

Abstract
Altered mitochondrial function and free radical-mediated tissue damage have been suggested as an important pathological event in isoproterenol (ISO)-induced cardiotoxicity. This study was undertaken to know the preventive effect of morin on mitochondrial damage in ISO-induced cardiotoxicity in male Wistar rats. Myocardial infarction (MI) in rats was induced by ISO (85 mg/kg) at an interval of 24 hours for 2 days. Morin was given to rats as pre-treatment for 30 days orally using an intragastric tube. ISO-treated rats showed a significant elevation of mitochondrial thiobarbituric acid reactive substances (TBARS) and hydrogen peroxide (HP) level and pre-treatment with morin significantly prevented the increase of TBARS and HP level to near normality. The level of enzymic and non-enzymic antioxidants was decreased significantly in ISO-treated rats and pre-treatment with morin significantly increased the levels of superoxide dismutase, catalase, glutathione peroxidase, glutathione-S-transferase, glutathione reductase, and reduced glutathione to normality. The activities of mitochondrial enzymes such as isocitrate dehydrogenase, alpha-ketoglutarate dehydrogenase, succinate dehydrogenase, and malate dehydrogenase were decreased significantly in ISO-treated myocardial ischemic rats and upon pre-treatment with morin restored these enzymes activity to normality. In addition, the decreased activities of cytochrome-C oxidase and NADH-dehydrogenases were observed in ISO-treated rats and pre-treatment with morin prevented the activities of cytochrome-C oxidase and NADH-dehydrogenase to normality. Pre-treatment with morin favorably restored the biochemical and functional parameters to near normal indicating morin to be a significant protective effect on cardiac mitochondrial function against ISO-induced MI in rats.
Keywords: Morin, Isoproterenol, Myocardial infarction, Mitochondrial enzymes, Lipid peroxidation
Introduction
Ischemic heart disease leading to myocardial infarction (MI) is a major clinical concern and remains as a clinical challenge and a problem of great importance, despite considerable advances in therapy and management that have been made over the past three decades. MI continues to be a major public health problem, not only in western countries but also increasingly in developing countries and makes significant contribution to the mortality statistics.1,2 MI is the acute condition of necrosis of the myocardium that occurs as a result of imbalance between coronary blood supply and myocardial demand. Isoproterenol (ISO), a synthetic catecholamine, causes severe stress in the myocardium, resulting in infarct-like necrosis of the heart muscle, by increasing lipid peroxidation through enhanced free radical formation.3,4 Experimental induction of MI by ISO in animal is a well-established model to study the protective role of various cardio-protective agents.5 Mitochondria are the main consumers of molecular oxygen in the cardiac cell, and this process functions as a transuding device to provide the energy required for ATP synthesis in the oxidative phosphorylation.6 Reactive oxygen species (ROS), including superoxide, hydroxyl radical, and hydrogen peroxide (HP), are generated by a number of cellular processes, including mitochondrial electron transport, NADPH oxidase, and xanthine oxidase.4 Prolonged oxidative stress in failing myocardium results in damage to mitochondrial DNA, ROS generation, and consequent cellular injury leading to functional decline. Thus, mitochondria serve as a both source and target of a ROS-mediated injury in failing heart.7 Studies have shown that oxidative stress in mice caused specific morphological changes in heart mitochondria with decreased ATP level, impaired contractility, and decreased survival.8 There is increasing evidence that defective mitochondrial energetics and abnormal substrate metabolism are fundamental characteristics of the failing heart.9 Understanding the molecular and biochemical mechanism(s) of cardioprotection and in particular the role that mitochondria play may have a significant impact in the clinical treatment of myocardial ischemia.
Flavonoids are a family of diphenylpropanes most commonly found in a variety of fruits, vegetables, juices, and components of herbal containing dietary supplements. The interest in the investigation of flavonoids stems from their biological properties, which include oxygen radical scavenging and antioxidant properties.10,11 Morin (3,5,7,2′,4′-pentahydroxyflavone) is a member of the flavonoid family which consists of a yellowish pigment found in mill (Prunus dulcis), (Chlorophora tinctoria), and other Moraceae used as food and herbal medicine (Fig. 1).12 Moreover, morin has been reported to possess a variety of biological properties against oxidative stress-induced damage, including the protection of cardiovascular cells,13,14 glomerular mesangial cells,15 hepatocytes,16,17 oligodendrocytes, and neurons18,19 damaged by oxidative stress. Previously, there was no report that the protective effect of mitochondrial damage on morin. Therefore, in the present study the main aim was to investigate the protective effect of morin on cardiac mitochondrial function during ISO-induced MI in rats.
