

Abstract
Objectives
Reactive oxygen species induce neurite degeneration before inducing cell death. However, the degenerative mechanisms have not yet been elucidated. While tocotrienols have a known neuroprotective function, the underlying mechanism remains unclear and may or may not involve antioxidant action. In this study, we hypothesize that free radical-derived membrane injury is one possible mechanism for inducing neurite degeneration. Therefore, we examined the potential neuroprotective effect of tocotrienols mediated through its antioxidant activity.
Methods
Mouse neuroblastoma neuro2a cells were used to examine the effect of the water-soluble free radical generator 2,2′-azobis(2-methylpropionamide) dihydrochloride (AAPH) on neurite dynamics. After 24 hours of AAPH treatment, cell viability, neurite number, and the number of altered neurites were measured in the presence or absence of α-tocotrienol.
Results
Treatment of neuro2a cells with a low concentration of AAPH induces neurite degeneration, but not cell death. Treatment with 5 µM α-tocotrienol significantly inhibited neurite degeneration in AAPH-treated neuro2a cells. Furthermore, morphological changes in AAPH-treated neuro2a cells were similar to those observed with colchicine treatment.
Conclusions
α-Tocotrienol may scavenge AAPH-derived free radicals and alkoxyl radicals that are generated from AAPH-derived peroxyl radicals on cell membranes. Therefore, α-tocotrienol may have a neuroprotective effect mediated by its antioxidant activity.
Keywords: AAPH, Tocotrienol, Neurite degeneration, Oxidative stress
Introduction
Reactive oxygen species (ROS) are continually produced in living tissues. Increased oxidative stress, for instance through the overproduction of ROS and/or attenuation of antioxidant defense systems, increases the risk of development of serious diseases and accelerates their progression.1 At the neuronal level, overproduction of ROS induces cell death and initiates various neurodegenerative disorders, including Alzheimer's,2–4 Parkinson's,5 and other severe diseases.6 In 1956, Harman7 reported that a relationship exists between ROS-related oxidative injury and aging (i.e. the free radical theory of aging). Neurons are known to play a crucial role in the central nervous system. If neurons have been attacked by ROS, recovery of brain function becomes difficult because many neurons may have been severely damaged or may have died. In order to prevent ROS-induced neuronal injury, it is necessary to identify early signs of neuronal change, prior to the induction of cell death.
To identify early events that may signal ROS-induced neuronal cell death, we are studying the function of neurites because they play an important role in neurotransmission and are characterized by expansion and contraction. A number of studies have examined neurite degeneration. In slow Wallerian degeneration model mice (WldS), neurite degeneration occurs in an anterograde manner.8 In contrast, nerve growth factor-free medium induces retrograde neurite degeneration (i.e. a dying-back process).9 Previously, we found that low concentrations of hydrogen peroxide induce axonal and dendrite degeneration in cerebellar granule cells (CGCs).10,11 Furthermore, using silver staining, we observed axonal degeneration in the hippocampal CA1 region of vitamin E-deficient young mice and normal aged mice.12
Vitamin E, which was discovered by Bishop and Evans in the 1920s,13 is one of the most common natural lipophilic vitamins, and its best-known function is as an antioxidant. Vitamin E scavenges free radicals and protects cells from lipid oxidation. Two vitamin E subtypes have been identified, tocopherols and tocotrienols, with each subtype having four isoforms (α, β, γ, and δ). Several lines of evidence have shown that α-tocopherol has the strongest antioxidant activity within the vitamin E family. Recently, other beneficial functions of vitamin E have been described, including prevention of neurodegenerative disorders4,14 and enhancement of immune responses.15,16 Some reports suggest that tocotrienols have neuroprotective activity; however, the detailed mechanism of this activity has yet to be elucidated. In addition, it is not clear whether the neuroprotective effect associated with tocotrienols is due to antioxidant or non-antioxidant activity. In this study, we used 2,2′-azobis(2-methylpropionamide) dihydrochloride (AAPH), a free radical generator, to examine changes in neurite morphology in the presence or absence of α-tocotrienol. Our hypothesis was based upon the fact that AAPH-derived peroxyl radicals attack cell membranes. Thus, if α-tocotrienol were found to protect against AAPH-induced neurite degeneration, the neuroprotective effect of α-tocotrienol would be due to an antioxidant activity (Fig. 1).
