3, DCM, reflux; h) CH3I, Mg, Et2O, N2, HCl; i) MnO2, DCM, reflux; j) AlCl3, CH3CN; (v) k) KCN, NCCH2COOEt, KOH, DMF, 60 °C, NaOH–H2O (5%), reflux; l) Mg, CH3I, Et2O, N2, HCl.

2.2. CYP1 inhibitory activity
These synthesized benzochalcones were evaluated for their capacity to suppress recombinant human CYP1 enzymes using the EROD (7-ethoxyresorufin O-deethylase) assay. The results are summarized in Tables 1 and 2. As a general trend, these benzochalcone derivatives displayed a significantly increased selectivity toward CYP1B1 over CYP1A2 when compared to ANF, because the CYP1A2 inhibitory activity of all the compounds were sharply decreased after the cleavage of ring-C of ANF, while their CYP1B1 inhibitory activity showed a slower decline, and two of them possessed similar levels of CYP1B1 inhibition as compared with ANF. Therefore, we can conclude that CYP1A2 is much more susceptible to the planarity of molecules. It was observed that compound 1 showed mild inhibitory potency on the CYP1B1 enzyme (IC50 = 157.7 nM), which was 26-fold less potent than the lead compound ANF (IC50 = 5.9 nM). However, its CYP1B1 inhibitory activity was slightly increased by introduction of a substituent to the B ring (2–9), and substitution at the para position of the B ring generally provided superior activity. To investigate the effect of the type of B ring on the CYP1B1 inhibitory ability, we changed the benzene ring with some aromatic heterocyclic rings. Among compounds 10–13, 3-pyridine ring substituted derivative 11 showed a remarkable CYP1B1 inhibitory effect (IC50 = 16.7 nM). With an aim of enhancing the hydrophobic interaction with the heme pocket, we extended the benzene ring with the methoxyl substituted naphthalene ring resulting in a significant loss of its CYP1B1 inhibitory activity (IC50 > 1000 nM), as seen with compound 14, suggesting that larger functional groups were not tolerated for the B ring.
Table 1. Inhibitory potency of B ring modified benzochalcones against CYP1 enzymes.
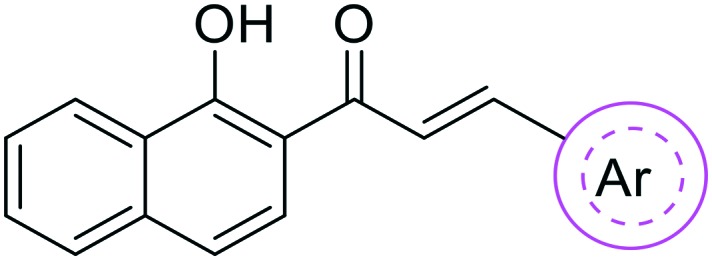
| ||||||
| Cmpd. | Ar | IC50 values (nM) |
IC50 ratio |
|||
| 1B1 | 1A1 | 1A2 | 1A1/1B1 | 1A2/1B1 | ||
| ANF | — | 5.9 ± 1.3 | 80.3 ± 10.8 | 18.0 ± 3.6 | 13.6 | 3.1 |
| 1 | Phenyl | 157.7 ± 18.5 | >1000 | >1000 | >6.3 | >6.3 |
| 2 | o-Fluorophenyl | 90.2 ± 11.4 | 330.0 ± 30.2 | >1000 | 3.7 | >11.1 |
| 3 | m-Fluorophenyl | 81.9 ± 14.7 | 499.0 ± 18.9 | >1000 | 6.1 | >12.2 |
| 4 | p-Fluorophenyl | 48.31 ± 8.2 | >1000 | >1000 | >20.7 | >20.7 |
| 5 | o-Chlorophenyl | 55.4 ± 7.8 | 529.0 ± 28.2 | >1000 | 9.5 | >18.1 |
| 6 | m-Chlorophenyl | 46.6 ± 8.7 | 434.9 ± 25.8 | >1000 | 9.3 | >21.5 |
| 7 | p-Chlorophenyl | 40.2 ± 6.3 | 401.2 ± 33.1 | >1000 | 10.0 | >24.9 |
| 8 | o-Hydroxyphenyl | 76.9 ± 3.9 | 364.7 ± 17.2 | >1000 | 4.7 | >13.0 |
| 9 | o-Methoxyphenyl | 37.8 ± 5.8 | 556.6 ± 28.7 | >1000 | 14.7 | >26.5 |
| 10 | 2-Pyridyl | 135.0 ± 14.8 | >1000 | >1000 | >7.4 | >7.4 |
| 11 | 3-Pyridyl | 16.7 ± 3.7 | 117.1 ± 18.9 | 227.0 ± 25.2 | 7.0 | 13.6 |
| 12 | 4-Pyridyl | 90.2 ± 6.2 | 300.9 ± 21.7 | >1000 | 3.3 | >11.1 |
| 13 | 2-Pyrrolyl | 51.8 ± 7.4 | 250.0 ± 25.9 | >1000 | 4.8 | >19.3 |
| 14 | 1,4,5,8-Tetramethoxynaphthalen-2-yl | >1000 | 532.6 ± 37.4 | >1000 | <0.5 | >1.0 |
Table 2. Inhibitory potency of naphthalene ring and linker modified benzochalcones against CYP1 enzymes.