Figure 1.
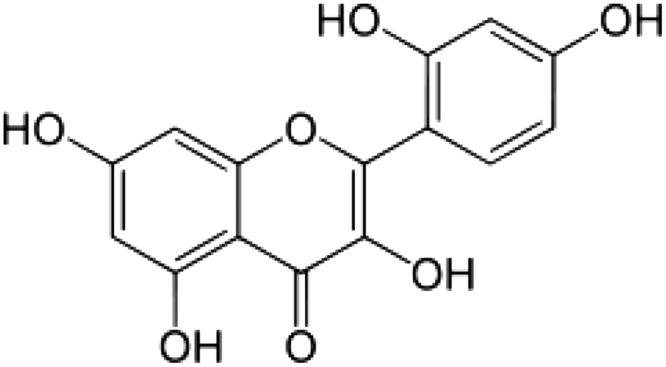
Structure of morin.
Materials and methods
Experimental animals
Male albino rats of Wistar strain of body weight (BW) ranging from 160 to 180 g were procured from Central Animal House, King Saud University, and they were maintained in an air-conditioned room (25 ± 1°C) with a 12-hour light/12-hour dark cycle. The animals were fed ad libitum with normal laboratory pellet diet used in the study was purchased from Grain Silos and Flour Mills Organization, Riyadh, Saudi Arabia and procedures involving animals and their care were accordance with the Policy of Research Centre, King Saud University.
Drugs and chemicals
ISO hydrochloride and morin hydrate (3,5,7,2′,4′-pentahydroxyflavone) of 95% purity were purchased from Sigma-Aldrich (St Louis, MO, USA). All other chemicals were of analytical grade.
Induction of experimental myocardial infarction
Myocardial ischemia was induced by subcutaneous injection (s.c.) of ISO hydrochloride (85 mg/kg BW, twice at an interval of 24 hours) for two consecutive days.5
Experimental design
The animals were randomly divided into six groups of six animals each. Group 1: control rats; Groups 2: normal rats treated with morin (40 mg/kg BW); Group 3: ISO control rats (85 mg/kg BW); Groups 4: rats were pre-treated with morin (40 mg/kg) and then subcutaneously injected with ISO. We have previously reported the effective dose for morin in ISO-induced rats to be 40 mg/kg BW, hence we have selected this effective dose (40 mg/kg BW) for the present study.5 Morin was dissolved in water and administered to rats orally using an intragastric tube daily for a period of 30 days and subsequently treated with ISO (85 mg/kg, s.c.) on 29th and 30th day in normal saline.
At the end of experiment period, rats were anesthetized with an intramuscular injection of ketamine hydrochloride (24 mg/kg BW), and sacrificed by cervical dislocation. The heart tissue was excised, washed in ice-cold isotonic saline, and blotted with a filter paper. The heart mitochondria were isolated from cell debris, nuclei, microsomes, soluble components, and contaminant red blood cells (RBC) using differential centrifugation.
Estimation of biochemical parameters
The heart mitochondria were isolated from cell debris, nuclei, microsomes, soluble components, and contaminant RBC using differential centrifugation by the method of Johnson and Lardy.20
Estimation of thiobarbituric acid reactive substances
The concentration of thiobarbituric acid reactive substances (TBARS) in heart mitochondria was estimated by the method of Niehaus and Samuelson.21 In brief, 0.1 ml of the mitochondrial suspension was treated with 2 ml of (1:1:1 ratio) TBA-tricarboxylic acid (TCA)-HCl reagent (0.37% thiobarbituric acid, 0.25 N HCl, and 15% TCA), placed in water bath for 15 minutes, cooled, and centrifuged at room temperature for 10 minutes. The absorbance of clear supernatant was measured against reference blank at 535.The values were expressed as nmol/mg of protein.