Figure 1.
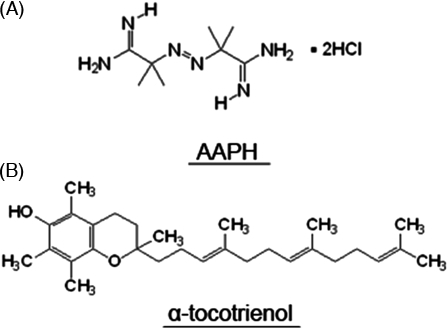
Chemical structure of AAPH (A) and α-tocotrienol (B).
Materials and methods
Cell culture and reagents
Neuro2a cells, which are derived from a mouse neuroblastoma C1300 tumor, were originally obtained from the Human Science Research Resources Bank (#IFO50081) (Osaka, Japan). The cells were grown in minimum essential medium containing 5% heat-inactivated fetal calf serum (FCS) (Biological Industries, Beit Haemek, Israel), 50 U/ml of penicillin, and 50 µg/ml of streptomycin, and were plated in the wells of polyethyleneimine-coated plates. In order to elicit neurite extension, the FCS concentration was decreased to 1%. After 72 hours, the cells were used in experiments.
All other chemical agents were obtained from either Wako Pure Chemical Industries Ltd. (Osaka, Japan) or Sigma-Aldrich Corp. (St Louis, MO, USA). All animal experiments were performed with the approval of the Animal Protection and Ethics Committee of the Shibaura Institute of Technology. All tissue culture plates and dishes were purchased from Becton Dickinson and Company (Franklin Lakes, NJ, USA).
Determination of cell death
Cell survival was assessed using the trypan blue dye exclusion assay. Cells at a density of 2.0–4.0 × 104 cells/well (24-well plate) were treated with various concentrations of AAPH. After 24 hours of AAPH treatment, an equal volume of 0.8% (w/v) trypan blue in phosphate-buffered saline (PBS) was added to the wells and the plate was incubated for 20 minutes in a CO2 incubator. The cells were then washed with PBS at least three times. The number of dead cells per unit of area was counted, and the data were presented as a percentage of the total number of cells. At least three wells were analyzed for all experimental conditions, and each experiment was repeated three times. Counts were independently confirmed by a student not involved in this study.
Neurite degeneration
Neurite degeneration was evaluated by monitoring morphological hallmarks, such as bead formation and fragmentation, as described previously, with some modifications.17 Twenty-four hours after treatment with each concentration of AAPH, neuro2a cells were fixed for 15 minutes at 4°C with 4% (w/v) paraformaldehyde in PBS. Photomicrographs of the cells were collected on an Olympus IX81 phase-contrast microscope (Olympus Corp., Tokyo, Japan) equipped with a DP71 digital camera (Olympus), after which the images were stored and processed on a personal computer. At least 60 neurites were evaluated for neurite formation per exposure. The cells were treated with 1 mM AAPH in the presence or absence of 5 µM α-tocotrienol (α-tocotrienol was kindly supplied by Eisai Food & Chemical Co. Ltd., Tokyo, Japan).
Statistical analysis
Data were plotted as the mean ± SD of the results of three independent experiments. Data were analyzed using the Student's t-test, with P < 0.05 considered significant. Furthermore, data were analyzed using the one-way analysis of variance (ANOVA).
