

ABSTRACT
Atherosclerosis is the main pathophysiological process underlying coronary artery disease (CAD). Acute complications of atherosclerosis, such as myocardial infarction, are caused by the rupture of vulnerable atherosclerotic plaques, which are characterized by thin, highly inflamed, and collagen-poor fibrous caps. Several lines of evidence mechanistically link the heme peroxidase myeloperoxidase (MPO), inflammation as well as acute and chronic manifestations of atherosclerosis. MPO and MPO-derived oxidants have been shown to contribute to the formation of foam cells, endothelial dysfunction and apoptosis, the activation of latent matrix metalloproteinases, and the expression of tissue factor that can promote the development of vulnerable plaque. As such, detection, quantification and imaging of MPO mass and activity have become useful in cardiac risk stratification, both for disease assessment and in the identification of patients at risk of plaque rupture. This review summarizes the current knowledge about the role of MPO in CAD with a focus on its possible roles in plaque rupture and recent advances to quantify and image MPO in plasma and atherosclerotic plaques.
KEYWORDS: Atherosclerosis, cardiovascular disease, coronary artery disease, imaging, myeloperoxidase, plaque rupture, reactive oxygen species, vulnerable plaques
Introduction
Coronary artery disease (CAD) is the single largest cause of mortality worldwide. The majority of CAD is caused by atherosclerosis, a progressive, multi-factorial, inflammatory disease of the arterial wall. The disease culminates in the rupture of atherosclerotic plaques, which may lead to arterial occlusion and life-threatening acute coronary syndromes (ACS). Atherosclerotic plaque rupture can occur as the first manifestation of the disease in otherwise asymptomatic subjects. Therefore, being able to predict the level of plaque instability could mean preventing new cardiovascular events and optimizing medical care for a vast number of patients.
Numerous clinical studies have assessed the utility of inflammatory biomarkers for characterization cardiovascular disease (CVD) severity and thus the risk of plaque rupture. Early epidemiological studies evaluated the clinical usefulness of inflammatory markers such as C-reactive protein, adhesion molecules, cytokines, and white cell count [1]. Currently, high sensitivity C-reactive protein and troponins are the only plasma biomarkers in routine use, but both have significant limitations. The detection of circulating myocardial troponin is useful for detecting cardiac injury in patients with advanced coronary disease but cannot identify vulnerable patients at risk of (but prior to) a plaque rupture event. The acute phase reactant CRP is an inflammatory biomarker with a definite association with cardiovascular events, however its value in guiding treatment decisions remains unproven [2]. Recent research has focused on novel inflammatory markers of which myeloperoxidase (MPO), released systemically and locally by activated leukocytes, has shown great promise [3]. It is currently hypothesized that MPO can be used as a diagnostic aid and risk stratification tool in patients who present to the emergency department with ACS [4].
This review aims to give an update on current knowledge about the role of MPO in human CAD in the context of thrombosis and sudden coronary death as a consequence of plaque rupture.
Atherosclerotic plaque rupture
Post-mortem studies have established atherosclerotic plaque rupture to be responsible for up to 75% of episodes of ACS [5]. Disrupted plaques are characterized by a high degree of inflammation, a large necrotic core and a thin fibrous cap. These plaque characteristics have been identified as important determinants of plaque vulnerability where fibrous cap thickness has been shown to be the best discriminator [5].
The exact molecular and cellular mechanisms underlying plaque destabilization have yet to be fully elucidated, however several key processes have been identified (Table 1).
Table 1. Characteristics of vulnerable atherosclerotic plaques and putative underlying processes.
| Key features of unstable plaques | Putative underlying processes |
|---|---|
| Fibrous cap thinning | Increase in apoptosis of VSMCs; decrease in the production of ECM; infiltration of macrophages and neutrophils and associated release of pro-inflammatory cytokines and MMPs |
| Intra-plaque inflammation | Accumulation of apolipoprotein B-containing lipoproteins in the sub-endothelial space; intra-plaque neovascularization or neoangiogenesis; increased endothelial permeability; increased mast cell activation |
| Increased necrotic core size | Death of macrophages and VSMCs; impaired clearance of apoptotic cells |
First, accelerated apoptosis of vascular smooth muscle cell (VSMC) is a major contributor to plaque instability. VSMCs increase the thickness and stability of the fibrous cap by producing extracellular matrix (ECM) proteins such as elastin and collagen [6]. However, the infiltration of macrophages, mast cells, and cytotoxic T lymphocytes into the plaque induces VSMC apoptosis through the release of pro-inflammatory cytokines and proteases [6]. This results in a thinning of the fibrous cap, exacerbated inflammation, and expansion of the necrotic core.
Second, macrophage death is thought to make a significant contribution to plaque rupture, as apoptotic macrophages are more prevalent in the fibrous cap of ruptured plaques than stable plaques [7]. Macrophages engulf apolipoprotein B-containing lipoproteins deposited in the sub-endothelial space, which are hydrolyzed in late endosomes to free cholesterol that is then re-esterified in the endoplasmic reticulum into the characteristic lipid droplets of foam cells [8]. In advanced atherosclerosis, stressors to the endoplasmic reticulum, such as impaired esterification and exposure to oxidized cholesterol, triggers apoptosis [8]. In vitro studies suggest that growth factor deprivation, oxidative stress, and death receptor activation by ligands that exist in advanced atherosclerosis also contribute to macrophage death [8]. Further, in atherosclerotic lesions, clearance of apoptotic cells by macrophages is impaired, possibly due to cytoplasmic overload of ingested material, oxidative stress, and competitive inhibition of receptors [9]. This exacerbates the accumulation of apoptotic cells, leading to an expansion of the necrotic core.
Third, macrophages and neutrophils may play a substantial role in the mechanical weakening of the fibrous cap, which inevitably precedes plaque rupture. Macrophages and neutrophils are attracted by chemokines produced by the inflammatory milieu and express members of the matrix metalloproteinase (MMP) family such as MMP-8, -9, -12, and -13, which are implicated in the degradation of ECM proteins [6].
Fourth, neoangiogenesis is a signature feature of advanced atherosclerotic plaques and is widely considered to be a major contributor to plaque weakening. Hypoxia, due to intimal thickening causing reduced oxygen diffusion, combined with higher oxygen requirements of metabolically active inflammatory cells, is believed to trigger neovascularization by inducing the expression of hypoxia-inducible transcription factor and vascular endothelial growth factor in macrophages [10]. However, these new vessels are structurally immature, leaky, and prone to rupture [11]. This promotes leukocyte infiltration and predisposition to intra-plaque hemorrhage, which is an independent predictor of future cardiovascular outcomes [12].
The enzymology of MPO
To contextualize the role of MPO in CAD and atherosclerosis, the cellular origins, structure, and basic mechanism of MPO enzymology must first be described.
Cellular origins
MPO is a major neutrophil effector protein, accounting for ∼5% of the cell’s dry mass and a concentration of ∼10 × 10−6 μg/cell [13]. MPO is stored in large amounts in the azurophilic granules from where it is released into the phagosome along with a plethora of anti-microbial agents after phagocytosis of pathogens. While the majority of MPO remains in the phagosome, up to 30% of total cellular MPO can be released as active enzyme into the extracellular space via degranulation or by association with neutrophil extracellular traps [14]. MPO is also present in monocytes, comprising one-third of the MPO content found in neutrophils. Differentiation of monocytes into macrophages is generally thought to be associated with a loss of MPO. However, granulocyte macrophage colony-stimulating factor has been identified as an endogenous regulator of macrophage MPO expression in human atherosclerosis, in support of a role for MPO-expressing macrophages in atheroma complication and ACS [15].
NADPH oxidase 2 assembled at the plasma or phagosomal membrane catalyzes the oxidation of NADPH to NADP+ with concomitant reduction of molecular oxygen (O2) to superoxide anion radical (O2•−). Dismutation of O2•− results in the formation of hydrogen peroxide (H2O2) that acts as an essential cofactor for the MPO catalytic cycle.
Structure
The three-dimensional structure of MPO encompasses a highly cationic surface (pI 10), abundant in lysine and arginine residues. This facilitates interaction of MPO with a variety of negatively charged surfaces and proteins, including bacterial and endothelial cell surfaces, ECM [16], low-density lipoprotein [17], apolipoprotein A-I [18], and ceruloplasmin [19]. MPO is 146 kDa homodimeric protein composed of functionally independent monomeric units joined by a single disulfide bond at Cys153. Each monomer is made up of a heavy glycosylated chain (58.5 kDa, 467 amino acids) and a light chain (14.5 kDa, 106 amino acids). Glycosylation of the heavy chain is important for MPO enzymatic activity, de-glycosylated MPO has significantly decreased chlorinating activity [20]. The heavy chain within each monomer contains a protoporphyrin IX heme derivative and a calcium-binding site essential for chain interaction.
The heme is linked to MPO by three covalent bonds, i.e. two ester bonds between the methyl groups of pyrrole rings A and C with Glu242 of the heavy chain and Asp94 of the light chain, respectively, and a sulfonium bond between pyrrole ring A and Met243 [21,22]. The sulfonium linkage causes planar distortion and asymmetry of the heme group, thereby contributing to the unique spectral properties and characteristic green color of MPO [21]. The heme is located within a deep crevice (15–20 Å) [23] limiting its access to small ions and H2O2 [24]. MPO activity is dependent on Asn421, ligated proximal to the heme group and His95 located distal to the heme. Asn421 serves as a hydrogen bond acceptor and facilitates the Fe(III)/Fe(II) reduction essential for compound I formation [25]. His95 accepts protons from H2O2 to initiate compound I formation and, upon cleavage of the oxygen–oxygen bond, it donates a proton to form water [26]. Located 3.5 Å from His95 is a hydrophobic pocket that interacts with most substrates that are oxidized by compound I. Within this vicinity the substrates can interfere with H2O2 binding and facilitate protonation of His95, which in turn stabilizes the substrate-bound complex at acidic pH. The following reference is a comprehensive review of the nature of the MPO active site [27].
Catalytic cycle
The catalytic cycle is initiated by the rapid two-electron oxidation of native Fe(III)-MPO by H2O2 (k ∼ 1.4 × 107 M−1s−1) to form water and compound I, a Fe(IV)-oxo porphyrin radical-cation species (Fe4+=O + porphyrin•+) (Figure 1) [27]. Native MPO is regenerated via reduction of compound I by either the ‘halogenation cycle’, a direct two-electron reduction by pseudo(halides), or by the ‘peroxidase cycle’, a two-step one-electron reduction via the formation of the redox intermediate, compound II (which retains the Fe4+=O center) [28]. Due to the high reduction potentials of compound I/native enzyme (1.16 V) [29], compound I/compound II (1.35 V), and compound II/native enzyme (0.97) [30], MPO can oxidize a myriad of substrates via both catalytic cycles. These reduction potentials, and hence the rate of substrate oxidation, are pH-dependent. For example, compound I is reduced readily to compound II at pH 7.8 and low concentrations of (pseudo)halide ions. At lower pH and in the presence of high concentrations of (pseudo)halide ions, the turnover of compound II is enhanced, thereby favoring the halogenation cycle. In vivo, MPO is expected to operate between pH 6.5 (i.e. at sites of inflammation) and pH 7.8 (inside the phagosome) [31].
Figure 1.
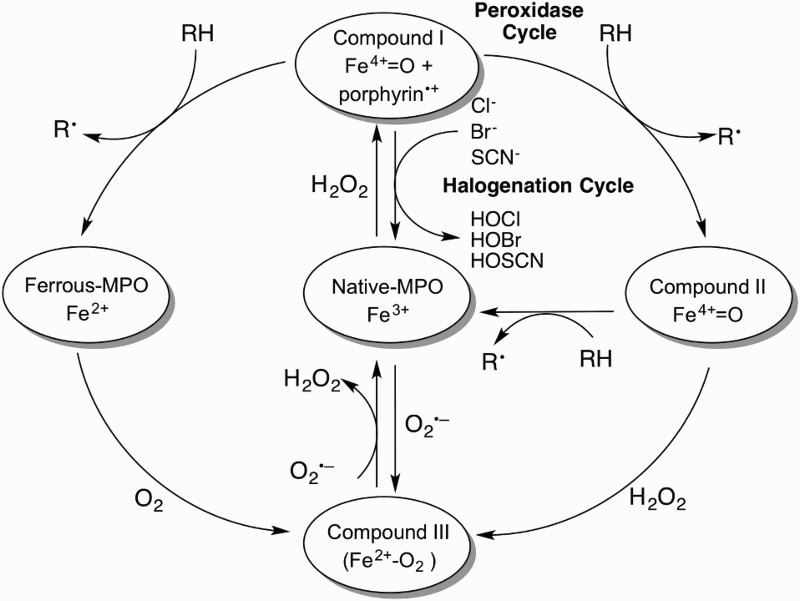
The MPO catalytic cycle. Native MPO reacts with H2O2 to form compound I. Compound I can be converted back to native MPO either by the halogenation cycle or by the two-step one-electron reduction in the peroxidase cycle. Ferrous-MPO and compound III are redox forms of MPO that exist outside the two catalytic cycles.
During the halogenation cycle (pseudo)halides, Cl−, Br−, and SCN− donate two electrons to compound I to form the corresponding reactive (pseudo)hypohalous acids: hypochlorous acid (HOCl), hypobromous acid (HOBr), and hypothiocyanous acid (HOSCN), respectively. The pKa-values for HOCl, HOBr, and HOSCN are 7.6, 8.7, and 3.5–4.9, respectively. At physiological pH therefore, the concentrations of HOCl and −OCl will be similar, while HOBr will predominate over −OBr and −OSCN will predominate over HOSCN [32–34]. In this review, the physiological mixtures of these species will be referred to as HOCl, HOBr, and HOSCN.
At neutral pH and physiological concentrations of the (pseudo)halides ions, Cl− serves as a major substrates for MPO, largely due to the greater abundance of Cl− relative to Br− and SCN− (Cl−, 100 −140 mM, Br− and SCN−, 20–100 μM), making HOCl the major MPO-derived oxidant. Factors that increase SCN− concentrations, such as diet and smoking, allow SCN− to be a competitive substrate for MPO [35]. Reaction with SCN− is afforded further by its large specificity constant (730-fold higher than Cl−) [36] and faster rate constant for compound I (9.6 × 106 versus 2.5 × 104 and 1.1 × 106 M−1s−1 for SCN−, Cl−, and Br−, respectively) [37]. Furthermore, HOCl and HOBr can oxidize SCN−, thereby increasing yields of HOSCN [38,39].
The peroxidase cycle is defined by the conversion of compound I to native MPO by a two-step one-electron reduction via compound II by radicals (nitric oxide, •NO, and O2•−), organic (including tyrosine, ascorbate, steroidal hormones and urate, as well as xenobiotics and drugs) and inorganic substrates (nitrite, NO2−). In certain cases, the resulting radical species can yield dimers and higher polymers, or react with other biomolecules such as lipids and proteins [28]. Radicals formed from amino/phenol derivatives can be reduced further to form electrophilic imino/quinone species that covalently react with thiol derivatives and other biomolecules [40].
Redox forms of MPO outside the halogenation and peroxidase cycles include Fe(II)-MPO, formed by the one-electron of reduction of native MPO and compound III. The latter is considered a ‘catalytically inactive’ ferrous dioxy intermediate formed either by the reaction of Fe(II)-MPO with O2, native MPO with O2•− or compound II with H2O2 [41]. Although considered ‘catalytically inactive’, ascorbate, O2•−, and paracetamol slowly reduce compound III to native MPO [42,43].
Reactivity of HOCl
Under physiological conditions HOCl is the most reactive two-electron oxidant, exceeding the reactivity of H2O2, peroxynitrite, and hydroperoxides [44]. HOCl reacts avidly with nucleophiles, with its fastest reactions occurring with thiols and thiolethers (e.g. Cys and Met residues in proteins, and reduced glutathione) (k ∼ 108 M−1s−1). Its reaction with other nucleophiles, including tryptophan, lysine, histidine, and terminal amines, occurs at k-values of ∼104–105 M−1s−1 [45,46]. HOCl reacts at much slower rates with double bonds, aromatic rings (including Tyr and Trp), fatty acid side chains and nucleobases (reviewed in reference [47]). Reactions with Tyr residues yield 3-chlorotyrosine, a specific biomarker for HOCl identified in human atherosclerotic lesions [48], together with chlorinated lipids [49,50]. The antioxidants ascorbate and urate react with HOCl with second-order rate constants of 6 × 106 and 2 × 105 M−1s−1, respectively [45,51]. Reaction of HOCl with amines yields chloramines, which are weaker oxidants, but more selective towards thiol residues than HOCl [52,53]. Once formed on proteins, chloramines and dichloramines can breakdown to form reactive carbonyls in addition to ammonia monochloramines that have a similar bactericidal potency to HOCl [54].