Estimation of lipid HP
The estimation of HP was done by the method of Jiang et al.22 0.1 ml of the mitochondrial suspension was treated with 0.9 ml of Fox reagent (88 mg of butylated hydroxytoluene, 7.6 mg of xylenol orange, and 9.8 mg of ammonium iron sulfate added to 90 ml of methanol and 10 ml of 250 mM sulfuric acid) and incubated at 37°C for 30 minutes. The color developed was read at 560 nm colorimetrically. Lipid hydroperoxides were expressed as nmol/100 mg of protein.
Reduced glutathione
GSH was determined by the method of Ellman.23 One milliliter of mitochondrial suspension was treated with 0.5 ml of Ellman's reagent (19.8 mg of 5,5′-dithiobisnitro benzoic acid in 100 ml of 0.1% sodium citrate) and 3.0 ml of phosphate buffer (0.2 M, pH 8.0). The absorbance was read at 412 nm in spectrophotometer. To prevent the auto-oxidation of GSH, the samples were reduced with potassium borohydride prior to analysis. GSH activity was expressed as nmol/100 mg of protein.
Superoxide dismutase activity
The activity of mitochondrial fraction of superoxide dismutase (SOD) was assayed by the method of Kakkar et al.24 0.5 ml of mitochondrial suspension was diluted with 1 ml of water. In this mixture, 2.5 ml of ethanol and 1.5 ml of chloroform (all reagents chilled) were added and shaken for 1 minute at 4°C then centrifuged. The enzyme activity in the supernatant was determined. The assay mixture contained 1.2 ml of sodium pyrophosphate buffer (0.025 M, pH 8.3), 0.1 ml of 186 µM N-methyl dibenzopyrazine methyl sulphate (PMS), 0.3 ml of 30 µM nitro blue tetrazolium (NBT), and 0.2 ml of 780 µM NADH, appropriately diluted enzyme preparation and water in a total volume of 3 ml. Reaction was started by the addition of NADH. After incubation at 30°C for 90 seconds the reaction was stopped by the addition of 1 ml glacial acetic acid. The reaction mixture was stirred vigorously and shaken with 4 ml of n-butanol. The intensity of the chromogen in the butanol layer was measured at 560 nm against butanol blank. A system devoid of enzyme served as control. One unit of the enzyme activity is defined as the enzyme reaction, which gave 50% inhibition of NBT reduction in 1 minute under the assay conditions.
Catalase activity
The activity of catalase (CAT) was measured by the method of Sinha.25 The reaction mixture (1.5 ml, vol) contained 1.0 ml of 0.01 M phosphate buffer (pH 7.0), 0.1 ml of mitochondrial suspension, and 0.4 ml of 2 M H2O2. The reaction was stopped by the addition of 2.0 ml of dichromate-acetic acid reagent (5% potassium dichromate and glacial acetic acid were mixed in 1:3 ratio). Then the absorbance was read at 620 nm; CAT activity was expressed as nmol of H2O2 consumed/minute/mg protein.
Glutathione peroxides and glutathione reductase activity
The activities of glutathione peroxidase (GPx) and glutathione reductase (GR) were determined by the method of Rotruck et al.26 Briefly, the reaction mixture contained 0.2 ml 0.4 M phosphate buffer (pH 7.0), 0.1 ml 10 mM sodium azide, 0.2 ml mitochondrial suspension homogenized in 0.4 M, phosphate buffer, pH 7.0, 0.2 ml glutathione, and 0.1 ml 0.2 mM HP. The contents were incubated for 10 minutes at 37°C, 0.4 ml 10% TCA was added to stop the reaction and centrifuged at 3200 × g for 20 minutes. The supernatant was assayed for glutathione content using Ellman's reagent (19.8 mg 5,5′-dithiobisnitrobenzoic acid in 100 ml 0.1% sodium nitrate).