Results
Optimization of AAPH concentration in neuro2a cells
Neuro2a cells, which are derived from a neuroblastoma cell line, are typically round in shape and do not have long neurites. This study involved assessment of neurite morphology under conditions of oxidative stress; therefore, we reduced the FCS concentration in the medium from 5 to 1%. After 72 hours, neuro2a cells showed long neurites (Fig. 2A) and were then treated with various concentrations of AAPH (0.2, 0.5, 1, 2, 3, 4, and 5 mM) for 24 hours to induce oxidative stress. As shown in Fig. 2B, treatment with AAPH resulted in the death of neuro2a cells in a concentration-dependent manner. In cultures treated with 2 mM AAPH, the ratio of dead to total cells (dead and living cells) was less than 15%. However, the ratio of dead to total cells was over 70% in cultures treated with 3 mM AAPH. Since the purpose of this study was to assess the condition of neurites before the induction of cell death, we chose a concentration of AAPH of less than 2 mM for subsequent experiments.
Figure 2.
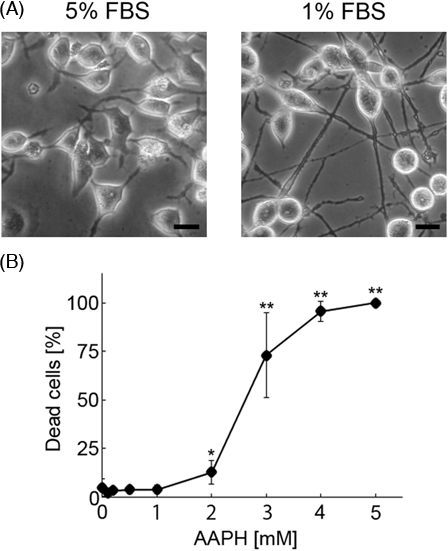
Photomicrographs showing neuro2a cells (A). In order to determine the condition of neurites, phase-contrast photomicrographs of the cells were taken. The scale bar represents 10 µm. AAPH induces death of neuro2a cells (B). Neuro2a cells were treated with various concentrations of AAPH. After 24 hours, the number of dead neuro2a cells was counted using the trypan blue dye exclusion assay. Each point represents the mean of three independent experiments. At least three wells were counted per experiment. Data were analyzed using the Student's t-test (*P < 0.05, **P < 0.01) and one-way ANOVA.
AAPH induces neurite degeneration in neuro2a cells
In a cell survival assay, analysis of the total number of cells showed that at concentrations less than 2 mM, AAPH had no significant impact on neuro2a cell numbers (Fig. 2B, from ANOVA) Furthermore, the ratio of neuro2a cells with neurites was significantly decreased following 2 mM AAPH treatment. As the purpose of this study was to identify signs of cell degeneration that occur before the induction of cell death in AAPH-treated neuro2a cells, we focused on changes in neurite morphology. AAPH induced neurite degeneration in a concentration-dependent manner (Fig. 3A). However, there was no significant difference in the total number of untreated neuro2a cells and cells treated with 0.2, 0.5, 0.7, and 1 mM AAPH (Fig. 3B) (from ANOVA). At concentrations over 0.5 mM, AAPH induced significant morphological abnormalities in neurites, such as the formation of neurite beads. Conversely, treatment with 2 mM AAPH resulted in a significant decrease in the total number of neurites and the number of altered neurites compared to non-treated neurites. Collectively, these results indicate that low concentrations of AAPH induce neurite degeneration before inducing cell death.
Figure 3.
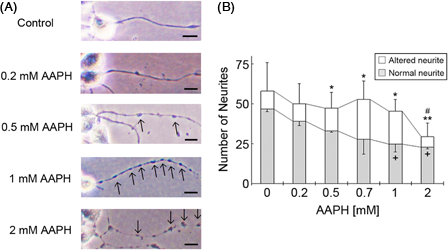
Treatment of neuro2a cells with low concentrations of AAPH induces neurite degeneration. Neuro2a cells were treated with various concentrations of AAPH. After 24 hours, the cells were fixed with 4% paraformaldehyde (PFA) in PBS. Photomicrographs of the cells were collected and analyzed on a personal computer. The scale bar represents 10 µm. Arrows indicate bead formation on the degenerating neurites of neuro2a cells (A). The results of quantitative analysis of neurite degeneration are shown (B). Each column represents the mean of three independent experiments. At least three wells were examined per experiment. Data were analyzed using the Student's t-test (+P < 0.05 between normal neurites at 0 and 1 mM AAPH, and 0 and 2 mM AAPH, respectively. *P < 0.05, **P < 0.01 between altered neurites at 0 and 0.5 mM AAPH, 0 and 0.7 mM AAPH, 0 and 1 mM AAPH, and 0 and 2 mM AAPH. #P < 0.05 between the total number of neurites in neurons treated with 0 and 2 mM AAPH) and one-way ANOVA.