When HOCl is generated in excess, its rapid reaction with thiols and thiolethers can significantly affect cellular homeostasis. Perturbations of the redox balance of reduced and oxidized glutathione by HOCl can imbalance carefully regulated redox pathways. Furthermore, HOCl can modulate protein function by reacting with Cys-rich active sites of proteins. For example, HOCl readily inactivates endothelial nitric oxide synthase (eNOS), creatine kinase, and glyceraldehyde-3-phosphate dehydrogenase, with the loss of activity correlating with thiol oxidation [53,55,56]. Conversely, HOCl can activate MMPs, e.g. MMP-7, by oxidizing a key Cys residue in the cysteine switch domain [57].
Potential roles of MPO in atherosclerosis and atherosclerotic plaque instability
The involvement of MPO in human atherosclerotic plaque progression has been well established. Enzymatically active MPO is present in atherosclerotic lesions, with higher concentrations around their shoulder region [55]. Further, advanced atheromatous plaques demonstrate higher numbers of intimal MPO-expressing macrophages than early stage lesions [15]. Compared with humans, the contribution of MPO/MPO-derived oxidants to atherosclerosis in mouse models of the disease is less clear. For example, mice deficient in LDL receptor and MPO (Ldlr–/– Mpo–/–) develop unexpectedly larger lesions (>50%) than Ldlr–/– animals [58]. Importantly however, atherosclerotic lesions in Ldlr–/– mice did not exhibit measurable MPO activity as assessed by the presence of 3-chlorotyrosine [58], whereas human atherosclerotic lesions contain 3-chlorotyrosine (see below). While the precise role of MPO in murine atherosclerosis remains unclear, MPO and MPO-derived oxidants have numerous potential pro-atherogenic properties (Table 2).
Table 2. Potential pro-atherogenic actions of MPO and putative underlying processes.
| Pro-atherogenic actions of MPO | Putative underlying processes |
|---|---|
| Atherogenic LDL | Chlorination of apolipoprotein B-100 by MPO-derived HOCl and halogenating species; oxidation by MPO-derived reactive nitrogen species; thiocyanate-mediated carbamylation catalyzed by MPO |
| Dysfunctional HDL | Oxidation of tyrosine residues on apolipoprotein A-I by MPO-derived HOCl and reactive nitrogen species |
| Decreased NO bioavailability and endothelial dysfunction | Inhibition of eNOS by chlorination of its substrate arginine; uncoupling of eNOS by direct oxidation; intracellular dissociation of eNOS from the plasma membrane; decreased eNOS expression caused by chlorinated HDL |
| Generation of thrombogenic environment | Promotion of endothelial cell apoptosis and detachment by HOCl; induction of tissue factor activity in endothelial cells by HOCl |
| Thinning of plaque fibrous cap | Activation of MMPs by HOCl-mediated oxidation; inhibition of tissue inhibitors of MMPs (TIMPs) by MPO-derived oxidants |
Formation of atherogenic LDL
MPO has been shown to participate in multiple pathways of LDL oxidation, mediated by both radical 1e-oxidation and non-radical 2e-oxidation [59]. Compared with native LDL, oxidized LDL is more avidly taken up by macrophages, and this can lead to foam cell formation in vitro, and could conceivably contribute to an enlarged lipid core and exacerbated stress on the fibrous cap matrix, making atherosclerotic plaques more prone to rupture [60].
As mentioned earlier, MPO forms the highly reactive 2e-oxidant HOCl, which preferentially reacts with amino acids rather than lipids [59]. HOCl chlorinates electron-rich substrates on apolipoprotein B-100 such as Lys residues [61] and Tyr, forming MPO-specific 3-chlorotyrosine [3]. Atherosclerotic plaque is enriched in 3-chlorotyrosine and circulating LDL of patients with CVD contains 3-chlorotyrosine [48]. Exposure of mouse peritoneal macrophages to HOCl-oxidized LDL resulted in increased intracellular concentrations of cholesterol and cholesteryl esters, with Lys residues of apoliopoprotein B-100 representing the major oxidative target [62]. Competition studies revealed that this increased uptake is due to recognition of HOCl-modified LDL by the macrophage class B scavenger receptors CD36 and SR-BI [63]. Further, the MPO/HOCl system can generate a series of secondary oxidation products, such as tyrosine radicals, p-hydroxyphenylacetaldehyde and highly reactive unsaturated aldehydes-glyceraldehyde, 2-hydroxypropanal, and acrolein [59]. These products can themselves take part in oxidation reactions to generate high-uptake LDL. For example, LDL incubated with glyceraldehyde and glycolaldehyde was consumed by human monocyte-derived macrophages to a greater extent than native LDL [64], an activity mediated by the macrophage acetyl LDL receptor [65], now known as a class A scavenger receptor [63]. In addition to enhancing foam cell formation, LDL modified by HOCl may contribute to the development of atherosclerosis in several ways (Table 3). Importantly, the involvement of HOCl-mediated oxidation of LDL in atherogenesis could explain the poor outcome of antioxidant trials in the treatment of atherosclerosis, particularly α-tocopherol (vitamin E), as these antioxidants are effective only against 1e-oxidation reactions [59].
Table 3. Potential contributions of HOCl-modified LDL to the development of atherosclerosis.
| Induction of chemokine release by monocytes and chemotactic migration of neutrophils |
| Activation of the respiratory burst in macrophages and neutrophils, with resultant increase in O2•– (and H2O2) |
| Inactivation of macrophage lysosomal proteases, leading to accumulation of intracellular lipids |
| Decreased •NO synthesis by endothelial cells |
| Endothelial leakage and stimulation of leukocyte adherence to, and migration into, the sub-endothelial space |
| Enhanced platelet reactivity and release reaction |
Moreover, MPO/H2O2 oxidizes nitrite (NO2−) to nitryl chloride (NO2Cl) and reactive nitrogen dioxide radicals (•NO2), both of which are capable of oxidizing Tyr residues in apolipoprotein B-100 as well as lipids [59]. The resulting product, NO2LDL, is recognized by the macrophage scavenger receptor CD36 [66]. LDL from aortic atherosclerotic intima was found to contain 90-fold higher levels of protein standardized 3-nitrotyrosine than plasma LDL [67]. However, while Tyr nitration is characteristic of LDL exposed to MPO/H2O2/NO2−, competition studies suggest that oxidized lipids, rather than modified proteins or lipid-protein adducts, are the essential moiety in NO2-LDL that confers CD36-dependent receptor recognition [66]. In addition to nitrating LDL protein, •NO2 also initiates LDL lipid peroxidation [68], a process that is characteristic of oxidation by radicals and may result in binding to macrophages and foam cell formation in vitro [69]. This 1e-oxidation reaction can be inhibited by LDL associated α-tocopherol and is independent of the chloride ion, unlike HOCl-induced oxidation of LDL’s tyrosine and lysine residues [59].
As we have learned, MPO/H2O2 also catalyzes the oxidation of thiocyanate (SCN−, a pseudohalide present at elevated concentrations in smokers), resulting in the production of cyanate (OCN–), which carbamylates nucleophilic groups such as Lys residues on LDL into homocitrulline [70]. Carbamylated LDL binds to the scavenger receptor SRA−1 on macrophages, is avidly ingested and leads to foam cell formation [70]. The feasibility of such a biological mechanism was supported by the results of clinical studies, revealing that plasma concentrations of protein-bound homocitrulline were independently associated with the presence CAD, and was independently predictive of future myocardial infarction (MI), stroke and death at 3 years follow-up [70].
Attenuation of the anti-atherogenic properties of high-density lipoproteins (HDL)
Clinical and epidemiological studies consistently show that low-plasma concentrations of HDL are strongly and independently associated with an increased risk of CAD [71]. The major mechanism by which HDL is thought to exert its cardio-protective effects is by removing cholesterol from the artery wall through a process referred to as reverse cholesterol transport, involving the interaction of apolipoprotein A-I with ATP-binding cassette transporters ABCA1 and ABCG1 [72]. Further, HDL and its subpopulations have anti-oxidative, anti-inflammatory, cytoprotective, vasodilatory, and anti-thrombotic activities, all of which may slow or prevent the development of atherosclerosis [73]. One of MPO’s putative contributions to atherosclerosis involves site-specific oxidative modification of HDL, thereby attenuating its anti-atherogenic properties.
MPO binds to helix 8 on apolipoprotein A-I [74], the major protein associated with HDL. In the presence of H2O2, Cl− and/or NO2−, MPO may decrease the ability of HDL to facilitate cholesterol efflux from lipid-laden cells [75,76]. The potential biological relevance of such MPO-mediated modification of HDL is supported by multiple findings: (i) HOCl-modified proteins co-localize with apolipoprotein A-I in human plaques and endothelial cells overlaying atherosclerotic lesions [77]; (ii) apolipoprotein A-I recovered from human atherosclerotic lesions is enriched in nitrotyrosine and 3-chlorotyrosine [18]; (iii) there is an inverse correlation between plasma apolipoprotein A-I’s content of 3-chlorotyrosine (a marker of MPO activity – see below) and its ability to accept cholesterol foam cells [18,78]; and (iv) the 3-chlorotyrosine content of apolipoprotein A-I are greater in subjects with CAD than in control subjects [74]. However, it is important to note that while MPO-modified apolipoprotein A-I may reduce the ability of HDL to perform reverse cholesterol transport, cholesterol efflux capacity is not fully dependent on circulating levels of functional apolipoprotein A-I [72].
In addition, oxidized HDL competes with native HDL as a ligand for the scavenger receptor BI, and this may further interfere with mobilization of cholesterol from peripheral tissues to the liver [79]. Also, in vitro studies revealed that incubating reconstituted HDL with increasing amounts of MPO not only rendered the lipoprotein ‘dysfunctional’, but resulted in a pro-inflammatory particle that induced the expression of vascular cell adhesion molecule 1 in endothelial cells, and hence may promote the entry of circulating monocytes into the vessel wall [80].
Promotion of endothelial dysfunction
MPO limits the bioavailability of nitric oxide (•NO), leading to endothelial dysfunction. MPO disrupts NO metabolism by at least two pathways: the consumption by reactive species formed by MPO and the interruption of endogenous NO synthesis [3].
MPO and its reaction products can potentially disrupt •NO formation/bioavailability in several ways: (i) HOCl can chlorinate arginine, the substrate of eNOS, thereby limiting arginine bioavailability and forming chlorinated arginine that can also directly inhibit eNOS [81]; (ii) HOCl can directly oxidize eNOS, leading to monomerization and uncoupling of the synthase [59]; and (iii) lipoproteins modified in vitro by the MPO/H2O2/NO2− system may cause dissociation of eNOS from the plasma membrane of endothelial cells and decrease eNOS expression [82].
The potential pathophysiological relevance of the above findings is supported by the observation that individuals with high-plasma MPO content more likely demonstrate endothelial dysfunction compared with subjects with low MPO, even after adjusting for traditional cardiovascular risk factors and medications [83]. Likewise, in patients with symptomatic CAD forearm blood flow in response to •NO-liberating acetylcholine inversely correlates with MPO plasma concentrations [84]. Suggesting that MPO promotes endothelial dysfunction in inflammatory vascular disease by enhancing the catabolism of •NO.
Promotion of endothelial cell apoptosis and thrombosis
In vitro evidence suggests that MPO may provoke superficial plaque erosion and increase the propensity for thrombus formation in the setting of plaque instability/rupture (see below) (Figure 2). Sugiyama et al. demonstrated that exposure of cultured human endothelial cells to MPO-expressing macrophages and concentrations of HOCl achieved in vivo resulted in endothelial cell death by apoptosis and subsequent desquamation, whereas sub-lethal HOCl concentrations induced tissue factor activity in endothelial cells, which may in turn promote thrombosis [85].
Figure 2.
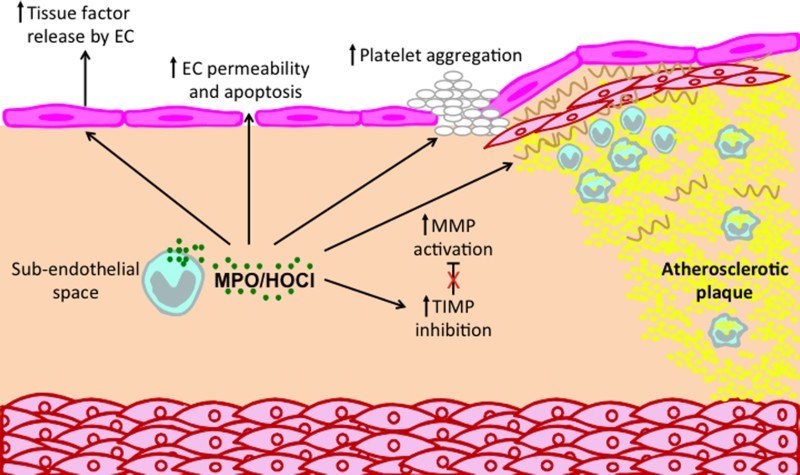
Potential roles of MPO and MPO-derived oxidants in promoting atherosclerotic plaque instability. MPO can affect a number of processes that contribute to plaque instability and possible plaque rupture. MPO released by monocyte/macrophages and neutrophil can activate MMPs and inhibit TIMPs, leading to reduction in ECM, especially in the fibrous cap. In addition, MPO may contribute to a thrombogenic environment by inducing endothelial cells to release tissue factor and by priming of platelet aggregation. Finally, MPO can increase endothelial cell (EC) permeability and apoptosis, thereby increasing the leakiness of the endothelium.
Activation of matrix MMPs
As indicated earlier, the thickness of the fibrous cap is a key determinant of the relative stability of an atherosclerotic plaque [5]. MPO may contribute to plaque destabilization through the activation of MMPs and inactivation of their inhibitors, resulting in degradation of the fibrous cap matrix. Activation of human neutrophils can result in the simultaneous release of MPO and active MMP-9 [86], suggesting that MPO may activate MMPs upon degranulation. Indeed, low concentrations of HOCl have been reported to directly activate MMP-7 in vitro by oxidation of the thiol moiety of the MMP cysteine switch domain [57]. Moreover, MPO-generated oxidants can inactivate tissue inhibitor of metalloproteinase-1 that otherwise counteracts the action of MMPs [87], thereby overall weakening ECM.
Evidence for association between circulating MPO and CAD
Numerous primary studies have investigated the association of circulating concentration of MPO protein with CAD. Of the 20 studies assessed, 15 found a positive association between elevated MPO protein concentration and CAD presence and/or severity, while five studies found no association (Table 4). Zhang et al. were the first to report a positive association between plasma MPO and the presence of CAD, diagnosed by coronary angiography and defined as previous MI, previous percutaneous coronary intervention (PCI) or >50% stenosis in at least one coronary artery [1]. They observed odds ratios for the highest versus lowest MPO quartile, and second and third versus lowest MPO quartile, to remain associated with CAD after adjustment for CAD risk factors, white cell count and Framingham Global Risk Score. LaFramboise et al. also reported serum MPO to be 1.2- to 3.1-fold higher in CAD patients than patients with non-significant CAD, using the same definition of CAD [88]. Kaya et al. observed plasma MPO to be significantly higher in 73 patients with a heart attack (ST Segment Elevation Myocardial Infarction, STEMI) than in 46 age- and sex-matched healthy controls [89]. While these studies collected venous blood to measure MPO, Alipour et al. found a positive association between MPO concentration and stable CAD in blood samples collected from the abdominal aorta and left and right coronary arteries [90].