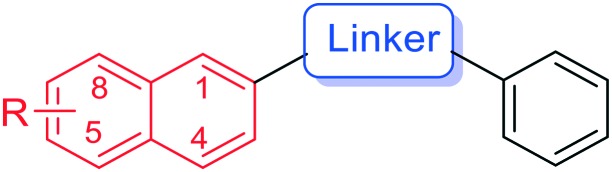
| |||||||
| Cmpd. | R | Linker | IC50 values (nM) |
IC50 ratio |
|||
| 1B1 | 1A1 | 1A2 | 1A1/1B1 | 1A2/1B1 | |||
| 15 | H |
|
95.5 ± 7.4 | >1000 | >1000 | >10.5 | >10.5 |
| 16 | 1-OCH3 |
|
51.4 ± 8.9 | 220.5 ± 32.3 | >1000 | 4.3 | >19.5 |
| 17 | 1-NH2 |
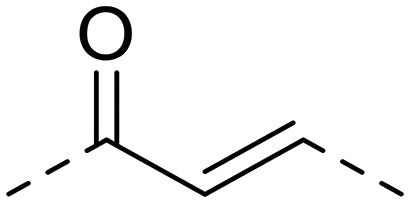
|
112.4 ± 7.3 | 285.8 ± 25.7 | >1000 | 2.5 | >8.9 |
| 18 | 1-OH-4,5,8-TriOCH3 |
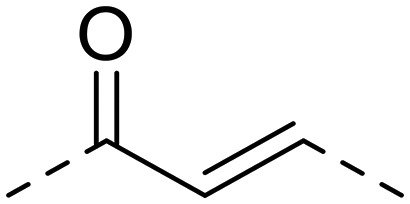
|
4.9 ± 0.6 | 161.3 ± 17.4 | 734.7 ± 31.2 | 32.9 | 149.9 |
| 19 | 1-OH |
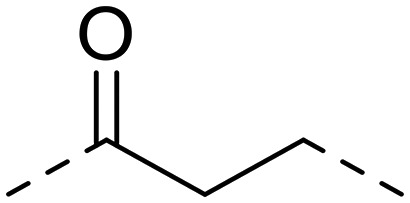
|
151.3 ± 25.8 | 306.4 ± 11.3 | >1000 | 2.0 | >6.6 |
| 20 | H |
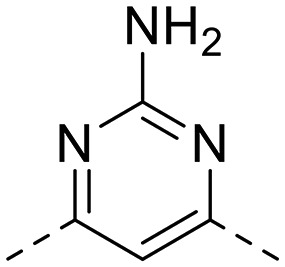
|
4.8 ± 0.5 | 51.8 ± 9.7 | >1000 | 10.8 | >208.3 |
In order to obtain more information about the relationship between the structure and CYP1 inhibitory effects, we therefore turned our attention to the naphthalene and linker moieties. As shown in Table 2, methylation of the hydroxyl group of compound 1 led to 16, which exhibited an increased inhibitory effect on CYP1B1 (IC50 values of 51.4 vs. 157.7 nM). In addition, the unsubstituted analogue 15 was also more potent than the hydrophilic group substituted compounds 1 and 17 in blocking the CYP1B1 enzyme. We hypothesized that the hydrophobic group would be beneficial for inhibiting CYP1B1. To test this hypothesis, we introduced three methoxyl groups to the naphthalene moiety (18). As expected, this compound exhibited a dramatic increase in the CYP1B1 inhibitory activity with an IC50 value of 4.9 nM, which is equipotent to that of ANF (IC50 value of 5.9 nM against CYP1B1). Further confirmation of the importance of α,β-unsaturated ketone (Michael reaction acceptor) was also conducted. Compound 19, obtained by selective reduction of the carbon–carbon double bond of compound 1, retained its inhibitory activity against CYP1B1 despite lacking a Michael reaction acceptor. However, a significantly increased effect was observed from substitution with aminopyrimidine (20), resulting in a 20-fold increase in enzymatic potency of CYP1B1 as compared with compound 15 (IC50 values of 4.8 and 95.5 nM, respectively). The results suggested that α,β-unsaturated ketone is not a necessary structure for CYP1 inhibitory potency and a rigid functional group is more favorable.
2.3. Cytotoxic activity on cancer cells
To explore the potential anticancer activity of these synthesized compounds, we evaluated their cytotoxic activity against four selected cancer cell lines, MCF-7 (estrogen receptor positive tumor cell line), MDA-MB-231(estrogen receptor negative tumor cell line), LCC6/P-gp and MCF-7/1B1. Among them, LCC6/P-gp cells were obtained by transfection of the mdr1 gene that can encode P-glycoprotein (P-gp), while the CYP1B1 overexpressed MCF-7 cell line (referred to as MCF-7/1B1) was obtained by the induction of TCDD (2,3,7,8-tetrachlorodibenzo-p-dioxin) in MCF-7 cells. It should be noted that LCC6/P-gp and MCF-7/1B1are drug resistant cancer cell lines, which were characterized by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay (data not shown).
The cytotoxic effect of these benzochalcones on selected tumour cells was determined by the standard MTT cell viability assay. 5-Fluorouracil (5-FU) and ANF were used as positive control drugs. As the data presented in Table 3, these compounds exhibited varying degrees of activities, and most of them were more potent than 5-FU and ANF. In terms of selectivity, they were found to be superior in their activity against MCF-7 cells. As illustrated by compounds 1 to 9, variations of the substituents on the phenyl ring did not result in any obvious change in activity. However, a substantial difference was observed when the benzene ring was replaced with an aromatic heterocyclic ring (10–13). Among them, the pyridyl substituted benzochalcones 10–12 generally showed higher cytotoxic activity against the tested cells than the other compounds. In particular, the 2-pyridyl substituted chalcone derivative 10, the most vigorous one in this series, was found to have a broad spectrum of anticancer activities that showed a strong growth inhibitory effect not only on wild type cells (MCF-7 and MDA-MB-231, IC50 values of 1.8 and 14.3 μM, respectively), but also on drug resistant cells (LCC6/P-gp and MCF-7/1B1, IC50 values of 7.7 and 11.3 μM, respectively). To our surprise, replacement of the benzene ring with the tetramethoxy substituted naphthalene ring (14) led to a dramatic drop of its CYP1B1 enzyme activity, while the cytotoxic activities against tumor cells MCF-7 and LCC6/P-gp with IC50 values of 1.9 and 3.9 μM increased, respectively. It was speculated that the enhanced cellular activity of compound 14 may be partly due to the improved lipophilicity and membrane permeability as well as the nature of cells.