Tocotrienol prevents AAPH-induced neurite degeneration in neuro2a cells
The total number of cells did not differ in the presence or absence of 1 mM AAPH (Fig. 4B). However, the number of normal healthy neurites was significantly lower in neuro2a cells treated with 1 mM AAPH (Fig. 3B). Treatment of AAPH-treated neuro2a cells with 5 µM α-tocotrienol significantly inhibited neurite degeneration, indicating that α-tocotrienol exhibits antioxidant activity that prevents AAPH-induced neurite degeneration.
Figure 4.
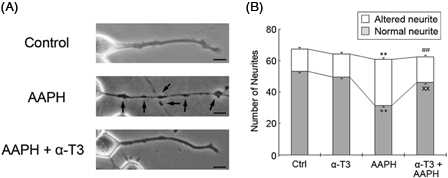
α-Tocotrienol prevents AAPH-induced neurite degeneration. Neuro2a cells were treated with α-tocotrienol (5 µM) in the presence or absence of 1 mM AAPH. After 24 hours, the cells were fixed with 4% PFA in PBS. Photomicrographs of the cells were collected and analyzed on a personal computer. The scale bar represents 10 µm. Arrows indicate bead formation on the degenerating neurites of neuro2a cells (A). The results of quantitative analysis of neurite degeneration are shown (B). Each column represents the mean of three independent experiments. At least three wells were examined per experiment. Data were analyzed using the Student's t-test. ++P < 0.01 between normal control neurites and neurites of neurons treated with 1 mM AAPH. xxP < 0.01 between normal neurites of neurons treated with 5 µM α-tocotrienol in the presence or absence of 1 mM AAPH. **P < 0.01 between altered neurites of control neurons and those of neurons treated with 1 mM AAPH. ##P < 0.01 between altered neurites at 5 µM α-tocotrienol in the presence or absence of 1 mM AAPH.
Discussion
AAPH induces neurite degeneration before inducing death in neuro2a cells
A variety of free radical generators, such as AAPH, 2,2′-azobis[2-methyl-N-(2-hydroxyethyl)-propionamide] (AMPH), 2,2′-azobis(isobutyronitrile) (AIBN), and others have been used in free radical research.18 We used AAPH in this study because it is one of the most commonly used water-soluble free radical generators for research purposes and is used in the oxygen radical absorbance capacity assay to determine antioxidant scavenging capacity.19,20 The peroxyl radical produced by AAPH is known to directly attack lipids and proteins,21 and induces the formation of lipid hydroperoxides in cell membranes.22 In this study, treatment with AAPH induced cell death in a concentration-dependent manner (Fig. 2). However, treatment of neuro2a cells with low concentrations of AAPH (less than 1 mM) induced neurite degeneration, but not cell death (Fig. 3). The appearance of significant beading, which is a hallmark of neurite degeneration,8,17 was observed in AAPH-treated neuro2a cells. The number of damaged neurites was highest in cultures of neuro2a cells treated with 0.7 mM AAPH, and lowest in cultures of neuro2a cells treated with 2 mM AAPH. These results indicated that neurite degeneration occurs prior to AAPH-induced cell death. Previously, we found induction of apoptosis, reduced antioxidant enzyme activity, enhancement of thiobarbituric acid reactive substances level, and the appearance of axonal degeneration in the hippocampal CA1 region of normal old mice and vitamin E-deficient mice.2,11,12,23 We also found that cognitive function, as assessed by maze tasks, is significantly reduced in vitamin E-deficient rats and normal old rats.24,25 Collectively, these data suggest the possibility that neurite degeneration via oxidative injury is responsible, at least in part, for cognitive dysfunction associated with aging. This hypothesis is consistent with the free radical theory of aging7; however, further experiments are needed to more clearly define the relationship between ROS-induced neurite degeneration and cognitive dysfunction during aging.