Table 4. Association between circulating MPO protein and presence/severity of CAD.
| Studies supporting | ||||||
|---|---|---|---|---|---|---|
| Publication | Patient population | Number of participants | CAD definition | Main outcome | P value | References |
| Zhang et al. (2001) | Patients identified to have CAD in the cardiology clinic or undergoing coronary catheterization | 158 patients with established CAD (cases) and 175 patients without angiographically significant CAD (controls) | Previous MI, previous PCI or >50% stenosis in at least one coronary artery | MPO activity per mg of neutrophil protein was significantly greater for CAD cases than controls | P < 0.001 | [1] |
| Ruef et al. (2006) | Patients with documented CAD presenting with stable angina pectoris or ACS | 54 patients with ACS, 108 with stable angina pectoris and 46 controls without CAD | At least one stenosis >20% in at least one coronary artery segment | MPO significantly correlated with the presence and severity of CAD in the order control < stable angina pectoris < ACS | P < 0.05 | [91] |
| Meuwese et al. (2007) | EPIC-Norfolk population study patients who developed CAD during follow-up | 1138 cases and 2237 controls |
Code 410–414 according to the International Classification of Diseases-9th revision | Median serum MPO levels were higher in cases than controls Baseline MPO levels were higher in subjects with compared with non-fatal CAD Risk of future CAD increased in consecutive MPO quartiles |
P = 0.001 P = 0.001 P = 0.001 |
[100] |
| Ndrepepa et al. (2008) | Patients undergoing coronary angiogram because of clinical symptoms of CAD | 680 cases (382 patients with stable CAD, 64 with unstable angina, 43 with Non-STEMI, 191 with STEMI) and 194 controls | CAD was confirmed by the presence of coronary stenosis ≥ 50% lumen obstruction in at least one of the three main coronary | Higher levels of MPO were found with progression of CAD from stable CAD to non-ST-segment elevation ACS and acute MI | P < 0.001 | [92] |
| Goldmann et al. (2009) | Patients admitted with acute MI who underwent coronary angiography and primary PCI | 38 acute MI patients with symptom onset of ≤ 2 hours (cases) and 50 patients with stable CAD (controls) | Controls had angiographically documented CAD | Compared to patients with stable CAD, those finally diagnosed for AMI exhibited significantly elevated MPO plasma levels | P < 0.01 | [97] |
| Heslop et al. (2010) | Patients who had coronary angiography for indications other than ACS | 705 patients had CAD on angiogram (cases) and 180 patients had no angiographic evidence of CAD (controls) | Presence of CAD was defined by the presence of any lesion >20% stenosis, and severe CAD was defined by presence of any lesion >50% stenosis | Patients with severe lesions had higher MPO MPO predicted angiographic CAD and severe CAD in covariate adjusted analysis |
P = 0.022 P = 0.03 P = 0.021 |
[98] |
| Pawlus et al. (2010) | Patients with symptoms of CAD who underwent coronary angiography | 31 patients with MI, 17 patients with unstable angina, 31 patients with stable angina (SA) and 21 controls | Significant CAD was defined as >50% stenosis in at least one coronary artery | In the unstable angina and MI groups, the concentration of MPO was significantly higher than in the control and SA group | P < 0.001 | [93] |
| Khan et al. (2011) | Patients aged 35–80 who presented to the emergency department with recent onset of chest pain | 61 patients with AMI, 58 patients with unstable angina, and 61 patients with stable CAD | Stable CAD was diagnosed if angiography results revealed >70% stenosis in one of the main coronary arteries | Patients with AMI had significantly higher median MPO levels than those with unstable angina or stable CAD | P < 0.001 | [96] |
| Samsamshariat et al. (2011) | Patients who had consecutively undergone coronary angiography owing to clinical manifestations of CAD or suspected changes on electrocardiography | 50 stable CAD patients, 50 unstable CAD patients and 50 control subjects | CAD patients had a ≥50% stenosis in one of the main coronary arteries | Plasma MPO levels were significantly higher in unstable CAD patients than in stable CAD patients and control subjects, and significantly higher in stable CAD patients than in control subjects | P < 0.001 | [94] |
| Rebeiz et al. (2011) | Patients presenting with chest pain and negative cardiac troponin-T concentration and undergoing coronary angiography | 389 patients presenting with chest pain and negative cardiac troponin-T concentration and undergoing coronary angiography | At least one coronary stenosis causing a 70% or more diameter reduction | Increasing quartiles of MPO concentration strongly associated with a >70% coronary stenosis, coronary thrombus and plaque ulceration assessed by angiography | P < 0.0001 | [101] |
| Tretjakovs et al. (2012) | Non-diabetic patients with CAD | 22 patients with stable angina, 22 patients with unstable angina and 22 healthy controls | CAD diagnosed by stenosis ≥50% of at least one of the three main epicardial branches of coronary arteries | MPO levels in patients with stable angina pectoris or unstable angina pectoris were higher than those in healthy controls | P < 0.05 | [95] |
| LaFramboise et al. (2012) | Patients who presented with symptoms of heart disease and were referred for cardiac catheterization | 209 patients required coronary revascularization (cases) and 150 patients without flow-limiting CAD who did not require percutaneous intervention (controls) | Significant CAD was defined as ≥50% obstruction in any epicardial vessel | MPO was 1.2–3.1 times higher in cases than controls | P < 0.001 | [88] |
| Kaya et al. (2012) | Patients admitted to the intensive coronary care unit with a diagnosis of ST-elevation MI (STEMI) | 73 patients with STEMI and 46 healthy controls | Controls did not have any documented CAD or pathological findings on ECG | Plasma MPO levels were higher in patients than in controls | P = 0.001 | [89] |
| Alipour et al. (2013) | Patients undergoing diagnostic coronary angiography, indicated by stable angina, analysis of ventricular arrhythmia’s and valve disease | 52 patients with multi-vessel CAD and 25 healthy controls | Multi-vessel CAD defined as at least one wall irregularity in both left and right coronary systems. Wall irregularities were defined as observed plaques in the coronaries < 50% diameter stenosis of the lumen | MPO was higher in patients with CAD than in controls | P < 0.05 | [90] |
| Baseri et al. (2014) | Patients who had the clinical features of stable CAD and were enrolled for elective diagnostic coronary angiography | 68 patients with angiographic CAD (cases) and 66 patients with normal angiography (controls) | CAD defined as ‘stenosis showed angiographically’ | Plasma concentrations of MPO were significantly higher in the CAD patients than in controls | P < 0.001 | [99] |
| Studies contradicting | ||||||
| Uydu et al. (2013) | Patients with symptoms of stable CAD who were scheduled for coronary angiography | 111 patients with stable CAD (cases) and 66 healthy subjects (controls) | CAD was diagnosed if there was at least one lesion with > 50% stenosis in luminal diameter on coronary angiography | No significant difference in MPO levels between cases and controls | P > 0.05 | [102] |
| Kubala et al. (2008) | Patients scheduled for diagnostic coronary angiography | 289 patients with stable CAD and 268 patients with no CAD | Diagnosis of CAD was defined as a luminal narrowing of at least 50% of a vessel diameter in any of the analyzed 15 coronary artery segments | There was no significant difference in the MPO plasma levels between subjects with and without stable CAD | P > 0.05 | [103] |
| Chen et al. (2011) | Patients aged 30–65 from the Dallas Heart Study population sample were invited to participate after an initial home visit for collection of survey data | 3294 | Coronary artery calcification (CAC) was determined as the average score on two consecutive electron beam computed tomography scans | MPO was not associated with coronary artery calcification in either crude or adjusted analyses | P > 0.05 | [107] |
| Scharnagl et al. (2014) | Patients hospitalized for coronary angiography (LURIC study) | 2391 patients with angiographic CAD and 645 patients with no angiographic CAD | CAD defined as at least one > 20% stenosis in at least one of 15 coronary arterial segments | MPO levels did not differ between CAD and non-CAD patients and quartiles of MPO were not associated with the severity of CAD as determined by the Friesinger coronary score |
P = 0.837 (stable CAD vs controls) P = 0.992 (unstable angina troponin T positive vs controls) P = 0.380 (4th quartile vs 1st quartile Friesinger score) |
[105] |
| Wainstein et al. (2010) | Patients who were scheduled for an elective coronary angiography | 135 | CAD severity was assessed angiographically using a six-level score based on degree of stenosis and number of diseased vessels | Insignificant trend towards elevated MPO plasma levels in patients with higher CAD severity score | P = 0.15 | [104] |
There is considerable evidence that circulating MPO protein concentrations not only correlate with the presence of CAD, but also with disease severity. Thus, several studies reported a significant positive association between MPO and severity of CAD patients divided into three groups: (1) controls; (2) stable angina; and (3) ACS [91–95].
A number of studies correlated circulating concentrations of MPO with clinical and physical measures of CAD severity. Thus, MPO was reported to be higher in patients diagnosed with MI than in patients with angina [96] or stable CAD [97], and there is a significant association between MPO concentrations and angiographically measured coronary artery narrowing [98,99]. The findings from a large prospective study consisting of 3375 healthy patients suggest that MPO can be used to predict future risk of CAD after adjusting for traditional cardiovascular risk factors [100]. Notably, this association was substantially stronger for subjects who experienced fatal rather than non-fatal CAD, supporting the notion that MPO correlates with CAD severity. Interestingly, Rebeiz et al. reported elevated plasma MPO to be useful to identify coronary stenosis in 398 troponin-negative patients with chest pain. In this cohort, increasing quartiles of MPO concentration strongly associated with >70% coronary stenosis, coronary thrombus, and plaque ulceration assessed by angiography [101].
There are also studies reporting a lack of association between circulating MPO and CAD. Two studies reported plasma MPO concentration not to be elevated in patients with stable CAD [102,103]. This suggests that systemic release of MPO is not a characteristic feature of asymptomatic CAD, although a possible confounding factor in one of these two studies is that the absence of CAD in the control group of volunteers was not ruled out angiographically [102]. Two other studies reporting a lack of significant association between MPO and CAD [104,105] used unusual definitions of disease severity. Specifically, in a study consisting of 3036 patients hospitalized for coronary angiography, CAD was diagnosed as >20% stenosis [105] rather than the 50% benchmark considered to be relevant for CAD outcome [106]. Similarly, Wainstein et al. determined CAD severity using a six-tier scoring system. They defined non-significant CAD as <70% stenosis in ≥1 epicardial vessel and <50% stenosis of the left main coronary [104], whereas most studies considered >50% stenosis in an epicardial vessel to be significant disease [1,88,92–95,98,102,103]. MPO was reported in one study not to correlate with coronary artery calcification as determined by electron beam computed tomography [107]. That study collected blood from community volunteers, and the samples were kept on ice for up to 4 hours before centrifugation and freezing, which is not ideal for MPO quantification. They also assessed for correlation between MPO and aortic atherosclerosis, and only found increased MPO in the highest quartile of aortic disease. Heavy calcification is a late manifestation of atherosclerosis and is often associated with stable rather than vulnerable plaques [5]. Importantly, coronary plaques with thin fibrous caps and larger necrotic cores, thus increasing vulnerability, have an inverse correlation with the size of spotty calcification [108] Thus, increasing coronary calcification does not necessarily indicate the presence of vulnerable inflamed plaques. Furthermore, the finding of Chen et al. [107] does not exclude an association of MPO with non-calcified coronary plaques.
A possible contributing factor to the different research findings is the inconsistency of enzyme-linked immunosorbent assay (ELISA) used to measure MPO protein. Most studies used variations of sandwich ELISA, with intra- and inter-assay coefficients of variation ranging between 1.2–10.4 and 5–13% respectively. Unfortunately, it is difficult to quantitatively compare these discrepancies due to the considerable disparity in the amount of information provided about the ELISA used across studies.
Utility of MPO to predict major adverse coronary events (MACEs)
Given its potentially important role in the pathology of CAD, acute elevations of MPO may represent the activation of neutrophils and macrophages immediately prior to plaque rupture. Goldmann et al. [97] observed MPO to be grossly elevated in patients with acute MI as early as two hours after onset of symptoms, and possibly even before the occurrence of myocardial injury. Several studies have evaluated the usefulness of MPO as a risk assessment tool in predicting MACEs (i.e. MI, re-infarction, the need for revascularization, or cardiac death) over time periods ranging from length of hospitalization to four years. The bulk of evidence strongly reinforces the predictive value of circulating MPO protein as a biomarker for MACEs in patients with ACS (Table 5).
Table 5. Association between MPO and MACE prediction.
| Studies supporting | ||||||
|---|---|---|---|---|---|---|
| Publication | Patient population | Number of participants | Follow-up period | Main outcome | P value | References |
| Brennan et al. (2003) | Patients presenting to the emergency department within 24 hours of chest pain | 604 | 30 days and 6 months | MPO is an independent predictor of increased risk of MI, the need for revascularization, and major adverse coronary outcomes within 30 days and 6 months after presentation with chest pain | P < 0.001 | [109] |
| Cavusoglu et al. (2007) | Patients who underwent diagnostic coronary angiography for evaluation of ACS | 182 | 2 years | MPO was found to be an independent predictor of MI on long-term follow-up | P = 0.0172 | [113] |
| Morrow et al. (2008) | Patients presenting with non-ST elevation ACS, within 24 hours of symptom onset | 1524 | 30 days and 6 months | Elevated baseline concentration of MPO was independently associated with a nearly two-fold higher risk of non-fatal MI or recurrent ACS at 30 days The association between MPO and risk of non-fatal MI or recurrent ACS was attenuated by 6 months |
P = 0.001 P = 0.14 |
[111] |
| Wong et al. (2009) | Asymptomatic adults without known CVD | 1302 | 3.8 years | Having MPO concentrations above the median was independently predictive of CVD events | P = 0.034 | [117] |
| Roman et al. (2010) | Patients presenting to ED with ACS and patients with stable CAD from an outpatient clinic | 130 patients with ACS and 178 patients with stable angina | Length of hospitalization | Among patients with ACS, baseline MPO level was an independent predictor of major adverse cardiac events during hospitalization | Not given | [112] |
| Tang et al. (2011) | Patients undergoing elective diagnostic coronary angiography with evidence of significant atherosclerotic burden, but without evidence of MI | 1895 | 3 years | MPO is an independent predictor of and 3-year incident risk of non-fatal MI | P < 0.036 | [116] |
| Kaya et al. (2012) | Patients admitted to the intensive coronary care unit with a diagnosis of STEMI | 73 patients with STEMI and 46 healthy controls | 25 ± 16 months | High-plasma MPO levels were independent predictors of MACE | P = 0.003 | [89] |
| Tang et al. (2013) | Patients who underwent elective cardiac catheterization without prior ACS | 3635 | 3 years | The predictive value of MPO for future risk of incident MACEs is enhanced when combined with high sensitivity C-reactive protein and type B natriuretic peptide | P < 0.001 | [174] |
| Liu et al. (2015) | Patients admitted to hospital and undergoing diagnostic coronary angiography | 201 ACS patients and 210 non-ACS patients | 4 years | Patients with elevated baseline MPO concentrations had higher incidences of MACEs compared to CHD patients with normal-low baseline MPO values | P < 0.001 | [114] |
| Studies contradicting | ||||||
| Apple et al. (2007) | Patients presenting with symptoms suggestive of ACS | 457 | 4 months | There was a trend between elevated baseline MPO and higher incidence of MACEs | P = 0.09 | [115] |
In 604 patients presenting with chest pain, Brennan et al. observed elevated levels of MPO to be independently associated with increased risk of MACEs at 30 days and 6 months follow-up, after adjusting for risk factors [109]. Specifically, at 30 days cardiac troponin alone predicted 58% of MACEs compared with 84.5% with the addition of baseline MPO measurements. In 462 of these patients who were consistently negative for troponin, MPO remained a strong, independent predictor of MACEs at 30 days and 6 months [109]. This suggests that MPO is a predictor of the presence of vulnerable plaque rather than only a marker of inflammation in response to myocardial necrosis. Likewise, the CAPTURE trial, involving 547 patients with recurrent chest pain at rest, observed MPO serum levels >350 ng/mL to be highly predictive for non-fatal MI within 72 hours [110]. This association remained strong even in troponin-negative patients. Similarly, Morrow et al. reported elevated baseline MPO in patients presenting with non-ST elevation ACS to be associated with a higher risk of non-fatal MI or recurrent ACS within 30 days, and inclusion of MPO with B-type natriuretic peptide and cardiac troponin to improve short-term risk assessment and patient stratification [111]. Moreover, MPO was observed to be an independent predictor of MACEs in ACS patients during hospitalization [112].