Table 3. Cytotoxic activities of benzochalcones against MCF-7, MDA-MB-231, LCC6/P-GP and MCF-7 cell lines.
| Cmpd. | IC50 values (μM) |
|||
| MCF-7 | MDA-MB-231 | LCC6/P-gp | MCF-7/1B1 | |
| 1 | 25.9 ± 3.2 | 46.2 ± 5.3 | >100 | 32.3 ± 4.5 |
| 2 | 25.8 ± 4.1 | 37.8 ± 2.8 | 29.8 ± 2.6 | 65.1 ± 4.1 |
| 3 | 29.1 ± 3.9 | 66.4 ± 4.4 | 67.3 ± 3.9 | 33.8 ± 4.1 |
| 4 | 48.1 ± 3.7 | 79.6 ± 4.9 | 48.6 ± 2.9 | >100 |
| 5 | 20.6 ± 2.2 | 43.5 ± 3.6 | 12.2 ± 2.7 | 42.4 ± 4.2 |
| 6 | 22.3 ± 1.2 | 37.6 ± 1.8 | 20.9 ± 2.0 | 41.3 ± 3.2 |
| 7 | 38.5 ± 2.9 | 65.3 ± 4.2 | 45.7 ± 5.3 | 48.4 ± 4.2 |
| 8 | 26.9 ± 3.1 | 43.9 ± 4.0 | 25.1 ± 3.8 | 26.4 ± 4.5 |
| 9 | 43.4 ± 1.6 | 75.3 ± 6.8 | 43.9 ± 4.4 | >100 |
| 10 | 1.8 ± 0.3 | 14.3 ± 0.8 | 7.7 ± 0.9 | 11.3 ± 0.8 |
| ANF | 80.7 ± 7.6 | >100 | 82.3 ± 8.5 | >100 |
| 11 | 7.6 ± 0.7 | 19.8 ± 2.8 | 8.0 ± 2.3 | 12.7 ± 1.7 |
| 12 | 20.5 ± 1.3 | 25.7 ± 2.5 | 15.8 ± 2.4 | 14.8 ± 1.9 |
| 13 | 39.6 ± 4.1 | 48.7 ± 3.3 | >100 | >100 |
| 14 | 1.9 ± 0.4 | 62.2 ± 5.7 | 3.9 ± 0.4 | 43.8 ± 6.3 |
| 15 | 24.7 ± 2.1 | 34.1 ± 3.0 | 19.4 ± 2.7 | 18.2 ± 1.8 |
| 16 | 22.5 ± 2.8 | 35.2 ± 2.2 | 30.8 ± 1.9 | 45.5 ± 3.5 |
| 17 | 20.3 ± 1.2 | 35.5 ± 3.4 | 80.9 ± 6.2 | 23.8 ± 3.1 |
| 18 | 6.3 ± 1.4 | 15.8 ± 2.2 | 9.6 ± 1.3 | 15.1 ± 1.8 |
| 19 | >100 | >100 | >100 | >100 |
| 20 | 46.8 ± 4.7 | 48.9 ± 4.1 | >100 | 20.4 ± 3.3 |
| 5-FU | >100 | 93.8 ± 6.1 | >100 | >100 |
Modification of naphthalene and linker parts could also affect the activities of 15–20. A remarkable change was not observed for the removal of the free hydroxyl group on the naphthalene part of compound 1 (15), conversion of this free hydroxyl group to an ether (16), or replacement of this substituent with an amino group (17). However, introduction of three methoxyl groups to the naphthalene moiety (18) led to a significant increase in cytotoxic activities against these selected cancer cells, with IC50 values ranging from 6.3 to 15.8 μM. Coincidentally, as discussed above, this compound also exerted a strong CYP1B1 inhibitory ability, suggesting that compound 18 could be considered as a potential lead compound for developing multifunctional drugs with a broad range of applications for cancer prevention and treatment. It is interesting to note that both 14 and 18 bearing multiple methoxyl groups showed promising cytotoxicity, which is in agreement with the previous study made by Valdameri et al.22 As seen with compound 19, reduction of the carbon–carbon double bond of compound 1 resulted in a significant loss of its activity. Additionally, pyrimidine ring connected derivative 20 was also less active as compared to the corresponding α,β-unsaturated ketone connected derivative 15. Built upon these phenomena, we can conclude that the α,β-unsaturated ketone moiety of chalcones may serve as an important pharmacophore for cytotoxic activity.
2.4. Molecular docking
The crystal structure of CYP1B1 in complex with ANF is also known (PDB 3PM0). To gain insight into the potential binding modes and rationalize the observed efficiency of CYP1B1 inhibition by these chalcones, compound 18 as a potent CYP1B1 inhibitor was selected for molecular docking studies based on ; 3PM0. As shown in Fig. 2, 18 tightly fitted the active site of CYP1B1, and its naphthalene part was surrounded by hydrophobic residues such as Val126, Phe268, Leu264, Thr325 and Gly329, as well as was covered by the phenyl ring of Phe231. In this case, introduction of three methoxyl groups to the naphthalene moiety could increase the hydrophobicity and electron density, which provided a favorable hydrophobic contact with these hydrophobic residues and an enhanced π–π stacking interaction with Phe231. The distance between the closest carbon atom on the phenyl ring of ANF and heme iron is 4.2 Å, while the corresponding part of 18 is much closer to the heme iron, with a distance of 3.5 Å, which may play a more important role in affecting the formation of the reactive heme iron-oxo intermediate during catalysis. Alternatively, the presence of a hydroxyl group provided an additional interaction with Gly329 via formation of a hydrogen bond, with a distance of 2.1 Å. All of these factors could contribute to the improvement in the enzyme inhibition activity of CYP1B1.
Fig. 2. The binding model of compound 18 with CYP1B1 (hydrogen bonding is depicted by red dashed lines, whereas yellow dashed lines represent distance).

3. Conclusions
In summary, structural modification of ANF gave a series of benzochalcone derivatives, which exhibited varied levels of inhibitory potency on CYP1 enzymes. Compared with ANF, almost all compounds presented an enhanced selectivity for inhibition of CYP1B1 over CYP1A2. It was clearly observed that among all benzochalcones, compounds 18 and 20 were identified as the most potent CYP1B1 inhibitors, with IC50 values in the single-digit nanomolar range. In addition, the cytotoxic activity of these benzochalcones against four selected cancer cell lines was also determined, and compounds 10, 11 and 14 showed remarkable growth inhibitory effects not only on the wild-type MCF-7 cell line, but also on the drug resistant LCC6/P-gp cell line. Notably, compounds 10 and 11 were also found to be effective for the drug resistant MCF-7/1B1 cell line, which provide an additional clue for developing drugs to overcome drug resistance. Consistent with the aim of our research, compound 18 exhibited good biological activities in both enzymatic and cellular assays, which provides an important lead compound for developing multi-functional agents to surmount drug–drug interactions that frequently occur during the combined administration of drugs. Based on this observation, the identification of compounds with more potent inhibitory activity for both enzymes and cells is continuing and will be reported in due course.
