

ABSTRACT
Objectives: In tissue samples from patients with colorectal cancer (CRC), oxidation of C420 and C457 of plakoglobin (Pg) within tumor tissue was identified by proteomic analysis. The aim of this study was to identify the roles of Pg C420 and C457.
Methods: Human CRC tissues, CRC and breast cancer cells, and normal mouse colon were prepared to validate Pg oxidation. MC38 cells were co-transfected with E-cadherin plus wild type (WT) or mutant (C420S or C457S) Pg to evaluate protein interactions and cellular localization, proliferation, and migration.
Results: Pg was more oxidized in stage III CRC tumor tissue than in non-tumor tissue. Similar oxidation of Pg was elicited by H2O2 treatment in normal colon and cancer cells. C457S Pg exhibited diminished binding to E-cadherin and α-catenin, and reduced the assembly of E-cadherin–α-/β-catenin complexes. Correspondingly, immunofluorescent analysis of Pg cellular localization suggested impaired binding of C457S Pg to membranes. Cell migration and proliferation were also suppressed in C457S-expressing cells.
Discussion: Pg appears to be redox-sensitive in cancer, and the C457 modification may impair cell migration and proliferation by affecting its interaction with the E-cadherin/catenin axis. Our findings suggest that redox-sensitive cysteines of Pg may be the targets for CRC therapy.
KEYWORDS: Colorectal cancer, cysteine, E-cadherin, oxidation, plakoglobin
Introduction
Colorectal cancer (CRC) is the third most common cancer in the world,[1] and it is characterized by a high recurrence rate and resistance to currently available therapies in most patients with metastatic CRC. This makes understanding the molecular mechanisms and genetic changes associated with CRC progression crucially important for the discovery of new therapeutic targets as well as the identification of patients who are at high risk.[2]
During tumor progression, cancer cells modify their stromal environment by generating a range of growth factors and proteases.[3] The tumor microenvironment is characterized by hypoxia, low pH, and nutrient deprivation, which together cause severe alterations in cell metabolism and physiology, including hypoxia–reoxygenation injury, inflammatory cell activation, and the induction of oxidant-generating enzymes.[4,5] In addition, pre-neoplastic and cancer cells require high levels of adenosine triphosphate to maintain their proliferation rate, which leads to increases in the production of reactive oxygen species (ROS) via the mitochondrial respiratory chain.[6] The ROS produced in the tumor environment then exert local mutagenic effects, leading to the modification of critical proteins.[7,8]
The predominant cellular targets of ROS are the amino acids of proteins.[9] ROS modify important cysteines in numerous redox-sensitive proteins, often causing inactivation of the affected proteins.[10–12] Although oxidative modification of redox-sensitive cysteines is recognized to be a central mechanism for dynamic post-translational regulation in many diseases,[9,13] the physiological and pathophysiological functions of cysteine oxidation have not been well characterized. For example, the specific sites of modification often remain unknown due to the difficulties in selectively detecting specific thiol modifications. However, recent advances have enabled the detection and quantification of protein cysteine oxidation.[14] Using proteomics, we previously identified proteins containing oxidation-sensitive cysteines within tumors and the surrounding normal tissues from patients with CRC.[15] Plakoglobin (Pg) (also known as Junction Plakoglobin or γ-catenin) was identified as containing redox-sensitive cysteines (unpublished data), and the locations of the specific oxidatively modified cysteine residues within its amino acid sequence were determined. Pg interacts with the cytoplasmic domain of cadherins and a number of other intracellular partners and is typically associated with tumor/metastasis suppressor activity.[16–18] Our aim was to determine whether, by affecting the interaction of Pg with cadherins/catenins, the modification of oxidized target cysteines suppresses or stimulates oncogenic activity in CRC cells. Our findings suggest that the oxidation-sensitive cysteines of Pg may be potential targets for CRC therapy.
Material and methods
All chemicals were purchased from Sigma-Aldrich (St Louis, MO), unless noted otherwise.
CRC tissue samples
This study was approved by the Institutional Review Board of Chonnam National University, Hwasun Hospital, Korea. To validate protein oxidation, six tissue samples from patients with CRC, which included both tumor tissue (pT) and adjacent normal tissue (non-tumor, pN), were collected with informed consents from Hwasun Hospital-National Biobank of Korea between 2004 and 2009. Information about samples is summarized in Supplementary Table 1.
DNA cloning and stable transfection
Constructs encoding human E-cadherin and HA-tagged human Pg (wild type (WT), C420S, or C457S) were, respectively, cloned into the pCDNA3.1(+)/puro and pEGFP-N1/Neo vectors. MC38 murine colon carcinoma cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% (v/v) fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C under 5% CO2. Using Lipofectamine® 3000 (Invitrogen, Carlsbad, CA), plasmid DNA encoding WT, C420S, or C457S Pg was co-transfected into MC38 cells along with plasmid DNA encoding E-cadherin. Mock vectors without Pg and E-cadherin constructs were also co-transfected into MC38 cells and used as controls. Cells resistant to puromycin (1 μg/ml) and G418 (500 μg/ml) were isolated and expanded, after which they were screened for Pg and E-cadherin expression using western blotting.
Stimulation of cell–cell interaction with fibroblast growth factor-2
MC38 cells expressing a WT or mutant Pg were serum-starved for 24 h and then maintained for 3 h with FBS-reduced (1% FBS) media containing 50 ng/ml recombinant human fibroblast growth factor-2 (FGF-2) (Peprotech, Rocky Hill, NJ) and 1 μg/ml heparin to stimulate the cadherin/catenin axis.
Oxidation of Pg by H2O2
Human CRC (HT29, Caco2, SW480, DLD1, and HCT116), breast cancer cells (MCF-7), and murine CRC (MC38) cells transfected with mock vector or either WT or mutant Pg were grown in DMEM at 37°C under 5% CO2, and normal colon tissue was collected from C57BL6/J mice, after which the cells and colon tissue were lysed in chilled radioimmunoprecipitation assay buffer (50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 2 mM ethylenediaminetetraacetic acid, and protease inhibitors). The protein extracts were treated with 0, 1, or 5 mM H2O2 for 20 min at 50°C and the extent of Pg oxidation was analyzed by biotinylated iodoacetamide (BIAM)-streptavidin (SA) immunoprecipitation (IP) and immunoblotting with Pg antibody (Cell Signaling Technology (CST), Danvers, MA). In addition, MC38 cells transfected with mock vector or either WT or mutant Pg were treated with 0 or 1 mM H2O2 for 24 h at 37°C under 5% CO2 and the Pg protein level was evaluated to check the influences of different types of Pg on oxidative stress.