These above findings suggest that measurement of MPO may be useful for risk stratification of patients presenting to the emergency department with chest pain, and that elevated plasma MPO may be a marker of vulnerable plaque. Indeed, this predictive value may remain over longer follow-up periods. Thus, in ACS patients at 2 years follow-up, MPO persisted as an independent predictor of MI [113]. Similarly, in a study following 73 subjects diagnosed with anterior STEMI and 46 healthy controls over a mean period of 25 ± 16 months, patients with plasma MPO concentrations >68 ng/mL experienced a three-fold higher incidence of MACEs than the low MPO group [89]. At 4 years follow-up, ACS patients with baseline MPO activity >16.69 U/L had higher incidences of MACEs compared with those with normal or low baseline MPO activity [114]. Investigating the relationship between multiple biomarkers and MACEs at 4 months follow-up in 457 patients presenting with symptoms suggestive of ACS, a trend (P = 0.09) for higher cardiac events with elevated baseline MPO was reported [115]. In that study, patients in whom both cardiac troponin and MPO were increased had the highest MACEs.
MPO may also be useful to predict MACEs in asymptomatic patients. Thus, in 3635 subjects without prior ACS and undergoing elective cardiac catheterization, plasma MPO >27 ng/mL was a useful predictor of incident MACEs within the ensuing 3 years [116]. This predictive value increased further when MPO was combined with, high sensitivity C-reactive protein and type B natriuretic peptide as additional biomarkers [116]. Similarly, in 1302 asymptomatic adults followed for an average of 3.8 years, persons at or above the median MPO concentration had a 2-fold greater CVD event rate than subjects below the median MPO [117].
Evidence for MPO predicting cardiovascular mortality
There is strong evidence that plasma MPO may predict long-term risk of cardiovascular mortality in patients with CAD (Table 6). In 885 patients with stable severe angiographic CAD, mortality increased across MPO tertiles [98]. After 13 years follow-up and covariate analysis, patients with the highest MPO tertile were twice as likely to have died from a cardiovascular event than those in the lowest tertile [98]. In patients with angiographic CAD followed for nearly 8 years, those in the highest MPO quartile were 1.5 times more likely to die from cardiovascular causes than those in the lowest MPO quartile after fully adjusting for variables [105]. Interestingly, a separate study using a follow-up of only 3.5 years did not observe MPO to independently correlate with mortality in patients with stable CAD [118]. These findings suggest that the higher cardiovascular risk associated with MPO may be more in the medium to long term.
Table 6. Association between MPO and future cardiovascular mortality.
| Studies supporting | ||||||
|---|---|---|---|---|---|---|
| Publication | Patient population | Number of participants | Follow-up period | Main outcome | P value | References |
| Scharnagl et al. (2014) | Patients hospitalized for coronary angiography (LURIC study) | 645 | 7.75 years | High levels of MPO (4th quartile) is predictive of total and cardiovascular mortality in patients with CAD, independent of established cardiovascular risk factors | P = 0.009 | [105] |
| Heslop et al. (2010) | Patients who had coronary angiography for indications other than ACS | 885 | 13 years | After covariate analysis, patients with the highest MPO tertile were twice as likely to have died from a cardiovascular event than those in the lowest tertile | P < 0.001 | [98] |
| Tsimikas et al. (2010) | Healthy patients between 45 and 79 years of age from age-sex registers of general practices in Norfolk, United Kingdom | 25,663 | 6 years | Patients in the highest MPO tertile were nearly two-times more likely to have died from fatal CAD than those in the lowest tertile | Not given | [119] |
| Studies contradicting | ||||||
| Stefanescu et al. (2008) | Patients with stable CAD undergoing coronary angiography | 382 | 3.5 years | MPO does not independently correlate with mortality in patients with stable CAD | P = 0.77 | [118] |
The utility of MPO in predicting cardiovascular mortality in non-CAD patients remains unclear. In a large study in 25,663 healthy men and women followed for 6 years, subjects in the highest tertiles for both oxidized phospholipids on apolipoprotein B-100 and MPO were nearly two-times more likely to have died from fatal CAD than those in the lowest tertile [119]. In a comparatively much smaller study following 645 patients without CAD for 7.75 years, MPO levels were not significantly associated with cardiovascular mortality [105]. Additional large-scale studies are needed to establish whether circulating MPO can be predictive of cardiovascular mortality in healthy subjects.
Measurement of MPO and its activity
Measuring MPO protein and activity is crucial to understanding its role in atherosclerosis and CVD. Several assays have been reported for this purpose. The following discusses the various methods in the context of clinical studies and utility to predict MACEs or cardiovascular mortality.
Detection of MPO protein
The majority of clinical studies investigating a role of MPO in CVD have measured protein content in plasma/serum or circulating leukocytes (Tables 7–9). ELISA is commonly used to measure MPO concentration. This method uses a solid-phase enzyme immunoassay to detect an antigen usually present in a liquid sample. The ‘sandwich’ ELISA format is most popular, highlighted by the availability of several commercial kits that claim to detect MPO concentrations as low as 0.16 ng/mL. Sandwich ELISA is based on the concept of physically linking a capture antibody to a solid surface (e.g. 96-well plate), placing the sample onto the capture antibody to allow antigen binding, followed by the addition of a detection antibody, an enzyme-linked secondary antibody and finally a substrate that is then converted by the linked enzyme into a read-out for quantification (usually fluorescence or chemiluminescence).
Table 7. Association between circulating MPO protein and presence/severity of CAD.
| Studies supporting (References) | Sample type | MPO analysis method |
|---|---|---|
| [1] | EDTA plasma | Sandwich ELISA |
| Circulating leukocytes | Peroxidase activity by guaiacol oxidation | |
| [91] | Plasma | Sandwich ELISA |
| [100] | Serum | ELISA |
| [175] | Plasma | EIA |
| [92] | EDTA plasma | |
| [97] | Plasma | ELISA |
| [98] | Plasma | ELISA |
| [93] | Serum | Sandwich ELISA |
| [96] | EDTA plasma | Microparticle immunoassay |
| [101] | ||
| [94] | Plasma | ELISA |
| [89] | Plasma | ELISA |
| [88] | Serum | ELISA |
| [95] | Serum | Microparticle immunoassay |
| [90] | Isolated leukocytes | FACS |
| [99] | EDTA plasma | ELISA |
| Studies contradicting | ||
| [103] | EDTA plasma | ELISA |
| [104] | Heparin plasma | ELISA |
| [107] | EDTA plasma | Sandwich ELISA |
| [102] | Plasma | Sandwich ELISA |
| [105] | Plasma | ELISA |
Table 8. Association between MPO and MACE prediction.
| Studies supporting (References) | Sample type | MPO analysis method |
|---|---|---|
| [109] | Plasma | ELISA |
| [113] | Plasma | ELISA |
| [111] | Citrate plasma | ELISA |
| [117] | EDTA plasma | ELISA |
| [112] | ||
| [116] | EDTA plasma | Sandwich ELISA |
| [89] | Plasma | ELISA |
| [174] | EDTA plasma | Sandwich ELISA |
| [114] | Serum | Latex enhanced Immunoturbidimetric Assay |
| Studies contradicting | ||
| [115] | Heparin plasma | ELISA |
Table 9. MPO is associated with future cardiovascular mortality.
Although ELISA is easy to use, only few reports have compared the various kits to determine their accuracy and reliability. Comparisons of in-house MPO ELISA with commercial kits generally show strong positive correlations (Pearson correlation coefficient >0.9) [120,121]. However, there is concern around the accuracy of the assays given that different concentrations of MPO can be obtained in the same samples when applying different ELISA kits/methods [120]. As a consequence, it is often difficult to compare absolute values of MPO protein between different studies. Other general factors that can affect MPO ELISA measurements include sample handling, storage, and anti-coagulant used. For example, the plasma concentration of MPO increases with time if human whole blood is kept at room temperature prior to plasma preparation, collection, and storage [120]. In addition, several studies have reported heparin plasma to contain more MPO protein than EDTA plasma [120–123], possibly due to ex vivo ‘release’ of MPO from blood cells/matrix by heparin [124]. This interpretation is supported by the observation that intravenous administration of heparin mobilizes MPO from vascular compartments, yielding higher values compared to baseline plasma concentrations, especially in patients with CAD [125].
Thus, standardization of both sample collection and detection method is urgently needed, not least because many reports lack information on the anti-coagulant used (Tables 7–9). To date, only the CardioMPO ELISA (Cleveland Heart Lab) has obtained approval from the Food and Drug Administration (FDA) as a diagnostic blood test for MPO in the clinical setting.
Surrogate biomarkers
The measurement of MPO protein provides useful information on the abundance of the enzyme. While some studies have reported a high correlation between MPO protein mass and activity [1,126], others observed considerable variation in enzyme activity between individuals with similar plasma MPO protein content [127], in part due to effects of age and gender. In addition, several polymorphisms have been identified, associated with increased [127] or decreased [128] MPO activity relative to MPO protein. Two main strategies have been used for measuring MPO activity ex vivo or in vivo. One relies on the detection of ‘footprints’ of HOCl-mediated reactions (surrogate biomarkers) while the other uses a probe (usually with fluorescent or chemiluminescent property) to indirectly measure the peroxidase or chlorinating activity of MPO/HOCl.
Of the surrogate biomarkers used for HOCl/MPO activity, 3-chlorotyrosine, formed as the product of the reaction of HOCl with protein tyrosine residues and determined by mass spectrometry-based methods, is often considered the ‘gold standard’ [129]. Its suitability as a potential biomarker for CVD is supported by the demonstration of 30-fold higher amounts of 3-chlorotyrosine LDL present in human atherosclerotic lesions compared with that in circulating LDL [48]. However, there are limitations to using 3-chlorotyrosine as a biomarker for MPO activity. These include the slow rate of formation of 3-chlorotyrosine, its susceptibility to further oxidation [130,131], and possible in vivo metabolism [132]. Moreover, chlorinated tyrosine may not be completely specific for MPO, as non-enzymatic chlorination of tyrosine has been reported to occur with aqueous chlorine in rat stomach fluid [133].
Another practical limitation with measuring 3-chlorotyrosine in biological samples is that the signal is diluted out by tyrosine liberated from oxidized and non-oxidized proteins during sample preparation. To address this problem, certain studies have specifically sampled the site(s) where MPO is thought to be active, or have purified potential target proteins for MPO/HOCl oxidation from these sites. For example, Bergt el al. [68] reported high levels of 3-chlorotyrosine in HDL isolated from atherosclerotic lesions. The same investigators observed Tyr192 on apolipoprotein A-I of HDL to be a major site for chlorination [134]. Compared with control subjects, Tyr192 chlorination is increased in atherosclerotic lesions [135] as well as in circulating HDL of patients with CVD [136]. This highlights the potential utility of specific tyrosine residues in certain proteins as biomarkers for the role of MPO in CVD.
In addition to its reaction with proteins, HOCl can also oxidize and chlorinate biological lipids, with chlorinated plasmalogens arguably having received most attention. Plasmalogens are phospholipids found in many tissues, especially in the nervous, immune, and cardiovascular systems, including cardiomyocytes, endothelial, and VSMCs [137]. HOCl reacts with the vinyl ether group of plasmalogens, releasing α-chlorofatty aldehydes such as 2-chlorohexadecanal [138] which can be metabolized further to 2-chlorofatty acids and 2-chlorofatty alcohols [139]. Using mass spectrometry, several of these metabolites have been detected in human atheroma and LDL isolated from it [50,140], while further downstream metabolites such as 2-chloradipic acid have been detected in the urine of humans [141].
Another promising potential biomarker for in vivo HOCl/MPO activity is glutathione sulfonamide. Glutathione sulfonamide is formed by the fast reaction of HOCl with reduced glutathione (Glu–Cys–Gly) resulting in a covalent bond between the sulfur atom of cysteine and the amine moiety of glutamate [142,143]. In vitro, glutathione sulfonamide is produced when cells, such as neutrophils and endothelial cells, are treated with reagent HOCl [144]. Glutathione sulfonamide is also detected by liquid chromatography/mass spectrometry in the airways of children with cystic fibrosis [145] and in the lung lavage fluid of pre-term babies with respiratory infections, where its concentration correlates with MPO and protein 3-chlorotyrosine [144]. However, it should be noted that glutathione sulfonamide is not specific for HOCl, as it can also be formed by the reaction of glutathione with HOBr and peroxynitrite, albeit at lower yield than observed with HOCl [142].
Measuring MPO activity
Although surrogate biomarkers of HOCl are useful footprints for oxidative damage caused by HOCl/MPO activity, they do not provide information on the location or time of formation of HOCl. To obtain information on MPO activity at a particular point in time, various substrates or probes for the chlorinating (HOCl) or peroxidase activity of MPO have been developed and employed. Upon reaction with HOCl/MPO in vitro or in vivo, these ‘probes’ commonly give rise to stable products with chromogenic, fluorescent or chemiluminescent properties. Importantly, however, many of these probes (e.g. guaiacol, luminol, L-012, Amplex® Red, aminophenyl-fluorescein, hydroxyphenyl-fluorescein) are not specific for HOCl/MPO, as they can also react with other reactive oxygen species and peroxidases. This complicates the detection and quantification of HOCl/MPO activity in complex biological samples.
To overcome these issues, enrichment or ‘capture’ of MPO from the biological matrices (e.g. blood, plasma or tissue/cell lysates) prior to activity measurement has been utilized using different technologies [1,146] For example, Franck et al. [147] developed the ‘Specific Immunological Extraction followed by Enzymatic Detection’ (SIEFED) method to detect MPO activity in human plasma and carotid extracts with a limit of detection of 0.08 mU/mL. The assay essentially immobilizes MPO onto a solid surface using anti-MPO antibodies, followed by addition of Amplex® Red (also known as ADHP) in the presence of H2O2 and nitrite to detect MPO’s peroxidase activity. In this assay, H2O2 converts active MPO to compound I that itself oxidizes Amplex® Red to the fluorescent resorufin that is then detected, using 544 and 590 nm as excitation and emission wavelengths, respectively. The SIEFED assay has also been combined with an ELISA (SIEFED/ELISAcb) to determine the ‘specific’ activity of MPO by the ratio of active and total MPO. This assay has been applied to human plasma after stimulation of neutrophils in whole blood [148]. Others have utilized similar antibody-capture techniques of MPO from plasma or isolated leukocytes followed by detection of chlorinating activity with APF as a substrate for HOCl [146,149].
Goiffon et al. [126] described the MPO activity on polymer surface (MAPS) method. This assay ‘captures’ MPO based on its ability to non-specifically adsorb onto a solid surface in the presence of dilute human plasma. Once bound to the polymer surface, MPO is washed free of endogenous inhibitors (e.g. ceruloplasmin, ascorbate) and MPO activity then detected using the luminol analog L-012. To enhance the signal, L-012-dependent bioluminescence imaging is performed in the presence of 20 mM NaBr (instead of NaCl), as MPO utilizes NaBr more efficiently than NaCl, and the resulting HOBr also effectively oxidizes L-012. Using 72 samples from cardiac catheterization patients, MAPS data highly correlated with parallel ELISA assays including the FDA-approved test from the Cleveland Heart Lab referred to earlier with similar reproducibility and accuracy [126]. In fact, the MAPS and ELISA were more highly correlated than protocol-matched ELISAs performed at different analysis sites. The major advantage of MAPS is its low cost (<5% of the ELISA cost), shorter sample work-up time and lower sample volume requirements [126]. However, a disadvantage of MAPS is that L-012 used for activity is not specific for HOCl/MPO activity.