4. Experimental
4.1. Chemistry
Commercially available starting materials, reagents, and dry solvents were used as supplied. During the synthesis reaction, progress was monitored by thin-layer chromatography (TLC, silica gel GF254) and visualized with UV light (254 or 365 nm). Column chromatography was conducted on silica gel (300–400 mesh) from Qingdao Ocean Chemical Factory. 1H-NMR and 13C-NMR spectra were recorded on a Varian Mercury-300 (400 MHz) spectrometer using DMSO-d6 or CDCl3 as solvents and TMS as an internal standard. All chemical shifts are given in ppm and coupling constants are given in Hz. Mass spectra were recorded with HRMS (ESI, Agilent 6540 QTOF).
4.2. General procedure for the preparation of compounds 1–18
To a mixture of 2.5 mmol of selected naphthyl methyl ketone and 2.5 mmol of selected benzaldehyde in a 100 ml round bottom flask, 30 ml ethanol and 40 mmol of KOH were added. The mixture was stirred at room temperature for 24 h. After completion of the reaction as indicated by TLC, the mixture was poured in ice water and acidified with dilute HCl. Thr precipitated product was filtered and then was purified by flash chromatography on silica to give the corresponding title compound.
4.2.1. (E)-1-(1-Hydroxynaphthalen-2-yl)-3-phenylprop-2-en-1-one (1)
Yellow solid; yield 53%; 1H NMR (400 MHz, CDCl3) δ 14.85 (s, 1H), 8.50 (d, J = 8.3 Hz, 1H), 7.97 (d, J = 15.4 Hz, 1H), 7.83 (d, J = 8.8 Hz, 1H), 7.79–7.67 (m, 4H), 7.63 (t, J = 7.5 Hz, 1H), 7.53 (t, J = 7.6 Hz, 1H), 7.44 (s, 3H), 7.30 (d, J = 8.9 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 193.19, 164.39, 145.03, 137.37, 134.75, 130.81, 130.19, 129.01, 128.65, 127.38, 125.91, 125.47, 124.49, 123.89, 120.44, 118.21, 113.44. HRMS (ESI) calcd. for [C19H14O2 + H]+ 275.1072, found 275.10760.
4.2.2. (E)-3-(2-Fluorophenyl)-1-(1-hydroxynaphthalen-2-yl)prop-2-en-1-one (2)
Yellow solid; yield 55%; 1H NMR (400 MHz, CDCl3) δ 14.79 (s, 1H), 8.49 (d, J = 8.2 Hz, 1H), 8.00 (d, 15.8 Hz, 1H), 7.88–7.71 (m, 3H), 7.70–7.58 (m, 2H), 7.53 (t, J = 7.2 Hz, 1H), 7.44–7.33 (m, 1H), 7.29 (d, J = 9.0 Hz, 1H), 7.21 (t, J = 7.4 Hz, 1H), 7.18–7.08 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 193.20, 164.47, 163.12, 160.58, 141.11, 137.78, 137.41, 132.13, 132.05, 130.24, 130.06, 127.39, 125.92, 124.57, 124.54, 124.50, 123.88, 123.17, 123.10, 118.28, 116.47, 116.25. HRMS (ESI) calcd. for [C19H13FO2 + H]+ 293.0978, found 293.09768.
4.2.3. (E)-3-(3-Fluorophenyl)-1-(1-hydroxynaphthalen-2-yl)prop-2-en-1-one (3)
Yellow solid; yield 52%; 1H NMR (400 MHz, CDCl3) δ 14.74 (s, 1H), 8.50 (d, J = 8.3 Hz, 1H), 7.91 (d, J = 15.4 Hz, 1H), 7.85–7.75 (m, 2H), 7.71 (d, J = 15.5 Hz, 1H), 7.65 (t, J = 7.3 Hz, 1H), 7.54 (t, J = 7.6 Hz, 1H), 7.49–7.35 (m, 3H), 7.31 (d, J = 8.7 Hz, 1H), 7.14 (t, J = 7.8 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 192.88, 164.52, 161.83, 143.47, 137.44, 130.60, 130.52, 130.33, 127.40, 126.00, 124.74, 124.52, 123.76, 121.76, 118.34, 117.70, 117.49, 114.72, 114.50. HRMS (ESI) calcd. for [C19H13FO2 + H]+ 293.0978, found 293.09770.
4.2.4. (E)-3-(4-Fluorophenyl)-1-(1-hydroxynaphthalen-2-yl)prop-2-en-1-one (4)
Yellow solid; yield 55%; 1H NMR (400 MHz, CDCl3) δ 14.80 (s, 1H), 8.49 (d, J = 8.3 Hz, 1H), 7.93 (d, J = 16 Hz, 1H), 7.82 (d, J = 8.9 Hz, 1H), 7.77 (d, J = 8.1 Hz, 1H), 7.72–7.61 (m, 4H), 7.54 (t, J = 7.6 Hz, 1H), 7.30 (d, J = 8.9 Hz, 1H), 7.13 (t, J = 8.1 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 192.99, 165.47, 164.42, 163.00, 143.69, 137.37, 130.60, 130.52, 130.23, 127.37, 125.95, 124.49, 123.79, 120.18, 118.24, 116.31, 116.10, 114.96, 113.37. HRMS (ESI) calcd. for [C19H13FO2 + H]+ 293.0978, found 293.09751.
4.2.5. (E)-3-(2-Chlorophenyl)-1-(1-hydroxynaphthalen-2-yl)prop-2-en-1-one (5)
Yellow solid; yield 51%; 1H NMR (400 MHz, CDCl3) δ 14.74 (s, 1H), 8.50 (d, J = 8.3 Hz, 1H), 8.36 (d, J = 15.5 Hz, 1H), 7.87–7.74 (m, 3H), 7.71 (d, J = 15.5 Hz, 1H), 7.64 (t, J = 7.4 Hz, 1H), 7.54 (t, J = 7.5 Hz, 1H), 7.47 (d, J = 7.3 Hz, 1H), 7.39–7.27 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 192.93, 164.52, 140.72, 137.42, 135.70, 133.11, 131.38, 130.39, 130.30, 127.95, 127.38, 127.09, 125.97, 125.44, 124.54, 123.83, 123.12, 118.30, 113.37. HRMS (ESI) calcd. for [C19H13ClO2 + H]+ 309.0682, found 309.06844.