The neuroprotective effect of α-tocotrienol is associated with antioxidant activity
Several lines of evidence have shown that tocotrienols exert neuroprotective effects. Khana et al.26 reported that α-tocotrienol affects the activity of 12-lipoxygenase in glutamate-treated neurons. Treatment with very low concentrations of tocotrienols, in the nanomolar range, affects signal transduction pathways involved in neuroprotection.27 In Alzheimer's disease patients, the plasma concentrations of tocotrienols are lower than in normal healthy subjects.28 In rats, treatment with tocotrienols prevents alcohol-induced cognitive dysfunction by suppressing the production of inflammatory cytokines, such as tumor necrosis factor-α and interleukin-1β.29 Previously, we also examined the neuroprotective effects of α- and γ-tocotrienols on hydrogen peroxide-treated neuro2a cells and CGCs. Both tocotrienols had a stronger neuroprotective effect than α-tocopherol.10 However, we could not determine that the neuroprotective effects of the tocotrienol isoforms were due to antioxidant function. As hydrogen peroxide is capable of easily passing through the cell membrane, it can reach the intracellular region and be converted to hydroxyl radicals, thereby affecting intracellular organelles and various signal pathways. Conversely, AAPH-derived peroxyl radicals do not cross the cell membrane because of their large molecular weight, resulting in AAPH-derived radicals attacking the outer cell membranes. Our purpose in this study was to determine whether the neuroprotective effect of tocotrienols is associated with antioxidant activity, thereby differentiating it from previous research. We found that treating neuro2a cells with α-tocotrienol significantly inhibited AAPH-induced neurite degeneration (Fig. 4), indicating that α-tocotrienol has antioxidant activity.
Vitamin E is a naturally occurring lipophilic compound, and thus associates with the cell membranes, whereas AAPH is a well-known, water-soluble free radical generator. How does vitamin E scavenge AAPH-derived free radicals and alkoxyl radicals, generated from AAPH-derived peroxyl radicals, on the cell membrane? The localization of vitamin E on the cell membrane is important. Some reports indicate that the hydroxyl group (chroman ring) associates with the surface of the cell membrane, while the phospholipid carbonyl group (phytyl chain) embeds within the membrane.30,31 Association with the membrane in this manner could explain, in part, how α-tocotrienol prevents AAPH-induced neurite degeneration. However, tocotrienols also have non-antioxidant activity. Some reports emphasize that the concentration of tocotrienols is very important with regard to neuroprotective effects.32 At low concentrations (e.g. in the nanomolar range), the neuroprotective effect of tocotrienols derives from their non-antioxidant activity. In contrast, at high concentrations (e.g. in the micromolar range), tocotrienols exert an antioxidant effect. Saito et al.33 reported that there is no difference in the biochemical activity of tocopherols and tocotrienols. Rather, the speed with which they are taken into the cell membrane differs; tocotrienols enter the membrane faster than tocopherols. In addition, tocotrienols exert antioxidant biological activity. It is possible that the neuroprotective effect of tocotrienols may derive from both their antioxidant and non-antioxidant activities.
Potential for microtubule depolymerization in AAPH-treated neuro2a cells
Treatment with low concentrations of AAPH induces neurite degeneration, as evidenced by the formation of numerous large neurite beads. In order to verify that neurite beading in neuro2a cells was due to treatment with AAPH, we compared the neurite morphology of neuro2a cells treated with AAPH to that of cells treated with colchicine, an inhibitor of microtubule polymerization. Treatment with 0.5 nM colchicine for 24 hours induced neurite beading in neuro2a cells (Fig. 5). The morphology of neurites in colchicine-treated neurons was similar to that of AAPH-treated neurons. In neuro2a cells treated with 1 nM colchicine, the neurites were thinner than in cells treated with 0.5 nM colchicine. Furthermore, we previously reported that treatment of neuro2a cells and CGCs with a low concentration of hydrogen peroxide induced truncated forms of collapsing response mediator protein (CRMP)-2.10,11 CRMP-2 plays a crucial role in neurite function and binds to tubulin dimers. These results indicate that AAPH-induced neurite degeneration is associated with microtubule disruption in neuro2a cells. However, further investigation is needed to clarify the relationship between ROS-induced neurite degeneration and microtubule disruption in neuronal cells.