BIAM labeling
Unmodified cysteine residues are selectively carbamidomethylated by iodoacetamide (IAM), as oxidatively modified cysteines do not react with IAM.[19] IAM was therefore applied for the detection of oxidatively modified proteins as previously published.[15] Briefly, pT and pN samples from patients with CRC were homogenized in radioimmunoprecipitation assay buffer containing 40 μM BIAM (Molecular Probes, Eugene, OR) and incubated for 4 h at 4°C. In addition, 40 μM BIAM was also added to the protein extracts from MCF-7, HCT116, and normal mouse colon tissue (mColon) treated with H2O2 as described above. The labeling reaction was quenched by adding 4 mM dithiothreitol, and the samples were processed for IP using SA-conjugated Sepharose® beads (GE Healthcare, Little Chalfont, UK).
Immunoprecipitation and immunoblotting
All steps in the IP protocol were carried out at 4°C. To detect oxidized/reduced (ox/red) protein, proteins labeled with BIAM were incubated overnight with SA-conjugated Sepharose® beads in a rotator. The mixtures were then centrifuged at 800g for 10 min, and the resulting supernatants were transferred to new tubes for the detection of oxidized proteins as the fraction of total protein that was not bound to the Sepharose® beads. The pellets were washed three times for the preparation of reduced proteins. All samples were loaded onto 10% SDS–polyacrylamide gel electrophoresis (SDS–PAGE) gels followed by immunoblotting with an anti-Pg antibody (CST).
To assess the involvement of residues C420 and C457 in the binding activity of Pg, proteins extracted from cells expressing WT or mutant Pg were incubated with anti-Pg (BD Biosciences, San Jose, CA) or anti-GFP agarose (MBL, Woburn, MA) overnight in a rotator. Protein G agarose (Invitrogen) was then added to the solution containing lysate–Pg antibody complexes and incubated overnight. The precipitated proteins were loaded onto SDS–PAGE gels and immunoblotted with the following primary antibodies: anti-HA (CST), anti-GFP (Abfrontier, Seoul, Korea), anti-N-cadherin (CST), anti-α-catenin (BD Biosciences), and anti-β-catenin (Abcam, Cambridge, UK). This was followed by incubation with horseradish peroxidase-conjugated secondary antibody, which was then detected using an enhanced chemiluminescence system (iNtRON Biotechnology, Seongnam, Korea).
Two-dimensional gel electrophoresis
The two-dimensional gel electrophoresis (2-DE) method was performed as described previously [20] for protein extracts from pT and pN samples from patients with CRC and MCF-7 and HCT116 cells treated with H2O2. Briefly, protein mixed with DeStreak™ rehydration solution (GE Healthcare) was applied to gel strips (GE Healthcare, Immobiline™ DryStrip, pH 3-10 NL, 7 cm) to rehydrate them, after which isoelectric focusing was carried out in four steps as follows: 50 V for 12 h, 500 V for 30 min, 1000 V for 30 min, and 8000 V for a total of 20,000 V-h. After reduction and alkylation, second dimension electrophoresis was conducted on an SDS–PAGE gel. Pg oxidation was then evaluated based on the shift in the location of the protein spot on the gel after immunoblotting with anti-Pg antibody (CST).
Immunofluorescence analysis
Immunofluorescent labeling was carried out as described previously.[21] Cells expressing WT or mutant Pg were grown on poly ʟ-lysine-coated glass coverslips. After treatment with FGF-2 or vehicle only as a control, the cells were fixed with 4% (w/v) paraformaldehyde and permeabilized with 0.1% (v/v) Triton™ X-100. The fixed cells were then stained with anti-HA (CST), anti-GFP (Abfrontier), anti-α-catenin (BD bioscience), and anti-β-catenin (CST) antibodies, and then with Alexa Fluor® 555-(Invitrogen) or Alexa Fluor® 488-conjugated (Invitrogen) secondary antibodies. 4′,6-Diamidino-2-phenylindole (DAPI) was used to stain the cell nuclei. The immunostained cells were then washed, mounted, and examined using an LSM 710 laser scanning confocal microscope (Carl Zeiss, Jena, Germany).
RNA extraction and quantitative real-time polymerase chain reaction
MC38 cells transfected with mock vector or either WT or mutant Pg were treated with FGF-2 or vehicle only as a control. Total RNA was isolated using RNeasy® kits (Qiagen, Venlo, Netherlands) primed with random hexamer oligonucleotides and was reverse transcribed using a PrimeScript™ RT Reagent Kit (TaKaRa Bio, Shiga, Japan). Real-time quantitative polymerase chain reaction was performed using SYBR® Green Master Mix (TaKaRa Bio). All target gene expression data were normalized to the expression of Gapdh. Vim, c-Myc, Ccnd1, and Fn1.
Tumor cell migration and proliferation assay
Cell migration was assessed using scratch-wound healing assays. After growing cells expressing WT or mutant Pg to 80−90% confluence, the confluent cells were treated with serum-free media containing 0.5 μg/ml mitomycin C for 4 h prior to scratching. The cells were scratched using a 200-µl pipette tip, then rinsed twice with phosphate-buffered saline before incubation in DMEM. During the incubation, the wounds were photographed at 0, 24, and 48 h after scratching, and the areas of the wounds were quantified using the ImageJ software (http://rsb.info.nih.gov). Cell migration was expressed in terms of wound healing as follows [22]: % of wound closure = ((At=0 h – At=Δh)/At=0 h) × 100%, where At = 0 h is the area of the wound measured immediately after scratching and At=Δh is the wound area measured 24 or 48 h after scratching.