4.2.6. (E)-3-(3-Chlorophenyl)-1-(1-hydroxynaphthalen-2-yl)prop-2-en-1-one (6)
Yellow solid; yield 59%; 1H NMR (400 MHz, CDCl3) δ 14.73 (s, 1H), 8.52–8.44 (m, 1H), 7.88 (d, J = 15.5 Hz, 1H), 7.81 (d, J = 8.9 Hz, 1H), 7.79–7.71 (m, 2H), 7.70–7.61 (m, 3H), 7.56–7.51 (m, 2H), 7.41–7.38 (m, 1H), 7.31 (d, J = 8.9 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 192.81, 164.51, 143.21, 137.45, 136.59, 135.05, 130.55, 130.33, 130.24, 128.00, 127.40, 126.99, 125.99, 124.88, 124.53, 124.43, 123.77, 121.81, 118.34. HRMS (ESI) calcd. for [C19H13ClO2 + H]+ 309.0682, found 309.06790.
4.2.7. (E)-3-(4-Chlorophenyl)-1-(1-hydroxynaphthalen-2-yl)prop-2-en-1-one (7)
Yellow solid; yield 55%; 1H NMR (400 MHz, CDCl3) δ 14.77 (s, 1H), 8.49 (d, J = 8.2 Hz, 1H), 7.96–7.88 (m, 1H), 7.81 (d, J = 8.9 Hz, 1H), 7.77 (d, J = 8.1 Hz, 1H), 7.72 (s, 1H), 7.66–7.60 (m, 3H), 7.56–7.52 (m, 1H), 7.41 (d, J = 8.4 Hz, 2H), 7.30 (d, J = 9.0 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 192.91, 164.46, 143.47, 137.41, 136.73, 133.26, 130.28, 129.74, 129.30, 127.38, 125.97, 124.52, 123.76, 120.97, 118.28, 113.37, 109.98, 77.30, 76.98, 76.66. HRMS (ESI) calcd. for [C19H13ClO2 + H]+ 309.0682, found 309.0678.
4.2.8. (E)-1-(1-Hydroxynaphthalen-2-yl)-3-(2-hydroxyphenyl)prop-2-en-1-one (8)
Yellow solid; yield 52%; 1H NMR (400 MHz, DMSO-d6) δ 15.15 (s, 1H), 10.39 (s, 1H), 8.35 (d, J = 8.0 Hz, 1H), 8.28 (d, J = 15.4 Hz, 1H), 8.21 (d, J = 9.0 Hz, 1H), 8.09 (d, J = 15.2 Hz, 1H), 7.98 (d, J = 7.5 Hz, 1H), 7.92 (d, J = 7.6 Hz, 1H), 7.72 (t, J = 7.3 Hz, 1H), 7.59 (t, J = 7.6 Hz, 1H), 7.45 (d, J = 9.0 Hz, 1H), 7.29 (d, J = 7.2 Hz, 1H), 6.95 (d, J = 8.2 Hz, 1H), 6.90 (t, J = 7.3 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 194.16, 163.57, 158.06, 141.17, 137.44, 133.14, 130.82, 129.50, 128.07, 126.61, 125.28, 124.98, 124.97, 124.08, 121.67, 119.93, 118.68, 116.78, 113.79. HRMS (ESI) calcd. for [C19H14O3 + H]+ 291.1021, found 291.10289.
4.2.9. (E)-1-(1-Hydroxynaphthalen-2-yl)-3-(2-methoxyphenyl)prop-2-en-1-one (9)
Yellow solid; yield 54%; 1H NMR (400 MHz, CDCl3) δ 14.96 (s, 1H), 8.50 (d, J = 8.3 Hz, 1H), 8.29 (d, J = 15.5 Hz, 1H), 7.91–7.82 (m, 2H), 7.77 (d, J = 7.9 Hz, 1H), 7.68 (d, J = 7.8 Hz, 1H), 7.63 (t, J = 7.5 Hz, 1H), 7.53 (t, J = 7.5 Hz, 1H), 7.41 (t, J = 7.9 Hz, 1H), 7.30 (d, J = 8.8 Hz, 1H), 7.02 (t, J = 7.5 Hz, 1H), 6.97 (d, J = 8.3 Hz, 1H), 3.96 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 193.80, 164.27, 159.02, 140.70, 137.30, 132.06, 130.00, 129.57, 127.34, 125.79, 125.53, 124.46, 124.08, 123.83, 121.14, 120.79, 118.04, 113.61, 111.31, 55.59. HRMS (ESI) calcd. for [C20H16O3 + H]+ 305.1178, found 305.11768.
4.2.10. (E)-1-(1-Hydroxynaphthalen-2-yl)-3-(pyridin-2-yl)prop-2-en-1-one (10)
Yellow solid; yield 48%; 1H NMR (400 MHz, CDCl3) δ 14.75 (s, 1H), 8.83–8.63 (m, 1H), 8.58–8.43 (m, 1H), 8.42–8.28 (m, 1H), 8.05–7.85 (m, 2H), 7.82–7.69 (m, 2H), 7.62 (d, J = 6.9 Hz, 1H), 7.56–7.43 (m, 2H), 7.30–7.20 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 193.46, 164.55, 152.85, 150.05, 142.53, 137.54, 137.09, 130.29, 127.38, 125.85, 125.36, 124.77, 124.58, 124.51, 124.48, 124.30, 118.30, 113.65. HRMS (ESI) calcd. for [C18H13NO2 + H]+ 276.1025, found 276.10255.
4.2.11. (E)-1-(1-Hydroxynaphthalen-2-yl)-3-(pyridin-3-yl)prop-2-en-1-one (11)
Yellow solid; yield 49%; 1H NMR (400 MHz, DMSO-d6) δ 14.91 (s, 1H), 9.08 (s, 1H), 8.67–8.59 (m, 1H), 8.44 (d, J = 7.9 Hz, 1H), 8.39–8.26 (m, 3H), 8.01–7.90 (m, 2H), 7.73 (t, J = 7.6 Hz, 1H), 7.60 (t, J = 7.6 Hz, 1H), 7.54–7.48 (m, 1H), 7.46 (d, J = 9.1 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 193.78, 163.81, 151.81, 151.18, 142.28, 137.62, 135.95, 131.08, 130.77, 128.10, 126.72, 125.51, 124.85, 124.41, 124.14, 123.42, 118.81, 113.75. HRMS (ESI) calcd. for [C18H13NO2 + H]+ 276.1025, found 276.10353.