Figure 5.
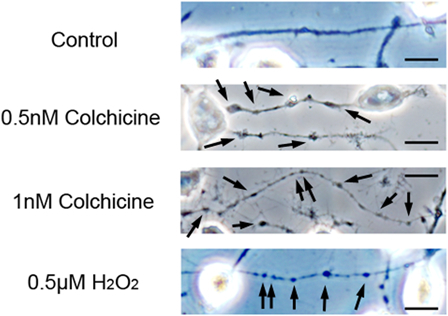
Colchicine induces neurite degeneration. Neuro2a cells were treated with various concentrations of colchicine. After 24 hours, the cells were fixed with 4% PFA in PBS. Photomicrographs of the cells were collected and analyzed on a personal computer. The scale bar represents 10 µm. Arrows indicate bead formation on the degenerating neurites of neuro2a cells. At least three wells were examined per experiment.
Conclusion
In this study, we found that α-tocotrienol prevents AAPH-induced neurite degeneration in neuro2a cells. Furthermore, our results indicate that the neuroprotective effect of α-tocotrienol may be related to its antioxidant activity. It is possible that tocotrienols play a beneficial role in neurodegenerative disorders via their neuroprotective functions. However, many of the details regarding the biological activities of tocotrienols, such as those related to their antioxidant and non-antioxidant activities, remain to be elucidated. We hope that our continuing investigations of the neuroprotective effect of tocotrienols will provide answers to some of these questions in the near future.
Acknowledgement
We would like to thank Prof. Tadashi Shinkai, Shibaura Institute of Technology, for his valuable advice regarding establishment of the culture samples.
Declaration of interest section
This work was supported by the Ministry of Education, Culture, Sports, Science, and Technology (MEXT)-Supported Program for the Strategic Research Foundation at Private Universities. This study was also supported by a grant-in-aid for Project Research from the Shibaura Institute of Technology (Tokyo, Japan).
References
- 1.Sies H. Oxidative stress: oxidants and antioxidants. Exp Physiol 1997;82(2):291–5. [DOI] [PubMed] [Google Scholar]
- 2.Fukui K, Takatsu H, Shinkai T, Suzuki S, Abe K, Urano S. Appearance of amyloid beta-like substances and delayed-type apoptosis in rat hippocampus CA1 region through aging and oxidative stress. J Alzheimers Dis 2005;8:299–309. [DOI] [PubMed] [Google Scholar]
- 3.Arikanoglu A, Akil E, Varol S, Yucei Y, Yuksel H, Cevik MU, et al.. Relationship of cognitive performance with prolidase and oxidative stress in Alzheimer disease. Neurol Sci 2013; doi:10.1007/s10072-013-1346-4. [DOI] [PubMed] [Google Scholar]
- 4.Chan A, Roqers E, Shea TB. Dietary difficiency in folate and vitamin E under conditions of oxidative stress increases phosphor-tau levels: potentiation by ApoE4 and alleviation by S-adenosylmethionine. J Alzheimer's Dis 2009;17(3):483–7. [DOI] [PubMed] [Google Scholar]
- 5.Seet RC, Lee CY, Lim EC, Tan JJ, Quek AM, Chong WL, et al.. Oxidative damage in Parkinson disease: measurement using accurate biomarkers. Free Radic Biol Med 2009;48(4):560–6. [DOI] [PubMed] [Google Scholar]