The proliferation of cells expressing WT or mutant Pg was quantified using thiazolyl blue tetrazolium bromide (MTT) assays. The cells were seeded in 96-well culture plates, incubated for 3 days, and stained every 24 h with MTT dye (0.5 mg/ml) for 4 h at 37°C. SDS-HCl (0.1 g SDS/ml of 0.01 M HCl) solution was then added to each well, and the plate was incubated for an additional 4 h at 37°C. The absorbance at 570 nm was then measured in each well, and a growth curve was expressed as the fold changes at 24 and 48 h compared with the value measured at 0 h. The 0 h value was designated as the absorbance at 12 h after cell seeding.
Statistics
Statistical analysis was performed using the SPSS 17.0 software (IBM, Armonk, NY). Differences in the ox/red protein ratio were analyzed using t tests. Cell migration and proliferation were evaluated using analysis of variance and post hoc Bonferroni’s multiple comparisons test. For all tests, values of P < 0.05 were considered significant.
Results
Oxidative modification of Pg in CRC
To identify oxidatively modified cysteines in CRC tissues, the tumor tissue (pT) and adjacent normal tissue (pN) from the 13 CRC patients were used for proteomic analysis. We focused on comparing Pg sequences containing differentially oxidized cysteines between pT and pN. We found that C420 and C457 of Pg were oxidatively modified to sulfinic acid in pT, but not in pN (Supplementary Table 2), indicating greater oxidation of C420 and C457 in pT than in pN. We further assessed the oxidation of Pg in pT by immunoblotting for Pg after BIAM-SA precipitation (Figure 1(a)) and then determining the ox/red ratio for Pg (Figure 1(b)). We found that the Pg ox/red ratio was higher in pT from stage III cancers than in pN, whereas there was no significant difference between the Pg ox/red ratios in pN and pT from stage II CRC (Figure 1(b)). This suggests that the level of Pg oxidation increases with CRC progression. A shift in Pg toward a more acidic form was also observed in pT from stage III CRC (Figure 1(c)).
Figure 1.
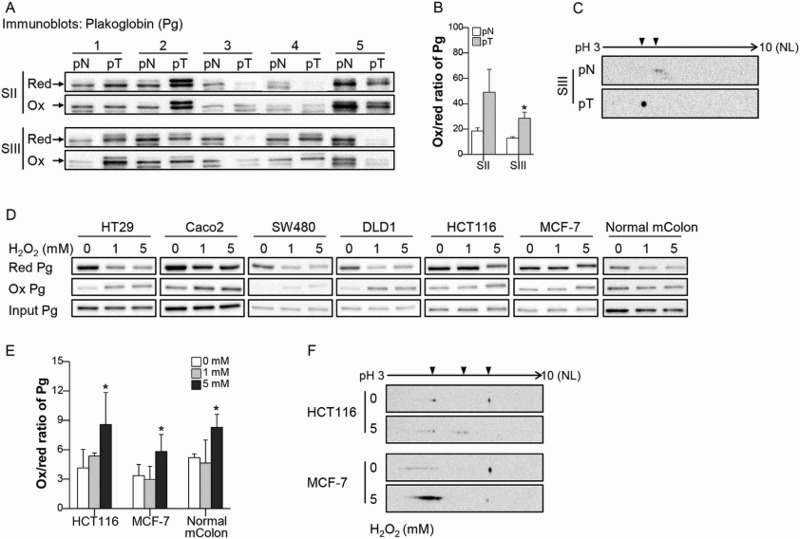
Pg is sensitive to oxidation in CRC. (a) Pg was detected after BIAM- SA precipitation in tumor tissue (pT) and adjacent normal tissue (pN). BIAM-labeled and unlabeled Pg were considered reduced (red) or oxidized (ox), respectively. Arrows indicate Pg. (b) The ox/red ratio was calculated based on the band densities of oxidized and reduced Pg. n = 5 for each group. (c) Pg oxidation was analyzed by 2-DE in pN and pT samples of patients with stage III CRC. Arrowheads indicate the location of the Pg spots. (d, e) After exposing CRC cells, MCF-7 cells, and normal mouse colon tissue (mColon) to H2O2, oxidation of Pg was detected using the BIAM-SA method and the ox/red ratio was calculated in HCT-116, MCF-7, and normal mColon. n = 3 for each group. (f) Pg oxidation by H2O2 treatment was confirmed using two-2-DE in HCT-116 and MCF-7 cells. Values are presented as mean ± standard error of the mean (SEM). *P < 0.05.
To assess the susceptibility of cellular Pg to oxidative stress related to tumor progression, different concentrations of H2O2 were administered to protein extracts of various cancer cells and normal mColon. We found that the level of BIAM-labeled (reduced) Pg was decreased in HT29, Caco2, SW480, and DLD1 cells treated with 1 mM H2O2 and HCT116, MCF-7, and normal mColon treated with 5 mM H2O2 (Figure 1(d)). The extent of Pg oxidation was also calculated in HCT116, MCF-7, and normal mColon, all of which showed an increase in the ox/red ratio of Pg after 5 mM H2O2 treatment (Figure 1(e)). In addition, Pg was shifted toward an acidic form in HCT116 cells and showed an increase in the abundance of the acidic form of Pg together with a decrease in the abundance of the basic form of Pg in MCF-7 cells exposed to 5 mM H2O2 (Figure 1(f)). Pg was generally oxidized in various cancer cells and healthy colon tissue. These findings support the idea that Pg is a redox-sensitive protein that can generally be oxidized under the conditions of oxidative stress found within tumors.
Sensitivity of Pg cysteine mutants to oxidative stress
MC38 cells transfected with mock vector or either WT, C420S, or C457S Pg were treated with 1 mM H2O2 and Pg protein levels were evaluated. The abundance of Pg protein was not significantly affected by H2O2 treatment in MC38 cells expressing WT, C420S, or C457S Pg (Figure 2(a,b)). Next, we estimated the extent of oxidation of Pg proteins with a mutated cysteine residue using the protein extracted from MC38 cells transfected with mock vector or either WT, C420S, or C457S Pg. C457S Pg protein showed less oxidation than WT and C420S Pg protein did, while C420S Pg protein was highly oxidized (Figure 2(c,d)). This result suggests that the mutation of C457 might protect Pg from oxidation and C457 could be a critical cysteine site related to Pg oxidation. However, all of WT, C420S, and C457S Pg protein showed an increase in the ox/red ratio of Pg after 1 mM H2O2 treatment and the abundance of C457S Pg oxidation was highly increased by 1 mM H2O2, indicating inevitable Pg protein oxidation under oxidative stress.