It should be noted, however, that measurement of the activity of ‘captured’ MPO does not necessarily reflect the in vivo activity of the enzyme. This is because the enzymatic activity of MPO is regulated by the local concentrations of H2O2, and low molecular weight (e.g. ascorbate and urate) and proteinaceous inhibitors (e.g. ceruloplasmin) [19]. Therefore, activity assays that retain MPO’s biological microenvironment may more truly reflect the in vivo activity of MPO. For this reason, we have developed a novel and highly sensitive assay to measure MPO activity in mice based on the in vivo conversion of hydroethidine to 2-chloroethidium, determined by liquid chromatography/mass spectrometry [150,151]. 2-Chloroethidium is as specific and more sensitive than 3-chlorotyrosine for measurement of MPO/HOCl activity in vivo. For example, in MPO-deficient mice subjected to vascular inflammation or tandem stenosis (as a model of atherosclerotic plaque rupture), neither 3-chlorotyrosine nor 2-chloroethidium was detected [150,151], confirming the high specificity of the assay for MPO. In wild type mice, inflammation significantly increased the arterial content of 2-chloroethidium in a femoral cuff model [149], whereas 3-chlorotyrosine did not change measurably, suggesting that 2-chloroethidium is more sensitive than 3-chlorotyrosine for MPO activity in vivo. This interpretation is further supported by the fact that 2-chloroethidium but not 3-chlorotyrosine was detected in stable plaques of the right carotid artery from the mouse model of atherosclerotic plaque rupture [151]. While in our studies [150] hydroethidine was administered to live mice prior to tissue collection, it is also feasible to use the conversion of hydroethidine to 2-chloroethidium to detect MPO activity ex vivo, e.g. in arteries from mouse models of atherosclerosis without a need for MPO capture [151].
Molecular imaging of MPO activity
Over the last 10 years numerous molecular imaging tracers have been developed that react with HOCl or MPO via different chemical mechanisms [152,153]. Many of the tracers utilize fluorescence properties, making them suitable for live cell imaging where they provide valuable spatial information on MPO activity. At the organism level, probes have been developed for use with non-invasive molecular imaging platforms allowing for in vivo localization of MPO activity. This requires administration of tracers with fluorescent, radioactive or paramagnetic properties that specifically bind to or react with target molecular structures. The tracers can then be localized using non-invasive imaging platforms such as fluorescence tomography, positron emission tomography and magnetic resonance imaging (MRI). In this way, molecular imaging provides insight into protein and cellular functions in native physiological environments, enabling a systems-level understanding of biological processes that contribute to different disease states.
As circulating MPO concentrations are associated with adverse outcomes in CVD, MPO has emerged as a potential biomarker of prognostic significance. Beyond the non-specific assessment of circulating MPO, molecular imaging of MPO activity has the capacity to directly implicate it in particular disease states through the localization of increased enzymatic activity in affected organs and tissues. If an association between tissue-specific MPO activity and CVD is identified, the clinical translation of such molecular imaging techniques could aid patient diagnosis and prognostication in addition to the development of novel therapeutics.
Several molecular imaging probes have been designed for the non-invasive localization of MPO activity. The majority of these tracers are activated on oxidation by MPO or HOCl, which results in either emission of light (fluorescence or luminescence imaging) or tissue accumulation of a conjugated contrast agent such as gadolinium. The mechanism and applications of current MPO probes is summarized in the following sections.
The potential utility of MPO for imaging of vulnerable atherosclerotic plaque
Limitations of current vascular imaging modalities
Post-mortem and serial intravascular ultrasound studies have identified features of atherosclerotic plaque that confer increased risk of rupture and adverse outcomes [154,155]. These include a thin fibrous cap, large necrotic core, high plaque volume with positive remodeling and high inflammatory cell content. Whilst X-ray contrast angiography, the current gold standard in coronary imaging, can accurately define arterial stenosis, it has limited ability to identify other features associated with high-risk plaque. At present, angiography serves to guide percutaneous and surgical coronary revascularization (i.e. angioplasty/stenting or bypass grafting). In the setting of acute MI, timely reperfusion results in substantial and sustained survival benefits. However, in the non-acute setting, this approach relieves symptoms but only prevents heart attack or prolongs life in selected subsets of patients [156–158]. More recently, intravascular ultrasound and optical coherence tomography have allowed for improved characterization of plaque composition. Importantly, these imaging modalities are invasive and remain based on contrast analysis (optical or ultrasound) but do not specifically inform on the active cellular and molecular processes that drive the evolution of atherosclerotic lesions.
As MPO is postulated to be a key driver of plaque instability molecular imaging of MPO activity may improve detection of high-risk plaque. To date numerous molecular imaging probes have been developed for the assessment of MPO enzymatic activity using a variety of imaging platforms such as MRI, optical fluorescence, and luminescence imaging.
MRI of MPO activity with bis-5HT-DTPA-gadolinium
Bis-5HT-DTPA-Gd (MPO-Gd) is a peroxidase-specific molecular imaging agent used for non-invasive localization of MPO activity with MRI. It links gadolinium-diethylenetriaminepentaacetic acid-gadolinium (DTPA-Gd) with two 5-hydroxytryptamine molecules via amide bonds. Peroxidase-mediated oxidation of the phenol moiety of 5-hydroxytryptamide in MPO-Gd leads to formation of a phenoxyl radical intermediate that can form oligomers (up to a pentamer) as well as cross-link to proteins via reaction with Tyr residues [159]. This results in the accumulation of gadolinium in tissues with peroxidase activity, giving rise to a >2-fold increase in T1-weighted MRI signal in in vivo imaging studies [160–162]. In vitro studies suggest that unlike MPO, eosinophil peroxidase is inefficient in oxidizing, and hence ‘activating’ MPO-Gd [162]. Moreover, in vivo studies employing MPO-deficient mice and models of MI and stroke showed no agent activation [160,163], indicating that MPO-Gd is a relatively specific imaging agent for assessment of MPO activity. Furthermore, MPO-Gd is highly sensitive and able to detect MPO concentrations as low as 0.005 U/mg, which is substantially lower than levels present in many pathological conditions (e.g. carotid plaque ∼250 U/mg) [55].
MPO-Gd has successfully been used in vivo for the localization of MPO activity and inflammation in animal models of CVDs such as atherosclerosis [161], vasculitis [164], and MI [160] Ronald et al. showed that MPO-Gd accumulates in aortic atherosclerotic plaque present in New Zealand White rabbits fed a high-fat diet [161]. The signal generated on T1-weighted MRI in diseased aortic segments was 2-fold higher than in normal segments and histological correlation demonstrated co-localization of MPO-rich areas with a high density of macrophages. Furthermore, the plaque retention times of MPO-Gd were significantly longer than non-targeted DTPA-Gd, suggesting that protein binding facilitates accumulation of the tracer. The imaging of MPO activity rather than phagocytes is likely to result in more specific localization of vulnerable plaques. MPO-Gd has also been shown to enhance regions aortic root inflammation in a mouse model of vasculitis [165]. Similarly, MPO-Gd has been successfully used to image aortic root inflammation in a mouse model of vasculitis [164]. Here, vascular inflammation was induced by intra-peritoneal injection of Candida albicans water-soluble fraction. Within the aortic root the mean contrast-to-noise ratio (CNR) following administration of MPO-Gd was 2.5-fold greater than vasculitis mice imaged with DTPA-Gd [164]. MPO-Gd has also been used to image myocardial inflammation in the LAD-ligation mouse model of MI [160]. Here administration of MPO-Gd resulted in 4-fold greater CNR compared to DTPA-Gd. The CNR peak for MPO-Gd (∼30 min) was later than DTPA-Gd (∼10 min) where the CNR had returned to baseline by 60 min.
Chemiluminescence resonance energy transfer (CRET) imaging with luminol
Luminol (5-amino-2,3-dihydro-1,4-phthalzine-dione) is a redox-sensitive chemiluminescent compound that emits blue light (λmax = 425 nm) on reaction with a number of oxidants, including the hydroxyl radical [166], O2•− and hydroperoxides/peroxidases [167]. Although luminol-derived chemiluminescence allows for the detection of very low concentrations of oxidizing substances, it is clearly not specific for HOCl [168]. Nonetheless, studies performed with neutrophils from humans with MPO deficiency suggest that the luminol reaction is partially dependent on MPO activity [169], although the exact nature of the oxidizing agent(s) and location (intra- versus extra-cellular) remains contentious.
In 2009 Gross et al. demonstrated that systemic administration of luminol enables non-invasive imaging of MPO activity in numerous models of inflammation including acute dermatitis, contact sensitivity, arthritis, and lymphocytic tumors, where bioluminescence correlated with areas of inflammation [170]. Interestingly, the observed signal was abolished in MPO gene knockout mice [170]. Whilst this method allowed for imaging of superficial inflammation, imaging of MPO in deeper tissues was not possible due to the limited tissue penetration of the emitted blue light. Following from this, Zhang et al. developed a CRET methodology in which blue light emitted from luminol excites concurrently administered nanoparticles that emit light in the near-infrared spectrum [171]. The longer wavelength emitted from the administered nanoparticles allowed for greater tissue penetration and detection of signal from deeper tissues. The methodology was successfully used to image MPO activity in a lipopolysaccharide-mediated lung injury model in mice [171]. Again, the CRET signal was abrogated in MPO gene knockout mice and was significantly attenuated after administration of the MPO inhibitor 4-aminobenzoic acid hydrazide.
MMSiR
MMSiR is a near-infrared probe based on Si-rhodamine that has been designed for the sensitive and selective detection of HOCl in real time [172]. It has multiple properties that make it suitable for in vivo imaging including pH-independence of fluorescence, resistance to photobleaching and autoxidation, good tissue penetration associated with emission of light in the near-infrared range [172]. The HOCl detection mechanism involves oxidation of the S-atom of MMSiR with release of the spiro-cyclized form and generation of its oxidized product SMSiR, which is highly fluorescent with an emission spectra in the far-red to near-infrared range. The probe has been successfully used to demonstrate inflammation in a mouse peritonitis model and in zymosan-activated neutrophils in vitro [172].
Sulfonaphthoaminophenyl fluorescein (SNAPF)
SNAPF selectively reacts with HOCl and has absorption and emission spectra in the far-red range [152]. The reaction of SNAPF with strong oxidants results in oxidative cleavage of the 4-aminophenyl moiety, generating highly fluorescent deprotonated dye with an absorption maximum of 614 nm and emission at 676 nm. The probe has successfully been used to image HOCl in stimulated primary human macrophages and in vivo in a murine model of peritonitis [152].
In summary, the in vivo assessment of atherosclerotic plaque MPO activity using molecular imaging techniques may provide further support for the involvement of this enzyme in the pathogenesis of vulnerable atherosclerotic plaque. The validation of such imaging techniques in models of high-risk plaque and plaque rupture would provide a basis for subsequent translational studies with the aim of improved identification of high-risk individuals.
Conclusions
Current evidence supports a causative role of MPO in coronary plaque rupture. Clinical studies strongly indicate that plasma concentrations of MPO correlate with the presence and severity of CAD. Importantly, increased plasma MPO has been shown to predict future adverse coronary events in many studies, confirming its potential role as a useful clinical marker. Unlike CRP, MPO is less involved in general systemic inflammation, thereby making it a more specific coronary marker than CRP. There are, however, significant technical hurdles to it being adopted for clinical use. There are multiple available assays for MPO protein and activity, and it is unclear which or if any of these is/are ideal for clinical use. The current blood collection requirement to analyze plasma MPO, i.e. that the blood needs to be stored cold and centrifuged and aliquoted immediately, is also impractical and near impossible for routine clinical use. The effect of heparin on plasma MPO content is a further significant barrier to its use in the higher risk patients with unstable coronary syndromes. This is because most of these patients receive heparin therapy on hospital admission, so that the only heparin-free blood sample consistently available will be the first set of bloods drawn when patient arrives. Molecular imaging of MPO, on the other hand, has shown promising findings in identifying at risk patients with vulnerable plaques, and the potential burden of such plaques. Future research will clarify if these techniques will find practical clinical use.
Notes on contributors
Nathaniel Teng is a medical student at the University of New South Wales. He undertook a research year with the Vascular Biology Division at the Victor Chang Cardiac Research Institute and the Cardiology Department at Prince of Wales Hospital, investigating the role of myeloperoxidase in acute coronary syndrome.
Ghassan J. Maghzal obtained his PhD in biochemistry/pathology from the University of Otago, Christchurch, New Zealand. His initial postdoctoral training was in Prof. Christine Winterbourn’s laboratory, establishing mass spectrometry methods for oxidative stress markers. Subsequently, he joined Prof. Roland Stocker’s laboratory in Australia where he investigated the role of superoxide in the activation of indoleamine 2,3-dioxygenase. His more recent work focuses on developing novel mass spectrometry methods to detect various reactive species in vivo. He is currently a senior staff scientist at the Victor Chang Cardiac Research Institute (Sydney, Australia) and conjoint senior lecturer at the University of New South Wales.
Jihan Talib is a biochemist at the Victor Chang Cardiac Research Institute where she works with Prof. Roland Stocker, studying the role of myeloperoxidase in heart disease. She completed her undergraduate degree in Advanced Medicinal Chemistry at the University of Wollongong in 2004 before pursuing her PhD on the binding of small molecules to DNA. Following the completion of her PhD in 2009, she joined Prof. Michael Davies at the Heart Research Institute in Sydney where she commenced investigating the role of myeloperoxidase in atherosclerosis.
Imran Rashid is a cardiologist and post-doctoral scientist with an interest in molecular and magnetic resonance imaging of atherosclerotic disease. He obtained PhD at the Heart Research Institute (Sydney) under the supervision of Prof. Michael Davies on the role of LDL glycation in foam cell formation. He underwent Cardiology training and a fellowship in cardiovascular MRI at Royal Prince Alfred Hospital and Cardiovascular Magnetic Resonance Sydney (CMRS). He has since undertaken post-doctoral studies at Victor Chang Cardiac Research Institute (VCCRI) where, in his project, he assessed the utility of molecular magnetic resonance imaging of inflammation (myeloperoxidase activity) for the identification of unstable atherosclerotic plaque.
Antony K. Lau is a senior staff specialist and interventional cardiologist at the Prince of Wales Hospital, Sydney, and a conjoint senior lecturer at the University of New South Wales. His PhD research investigated the roles of antioxidants in atherosclerosis and restenosis. His ongoing research looks at ways to improve screening for coronary artery disease, including imaging methods such as cardiac CT, invasive angiographic techniques with IVUS and OCT, and novel blood tests. His clinical interests include difficult-to-control hypertension and hyperlipidemia, and managing complex coronary artery disease and coronary interventions.
Roland Stocker is a biochemist trained at the Federal Institute of Technology (Zurich, Switzerland), Australian National University (Canberra, Australia), and University of California, Berkeley, CA. He has been an independent investigator since 1988, focusing on the roles of oxidoreductases, antioxidants, and oxidants in vascular health and disease. Following appointments at the Heart Research Institute, the University of New South Wales and the University of Sydney, he moved to the Victor Chang Cardiac Research Institute in Sydney, Australia in 2012, where he heads the Vascular Biology Division.
Acknowledgements
We thank Dr. Anita Ayer for help with Figure 2, and the Victor Chang Cardiac Research Institute for infrastructure support. RS also acknowledges support from Senior Principal Research Fellowship 1111632 from the National Health and Medical Research Council of Australia.
Disclosure statement
The laboratory of RS receives research material support from AstraZeneca who are developing inhibitors of myeloperoxidase.