4.2.12. (E)-1-(1-Hydroxynaphthalen-2-yl)-3-(pyridin-4-yl)prop-2-en-1-one (12)
Yellow solid; yield 46%; 1H NMR (400 MHz, CDCl3) δ 14.61 (s, 1H), 8.71 (dd, J = 4.5, 1.6 Hz, 2H), 8.49 (dd, J = 8.3, 0.6 Hz, 1H), 7.84 (d, J = 1.7 Hz, 2H), 7.80–7.74 (m, 2H), 7.69–7.62 (m, 1H), 7.57–7.52 (m, 1H), 7.50 (dd, J = 4.5, 1.6 Hz, 2H), 7.31 (d, J = 8.9 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 192.47, 164.78, 150.66, 141.87, 141.68, 137.54, 130.60, 127.45, 126.15, 125.34, 124.76, 124.58, 123.61, 122.09, 118.55, 113.25. HRMS (ESI) calcd. for [C18H13NO2 + H]+ 276.1025, found 276.10329.
4.2.13. (E)-1-(1-Hydroxynaphthalen-2-yl)-3-(1H-pyrrol-2-yl)prop-2-en-1-one (13)
Yellow solid; yield 51%; 1H NMR (400 MHz, CDCl3) δ 15.06 (s, 1H), 8.76 (brs, 1H), 8.48 (d, J = 8.7 Hz, 1H), 7.88 (d, J = 15.1 Hz, 1H), 7.77 (dd, J = 13.6, 8.4 Hz, 2H), 7.62 (t, J = 7.2 Hz, 1H), 7.52 (t, J = 7.4 Hz, 1H), 7.30 (d, J = 16.7 Hz, 2H), 7.03 (s, 1H), 6.78 (s, 1H), 6.37 (s, 1H); 13C NMR (101 MHz, CDCl3) δ 192.60, 164.01, 137.16, 134.32, 129.87, 129.37, 127.29, 125.77, 125.59, 124.40, 123.74, 123.57, 117.94, 116.04, 113.80, 113.45, 111.85. HRMS (ESI) calcd. for [C17H13NO2 + H]+ 264.1025, found 264.10236.
4.2.14. (E)-1-(1-Hydroxynaphthalen-2-yl)-3-(1,4,5,8-tetramethoxynaphthalen-2-yl)prop-2-en-1-one (14)
Yellow solid; yield 45%; 1H NMR (400 MHz, CDCl3) δ 14.93 (s, 1H), 8.49 (d, J = 14.6 Hz, 2H), 7.91 (d, J = 8.3 Hz, 1H), 7.80 (d, J = 18.1 Hz, 2H), 7.68–7.60 (m, 1H), 7.59–7.49 (m, 1H), 7.32 (d, J = 8.4 Hz, 1H), 7.14 (s, 1H), 6.93 (q, J = 8.2 Hz, 2H), 4.04 (s, 3H), 3.98 (s, 3H), 3.93 (s, 3H), 3.86 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 193.43, 164.33, 153.46, 152.09, 151.37, 151.26, 140.11, 137.32, 130.07, 127.36, 125.85, 125.53, 124.99, 124.50, 124.07, 123.00, 122.24, 121.31, 118.07, 113.61, 110.87, 108.75, 104.95, 63.68, 57.91, 57.42, 57.09. HRMS (ESI) calcd. for [C27H24O6 + H]+ 445.1651, found 445.16585.
4.2.15. (E)-1-(Naphthalen-2-yl)-3-phenylprop-2-en-1-one (15)
Pale white solid; yield 72%; 1H NMR (400 MHz, CDCl3) δ 8.53 (s, 1H), 8.11 (d, J = 8.6 Hz, 1H), 7.98 (d, J = 7.8 Hz, 1H), 7.94–7.82 (m, 3H), 7.73–7.63 (m, 3H), 7.62–7.51 (m, 2H), 7.43 (d, J = 4.6 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 190.18, 144.71, 135.54, 135.48, 134.97, 132.57, 130.54, 129.95, 129.53, 128.97, 128.57, 128.51, 128.39, 127.83, 126.78, 124.49, 122.09. HRMS (ESI) calcd. for [C19H14O + H]+ 259.1123, found 259.11315.
4.2.16. (E)-1-(1-Methoxynaphthalen-2-yl)-3-phenylprop-2-en-1-one (16)
Pale white solid; yield 56%; 1H NMR (400 MHz, CDCl3) δ 8.31–8.23 (m, 1H), 7.87–7.71 (m, 3H), 7.67–7.50 (m, 6H), 7.40–7.29 (m, 3H), 3.92 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 192.68, 156.98, 143.99, 136.58, 134.99, 130.47, 128.96, 128.53, 128.17, 128.10, 128.06, 126.67, 126.50, 126.06, 124.11, 123.17, 64.03. HRMS (ESI) calcd. for [C20H16O2 + H]+ 289.1229, found 289.1232.
4.2.17. (E)-1-(1-Aminonaphthalen-2-yl)-3-phenylprop-2-en-1-one (17)
Brown solid; yield 46%; 1H NMR (400 MHz, CDCl3) δ 7.95 (d, J = 8.2 Hz, 1H), 7.88 (d, 1H), 7.79–7.71 (m, 3H), 7.69–7.64 (m, 2H), 7.60–7.55 (m, 1H), 7.53–7.47 (m, 1H), 7.45–7.33 (m, 3H), 7.10 (d, J = 8.9 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 191.11, 150.12, 142.35, 136.36, 135.49, 129.93, 129.01, 128.86, 128.45, 128.22, 126.47, 125.41, 123.75, 123.35, 121.75, 115.42, 112.33. HRMS (ESI) calcd. for [C19H15NO + H]+ 274.1232, found 274.12352.
4.2.18. (E)-1-(1-Hydroxy-4,5,8-trimethoxynaphthalen-2-yl)-3-phenylprop-2-en-1-one (18)
Brown solid; yield 43%; 1H NMR (400 MHz, CDCl3) δ 12.83 (s, 1H), 7.86 (d, J = 15.4 Hz, 1H), 7.75 (d, J = 15.8 Hz, 1H), 7.66 (d, J = 6.7 Hz, 2H), 7.46–7.38 (m, 3H), 7.27 (s, 1H), 6.98 (d, J = 8.6 Hz, 1H), 6.88 (d, J = 8.3 Hz, 1H), 4.03 (s, 3H), 3.94 (s, 3H), 3.91 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 191.93, 152.81, 151.22, 148.74, 143.32, 135.21, 130.29, 130.13, 128.87, 128.50, 128.42, 124.33, 118.52, 117.36, 112.02, 108.22, 107.46, 58.06, 57.80, 57.01. HRMS (ESI) calcd. for [C22H20O5 + H]+ 365.1389, found 365.13991.