Figure 2.
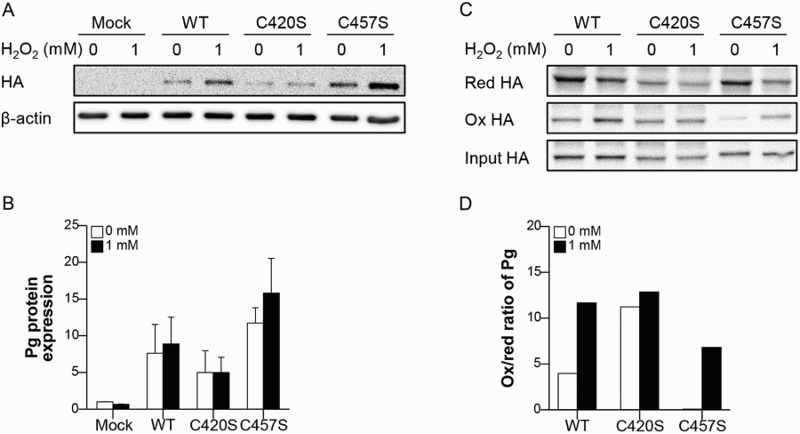
C457S of Pg is potentially redox-sensitive. (a, b) MC38 cells transfected with mock vector or either WT, C420S, or C457S Pg were treated with 1 mM H2O2 and the Pg protein level was evaluated. n = 2 for each group. (c, d) Proteins extracted from MC38 cells transfected with mock vector or WT, C420S, or C457S Pg were treated with 0 or 1 mM H2O2 and the oxidation of Pg was detected and analyzed using the BIAM-SA method. Values are presented as mean ± SEM.
Effect of Pg cysteine mutations on its binding ability
Cadherins signal indirectly by recruiting cytosolic proteins to the membrane that are then translocated to the nucleus, where they affect gene transcription. To investigate whether the mutation of Pg C420 and/or C457 affects the protein’s binding to cadherins/catenins, IP assays were performed using cells expressing a GFP-E-cadherin fusion protein plus WT, C420S, or C457S Pg. In addition, FGF-2 was used to stimulate upregulation of the cadherin/catenin system and enhance cell–cell contacts. The incubation time of the cells with FGF-2 was chosen based on the changes in Pg expression in an effort to select the most appropriate time for the assessment of the interaction between the Pg mutants and other molecules. Pg expression was increased after 3 h incubation with FGF-2, whereas it was decreased after 24 h incubation with FGF-2 (Figure 3(a) and Supplementary Figure 1). Although N-cadherin, α-catenin, and β-catenin levels were unaffected by FGF-2, E-cadherin expression was increased in cells incubated for 3 h with FGF-2 (Figure 3(a,b)). These conditions were therefore deemed appropriate for the evaluation of Pg recruitment by E-cadherin.
Figure 3.
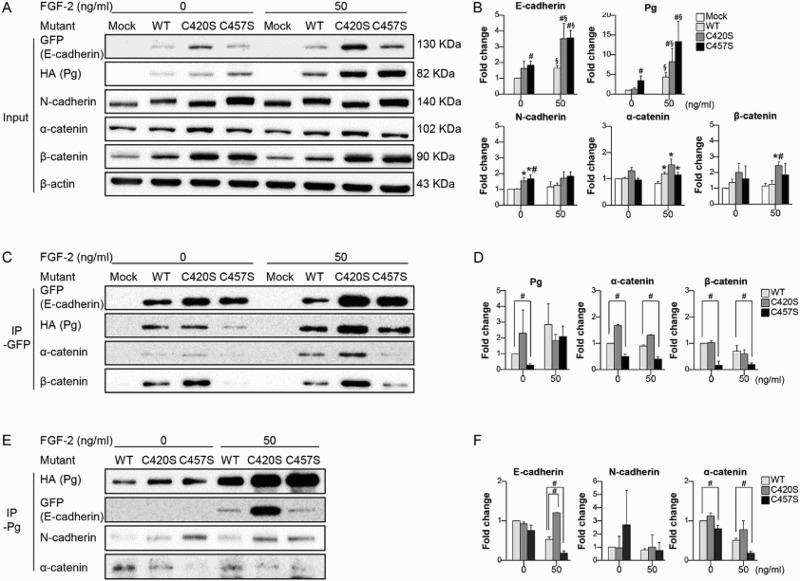
C457S influences E-cadherin/catenins interactions. (a, b) MC38 cells co-transfected with E-cadherin plus WT or mutant Pg were incubated with or without FGF-2 for 3 h, after which the levels of cadherin and catenin expression were evaluated. (c, d) Levels of catenin interacting with E-cadherin were analyzed using IP with GFP in the absence or presence of FGF-2. (e, f) Levels of cadherin/catenin interacting with Pg were analyzed using IP with Pg. Values are presented as mean ± SEM. *P < 0.05 vs. Mock, #P < 0.05 vs. WT, §P < 0.05 vs. without FGF-2.
The results of the IP assays with GFP showed that in cells expressing the C457S mutant, the binding of α- and β-catenins, and Pg to E-cadherin were suppressed as compared to the binding of these proteins in cells expressing WT Pg (Figure 3(c,d)). Levels of E-cadherin/catenin binding did not differ between cells expressing C420S or WT Pg, with or without FGF-2 treatment (Figure 3(c,d)). The suppressed binding of E-cadherin and α-catenin to Pg was also confirmed in cells expressing C457S Pg by reverse IP with Pg (Figure 3(e,f)). The binding of N-cadherin to Pg did not differ among cells expressing WT, C420S, and C457S Pg (Figure 3(e,f)), and binding between Pg and β-catenin was not observed (data not shown).