ORCID
Nathaniel Teng http://orcid.org/0000-0001-5596-6900
Ghassan J. Maghzal http://orcid.org/0000-0001-6652-8973
Jihan Talib http://orcid.org/0000-0003-2005-6573
Antony K. Lau http://orcid.org/0000-0002-8278-742X
Roland Stocker http://orcid.org/0000-0002-7414-6681
References
- [1].Zhang R, Brennan ML, Fu X, et al. Association between myeloperoxidase levels and risk of coronary artery disease. Jama. 2001;286:2136–2142. doi: 10.1001/jama.286.17.2136 [DOI] [PubMed] [Google Scholar]
- [2].Yousuf O, Mohanty BD, Martin SS, et al. High-sensitivity C-reactive protein and cardiovascular disease: a resolute belief or an elusive link? J Am Coll Cardiol. 2013;62:397–408. doi: 10.1016/j.jacc.2013.05.016 [DOI] [PubMed] [Google Scholar]
- [3].Koeth RA, Haselden V, Tang WH. Myeloperoxidase in cardiovascular disease. Adv Clin Chem. 2013;62:1–32. doi: 10.1016/B978-0-12-800096-0.00001-9 [DOI] [PubMed] [Google Scholar]
- [4].Dominguez-Rodriguez A, Abreu-Gonzalez P. Current role of myeloperoxidase in routine clinical practice. Exp Rev Cardiovascul Ther. 2011;9:223–230. doi: 10.1586/erc.11.2 [DOI] [PubMed] [Google Scholar]
- [5].Narula J, Nakano M, Virmani R, et al. Histopathologic characteristics of atherosclerotic coronary disease and implications of the findings for the invasive and noninvasive detection of vulnerable plaques. J Am Coll Cardiol. 2013;61:1041–1051. doi: 10.1016/j.jacc.2012.10.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Silvestre-Roig C, de Winther MP, Weber C, et al. Atherosclerotic plaque destabilization: mechanisms, models, and therapeutic strategies. Circ Res. 2014;114:214–226. doi: 10.1161/CIRCRESAHA.114.302355 [DOI] [PubMed] [Google Scholar]
- [7].Kolodgie FD, Narula J, Burke AP, et al. Localization of apoptotic macrophages at the site of plaque rupture in sudden coronary death. Am J Pathol. 2000;157:1259–1268. doi: 10.1016/S0002-9440(10)64641-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res. 2016;118:653–667. doi: 10.1161/CIRCRESAHA.115.306256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Schrijvers DM, De Meyer GR, Kockx MM, et al. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:1256–1261. doi: 10.1161/01.ATV.0000166517.18801.a7 [DOI] [PubMed] [Google Scholar]
- [10].Sluimer JC, Gasc JM, van Wanroij JL, et al. Hypoxia, hypoxia-inducible transcription factor, and macrophages in human atherosclerotic plaques are correlated with intraplaque angiogenesis. J Am Coll Cardiol. 2008;51:1258–1265. doi: 10.1016/j.jacc.2007.12.025 [DOI] [PubMed] [Google Scholar]
- [11].Sluimer JC, Daemen MJ. Novel concepts in atherogenesis: angiogenesis and hypoxia in atherosclerosis. J Pathol. 2009;218:7–29. doi: 10.1002/path.2518 [DOI] [PubMed] [Google Scholar]
- [12].Hellings WE, Peeters W, Moll FL, et al. Composition of carotid atherosclerotic plaque is associated with cardiovascular outcome: a prognostic study. Circulation. 2010;121:1941–1950. doi: 10.1161/CIRCULATIONAHA.109.887497 [DOI] [PubMed] [Google Scholar]
- [13].Schultz J, Kaminker K. Myeloperoxidase of the leucocyte of normal human blood. I. Content and localization. Arch Biochem Biophys. 1962;96:465–467. doi: 10.1016/0003-9861(62)90321-1 [DOI] [PubMed] [Google Scholar]
- [14].Parker H, Albrett AM, Kettle AJ, et al. Myeloperoxidase associated with neutrophil extracellular traps is active and mediates bacterial killing in the presence of hydrogen peroxide. J Leukoc Biol. 2012;91:369–376. doi: 10.1189/jlb.0711387 [DOI] [PubMed] [Google Scholar]
- [15].Sugiyama S, Okada Y, Sukhova GK, et al. Macrophage myeloperoxidase regulation by granulocyte macrophage colony-stimulating factor in human atherosclerosis and implications in acute coronary syndromes. Am J Pathol. 2001;158:879–891. doi: 10.1016/S0002-9440(10)64036-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Baldus S, Eiserich JP, Mani A, et al. Endothelial transcytosis of myeloperoxidase confers specificity to vascular ECM proteins as targets of tyrosine nitration. J Clin Invest. 2001;108:1759–1770. doi: 10.1172/JCI200112617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Carr AC, Myzak MC, Stocker R, et al. Myeloperoxidase binds to low-density lipoprotein: potential implications for atherosclerosis. FEBS Lett. 2000;487:176–180. doi: 10.1016/S0014-5793(00)02227-4 [DOI] [PubMed] [Google Scholar]
- [18].Zheng L, Nukuna B, Brennan ML, et al. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J Clin Invest. 2004;114:529–541. doi: 10.1172/JCI200421109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chapman AL, Mocatta TJ, Shiva S, et al. Ceruloplasmin is an endogenous inhibitor of myeloperoxidase. J Biol Chem. 2013;288:6465–6477. doi: 10.1074/jbc.M112.418970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Van AP, Slomianny MC, Boudjeltia KZ, et al. Glycosylation pattern of mature dimeric leukocyte and recombinant monomeric myeloperoxidase: glycosylation is required for optimal enzymatic activity. J Biol Chem. 2010;285:16351–16359. doi: 10.1074/jbc.M109.089748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kooter IM, Koehler BP, Moguilevsky N, et al. The Met243 sulfonium ion linkage is responsible for the anomalous magnetic circular dichroism and optical spectral properties of myeloperoxidase. J Biol Inorg Chem. 1999;4:684–691. doi: 10.1007/s007750050340 [DOI] [PubMed] [Google Scholar]
- [22].Kooter IM, Moguilevsky N, Bollen A, et al. Characterization of the Asp94 and Glu242 mutants in myeloperoxidase, the residues linking the heme group via ester bonds. Eur J Biochem. 1999;264:211–217. doi: 10.1046/j.1432-1327.1999.00606.x [DOI] [PubMed] [Google Scholar]
- [23].Zeng J, Fenna RE. X-ray crystal structure of canine myeloperoxidase at 3 Å resolution. J Mol Biol. 1992;226:185–207. doi: 10.1016/0022-2836(92)90133-5 [DOI] [PubMed] [Google Scholar]
- [24].Bolscher BG, Wever R. A kinetic study of the reaction between human myeloperoxidase, hydroperoxides and cyanide. Inhibition by chloride and thiocyanate. Biochim Biophys Acta. 1984;788:1–10. doi: 10.1016/0167-4838(84)90290-5 [DOI] [PubMed] [Google Scholar]
- [25].Dunford HB. Heme peroxidases. New York: John Wiley & Sons; 1999. [Google Scholar]
- [26].Fiedler TJ, Davey CA, Fenna RE. X-ray crystal structure and characterization of halide-binding sites of human myeloperoxidase at 1.8 A resolution. J Biol Chem. 2000;275:11964–11971. doi: 10.1074/jbc.275.16.11964 [DOI] [PubMed] [Google Scholar]
- [27].Furtmüller PG, Zederbauer M, Jantschko W, et al. Active site structure and catalytic mechanisms of human peroxidases. Arch Biochem Biophys. 2006;445:199–213. doi: 10.1016/j.abb.2005.09.017 [DOI] [PubMed] [Google Scholar]
- [28].Davies MJ, Hawkins CL, Pattison DI, et al. Mammalian heme peroxidases: from molecular mechanisms to health implications. Antiox Redox Signal. 2008;10:1199–1234. doi: 10.1089/ars.2007.1927 [DOI] [PubMed] [Google Scholar]
- [29].Arnhold J, Furtmüller PG, Regelsberger G, et al. Redox properties of the couple compound I/native enzyme of myeloperoxidase and eosinophil peroxidase. Eur J Biochem. 2001;268:5142–5148. doi: 10.1046/j.0014-2956.2001.02449.x [DOI] [PubMed] [Google Scholar]
- [30].Furtmüller PG, Arnhold J, Jantschko W, et al. Redox properties of the couples compound I/compound II and compound II/native enzyme of human myeloperoxidase. Biochem Biophys Res Commun. 2003;301:551–557. doi: 10.1016/S0006-291X(02)03075-9 [DOI] [PubMed] [Google Scholar]
- [31].Segal AW, Geisow M, Garcia R, et al. The respiratory burst of phagocytic cells is associated with a rise in vacuolar pH. Nature. 1981;290:406–409. doi: 10.1038/290406a0 [DOI] [PubMed] [Google Scholar]
- [32].Morris JC. The acid ionization constant of HOCl from 5 to 35°. J Phys Chem. 1966;70:3798–3805. doi: 10.1021/j100884a007 [DOI] [Google Scholar]
- [33].Prütz WA, Kissner R, Koppenol WH, et al. On the irreversible destruction of reduced nicotinamide nucleotides by hypohalous acids. Arch Biochem Biophys. 2000;380:181–191. doi: 10.1006/abbi.2000.1914 [DOI] [PubMed] [Google Scholar]
- [34].Nagy P, Jameson GN, Winterbourn CC. Kinetics and mechanisms of the reaction of hypothiocyanous acid with 5-thio-2-nitrobenzoic acid and reduced glutathione. Chem Res Toxicol. 2009;22:1833–1840. doi: 10.1021/tx900249d [DOI] [PubMed] [Google Scholar]
- [35].Morgan PE, Pattison DI, Talib J, et al. High plasma thiocyanate levels in smokers are a key determinant of thiol oxidation induced by myeloperoxidase. Free Radic Biol Med. 2011;51:1815–1822. doi: 10.1016/j.freeradbiomed.2011.08.008 [DOI] [PubMed] [Google Scholar]
- [36].van Dalen CJ, Whitehouse MW, Winterbourn CC, et al. Thiocyanate and chloride as competing substrates for myeloperoxidase. Biochem J. 1997;327:487–492. doi: 10.1042/bj3270487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Furtmüller PG, Burner U, Obinger C. Reaction of myeloperoxidase compound I with chloride, bromide, iodide, and thiocyanate. Biochemistry. 1998;37:17923–17930. doi: 10.1021/bi9818772 [DOI] [PubMed] [Google Scholar]
- [38].Ashby MT, Carlson AC, Scott MJ. Redox buffering of hypochlorous acid by thiocyanate in physiologic fluids. J Am Chem Soc. 2004;126:15976–15977. doi: 10.1021/ja0438361 [DOI] [PubMed] [Google Scholar]
- [39].Nagy P, Beal JL, Ashby MT. Thiocyanate is an efficient endogenous scavenger of the phagocytic killing agent hypobromous acid. Chem Res Toxicol. 2006;19:587–593. doi: 10.1021/tx050338c [DOI] [PubMed] [Google Scholar]
- [40].Bessems JG, Vermeulen NP. Paracetamol (acetaminophen)-induced toxicity: molecular and biochemical mechanisms, analogues and protective approaches. Crit Rev Toxicol. 2001;31:55–138. doi: 10.1080/20014091111677 [DOI] [PubMed] [Google Scholar]
- [41].Winterbourn CC, Hampton MB, Livesey JH, et al. Modeling the reactions of superoxide and myeloperoxidase in the neutrophil phagosome: implications for microbial killing. J Biol Chem. 2006;281:39860–39869. doi: 10.1074/jbc.M605898200 [DOI] [PubMed] [Google Scholar]
- [42].Marquez LA, Dunford HB. Reaction of compound III of myeloperoxidase with ascorbic acid. J Biol Chem. 1990;265:6074–6078. [PubMed] [Google Scholar]
- [43].Marquez LA, Dunford HB. Interaction of acetaminophen with myeloperoxidase intermediates: optimum stimulation of enzyme activity. Arch Biochem Biophys. 1993;305:414–420. doi: 10.1006/abbi.1993.1440 [DOI] [PubMed] [Google Scholar]
- [44].Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008;4:278–286. doi: 10.1038/nchembio.85 [DOI] [PubMed] [Google Scholar]
- [45].Folkes LK, Candeias LP, Wardman P. Kinetics and mechanisms of hypochlorous acid reactions. Arch Biochem Biophys. 1995;323:120–126. doi: 10.1006/abbi.1995.0017 [DOI] [PubMed] [Google Scholar]
- [46].Pattison DI, Davies MJ. Absolute rate constants for the reaction of hypochlorous acid with protein side chains and peptide bonds. Chem Res Toxicol. 2001;14:1453–1464. doi: 10.1021/tx0155451 [DOI] [PubMed] [Google Scholar]
- [47].Hawkins CL, Pattison DI, Davies MJ. Hypochlorite-induced oxidation of amino acids, peptides and proteins. Amino Acids. 2003;25:259–274. doi: 10.1007/s00726-003-0016-x [DOI] [PubMed] [Google Scholar]
- [48].Hazen SL, Heinecke JW. 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J Clin Invest. 1997;99:2075–2081. doi: 10.1172/JCI119379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Thukkani AK, McHowat J, Hsu FF, et al. Identification of alpha-chloro fatty aldehydes and unsaturated lysophosphatidylcholine molecular species in human atherosclerotic lesions. Circulation. 2003;108:3128–3133. doi: 10.1161/01.CIR.0000104564.01539.6A [DOI] [PubMed] [Google Scholar]
- [50].Messner MC, Albert CJ, McHowat J, et al. Identification of lysophosphatidylcholine-chlorohydrin in human atherosclerotic lesions. Lipids. 2008;43:243–249. doi: 10.1007/s11745-008-3151-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Winterbourn CC. Comparative reactivities of various biological compounds with myeloperoxidase-hydrogen peroxide-chloride, and similarity of oxidant to hypochlorite. Biochim Biophys Acta. 1985;840:204–210. doi: 10.1016/0304-4165(85)90120-5 [DOI] [PubMed] [Google Scholar]
- [52].Peskin AV, Winterbourn CC. Histamine chloramine reactivity with thiol compounds, ascorbate, and methionine and with intracellular glutathione. Free Radic Biol Med. 2003;35:1252–1260. doi: 10.1016/S0891-5849(03)00502-1 [DOI] [PubMed] [Google Scholar]
- [53].Peskin AV, Winterbourn CC. Taurine chloramine is more selective than hypochlorous acid at targeting critical cysteines and inactivating creatine kinase and glyceraldehyde-3-phosphate dehydrogenase. Free Radic Biol Med. 2006;40:45–53. doi: 10.1016/j.freeradbiomed.2005.08.019 [DOI] [PubMed] [Google Scholar]
- [54].Coker MS, Hu WP, Senthilmohan ST, et al. Pathways for the decay of organic dichloramines and liberation of antimicrobial chloramine gases. Chem Res Toxicol. 2008;21:2334–2343. doi: 10.1021/tx800232v [DOI] [PubMed] [Google Scholar]
- [55].Daugherty A, Dunn JL, Rateri DL, et al. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J Clin Invest. 1994;94:437–444. doi: 10.1172/JCI117342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Xu J, Xie Z, Reece R, et al. Uncoupling of endothelial nitric oxidase synthase by hypochlorous acid: role of NAD(P)H oxidase-derived superoxide and peroxynitrite. Arterioscler Thromb Vasc Biol. 2006;26:2688–2695. doi: 10.1161/01.ATV.0000249394.94588.82 [DOI] [PubMed] [Google Scholar]
- [57].Fu X, Kassim SY, Parks WC, et al. Hypochlorous acid oxygenates the cysteine switch domain of pro-matrilysin (MMP-7). A mechanism for matrix metalloproteinase activation and atherosclerotic plaque rupture by myeloperoxidase. J Biol Chem. 2001;276:41279–41287. doi: 10.1074/jbc.M106958200 [DOI] [PubMed] [Google Scholar]
- [58].Brennan ML, Anderson MM, Shih DM, et al. Increased atherosclerosis in myeloperoxidase-deficient mice. J Clin Invest. 2001;107:419–430. doi: 10.1172/JCI8797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Stocker R, Huang A, Jeranian E, et al. Hypochlorous acid impairs endothelium-derived nitric oxide bioactivity through a superoxide-dependent mechanism. Arterioscl Thromb Vasc Biol. 2004;24:2028–2033. doi: 10.1161/01.ATV.0000143388.20994.fa [DOI] [PubMed] [Google Scholar]
- [60].Prasad A, Tsimikas S. Candidate biomarkers for the detection of coronary plaque destabilization and rupture. Curr Atheroscler Rep. 2008;10:309–317. doi: 10.1007/s11883-008-0048-5 [DOI] [PubMed] [Google Scholar]