4.2.19. 1-(1-Hydroxynaphthalen-2-yl)-3-phenylpropan-1-one (19)
Compound 1 (0.5 mmol, 137 mg) was suspended in ethanol (50 ml), and a mixture of water solution (8 mL) containing 1.07 g (20 mmol) ammonium chloride at room temperature was added. The mixture is stirred vigorously, and 0.195 g (3 mmol) zinc powder added in three equal portions at intervals of 15 min. The progress of the reaction was monitored by TLC. When the reaction was finished, the suspended material was removed by filtration and the filtrate was evaporated under reduced pressure. The obtained residue was poured into water, and the precipitated crude product was filtered and recrystallized from ethanol to give pure compound 19 as pale white solid (116 mg, yield 84%). 1H NMR (400 MHz, CDCl3) δ 14.04 (s, 1H), 8.47 (d, J = 8.3 Hz, 1H), 7.75 (d, J = 8.2 Hz, 1H), 7.68–7.59 (m, 2H), 7.53 (t, J = 7.5 Hz, 1H), 7.35–7.27 (m, 4H), 7.27–7.21 (m, 3H), 3.40 (t, J = 7.7 Hz, 2H), 3.13 (t, J = 7.7 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 205.12, 162.57, 140.81, 137.27, 130.02, 128.58, 128.38, 127.36, 126.28, 125.92, 125.34, 124.44, 124.07, 118.34, 112.76, 40.34, 30.21. HRMS (ESI) calcd for [C19H16O2 + H]+ 277.1229, found 277.1220.
4.2.20. 4-(Naphthalen-2-yl)-6-phenylpyrimidin-2-amine (20)
To a mixture of compound 15 (1 mmol, 258 mg) and guanidine hydrochloride (2 mmol, 191 mg) in a 100 ml round bottom flask, 15 ml ethanol and 50% aqueous KOH solution (15 mL) were added. The mixture was stirred at reflux temperature for 1.5 h. Then, 30% aqueous H2O2 (5 ml) was added to the above mixture in small portions over a period of 1.5 h. After completion of the reaction as indicated by TLC, ethanol was removed under reduced pressure, and water was added to the residue. The precipitated title compound was filtered off, and washed with water several times. The crude solid was recrystallized from ethanol to give pure title compound 20 as a pale white solid (240 mg, yield 81%). 1H NMR (400 MHz, CDCl3) δ 8.58 (s, 1H), 8.16 (d, J = 8.5 Hz, 1H), 8.13–8.05 (m, 2H), 8.02–7.92 (m, 2H), 7.91–7.83 (m, 1H), 7.60 (s, 1H), 7.57–7.46 (m, 5H), 5.43 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 166.26, 166.04, 163.62, 137.73, 134.94, 134.44, 133.23, 130.49, 128.93, 128.78, 128.50, 127.72, 127.20, 127.17, 127.14, 126.48, 124.19, 104.50. HRMS (ESI) calcd. for [C20H15N3 + H]+ 298.1344, found 298.13599.
4.3. Enzyme assays
The recombinant human CYP1B1, CYP1A1, and CYP1A2 enzymes, each with P450 reductase (Supersomes),were purchased from BD Genetest. 7-Ethoxyresorufin (7-ER) was obtained from Sigma-Aldrich. Nicotinamide adenine dinucleotide phosphate (NADP+), d-glucose-6-phosphate (G-6-P) and glucose-6-phosphate dehydrogenase (G-6-PD) were purchased from Biosharp. The inhibitory activities of these prepared compounds against CYP1B1, CYP1A1, and CYP1A2 enzymes were determined using the EROD assay as reported earlier.19,23 Generally, a mixture with a final volume of 200 μl containing different concentrations (the final concentrations were 0.5, 1, 5, 10, 50, 100, 500 and 1000 nM, respectively) of tested compounds (except positive and negative control wells), an enzyme source (20 fmol CYP1B1, 10 fmol CYP1A1 or 60 fmol CYP1A2), 150 nM 7-ER,1.3 mM NADP+, 3.3 mM G-6-P, 0.5 U ml–1, G-6-PD and 3.3 mM MgCl2 was incubated in a black 96-well flat-bottomed microplate at 37 °C for varied time durations (incubation time for CYP1B1, CYP1A1, and CYP1A2 was 35, 15 and 50 min, respectively). Then, the reaction was stopped by the addition of 100 μl of methanol to all wells. Lastly, the fluorescence intensity was measured by FlexStation 3 apparatus with excitation and emission filters at 544 and 590 nm, respectively. For each concentration, three replicates were used and each test was repeated three times. The IC50 value for each compound was calculated with the GraphPad Prism Software (Version 5.0) using the non-linear regression formula, log (inhibitor) vs. normalized response-variable slope.
4.4. Cytotoxic activity assay
According to a reported procedure, MCF-7/1B1 cells were obtained by co-incubation of parental MCF-7 cells with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, 10 nM) for five days. The LCC6/P-gp cell line was obtained from The Hong Kong Polytechnic University. MCF-7, MDA-MB-231 and MCF-7/1B1 cells were grown in RPMI-1640 medium, while LCC6/P-gp was grown in DMEM/high glucose medium. Cell growth inhibition was determined by the MTT cytotoxicity assay. Generally, approximately 4000 cells, in suspended medium, were seeded into each well of a 96-well plate and incubated at 37 °C in a humidified atmosphere with 5% CO2 for 24 h. Then, different concentrations of synthesized compounds (the final concentrations were 0.5, 1, 5, 10, 25, 50 and 100 μM, respectively) were added (except the vehicle control group). The final volume in each well was 200 μl and the final concentration of DMSO in any assay system did not exceed 0.1% (v/v). After 48 h incubation, the culture medium was removed and refreshed with RPMI-1640 medium (200 μL). Next, MTT solution (20 μL, 5 mg mL–1) was added to each well and incubated for an additional 4 h. Medium was discarded and the resulting formazan crystals were dissolved in DMSO (150 μL per well). Absorbance values were determined using a microplate reader at 570 nm. Three replicates of each concentration were used in each assay and all assays were repeated three times. IC50 values were calculated with the GraphPad Prism Software (version 5.0) using the non-linear regression formula, log (inhibitor) vs. normalized response-variable slope.