Glutathione-S-transferase activity
Glutathione-S-transferase (GST) activity was determined spectrophotometrically by the method of Habig et al.27 The reaction mixture contained 1.0 ml 100 mM phosphate buffer (pH 6.5), 0.1 ml 30 mM 1-chloro-2, 4-dinitrobenzene (CDNB), and 0.7 ml double distilled water. After pre-incubating the reaction mixture for 5 minutes at 37°C, the reaction was started by the addition of 0.1 ml mitochondrial suspension and 0.1 ml of glutathione as substrate. After 5 minutes the absorbance was read at 340 nm. Reaction mixture without the enzyme was used as a blank. The activity of GST is expressed as nmol of CDNB conjugated/minute/100 mg protein.
Isocitrate dehydrogenase activity
Isocitrate dehydrogenase (ICDH) activity was assayed according to the method of Bell and Baron.28 Briefly, the re-action mixture consisting of 0.1 M Tris-HCl buffer, 0.1 M trisodium isocitrate, 0.015 M manganous chloride, 0.001 M NADP, and mitochondrial suspension was incubated at 37.80°C for 60 minutes. Added to this were 1.0 ml of 0.001 M DNPH and 0.5 ml of 5% EDTA. After 20-minute incubation, 10 ml of 0.4 N NaOH was added and the color intensity was measured at 390 nm in a Shimadzu UV-1601 (Tokyo, Japan) spectrometer. The activity of ICDH was expressed as NADH oxidized/hour/mg protein.
Alpha-keto glutaraldehyde dehydrogenase activity
The activity of alpha-keto glutaraldehyde dehydrogenase (α-KGDH) was estimated according to the method of Reed and Mukherjee.29 It is based on the calorimetric determination of ferrocyanide produced by the decarboxylation of alpha-ketoglutarate with ferricyanide as electron acceptor. To 0.15 ml of 0.1 M phosphate buffer, 0.1 ml each of 0.002 M thiamine pyrophosphate, 0.003 M magnesium sulfate, and 0.01 M potassium ferrocyanide was added. The total volume was made up to 1.2 ml with water. Mitochondrial suspension 0.2 ml was added and incubated at 30°C for 30 minutes. Aliquots of the supernatant after centrifugation were taken, 0.1 ml of 0.25 M potassium ferricyanide was added, and the volume was made up to 2.4 ml with water. One milliliter of 4% dupanol and 0.5 ml of ferric ammonium sulpfate dupanol reagent were added and incubated at 250°C for 30 minutes. The color intensity was measured at 540 nm in a Shimadzu UV-1601 (Tokyo, Japan) spectrophotometer. The activity of alpha-KDH was expressed as nmol of ferrocyanide formed/hour/mg protein.
Succinate dehydrogenase activity
Activity of succinate dehydrogenase (SDH) was estimated according to the method of Slater and Bonner.30 The rate of reduction of potassium ferricyanide was measured in the presence of sufficient potassium cyanide to inhibit cytochrome oxidase by following the rate of decrease in the optical density at 420 nm. Briefly, 0.2 ml of mitochondrial suspension was added to a reaction mixture containing 0.3 M phosphate buffer, 0.03 M EDTA, 0.4 M sodium succinate, 0.075 M potassium ferricyanide, bovine serum albumin, and 0.03 M potassium cyanide. The enzyme activity was measured at 420 nm in a Shimadzu UV-1601 spectrophotometer using UVPC Software package. The activity of SDH was expressed as nmol of succinate oxidized/minute/mg protein.
Malate dehydrogenase activity
Malate dehydrogenase (MDH) activity was assayed by the method of Mehler et al.31 The activity determination was based on the measurement of the rate of oxidation of NADH in the presence of the enzyme and excess of oxaloacetate. Briefly, 0.3 ml of 0.25 MTris-HCl buffer, 0.1 ml of NADH, and 0.1 ml of oxaloacetate were added and the total volume was made to 2.9 ml with water. The reaction was started by adding 0.1 ml of mitochondrial suspension. The enzyme activity was measured at 420 nm in a Shimadzu UV-1601 spectrophotometer using UVPC Software package. The activity of MDH was expressed as nmol of NADH oxidized/minute/mg protein.