- [61].Hazell LJ, Stocker R. Oxidation of low-density lipoprotein with hypochlorite causes transformation of the lipoprotein into a high-uptake form for macrophages. Biochem J. 1993;290:165–172. doi: 10.1042/bj2900165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hazell LJ, van den Berg JJM, Stocker R. Oxidation of low-density lipoprotein by hypochlorite causes aggregation that is mediated by modification of lysine residues rather than lipid oxidation. Biochem J. 1994;302:297–304. doi: 10.1042/bj3020297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Marsche G, Zimmermann R, Horiuchi S, et al. Class B scavenger receptors CD36 and SR-BI are receptors for hypochlorite-modified low density lipoprotein. J Biol Chem. 2003;278:47562–47570. doi: 10.1074/jbc.M308428200 [DOI] [PubMed] [Google Scholar]
- [64].Kawamura M, Heinecke JW, Chait A. Increased uptake of α-hydroxy aldehyde-modified low density lipoprotein by macrophage scavenger receptors. J Lipid Res. 2000;41:1054–1059. [PubMed] [Google Scholar]
- [65].Goldstein JL, Ho YK, Basu SK, et al. Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc Natl Acad Sci U S A. 1979;76:333–337. doi: 10.1073/pnas.76.1.333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Podrez EA, Febbraio M, Sheibani N, et al. Macrophage scavenger receptor CD36 is the major receptor for LDL modified by monocyte-generated reactive nitrogen species. J Clin Invest. 2000;105:1095–1108. doi: 10.1172/JCI8574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Leeuwenburgh C, Hardy MM, Hazen SL, et al. Reactive nitrogen intermediates promote low density lipoprotein oxidation in human atherosclerotic intima. J Biol Chem. 1997;272:1433–1436. doi: 10.1074/jbc.272.3.1433 [DOI] [PubMed] [Google Scholar]
- [68].Bergt C, Pennathur S, Fu X, et al. The myeloperoxidase product hypochlorous acid oxidizes HDL in the human artery wall and impairs ABCA1-dependent cholesterol transport. Proc Natl Acad Sci U S A. 2004;101:13032–13037. doi: 10.1073/pnas.0405292101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Podrez EA, Schmitt D, Hoff HF, et al. Myeloperoxidase-generated reactive nitrogen species convert LDL into an atherogenic form in vitro. J Clin Invest. 1999;103:1547–1560. doi: 10.1172/JCI5549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Wang Z, Nicholls SJ, Rodriguez ER, et al. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat Med. 2007;13:1176–1184. doi: 10.1038/nm1637 [DOI] [PubMed] [Google Scholar]
- [71].Di Angelantonio E, Sarwar N, Perry P, et al. Major lipids, apolipoproteins, and risk of vascular disease. Jama. 2009;302:1993–2000. doi: 10.1001/jama.2009.1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Khera AV, Cuchel M, de la Llera-Moya M, et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–135. doi: 10.1056/NEJMoa1001689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Rye KA, Barter PJ. Cardioprotective functions of HDLs. J Lipid Res. 2014;55:168–179. doi: 10.1194/jlr.R039297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Smith JD. Dysfunctional HDL as a diagnostic and therapeutic target. Arterioscler Thromb Vasc Biol. 2010;30:151–155. doi: 10.1161/ATVBAHA.108.179226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Shao B, Oda MN, Oram JF, et al. Myeloperoxidase: an oxidative pathway for generating dysfunctional high-density lipoprotein. Chem Res Toxicol. 2010;23:447–454. doi: 10.1021/tx9003775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Fisher EA, Feig JE, Hewing B, et al. High-density lipoprotein function, dysfunction, and reverse cholesterol transport. Arterioscler Thromb Vasc Biol. 2012;32:2813–2820. doi: 10.1161/ATVBAHA.112.300133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Malle E, Marsche G, Arnhold J, et al. Modification of low-density lipoprotein by myeloperoxidase-derived oxidants and reagent hypochlorous acid. Biochim Biophys Acta. 2006;1761:392–415. doi: 10.1016/j.bbalip.2006.03.024 [DOI] [PubMed] [Google Scholar]
- [78].Pennathur S, Bergt C, Shao B, et al. Human atherosclerotic intima and blood of patients with established coronary artery disease contain high density lipoprotein damaged by reactive nitrogen species. J Biol Chem. 2004;279:42977–42983. doi: 10.1074/jbc.M406762200 [DOI] [PubMed] [Google Scholar]
- [79].Nicholls SJ, Hazen SL. Myeloperoxidase, modified lipoproteins, and atherogenesis. J Lipid Res. 2009;50(Suppl):S346-S351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Undurti A, Huang Y, Lupica JA, et al. Modification of high density lipoprotein by myeloperoxidase generates a pro-inflammatory particle. J Biol Chem. 2009;284:30825–30835. doi: 10.1074/jbc.M109.047605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Zhang C, Reiter C, Eiserich JP, et al. L-arginine chlorination products inhibit endothelial nitric oxide production. J Biol Chem. 2001;276:27159–27165. doi: 10.1074/jbc.M100191200 [DOI] [PubMed] [Google Scholar]
- [82].Marsche G, Heller R, Fauler G, et al. 2-Chlorohexadecanal derived from hypochlorite-modified high-density lipoprotein-associated plasmalogen is a natural inhibitor of endothelial nitric oxide biosynthesis. Arterioscler Thromb Vasc Biol. 2004;24:2302–2306. doi: 10.1161/01.ATV.0000148703.43429.25 [DOI] [PubMed] [Google Scholar]
- [83].Vita JA, Brennan ML, Gokce N, et al. Serum myeloperoxidase levels independently predict endothelial dysfunction in humans. Circulation. 2004;110:1134–1139. doi: 10.1161/01.CIR.0000140262.20831.8F [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Baldus S, Heitzer T, Eiserich JP, et al. Myeloperoxidase enhances nitric oxide catabolism during myocardial ischemia and reperfusion. Free Radic Biol Med. 2004;37:902–911. doi: 10.1016/j.freeradbiomed.2004.06.003 [DOI] [PubMed] [Google Scholar]
- [85].Sugiyama S, Kugiyama K, Aikawa M, et al. Hypochlorous acid, a macrophage product, induces endothelial apoptosis and tissue factor expression: involvement of myeloperoxidase-mediated oxidant in plaque erosion and thrombogenesis. Arterioscler Thromb Vasc Biol. 2004;24:1309–1314. doi: 10.1161/01.ATV.0000131784.50633.4f [DOI] [PubMed] [Google Scholar]
- [86].Ehrenfeld P, Matus CE, Pavicic F, et al. Kinin B1 receptor activation turns on exocytosis of matrix metalloprotease-9 and myeloperoxidase in human neutrophils: involvement of mitogen-activated protein kinase family. J Leukoc Biol. 2009;86:1179–1189. doi: 10.1189/jlb.0109012 [DOI] [PubMed] [Google Scholar]
- [87].Wang Y, Rosen H, Madtes DK, et al. Myeloperoxidase inactivates TIMP-1 by oxidizing its N-terminal cysteine residue: an oxidative mechanism for regulating proteolysis during inflammation. J Biol Chem. 2007;282:31826–31834. doi: 10.1074/jbc.M704894200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].LaFramboise WA, Dhir R, Kelly LA, et al. Serum protein profiles predict coronary artery disease in symptomatic patients referred for coronary angiography. BMC Med. 2012;10:157. doi: 10.1186/1741-7015-10-157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Kaya MG, Yalcin R, Okyay K, et al. Potential role of plasma myeloperoxidase level in predicting long-term outcome of acute myocardial infarction. Tex Heart Inst J. 2012;39:500–506. [PMC free article] [PubMed] [Google Scholar]
- [90].Alipour A, Ribalta J, Njo TL, et al. Trans-vessel gradient of myeloperoxidase in coronary artery disease. Eur J ClinInvest. 2013;43:920–925. doi: 10.1111/eci.12121 [DOI] [PubMed] [Google Scholar]
- [91].Ruef J, März W, Winkelmann BR. Markers for endothelial dysfunction, but not markers for oxidative stress correlate with classical risk factors and the severity of coronary artery disease. (A subgroup analysis from the Ludwigshafen Risk and Cardiovascular Health Study). Scand Cardiovascul J. 2006;40:274–279. doi: 10.1080/14017430600925300 [DOI] [PubMed] [Google Scholar]
- [92].Ndrepepa G, Braun S, Mehilli J, et al. Myeloperoxidase level in patients with stable coronary artery disease and acute coronary syndromes. Eur J Clin Invest. 2008;38:90–96. doi: 10.1111/j.1365-2362.2007.01908.x [DOI] [PubMed] [Google Scholar]
- [93].Pawlus J, Holub M, Kozuch M, et al. Serum myeloperoxidase levels and platelet activation parameters as diagnostic and prognostic markers in the course of coronary disease. Int J Lab Hematol. 2010;32:320–328. doi: 10.1111/j.1751-553X.2009.01203.x [DOI] [PubMed] [Google Scholar]
- [94].Samsamshariat SZ, Basati G, Movahedian A, et al. Elevated plasma myeloperoxidase levels in relation to circulating inflammatory markers in coronary artery disease. Biomark Med. 2011;5:377–385. doi: 10.2217/bmm.11.28 [DOI] [PubMed] [Google Scholar]
- [95].Tretjakovs P, Jurka A, Bormane I, et al. Circulating adhesion molecules, matrix metalloproteinase-9, plasminogen activator inhibitor-1, and myeloperoxidase in coronary artery disease patients with stable and unstable angina. Clin Chim Acta. 2012;413:25–29. doi: 10.1016/j.cca.2011.10.009 [DOI] [PubMed] [Google Scholar]
- [96].Khan DA, Sharif MS, Khan FA. Diagnostic performance of high-sensitivity troponin T, myeloperoxidase, and pregnancy-associated plasma protein A assays for triage of patients with acute myocardial infarction. Korean J Lab Med. 2011;31:172–178. doi: 10.3343/kjlm.2011.31.3.172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Goldmann BU, Rudolph V, Rudolph TK, et al. Neutrophil activation precedes myocardial injury in patients with acute myocardial infarction. Free Radic Biol Med. 2009;47:79–83. doi: 10.1016/j.freeradbiomed.2009.04.004 [DOI] [PubMed] [Google Scholar]
- [98].Heslop CL, Frohlich JJ, Hill JS. Myeloperoxidase and C-reactive protein have combined utility for long-term prediction of cardiovascular mortality after coronary angiography. J Am Coll Cardiol. 2010;55:1102–1109. doi: 10.1016/j.jacc.2009.11.050 [DOI] [PubMed] [Google Scholar]
- [99].Baseri M, Heidari R, Mahaki B, et al. Myeloperoxidase levels predicts angiographic severity of coronary artery disease in patients with chronic stable angina. Adv Biomed Res. 2014;3:137. doi: 10.4103/2277-9175.135155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Meuwese MC, Stroes ES, Hazen SL, et al. Serum myeloperoxidase levels are associated with the future risk of coronary artery disease in apparently healthy individuals: the EPIC-Norfolk Prospective Population Study. J Am Coll Cardiol. 2007;50:159–165. doi: 10.1016/j.jacc.2007.03.033 [DOI] [PubMed] [Google Scholar]
- [101].Rebeiz AG, Tamim HM, Sleiman RM, et al. Plasma myeloperoxidase concentration predicts the presence and severity of coronary disease in patients with chest pain and negative troponin-T. Coron Artery Dis. 2011;22:553–558. doi: 10.1097/MCA.0b013e32834c5e98 [DOI] [PubMed] [Google Scholar]
- [102].Uydu HA, Bostan M, Yilmaz A, et al. Comparision of inflammatory biomarkers for detection of coronary stenosis in patients with stable coronary artery disease. Eur Rev Med Pharmacol Sci. 2013;17:112–118. [PubMed] [Google Scholar]
- [103].Kubala L, Lu G, Baldus S, et al. Plasma levels of myeloperoxidase are not elevated in patients with stable coronary artery disease. Clin Chim Acta. 2008;394:59–62. doi: 10.1016/j.cca.2008.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Wainstein RV, Wainstein MV, Ribeiro JP, et al. Association between myeloperoxidase polymorphisms and its plasma levels with severity of coronary artery disease. Clin Biochem. 2010;43:57–62. doi: 10.1016/j.clinbiochem.2009.07.022 [DOI] [PubMed] [Google Scholar]
- [105].Scharnagl H, Kleber ME, Genser B, et al. Association of myeloperoxidase with total and cardiovascular mortality in individuals undergoing coronary angiography—The LURIC study. Int JCardiol. 2014;174:96–105. doi: 10.1016/j.ijcard.2014.03.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Harris PJ, Lee KL, Harrell FE Jr, et al. Outcome in medically treated coronary artery disease. Ischemic events: nonfatal infarction and death. Circulation. 1980;62:718–726. doi: 10.1161/01.CIR.62.4.718 [DOI] [PubMed] [Google Scholar]
- [107].Chen LQ, Rohatgi A, Ayers CR, et al. Race-specific associations of myeloperoxidase with atherosclerosis in a population-based sample: the Dallas Heart Study. Atherosclerosis. 2011;219:833–838. doi: 10.1016/j.atherosclerosis.2011.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].van Velzen JE, de Graaf FR, de Graaf MA, et al. Comprehensive assessment of spotty calcifications on computed tomography angiography: comparison to plaque characteristics on intravascular ultrasound with radiofrequency backscatter analysis. J Nucl Cardiol. 2011;18:893–903. doi: 10.1007/s12350-011-9428-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Brennan ML, Penn MS, Van Lente F, et al. Prognostic value of myeloperoxidase in patients with chest pain. N Engl J Med. 2003;349:1595–1604. doi: 10.1056/NEJMoa035003 [DOI] [PubMed] [Google Scholar]
- [110].Baldus S, Heeschen C, Meinertz T, et al. Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation. 2003;108:1440–1445. doi: 10.1161/01.CIR.0000090690.67322.51 [DOI] [PubMed] [Google Scholar]
- [111].Morrow DA, Sabatine MS, Brennan ML, et al. Concurrent evaluation of novel cardiac biomarkers in acute coronary syndrome: myeloperoxidase and soluble CD40 ligand and the risk of recurrent ischaemic events in TACTICS-TIMI 18. Eur Heart J. 2008;29:1096–1102. doi: 10.1093/eurheartj/ehn071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Roman RM, Camargo PV, Borges FK, et al. Prognostic value of myeloperoxidase in coronary artery disease: comparison of unstable and stable angina patients. Coron Artery Dis. 2010;21:129–136. doi: 10.1097/MCA.0b013e328333f50d [DOI] [PubMed] [Google Scholar]
- [113].Cavusoglu E, Ruwende C, Eng C, et al. Usefulness of baseline plasma myeloperoxidase levels as an independent predictor of myocardial infarction at two years in patients presenting with acute coronary syndrome. Am J Cardiol. 2007;99:1364–1368. doi: 10.1016/j.amjcard.2006.12.060 [DOI] [PubMed] [Google Scholar]
- [114].Liu CN, Chen L, Yang Y, et al. Myeloperoxidase and high-sensitivity C-reactive protein for predicting major adverse cardiovascular events in patients with coronary heart disease. Int J Clin Med. 2015;6:262–270. doi: 10.4236/ijcm.2015.64033 [DOI] [Google Scholar]
- [115].Apple FS, Pearce LA, Chung A, et al. Multiple biomarker use for detection of adverse events in patients presenting with symptoms suggestive of acute coronary syndrome. Clin Chem. 2007;53:874–881. doi: 10.1373/clinchem.2006.080192 [DOI] [PubMed] [Google Scholar]
- [116].Tang WH, Wu Y, Nicholls SJ, et al. Plasma myeloperoxidase predicts incident cardiovascular risks in stable patients undergoing medical management for coronary artery disease. Clin Chem. 2011;57:33–39. doi: 10.1373/clinchem.2010.152827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Wong ND, Gransar H, Narula J, et al. Myeloperoxidase, subclinical atherosclerosis, and cardiovascular disease events. JACC Cardiovasc Imaging. 2009;2:1093–1099. doi: 10.1016/j.jcmg.2009.05.012 [DOI] [PubMed] [Google Scholar]