Effect of Pg cysteine mutations on its cellular localization
WT and C420S Pg (red fluorescence) localized with E-cadherin (green fluorescence) at the cell membrane even in unstimulated cells (Figure 4(a)). In contrast, C457S Pg was diffusely distributed throughout the cytoplasm in unstimulated cells. In FGF-2 stimulated cells, C457S Pg was partially localized at the cell membrane, but C457S Pg was still largely distributed throughout the cytoplasm. Thus, the interaction between the C457S mutant and membrane-bound proteins was not completely recovered despite upregulation of the cadherin/catenin system by FGF-2. Interestingly, α-catenin was more frequently observed in the nuclei of C457S Pg cells than in those of WT and C420S Pg cells (Figure 4(b)). β-Catenin localized in the cytoplasm and cell membrane with no significant differences among WT and mutant Pg cells, but exhibited a strong signal in the cell membranes of WT and C420S Pg cells compared with that of C457S Pg cells (Figure 4(c)). Overall, it appears that the C457S Pg mutation altered the assembly of the E-cadherin/catenins axis and disrupted the cellular localization of Pg.
Figure 4.
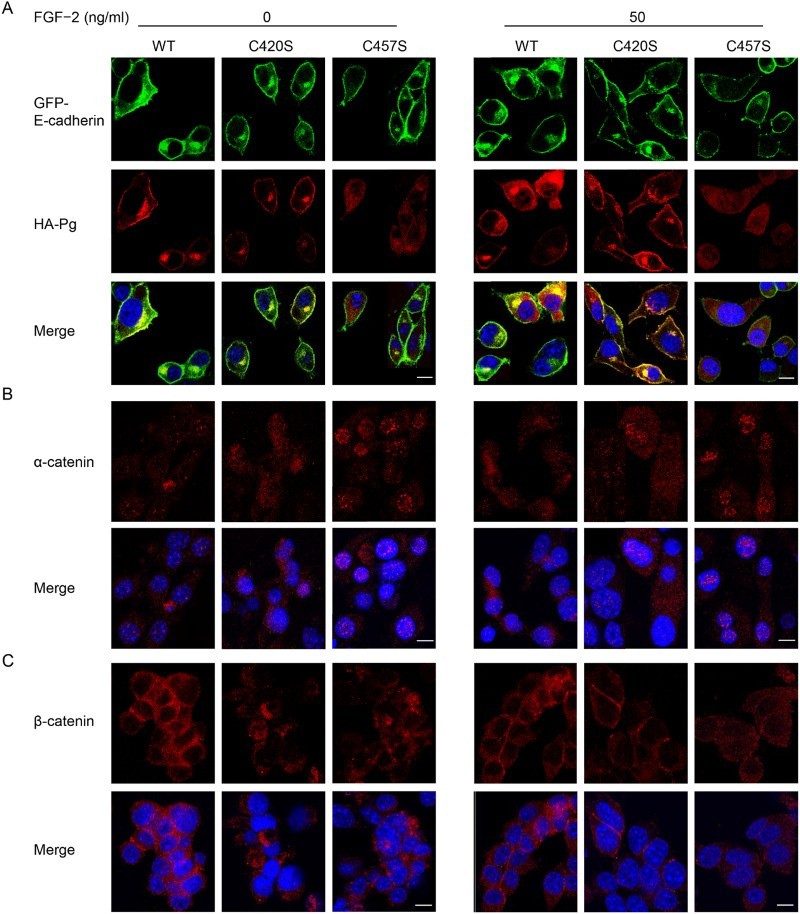
C457S alters the cellular localization of Pg. (a–c) Cellular distributions of E-cadherin (green), Pg (red), α-catenin (red), and β-catenin (red) were examined in MC38 cells expressing WT, C420S, and C457S Pg using immunofluorescent analysis. DAPI (blue) was used to stain cell nuclei. Scale bar = 10 μm.
The expression of β-catenin target genes and oncogenic activity in cysteine-mutated Pg cells
The genes Vim, c-Myc, Ccnd1, and Fn1 were analyzed as β-catenin target genes. Fn1 mRNA expression in C457S Pg cells decreased after treatment with both 0 and 50 ng/ml of FGF-2 compared with that in WT and C420S Pg cells (Figure 5). WT and C420S Pg cells showed large increases in Fn1 mRNA expression after being treated with FGF-2, whereas its expression was not stimulated by FGF-2 in C457S Pg cells.
Figure 5.
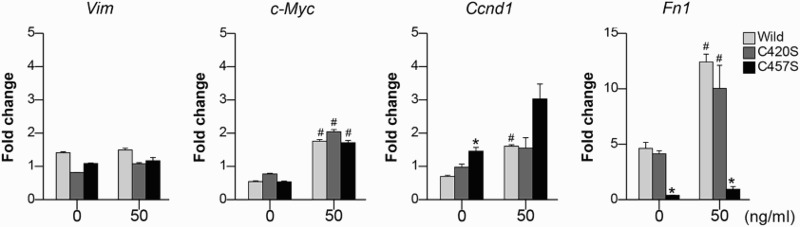
The expression in β-catenin target genes. The mRNA expression levels of Vim, c-Myc, Ccnd1, and Fn1 were assessed in MC38 cells transfected with mock vector or WT, C420S, or C457S Pg and treated with 0 or 50 ng/ml of FGF-2. Values are presented as mean ± SEM. *P < 0.05 vs. WT, #P < 0.05 vs. without FGF-2. n = 3 for each group.
The effects of Pg mutations on cancer cell migration and proliferation were evaluated using scratch-wound healing and MTT assays. Migration was monitored for 2 days after scratching cell monolayers (Figure 6(a)). C457S-expressing cells migrated slower than mock vector or WT-expressing cells, ultimately reducing wound healing as compared to that of the other groups at 24 and 48 h (Figure 6(b)). Proliferation was also lower in C457S-expressing cells than in the other groups at 24 h, but did not differ from WT at 48 h (Figure 6(c)). C475S-expressing cells appeared to show suppressed cell migration and reduced proliferation at early time points.
Figure 6.
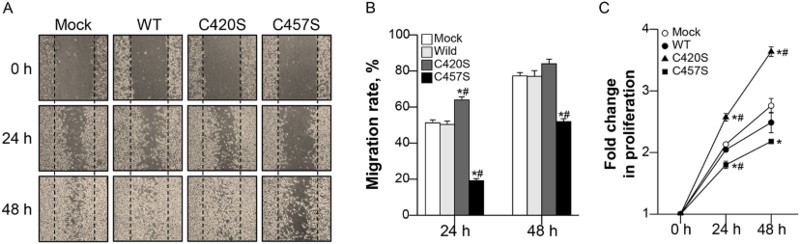
C457S Pg reduces MC38 cell migration and proliferation. (a, b) After scratch-wounding cell monolayers, photographs were taken at 0, 24, and 48 h. (c) The proliferation of cells expressing WT or mutant Pg was assayed using thiazolyl blue tetrazolium bromide (MTT) assay every 24 h for 2 days. Cell migration and proliferation levels are shown as the mean ± SEM of three independent experiments. *P < 0.05 vs. Mock, #P < 0.05 vs. WT.