Cytochrome-C-oxidase activity
Cytochrome-C-oxidase activity was assayed according to the method of Pearl et al.32 The enzyme activity was determined utilizing the accumulation of the free radical formed by the enzymatic univalent oxidation of a stable non-toxic substrate, N-phenyle-p-phenylene diamine. The reaction mixture consisted of 1.0 ml of the buffer, 0.2 ml of 0.2% solution of N-phenyle-p-phenylene diamine, 0.1 ml of cytochrome solution, and 0.5 ml of distilled water. The reaction mixture was incubated at 25°C for 5 minutes. Then added 0.2 ml of the enzyme preparation and the solution mixed well by inverting the cuvette. Blank containing water instead of enzyme, and control containing all the reagents except cytochrome-C, were also treated similarly. The change in optical density was measured in Shimadzu UV-1601 spectrophotometer (Tokyo, Japan) at 550 nm at an interval of 15 seconds for 5 minutes. The activity of the enzyme was expressed as nmol/minute/mg protein.
NADH dehydrogenase activity
NADH dehydrogenase activity was assayed according to the method of Minakami et al.33 The reaction mixture contained 1.0 ml of 0.1 M phosphate buffer, 0.1 ml of 0.03 M potassium ferricyanide, 0.1 ml of 0.1% NADH, and 1.6 ml of distilled water in a total volume of 3.0 ml. The temperature was brought to 30°C and 0.1% NADH was added just before the addition of the sample. A suitable aliquot of mitochondrial solution was added and the enzyme activity was measured at 420 nm in a Shimadzu UV-1601 spectrophotometer using UVPC Software package. The activity of NADH dehydrogenase was expressed as nmol of oxidized/minute/mg protein.
Statistical analysis
Statistical analysis was performed using one-way analysis of variance followed by Duncan's multiple range test (DMRT) using SPSS software package 10.0. Results were expressed as means ± SD from six rats in each group. P values <0.05 were considered as significant.
Results
Fig. 2 shows the level of TBARS and HP in the heart mitochondria of control and experimental rats. ISO-treated rats had showed a significant elevation of mitochondrial TBARS level and pre-treatment with morin significantly prevented the increase of lipid peroxidation to near normality. Rats pre-treated with morin decreased the levels of TBARS and HP to about 36 and 22%, respectively, compared to the ISO-alone-treated animals.
Figure 2.

Effect of morin on the levels of TBARS and HP in the heart mitochondrial of control and ischemic rats. Values are expressed as means ± SD for eight rats in each group. Values not sharing a common superscript in a column differ significantly at P < 0.05 (DMRT).
Fig. 3 represent the levels of SOD, CAT, GPx, GST, and GSH in the heart mitochondria of control and experimental rats. The level of enzymic and non-enzymic antioxidants levels were decreased significantly in ISO-treated rats and pre-treatment with morin precluded the decrease of SOD, CAT, GPx, GST, GR, and GSH to normality. Pre-treatment with morin significantly increased the activities of the antioxidants SOD, CAT, GPx, GST, GR, and GSH to 30, 58, 59, 19, 27, and 28%, respectively, compared to the ISO-treated animals.
Figure 3.

Effect of morin on the levels of SOD, CAT, GPx, GST, GR, and GSH in the heart mitochondrial of control and ischemic rats. Values are expressed as means ± SD for eight rats in each group. Values not sharing a common superscript (a–c) in a column differ significantly at P < 0.05 (DMRT). †CDNB: 1-chloro-2,4-dinitrobenzene. ‡One units is defined as the enzyme concentration required to inhibit the OD at 560 nm of chromogen production by 50% in 1 minute.
Fig. 4 shows the activities of mitochondrial TCA cycle enzymes, ICDH, alpha-KGDH, SDH, MDH, and respiratory chain enzymes of cytochrome-C oxidase and NADH-dehydrogenase in control and experimental animals. The activities of these enzymes had decreased significantly in ISO-treated myocardial ischemic rats and upon pre-treatment with morin restored these enzymes activity to normality. Morin pre-treatment significantly increased the activities of ICDH, alpha-KGDH, SDH, MDH, cytochrome-C oxidase, and NADH-dehydrogenase to 63, 30, 33, 35, 58, and 41%, respectively, compared to the ISO-treated animals.
Figure 4.