- 6.Pastor MD, Nogal A, Molina-Pinelo S, Meléndez R, Romero-Romero B, Mediano MD, et al.. Identification of oxidative stress related proteins as biomarkers for lung cancer and chronic obstructive pulmonary disease in bronchoalveolar Lavage. Int J Mol Sci 2013;14(2):3440–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harman D. Aging; a theory based on free radical and radiation chemistry. J Gerontrol 1956;11(3):298–300. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki K, Koike T. Mammalian Sir-2-related protein (SIRT) 2-mediated modulation of resistance to axonal degeneration in slow wallerian degeneration mice: a crucial role of tubulin deacetylation. Neuroscience 2007;147:599–612. [DOI] [PubMed] [Google Scholar]
- 9.Yang Y, Fukui K, Koike T, Zheng X. Induction of autophagy in neurite degeneration of mouse superior cervical ganglion neurons. Eur J Neurosci 2007;26(10):2979–88. [DOI] [PubMed] [Google Scholar]
- 10.Fukui K, Takatsu H, Koike T, Urano S. Hydrogen peroxide induces neurite degeneration: prevention by tocotrienols. Free Radic Res 2011;45(6):681–91. [DOI] [PubMed] [Google Scholar]
- 11.Fukui K, Ushiki K, Takatsu H, Koike T, Urano S. Tocotrienols prevent hydrogen peroxide-induced axon and dendrite degeneration in cerebellar granule cells. Free Radic Res 2012;46(2):184–93. [DOI] [PubMed] [Google Scholar]
- 12.Fukui K, Kawakami H, Honjo T, Ogasawara R, Takatsu H, Shinkai T, et al.. Vitamin E deficiency induces axonal degeneration in mouse hippocampal neurons. J Nutr Sci Vitaminol (Tokyo) 2012;58(6):377–83. [DOI] [PubMed] [Google Scholar]
- 13.Evans HM, Bishop KS. On the existence of a hitherto unrecognized dietary factor essential for reproduction. Science 1922;56:650–1. [DOI] [PubMed] [Google Scholar]
- 14.Sano M, Ernesto C, Thomas RG, Klauber MR, Schafer K, Grundman M, et al.. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer's disease. The Alzheimer's disease cooperative study. N Engl J Med 1997;336:1216–22. [DOI] [PubMed] [Google Scholar]
- 15.Adolfsson O, Huber BT, Meydani SN. Vitamin E enhanced IL-2 production in old mice: naive but not memory T cells show increased cell division cycling and IL-2-producing Capacity. J Immunol 2001;167:3809–17. [DOI] [PubMed] [Google Scholar]
- 16.Grammas P, Hamdheydari N, Benaksas EJ, Mou S, Pye QN, Wechter WJ, et al.. Anti-inflammatory effectrs of tocopherol metabolites. Biochem Biophys Res Commun 2004;319(3):1047–52. [DOI] [PubMed] [Google Scholar]
- 17.Ikegami K, Koike T. Non-apoptotic neurite degeneration in apoptotic neuronal death: pivotal role of mitochondrial function in neuritis. Neuroscience 2003;122:617–26. [DOI] [PubMed] [Google Scholar]
- 18.Yoshida Y, Itoh N, Saito Y, Hayakawa M, Niki E. Application of water-soluble radical initiator, 2,2′-azobis-[2-(2-imidazolin-2-yl9prppane)dihydrochloride], to a study of oxidative stress. Free Radic Res 2004;38(4):375–84. [DOI] [PubMed] [Google Scholar]
- 19.Kohri S, Fujii H, Oowada S, Endoh N, Sueishi Y, Kusakabe M, et al.. An oxygen radical absorbance capacity-like assay that directly quantifies the antioxidant's scavenging capacity against AAPH-derived free radicals. Anal Biochem 2009;386(2):167–71. [DOI] [PubMed] [Google Scholar]
- 20.Niki E, Omata Y, Fukuhara A, Saito Y, Yoshida Y. Assessment of radical scavenging capacity and lipid peroxidation inhibiting capacity of antioxidant. J Agric Food Chem 2008;56(18):8255–60. [DOI] [PubMed] [Google Scholar]