Discussion
The association between oxidative stress and CRC has been studied extensively and the importance of ROS in tumor initiation and development is well established.[23] ROS oxidize proteins and inhibit the proteolytic system, leading to alterations in the structure of proteins and changes to enzyme functions, such as the inhibition of enzymatic and binding activities.[24] The rapid division and high metabolic rate of CRC cells may be responsible for increased oxidation of both proteins and DNA.[25] However, the regulation of cysteine oxidation and the pathophysiologic role of redox-based thiol modification are still largely uncharacterized.[14] Our aim in the present study was to better understand the implications of the increased Pg oxidation that occurs with the progression of CRC. Increases in oxidative stress markers have also been reported in patients with progressive breast cancer.[26] These findings support the idea that quantifying the oxidation of redox-sensitive proteins could be a useful means of monitoring tumor progression.
It is noteworthy that Pg exhibited a slightly higher molecular weight in pT from patients with stage III CRC than in pN from the same patients (upper panel in Figure 1(a)). Pg is reportedly regulated in part through post-translational modification,[27] which is a widely observed mechanism for modulating the activity of proteins by changing the properties of proteins through modifications such as proteolytic cleavage or the addition of a modifying group to amino acids.[28] Although we have not confirmed the specific modification that resulted in the slightly higher molecular size of Pg in pT from patients with stage III CRC in this study, we identified a unique pattern of Pg oxidation that accompanied increases in oxidative stress via shifts in molecular size by the BIAM-SA system. The upward-shifted pattern of Pg molecular size was also observed in H2O2-treated HCT116 and MCF-7 protein extracts (Figure 1(d)).
Cysteines within proteins may be rapidly oxidized upon exposure to oxidative stress, resulting in the functional inactivation of the protein.[11] Our hypothesis was that the oxidation of Pg cysteines disrupts the cadherin/catenin axis by affecting the binding of Pg to E-cadherin, which would in turn alter the tumorigenic activity of cancer cells. Unfortunately, there was no direct way to oxidize specific target cysteines to prove our hypothesis. Therefore, to test the idea, we examined the effect of replacing target cysteines with serines, thereby representing alteration in Pg activity by cysteine oxidation. The MC38 cells used for this study were ideal for verifying the association between specific Pg cysteines and the E-cadherin/catenin axis because prior to transfection, they are negative for E-cadherin and Pg but positive for N-cadherin, α-catenin, and β-catenin. C420-expressing cells strongly expressed Pg and E-cadherin as compared to WT and C457-expressing cells, leading to strong interactions between Pg and the E-cadherin/catenin axis. Thus, the high proliferation and migration activities of C420S-expressing cells may reflect the interactions between the Pg mutant and the E-cadherin/catenin axis. In contrast, C457S showed a weaker binding activity despite C457S-expressing cells showing higher levels of Pg and E-cadherin expression than WT-expressing cells, and this weaker binding may result in decreased oncogenic activity as assessed by proliferation and migration assays. Tumor proliferation and migration consistently correlated with the interaction of Pg and E-cadherin, and apparently, C457 is central to the binding interaction between these proteins.
Pg consists of the N- and C-terminal domains, and a 560-amino-acid-long central region comprising 13 armadillo repeats.[29] Troyanovsky et al. [30] suggested that Pg has two functionally distinct major parts, namely, regions 1–5, which contain E-cadherin and α-catenin specific binding sites, and regions 6–13, which target Pg–cadherin complexes into junctional structures. They also showed that Pg with mutagenesis in regions 6–13 was exclusively localized in the cytoplasm and not at the cell–cell contacts. Other reports have explained the contribution of the domain near the C-terminus of Pg to its association with the classical cadherins such as E-cadherin [31] and the contribution of regions 6–13 to its affinity for E-cadherin.[32] These results support that C457, which is located in region 8, can play important roles in directly binding to E-cadherin or indirectly mediating E-cadherin/catenin assembly.
The primary function of Pg is to mediate cell–cell adhesion via cadherins,[33] which are transmembrane glycoproteins that interact with catenin family proteins. The cytoplasmic domain of E-cadherin interacts with β-catenin or Pg, which then interacts with α-catenin. Despite the reported tumor-suppressing activity of Pg,[17,18] several studies have suggested that Pg expression may lead to a transformed phenotype associated with increased oncogenic β-catenin signaling.[34,35] Our results from experiments on cells expressing the C457S mutant suggest that Pg indirectly affects the β-catenin/α-catenin axis, which is linked to E-cadherin, and this may affect CRC cell proliferation and migration. Interrupting the E-cadherin/β-catenin interaction and thus inhibiting oncogenic β-catenin signaling may suppress the oncogenic activity of C457S-expressing cells (Figure 5).
In addition, we checked the binding between Pg and E-cadherin in pN and pT of CRC stages II and III tissues to evaluate tumor development based on E-cadherin/Pg binding. However, we found that there was no significant difference in the interaction of Pg and E-cadherin between pT and pN, irrespective of disease stage, though there was a high degree of variation in the expression level of Pg among samples (data not shown). It is considered that pN also may have tumorigenic characteristics within the tumor microenvironment, which makes determination of the differences difficult. Unfortunately, the use of normal colon tissue from healthy subjects could not be used as a control for comparison with pT because the normal tissue lacked expression of Pg.[36]
In the present study, we determined that C457 in Pg may be important for mediating E-cadherin/catenin binding and regulating tumor activity in CRC. Our results suggest that C457 could be a useful therapeutic target for CRC treatment and imply that profiling and targeting redox-sensitive cysteines may be a promising approach for cancer diagnosis and therapy.