4.5. Molecular docking
The molecular docking experiment was carried out using the docking program in MOE 2008 based on the crystal structures of CYP1B1 (PDB ID, 3PM0). The initial 3D conformation of compound 18 was optimized in ChemBio3D Ultra using the MM2 energy minimization method. All molecules were removed from the crystal structures before the experiment. Hydrogens and partial charges were added with the protonate 3D application. The residues within a radius of 8.0 Å around the ligands were selected as docking sites.
Docking parameters were kept at default, except for the first scoring function, where ASE Scoring was used instead of the default London dG. The best pose was characterized by the scoring results.
Conflicts of interest
The authors declare no conflicts of interest.
Supplementary Material
Acknowledgments
The research reported in this publication was supported by the Fundamental Research Funds for the Central University (No. ZH2018QNB15), the Shanghai Natural Science Fund (No. 16ZR1418100), the Shanghai Science and Technology Innovation Program (No. 15431900700) and the National Natural Science Foundation of China (Grant No. 21602132). We want to express our gratitude to Dr. Iris L. K. Wong and Prof. Larry M. C. Chow in the Hong Kong Polytechnic University for the provision of the well-cultured LCC6 drug-resistant cancer cells.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c9md00258h
References
- Zhou S., Wang B., Yang L., Liu J. Drug Metab. Rev. 2010;42:268–354. doi: 10.3109/03602530903286476. [DOI] [PubMed] [Google Scholar]
- Nebert D. W., Shi Z., Galvez-Peralta M., Uno S., Dragin N. Mol. Pharmacol. 2013;84:304–313. doi: 10.1124/mol.113.086637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes C. L., Spink D. C., Spink B. C., Cao J. Q., Walker N. J., Sutter T. R. Proc. Natl. Acad. Sci. U. S. A. 1996;93:9776–9781. doi: 10.1073/pnas.93.18.9776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J., Zhang Q., Cui Q., Huang G., Pan X., Li S. ChemMedChem. 2016;11:2102–2118. doi: 10.1002/cmdc.201600316. [DOI] [PubMed] [Google Scholar]
- Dong J., Zhang Q., Wang Z., Huang G., Li S. ChemMedChem. 2018;13:1490–1507. doi: 10.1002/cmdc.201800253. [DOI] [PubMed] [Google Scholar]
- Pathania S., Bhatia R., Baldi A., Singh R., Rawal R. K. Biomed. Pharmacother. 2018;105:53–65. doi: 10.1016/j.biopha.2018.05.117. [DOI] [PubMed] [Google Scholar]
- Dutour R., Cortes-Benitez F., Roy J., Poirier D. ACS Med. Chem. Lett. 2017;8:1159–1164. doi: 10.1021/acsmedchemlett.7b00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray G. I., Taylor M. C., McFadyen M. C. E., McKay J. A., Greenlee W. F., Burke M. D., Melvin W. T. Cancer Res. 1997;57:3026–3031. [PubMed] [Google Scholar]
- McFadyen M. C., Murray G. I. Future Oncol. 2005;1:259–263. doi: 10.1517/14796694.1.2.259. [DOI] [PubMed] [Google Scholar]
- Sudhakaran M., Sardesai S., Doseff A. I. Antioxidants. 2019;8:103. doi: 10.3390/antiox8040103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J., Zhang Q., Meng Q., Wang Z., Li S., Cui J. Mini-Rev. Med. Chem. 2018;18:1714–1732. doi: 10.2174/1389557518666180515145633. [DOI] [PubMed] [Google Scholar]
- Shimada T., Yamazaki H., Foroozesh M., Hopkins N. E., Alworth W. L., Guengerich F. P. Chem. Res. Toxicol. 1998;11:1048–1056. doi: 10.1021/tx980090+. [DOI] [PubMed] [Google Scholar]
- Mateeva N., Eyunni S. V. K., Redda K. K., Ononuju U., Hansberry T. D., Aikens C., Nag A. Bioorg. Med. Chem. Lett. 2017;27:2350–2356. doi: 10.1016/j.bmcl.2017.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams I. S., Joshi P., Gatchie L., Sharma M., Satti N. K., Vishwakarma R. A., Chaudhuri B., Bharate S. B. Bioorg. Med. Chem. Lett. 2017;27:3683–3687. doi: 10.1016/j.bmcl.2017.07.010. [DOI] [PubMed] [Google Scholar]
- Horley N. J., Beresford K. J., Chawla T., McCann G. J., Ruparelia K. C., Gatchie L., Sonawane V. R., Williams I. S., Tan H. L., Joshi P., Bharate S. S., Kumar V., Bharate S. B., Chaudhuri B. Eur. J. Med. Chem. 2017;129:159–174. doi: 10.1016/j.ejmech.2017.02.016. [DOI] [PubMed] [Google Scholar]
- Boumendjel A., McLeer-Florin A., Champelovier P., Allegro D., Muhammad D., Souard F., Derouazi M., Peyrot V., Toussaint B., Boutonnat J. BMC Cancer. 2009;9:242. doi: 10.1186/1471-2407-9-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardim G. A. M., Guimarães T. T., Pinto M. C. F. R., Cavalcanti B. C., de Farias K. M., Pessoa C., Gatto C. C., Nair D. K., Namboothiri I. N. N., da Silva Júnior E. N. Med. Chem. Commun. 2015;6:120–130. [Google Scholar]
- Cui J., Zhou W., Li S. J. Chem. Res. 2012;36:264–265. [Google Scholar]
- Dong J., Wang Z., Meng Q., Zhang Q., Huang G., Cui J., Li S. RSC Adv. 2018;8:15009–15020. doi: 10.1039/c8ra00465j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Zhang Y., Ji Y. J. Chin. Chem. Soc. 2008;55:390–393. [Google Scholar]
- Rehman T. U., Khan I. U., Ashraf M., Tarazi H., Riaz S., Yar M. Arch. Pharm. 2017;350:1600304. doi: 10.1002/ardp.201600304. [DOI] [PubMed] [Google Scholar]
- Valdameri G., Gauthier C., Terreux R., Kachadourian R., Day B. J., Winnischofer S. M., Rocha M. E., Frachet V., Ronot X., Di Pietro A., Boumendjel A. J. Med. Chem. 2012;55:3193–3200. doi: 10.1021/jm2016528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J., Meng Q., Zhang X., Cui Q., Zhou W., Li S. J. Med. Chem. 2015;58:3534–3547. doi: 10.1021/acs.jmedchem.5b00265. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.