- [118].Stefanescu A, Braun S, Ndrepepa G, et al. Prognostic value of plasma myeloperoxidase concentration in patients with stable coronary artery disease. Am Heart J. 2008;155:356–360. doi: 10.1016/j.ahj.2007.10.017 [DOI] [PubMed] [Google Scholar]
- [119].Tsimikas S, Mallat Z, Talmud PJ, et al. Oxidation-specific biomarkers, lipoprotein(a), and risk of fatal and nonfatal coronary events. J Am Coll Cardiol. 2010;56:946–955. doi: 10.1016/j.jacc.2010.04.048 [DOI] [PubMed] [Google Scholar]
- [120].Chang P-Y, Wu T-L, Hung C-C, et al. Development of an ELISA for myeloperoxidase on microplate: normal reference values and effect of temperature on specimen preparation. Clin Chim Acta. 2006;373:158–163. doi: 10.1016/j.cca.2006.05.030 [DOI] [PubMed] [Google Scholar]
- [121].Mocatta TJ, Pilbrow AP, Cameron VA, et al. Plasma concentrations of myeloperoxidase predict mortality after myocardial infarction. J Am Coll Cardiol. 2007;49:1993–2000. doi: 10.1016/j.jacc.2007.02.040 [DOI] [PubMed] [Google Scholar]
- [122].Shih J, Datwyler SA, Hsu SC, et al. Effect of collection tube type and preanalytical handling on myeloperoxidase concentrations. Clin Chem. 2008;54:1076–1079. doi: 10.1373/clinchem.2007.101568 [DOI] [PubMed] [Google Scholar]
- [123].Scheffer PG, van der Zwan LP, Schindhelm RK, et al. Myeloperoxidase concentrations in EDTA-plasma of healthy subjects are discordant with concentrations in heparin-plasma and serum. Clin Biochem. 2009;42:1490–1492. doi: 10.1016/j.clinbiochem.2009.06.004 [DOI] [PubMed] [Google Scholar]
- [124].Léculier C, Couprie N, Adeleine P, et al. The effects of high molecular weight- and low molecular weight-heparins on superoxide ion production and degranulation by human polymorphonuclear leukocytes. Thromb Res. 1993;69:519–531. doi: 10.1016/0049-3848(93)90056-T [DOI] [PubMed] [Google Scholar]
- [125].Baldus S, Rudolph V, Roiss M, et al. Heparins increase endothelial nitric oxide bioavailability by liberating vessel-immobilized myeloperoxidase. Circulation. 2006;113:1871–1878. doi: 10.1161/CIRCULATIONAHA.105.590083 [DOI] [PubMed] [Google Scholar]
- [126].Goiffon RJ, Martinez SC, Piwnica-Worms D. A rapid bioluminescence assay for measuring myeloperoxidase activity in human plasma. Nat Commun. 2015;6:6271. doi: 10.1038/ncomms7271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Chevrier I, Tregouet DA, Massonnet-Castel S, et al. Myeloperoxidase genetic polymorphisms modulate human neutrophil enzyme activity: genetic determinants for atherosclerosis? Atherosclerosis. 2006;188:150–154. doi: 10.1016/j.atherosclerosis.2005.10.012 [DOI] [PubMed] [Google Scholar]
- [128].Piedrafita FJ, Molander RB, Vansant G, et al. An alu element in the myeloperoxidase promoter contains a vcmposite SP1-thyroid hormone-retinoic acid response element. J Biol Chem. 1996;271:14412–14420. doi: 10.1074/jbc.271.24.14412 [DOI] [PubMed] [Google Scholar]
- [129].Kettle AJ, Albrett AM, Chapman AL, et al. Measuring chlorine bleach in biology and medicine. Biochim Biophys Acta. 2014;1840:781–793. doi: 10.1016/j.bbagen.2013.07.004 [DOI] [PubMed] [Google Scholar]
- [130].Kettle AJ. Neutrophils convert tyrosyl residues in albumin to chlorotyrosine. FEBS Lett. 1996;379:103–106. doi: 10.1016/0014-5793(95)01494-2 [DOI] [PubMed] [Google Scholar]
- [131].Fu S, Wang H, Davies M, et al. Reactions of hypochlorous acid with tyrosine and peptidyl-tyrosyl residues give dichlorinated and aldehydic products in addition to 3- chlorotyrosine. J Biol Chem. 2000;275:10851–10858. doi: 10.1074/jbc.275.15.10851 [DOI] [PubMed] [Google Scholar]
- [132].Mani AR, Ippolito S, Moreno JC, et al. The metabolism and dechlorination of chlorotyrosine in vivo. J Biol Chem. 2007;282:29114–29121. doi: 10.1074/jbc.M704270200 [DOI] [PubMed] [Google Scholar]
- [133].Nickelsen MG, Nweke A, Scully FE Jr, et al. Reactions of aqueous chlorine in vitro in stomach fluid from the rat: chlorination of tyrosine. Chem Res Toxicol. 1991;4:94–101. doi: 10.1021/tx00019a013 [DOI] [PubMed] [Google Scholar]
- [134].Shao B, Fu X, McDonald TO, et al. Acrolein impairs ATP binding cassette transporter A1-dependent cholesterol export from cells through site-specific modification of apolipoprotein A-I. J Biol Chem. 2005;280:36386–36396. doi: 10.1074/jbc.M508169200 [DOI] [PubMed] [Google Scholar]
- [135].Shao B, Pennathur S, Heinecke JW. Myeloperoxidase targets apolipoprotein A-I, the major high density lipoprotein protein, for dite-dpecific oxidation in human atherosclerotic lesions. J Biol Chem. 2012;287:6375–6386. doi: 10.1074/jbc.M111.337345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Shao B, Tang C, Sinha A, et al. Humans with atherosclerosis have impaired ABCA1 cholesterol efflux and enhanced high-density lipoprotein oxidation by myeloperoxidase. Circ Res. 2014;114:1733–1742. doi: 10.1161/CIRCRESAHA.114.303454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Nagan N, Zoeller RA. Plasmalogens: biosynthesis and functions. Prog Lipid Res. 2001;40:199–229. doi: 10.1016/S0163-7827(01)00003-0 [DOI] [PubMed] [Google Scholar]
- [138].Albert CJ, Crowley JR, Hsu F-F, et al. Reactive chlorinating species produced by myeloperoxidase target the vinyl ether bond of plasmalogens: identification of 2-chlorohexadecanal. J Biol Chem. 2001;276:23733–23741. doi: 10.1074/jbc.M101447200 [DOI] [PubMed] [Google Scholar]
- [139].Wildsmith KR, Albert CJ, Anbukumar DS, et al. Metabolism of myeloperoxidase-derived 2-chlorohexadecanal. J Biol Chem. 2006;281:16849–16860. doi: 10.1074/jbc.M602505200 [DOI] [PubMed] [Google Scholar]
- [140].Thukkani AK, Albert CJ, Wildsmith KR, et al. Myeloperoxidase-derived reactive chlorinating species from human monocytes target plasmalogens in low density lipoprotein. J Biol Chem. 2003;278:36365–36372. doi: 10.1074/jbc.M305449200 [DOI] [PubMed] [Google Scholar]
- [141].Brahmbhatt VV, Albert CJ, Anbukumar DS, et al. ω-Oxidation of α-chlorinated fatty acids: identifiction of α-chlorinated dicarboxylic acids. J Biol Chem. 2010;285:41255–41269. doi: 10.1074/jbc.M110.147157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Harwood DT, Kettle AJ, Winterbourn CC. Production of glutathione sulfonamide and dehydroglutathione from GSH by myeloperoxidase-derived oxidants and detection using a novel LC-MS/MS method. Biochem J. 2006;399:161–168. doi: 10.1042/BJ20060978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Winterbourn CC, Brennan SO. Characterization of the oxidation products of the reaction between reduced glutathione and hypochlorous acid. Biochem J. 1997;326:87–92. doi: 10.1042/bj3260087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Harwood DT, Darlow BA, Cheah F-C, et al. Biomarkers of neutrophil-mediated glutathione and protein oxidation in tracheal aspirates from preterm infants: association with bacterial infection. Pediatr Res. 2011;69:28–33. doi: 10.1203/PDR.0b013e3181ff2378 [DOI] [PubMed] [Google Scholar]
- [145].Kettle AJ, Turner R, Gangell CL, et al. Oxidation contributes to low glutathione in the airways of children with cystic fibrosis. Eur Respir J. 2014;44:122–129. doi: 10.1183/09031936.00170213 [DOI] [PubMed] [Google Scholar]
- [146].Pulli B, Ali M, Forghani R, et al. Measuring myeloperoxidase activity in biological samples. PLoS ONE. 2013;8:e67976. doi: 10.1371/journal.pone.0067976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Franck T, Kohnen S, Boudjeltia KZ, et al. A new easy method for specific measurement of active myeloperoxidase in human biological fluids and tissue extracts. Talanta. 2009;80:723–729. doi: 10.1016/j.talanta.2009.07.052 [DOI] [PubMed] [Google Scholar]
- [148].Franck T, Minguet G, Delporte C, et al. An immunological method to combine the measurement of active and total myeloperoxidase on the same biological fluid, and its application in finding inhibitors which interact directly with the enzyme. Free Radic Res. 2015;49:790–799. doi: 10.3109/10715762.2015.1027197 [DOI] [PubMed] [Google Scholar]
- [149].Flemmig J, Remmler J, Zschaler J, et al. Detection of the halogenating activity of heme peroxidases in leukocytes by aminophenyl fluorescein. Free Radic Res. 2015;49:768–776. doi: 10.3109/10715762.2014.999676 [DOI] [PubMed] [Google Scholar]
- [150].Maghzal GJ, Cergol KM, Shengule SR, et al. Assessment of myeloperoxidase activity by the conversion of hydroethidine to 2-chloroethidium. J Biol Chem. 2014;289:5580–5595. doi: 10.1074/jbc.M113.539486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Talib J, Maghzal GJ, Cheng D, et al. Detailed protocol to assess in vivo and ex vivo myeloperoxidase activity in mouse models of vascular inflammation and disease using hydroethidine. Free Radic Biol Med. 2016;97:124–135. doi: 10.1016/j.freeradbiomed.2016.05.004 [DOI] [PubMed] [Google Scholar]
- [152].Shepherd J, Hilderbrand SA, Waterman P, et al. A fluorescent probe for the detection of myeloperoxidase activity in atherosclerosis-associated macrophages. Chem Biol. 2007;14:1221–1231. doi: 10.1016/j.chembiol.2007.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Hu JJ, Wong N-K, Gu Q, et al. HKOCl-2 series of green BODIPY-based fluorescent probes for hypochlorous acid detection and imaging in live cells. Org Lett. 2014;16:3544–3547. doi: 10.1021/ol501496n [DOI] [PubMed] [Google Scholar]
- [154].Stone GW, Maehara A, Lansky AJ, et al. A prospective natural-history study of coronary atherosclerosis. N Engl J Med. 2011;364:226–235. doi: 10.1056/NEJMoa1002358 [DOI] [PubMed] [Google Scholar]
- [155].Otsuka F, Joner M, Prati F, et al. Clinical classification of plaque morphology in coronary disease. Nat Rev Cardiol. 2014;11:379–389. doi: 10.1038/nrcardio.2014.62 [DOI] [PubMed] [Google Scholar]
- [156].Cameron A, Davis KB, Green G, et al. Coronary bypass surgery with internal-thoracic-artery grafts-effects on survival over a 15-year period. N Engl J Med. 1996;334:216–220. doi: 10.1056/NEJM199601253340402 [DOI] [PubMed] [Google Scholar]
- [157].Boden WE, O'Rourke RA, Teo KK, et al. Optimal medical therapy with or without PCI for stable coronary disease. N Engl J Med. 2007;356:1503–1516. doi: 10.1056/NEJMoa070829 [DOI] [PubMed] [Google Scholar]
- [158].De Bruyne B, Pijls NH, Kalesan B, et al. Fractional flow reserve-guided PCI versus medical therapy in stable coronary disease. N Engl J Med. 2012;367:991–1001. doi: 10.1056/NEJMoa1205361 [DOI] [PubMed] [Google Scholar]
- [159].Querol M, Chen JW, Bogdanov AA Jr. A paramagnetic contrast agent with myeloperoxidase-sensing properties. Org Biomol Chem. 2006;4:1887–1895. doi: 10.1039/b601540a [DOI] [PubMed] [Google Scholar]
- [160].Nahrendorf M, Sosnovik D, Chen JW, et al. Activatable magnetic resonance imaging agent reports myeloperoxidase activity in healing infarcts and noninvasively detects the antiinflammatory effects of atorvastatin on ischemia-reperfusion injury. Circulation. 2008;117:1153–1160. doi: 10.1161/CIRCULATIONAHA.107.756510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Ronald JA, Chen JW, Chen Y, et al. Enzyme-sensitive magnetic resonance imaging targeting myeloperoxidase identifies active inflammation in experimental rabbit atherosclerotic plaques. Circulation. 2009;120:592–599. doi: 10.1161/CIRCULATIONAHA.108.813998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Rodriguez E, Nilges M, Weissleder R, et al. Activatable magnetic resonance imaging agents for myeloperoxidase sensing: mechanism of activation, stability, and toxicity. J Am Chem Soc. 2010;132:168–177. doi: 10.1021/ja905274f [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Breckwoldt MO, Chen JW, Stangenberg L, et al. Tracking the inflammatory response in stroke in vivo by sensing the enzyme myeloperoxidase. Proc Natl Acad Sci U S A. 2008;105:18584–18589. doi: 10.1073/pnas.0803945105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Su HS, Nahrendorf M, Panizzi P, et al. Vasculitis: molecular imaging by targeting the inflammatory enzyme myeloperoxidase. Radiology. 2012;262:181–190. doi: 10.1148/radiol.11110040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Chen YC, Bui AV, Diesch J, et al. A novel mouse model of atherosclerotic plaque instability for drug testing and mechanistic/therapeutic discoveries using gene and microRNA expression profiling. Circ Res. 2013;113:252–265. doi: 10.1161/CIRCRESAHA.113.301562 [DOI] [PubMed] [Google Scholar]
- [166].Yildiz G, Demiryurek AT. Ferrous iron-induced luminol chemiluminescence: a method for hydroxyl radical study. J Pharmacol Toxicol Methods. 1998;39:179–184. doi: 10.1016/S1056-8719(98)00025-2 [DOI] [PubMed] [Google Scholar]
- [167].Yamamoto Y, Ames BN. Detection of lipid hydroperoxides and hydrogen peroxide at picamole levels by an HPLC and isoluminol chemiluminescence assay. Free Radic Biol Med. 1987;3:359–361. doi: 10.1016/S0891-5849(87)80048-5 [DOI] [PubMed] [Google Scholar]
- [168].Wardman P. Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: progress, pitfalls, and prospects. Free Radic Biol Med. 2007;43:995–1022. doi: 10.1016/j.freeradbiomed.2007.06.026 [DOI] [PubMed] [Google Scholar]
- [169].Gerber CE, Kuci S, Zipfel M, et al. Phagocytic activity and oxidative burst of granulocytes in persons with myeloperoxidase deficiency. Eur J Clin Chem Clin Biochem. 1996;34:901–908. [DOI] [PubMed] [Google Scholar]
- [170].Gross S, Gammon ST, Moss BL, et al. Bioluminescence imaging of myeloperoxidase activity in vivo. Nat Med. 2009;15:455–461. doi: 10.1038/nm.1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Zhang N, Francis KP, Prakash A, et al. Enhanced detection of myeloperoxidase activity in deep tissues through luminescent excitation of near-infrared nanoparticles. Nat Med. 2013;19:500–505. doi: 10.1038/nm.3110 [DOI] [PubMed] [Google Scholar]
- [172].Koide Y, Urano Y, Hanaoka K, et al. Development of an Si-rhodamine-based far-red to near-infrared fluorescence probe selective for hypochlorous acid and its applications for biological imaging. J Am Chem Soc. 2011;133:5680–5682. doi: 10.1021/ja111470n [DOI] [PubMed] [Google Scholar]
- [173].Stocker R, Keaney JF Jr. Role of oxidative modifications in atherosclerosis. Physiol Rev. 2004;84:1381–1478. doi: 10.1152/physrev.00047.2003 [DOI] [PubMed] [Google Scholar]
- [174].Tang WHW, Iqbal N, Wu Y, et al. Usefulness of cardiac biomarker score for risk stratification in stable patients undergoing elective cardiac evaluation across glycemic status. Am J Cardiol. 2013;111:465–470. doi: 10.1016/j.amjcard.2012.10.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Düzgünçinar O, Yavuz B, Hazirolan T, et al. Plasma myeloperoxidase is related to the severity of coronary artery disease. Acta Cardiol. 2008;63:147–152. doi: 10.2143/AC.63.2.2029520 [DOI] [PubMed] [Google Scholar]