Supplementary Material
Notes on contributors
Dr Suhee Kim is a research professor of the Department of Biochemistry at School of Dentistry in Chonnam National University. She graduated with BS/MS degree in College of Veterinary Medicine from Chonbuk National University and received her PhD in College of Veterinary Medicine from Chonbuk National University in 2009. During her PhD course, she studied on functional damages of sperm after cryopreservation and improvement of cryopreserved sperm quality in dogs. Following her graduate studies, she worked as a postdoctoral researcher from December 2010 to November 2012 at Department of Veterinary Pathobiology, University of Missouri-Columbia, studying rat sperm cryobiology. Recently, she joined Prof. Lee, Tae-Hoon's lab in Chonnam National University and is studying on ROS and defense mechanisms using animal disease model.
Dr Sun Hee Ahn is a research professor of the Department of Biochemistry at School of Dentistry in Chonnam National University. She graduated with BS/MS degree in Biotechnology and Bioengineering from Pukyong National University. She received her PhD in Microbial Engineering from Pukyong National University in 2006. While earning her PhD, she studied various virulence factors such as (metalloprotease, phospholipase, hemolysin, phosphomannomutase (PMM), and flagella motor protein (MotX)), using knock-out mutations and animal models. Through these studies, she developed a keen insight into the molecular mechanism of bacterial pathogenesis. Following her graduate studies, she worked as a postdoctoral researcher from September 2006 to May 2008 at Case Western Reserve University studying receptor proteins in oral epithelial cells which interact with adhesin proteins of Fusobacterium nucleatum. This further sparked her interest in researching the host-pathogen interaction, and brought her to Duke. During her time at Duke (2008–2012), Dr. Ahn has produced a number of interesting results while studying murine host susceptibility to S. aureus by microarray and QTL analyses. She published these results in the peer-reviewed journal. She was also Principal Investigator on a competitive grant from Pfizer Inc. Recently, she joined Prof. Lee, Tae-Hoon's lab in Chonnam National University and is studying Host Redox Signaling in bacterial infectious disease model.
Hee-Young Yang received MS/PhD degree in Chonnam National University. Now she is working in the Daegu Gyeongbuk Medical Innovation Foundation (DFMIF) as researcher.
Jin-Sil Lee is a student in the master's course of School of Dentistry at Chonnam National University.
Hyang-Gi Choi is a student in the master's course of School of Dentistry at Chonnam National University.
Dr Young-Kyu Park is a professor of division of gastrointestinal surgery, department of surgery at Chonnam National University Hwasun Hospital. He received MD/PhD in Chonnam National University Medical School. He studied on development of GI tract cancer at Vanderbilt University in USA during postdoctoral course. Now he is working in the translational research field of stomach and colon cancer and also many clinical researches as surgical oncologist.
Tae-Hoon Lee is a professor of Biochemistry, School of Densitry at Chonnam National University. He is a Biochemist in Dental Science. Scientific Interest: Redox regulation in protein. Experimental Models: Disease model mice as osteoporosis, bone defect and cancer.
Funding Statement
This work was supported by Korea Mouse Phenotyping Project (NRF-2014M3A9D5073721) of the Ministry of Science, ICT and Future Planning through the National Research Foundation and by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (2011-0030121).
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCiD
Suhee Kim http://orcid.org/0000-0002-9315-0360
Sun Hee Ahn http://orcid.org/0000-0002-0772-9588
Hee-Young Yang http://orcid.org/0000-0001-7654-8708
Jin-Sil Lee http://orcid.org/0000-0002-9795-3128
Hyang-Gi Choi http://orcid.org/0000-0001-7487-4585
Young-Kyu Park http://orcid.org/0000-0002-3009-404X
Tae-Hoon Lee http://orcid.org/0000-0003-0105-1750
References
- [1].Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet. 2014;383(9927):1490–1502. doi: 10.1016/S0140-6736(13)61649-9 [DOI] [PubMed] [Google Scholar]
- [2].Johnston PG. Identification of clinically relevant molecular subtypes in colorectal cancer: the dawning of a new era. Oncologist. 2014;19(5):568–573. doi: 10.1634/theoncologist.2014-038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mueller MM, Fusenig NE. Friends or foes — bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4(11):839–849. [DOI] [PubMed] [Google Scholar]
- [4].Roessner A, Kuester D, Malfertheiner P, Schneider-Stock R. Oxidative stress in ulcerative colitis-associated carcinogenesis. Pathol Res Pract. 2008;204(7):511–524. doi: 10.1016/j.prp.2008.04.011 [DOI] [PubMed] [Google Scholar]
- [5].Yuan J, Glazer PM. Mutagenesis induced by the tumor microenvironment. Mutat Res. 1998;400(1–2):439–446. doi: 10.1016/S0027-5107(98)00042-6 [DOI] [PubMed] [Google Scholar]
- [6].Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat. 2004;7(2):97–110. doi: 10.1016/j.drup.2004.01.004 [DOI] [PubMed] [Google Scholar]
- [7].Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15(2):247–254. doi: 10.1016/S0955-0674(03)00002-4 [DOI] [PubMed] [Google Scholar]
- [8].Veal EA, Day AM, Morgan BA. Hydrogen peroxide sensing and signaling. Mol Cell. 2007;26(1):1–14. doi: 10.1016/j.molcel.2007.03.016 [DOI] [PubMed] [Google Scholar]
- [9].Kumsta C, Thamsen M, Jakob U. Effects of oxidative stress on behavior, physiology, and the redox thiol proteome of Caenorhabditis elegans. Antioxid Redox Signal. 2011;14(6):1023–1037. doi: 10.1089/ars.2010.3203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Stadtman ER. Protein oxidation in aging and age-related diseases. Ann N Y Acad Sci. 2001;928:22–38. doi: 10.1111/j.1749-6632.2001.tb05632.x [DOI] [PubMed] [Google Scholar]