- 21.Piga R, Saito Y, Yoshida Y, Niki E. Cytotoxic effects of various stressors on PC12 cells: involvement of oxidative stress and effect of antioxidants. Neurotoxicology 2007;28:67–75. [DOI] [PubMed] [Google Scholar]
- 22.Takahashi M, Shibata M, Niki E. Estimation of lipid peroxidation of live cells using a fluorescent probe, diphenyl-1-pyrenylphosphine. Free Radic Biol Med 2001;31(2):164–74. [DOI] [PubMed] [Google Scholar]
- 23.Onodera K, Omoi N, Fukui K, Hayasaka T, Shinkai T, Suzuki S, et al.. Oxidative damage of rat cerebral cortex and hippocampus, and changes in antioxidative defense systems caused by hyperoxia. Free Radic Res 2003;37:367–72. [DOI] [PubMed] [Google Scholar]
- 24.Fukui K, Omoi N, Hayasaka T, Shinkai S, Suzuki S, Abe K, et al.. Cognitive impairment of rats caused by oxidative stress and aging, and its prevention by vitamin E. Ann N.Y. Acad Sci 2002;959:275–84. [DOI] [PubMed] [Google Scholar]
- 25.Fukui K, Onodera K, Shinkai T, Suzuki S, Urano S. Impairment of learing and memory in rat caused by oxidative stress and aging, and changes in antioxidative defense systems. Ann N.Y. Acad Sci 2001;928:169–76. [DOI] [PubMed] [Google Scholar]
- 26.Khana S, Roy S, Ryu H, Bahadduri P, Swaan PW, Ratan RR, et al.. Molecular basis of vitamin E action: tocotrienol modulates 12-lipoxygenase, a key mediator of glutamate-induced neurodegeneration. J Biol Chem 2003;278(44):43508–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frank J, Chin XW, Schrader C, Eckrt GP, Rimbach G. Do tocotrienols have potential as neuroprotective dietary factors? Aging Res Rev 2012;11(1):163–80. [DOI] [PubMed] [Google Scholar]
- 28.Mangialasche F, Xu W, Kivipelto M, Constanzi E, Ercolani S, Pigliautile M, et al.. AddNeuroMed Consortium. Tocopherols and tocotrienols plasma levels are associated with cognitive impairment. Neurobiol Aging 2012;33(10):2282–90. [DOI] [PubMed] [Google Scholar]
- 29.Tiwari V, Kuhad A, Chopra K. Suppression of neuro-inflammatory signaling cascade by tocotrienol can prevent chronic alcohol-induced cognitive dysfunction in rats. Behav Brain Res 2009;203(2):296–303. [DOI] [PubMed] [Google Scholar]
- 30.Niki E. Function of vitamin E as antioxidant in the membranes. In: , Mino M, Nakamura H, Diplock TA, (eds.) Vitamin E its usefulness in health and in curing diseases. Basel, Switzerland: S. Karger AG; 1993. p. 23–30. [Google Scholar]
- 31.Gomez-Fernandez JC, Villalain J, Aranda FJ, Ortiz A, Micol V, Coutinho A, et al.. Localization of alpha-tocopherol in membranes. Ann N.Y. Acad Sci 1989;570:109–20. [DOI] [PubMed] [Google Scholar]
- 32.Sen CK, Khana S, Roy S, Packer L. Molecular basis of vitamin E action. Tocotrienol potently inhibits glutamate-induced pp60 (c-Src) kinase activation and death of HT4 neuronal cells. J Biol Chem 2000;270:30749–54. [DOI] [PubMed] [Google Scholar]
- 33.Saito Y, Nishio K, Akagawa YO, Yamanaka K, Miyama A, Yoshida Y, et al.. Cytoplasmic effect of vitamin E homologues aainst glutamate-induced cell death in immature primary cortical neuron cultures: Tocopherols and tocotrienols exert similar effects by antioxidant function. Free Radic Biol Med 2010;49(10):1542–9. [DOI] [PubMed] [Google Scholar]