- [11].Sohal RS. Role of oxidative stress and protein oxidation in the aging process. Free Radic Biol Med. 2002;33(1):37–44. doi: 10.1016/S0891-5849(02)00856-0 [DOI] [PubMed] [Google Scholar]
- [12].Brandes N, Schmitt S, Jakob U. Thiol-based redox switches in eukaryotic proteins. Antioxid Redox Signal. 2009;11(5):997–1014. doi: 10.1089/ars.2008.2285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Paulsen CE, Carroll KS. Orchestrating redox signaling networks through regulatory cysteine switches. ACS Chem Biol. 2010;5(1):47–62. doi: 10.1021/cb900258z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Leonard SE, Carroll KS. Chemical ‘omics’ approaches for understanding protein cysteine oxidation in biology. Curr Opin Chem Biol. 2011;15(1):88–102. doi: 10.1016/j.cbpa.2010.11.012 [DOI] [PubMed] [Google Scholar]
- [15].Yang HY, Chay KO, Kwon J, Kwon SO, Park YK, Lee TH. Comparative proteomic analysis of cysteine oxidation in colorectal cancer patients. Mol Cells. 2013;35(6):533–542. doi: 10.1007/s10059-013-0058-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Parker HR, Li Z, Sheinin H, Lauzon G, Pasdar M. Plakoglobin induces desmosome formation and epidermoid phenotype in N-cadherin-expressing squamous carcinoma cells deficient in plakoglobin and E-cadherin. Cell Motil Cytoskeleton. 1998;40(1):87–100. doi: [DOI] [PubMed] [Google Scholar]
- [17].Simcha I, Geiger B, Yehuda-Levenberg S, Salomon D, Ben-Ze’ev A. Suppression of tumorigenicity by plakoglobin: an augmenting effect of N-cadherin. J Cell Biol. 1996;133(1):199–209. doi: 10.1083/jcb.133.1.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Charpentier E, Lavker RM, Acquista E, Cowin P. Plakoglobin suppresses epithelial proliferation and hair growth in vivo. J Cell Biol. 2000;149(2):503–520. doi: 10.1083/jcb.149.2.503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bardwell J. Thiol modifications in a snapshot. Nat Biotechnol. 2005;23(1):42–43. doi: 10.1038/nbt0105-42 [DOI] [PubMed] [Google Scholar]
- [20].Yang H-Y, Lee T-H. The oxidative modification of COL6A1 in membrane proteins of ovarian cancer patients. Reprod Dev Biol. 2012;36(1):39–47. doi: 10.1095/biolreprod.112.099234 [DOI] [Google Scholar]
- [21].Ahn SH, Yang HY, Tran GB, et al. . Interaction of peroxiredoxin V with dihydrolipoamide branched chain transacylase E2 (DBT) in mouse kidney under hypoxia. Proteome Sci. 2015;13:32. doi: 10.1186/s12953-014-0061-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yue PY, Leung EP, Mak NK, Wong RN. A simplified method for quantifying cell migration/wound healing in 96-well plates. J Biomol Screen. 2010;15(4):427–433. doi: 10.1177/1087057110361772 [DOI] [PubMed] [Google Scholar]
- [23].Chang D, Wang F, Zhao YS, Pan HZ. Evaluation of oxidative stress in colorectal cancer patients. Biomed Environ Sci. 2008;21(4):286–289. doi: 10.1016/S0895-3988(08)60043-4 [DOI] [PubMed] [Google Scholar]
- [24].Perse M. Oxidative stress in the pathogenesis of colorectal cancer: cause or consequence? Biomed Res Int. 2013;2013:1–9. doi: 10.1155/2013/769295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Foksinski M, Rozalski R, Guz J, et al. . Urinary excretion of DNA repair products correlates with metabolic rates as well as with maximum life spans of different mammalian species. Free Radic Biol Med. 2004;37(9):1449–1454. doi: 10.1016/j.freeradbiomed.2004.07.014 [DOI] [PubMed] [Google Scholar]
- [26].Tesarova P, Kalousova M, Trnkova B, et al. . Carbonyl and oxidative stress in patients with breast cancer–is there a relation to the stage of the disease? Neoplasma. 2007;54(3):219–224. [PubMed] [Google Scholar]
- [27].Wickline ED, Awuah PK, Behari J, Ross M, Stolz DB, Monga SP. Hepatocyte gamma-catenin compensates for conditionally deleted beta-catenin at adherens junctions. J Hepatol. 2011;55(6):1256–1262. doi: 10.1016/j.jhep.2011.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mann M, Jensen ON. Proteomic analysis of post-translational modifications. Nat Biotechnol. 2003;21(3):255–261. doi: 10.1038/nbt0303-255 [DOI] [PubMed] [Google Scholar]
- [29].Cowin P, Burke B. Cytoskeleton—membrane interactions. Curr Opin Cell Biol. 1996;8(1):56–65. doi: 10.1016/S0955-0674(96)80049-4 [DOI] [PubMed] [Google Scholar]
- [30].Troyanovsky RB, Chitaev NA, Troyanovsky SM. Cadherin binding sites of plakoglobin: localization, specificity and role in targeting to adhering junctions. J Cell Sci. 1996;109(Pt 13):3069–3078. [DOI] [PubMed] [Google Scholar]
- [31].Wahl JK, Sacco PA, McGranahan-Sadler TM, Sauppe LM, Wheelock MJ, Johnson KR. Plakoglobin domains that define its association with the desmosomal cadherins and the classical cadherins: identification of unique and shared domains. J Cell Sci. 1996;109(Pt 5):1143–1154. [DOI] [PubMed] [Google Scholar]
- [32].Chitaev NA, Leube RE, Troyanovsky RB, Eshkind LG, Franke WW, Troyanovsky SM. The binding of plakoglobin to desmosomal cadherins: patterns of binding sites and topogenic potential. J Cell Biol. 1996;133(2):359–369. doi: 10.1083/jcb.133.2.359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Aktary Z, Pasdar M. Plakoglobin: role in tumorigenesis and metastasis. Int J Cell Biol. 2012;2012:1–14. doi: 10.1155/2012/189521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Li L, Chapman K, Hu X, Wong A, Pasdar M. Modulation of the oncogenic potential of β-catenin by the subcellular distribution of plakoglobin. Mol Carcinog. 2007;46(10):824–838. doi: 10.1002/mc.20310 [DOI] [PubMed] [Google Scholar]
- [35].Teuliere J, Faraldo MM, Shtutman M, et al. . Beta-catenin-dependent and -independent effects of DeltaN-plakoglobin on epidermal growth and differentiation. Mol Cell Biol. 2004;24(19):8649–8661. doi: 10.1128/MCB.24.19.8649-8661.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yang HY, Kwon J, Park HR, et al. . Comparative proteomic analysis for the insoluble fractions of colorectal cancer patients. J Proteomics. 2012;75(12):3639–3653. doi: 10.1016/j.jprot.2012.04.018 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.