Effect of morin on the levels of ICDH, alpha-KGDH, SDH, MDH, cytochrome-C oxidase, and NADH-dehydrogenase in the heart mitochondrial of control and ischemic rats. Values are expressed as means ± SD for eight rats in each group. Values not sharing a common superscript in a column differ significantly at P < 0.05 (DMRT).
Discussion
Isoproterenol (ISP), a synthetic beta-adrenergic agonist which increases the myocardial oxygen demand that leads to ischemic necrosis of myocardium in rats similar to that seen in human MI.1,34 Lipid peroxidation has been hypothesized to be a major mechanism of free radical-mediated cell damage. The process of lipid peroxidation is one of the oxidative conversion of polyunsaturated fatty acids (PUFAs) to products known as malondialdehyde, which is usually measured as TBARS or lipid peroxides, which is the most studied, biologically relevant, free radical reaction.35 An increase in the concentrations of mitochondrial TBARS and HP in ISO-treated rats indicates increased lipid peroxidation, which could be attributed to a deficiency of antioxidant defense mechanism.36 Increased mitochondrial lipid peroxides levels are in concert with a decreased mitochondrial antioxidant status in the ISO-intoxicated group observed herein.
In our study, ISO-induced rats exhibited decreased activities of SOD and CAT in the heart. SOD plays an important role in protecting the cells from oxidative damage by converting superoxide radicals into HP, which is further metabolized by CAT to molecular oxygen and water. The decrease in the activities of these antioxidant enzymes might be due to myocardial cell damage. Superoxide radicals generated at the site of damage modulates SOD and CAT resulting in the decreased activities of these enzymes and accumulation of superoxide anion, which also damages the myocardium. A significant decrease in the activities of antiperoxidative enzymes, SOD and CAT may be due to diminished scavenging of HO• formed by ISO-induced lipid peroxidation.37 A decrease in the activity of GPx makes mitochondria susceptible to ISO-induced myocardial damage that leads to mitochondrial dysfunction.38 The decrease in the activities of GPx and GST may be due to a reduced availability of their substrate, GSH.39 GSH protects mitochondrial membrane from the damaging action of lipid peroxidation. Depletion in the levels of mitochondrial GSH seems to be a major mechanism for inducing an imbalance of mitochondrial function.39 In the present study activity of GR was increased in ISO-induced rats. GR is an important enzyme for the maintenance of intracellular concentrations of GSH, which plays a major role as a substrate for GPx and GST. Pre-treatment with morin significantly improved the mitochondrial levels of SOD, CAT, GSH, GPx, GST, GR; and concomitantly decreased lipid peroxide levels. Morin might prevent the initiation and propagation of the lipid peroxidation process by scavenging free radicals,13,14,17 thereby might have improved the antioxidants levels. Inhibition of mitochondrial lipid peroxidation and augmentation of antioxidants by morin are proposed to play a role in preventing myocardial damage during ischemia. Hence the reduction in lipid peroxides with an increase in GSH on morin pre-treatment to ISO-induced rats could be due to the antioxidant effect of citrate cycle intermediates.
Ischemia is associated with an impairment of myocardial energy production, mitochondrial respiration, and diminished oxygen uptake. The mechanisms underlying these alterations are studied by assessing the activities of dehydrogenases of the TCA cycle and respiratory enzymes like, cytochrome-C oxidase and NADH dehydrogenase. ISO has been reported to cause tissue hypoxia where there is an oxygen demand.40 In hypoxia, TCA cycle enzymes activities are expected to be low.41 In the present study, the activities of TCA cycle enzymes were decreased in ISO-administered rats. The decreased activities of TCA cycle enzymes which are located in the outer membrane could be due to the free radicals produced by ISO. Numerous studies have shown the excessive generation of free radicals during myocardial ischemia. The increased oxygen free radical have attributed to myocardial ischemia and/or reperfusion.42 Free radicals and oxidants triggers oxidation of DNA, protein and lipids, especially oxidize PUFA present in the membrane, culminating in loss of cellular integrity.43,44 Pre-treatment with morin maintained the activities of TCA cycle enzymes in ISO-induced rats. This could be due to the increased availability of these free amino acids and entry into TCA cycle via the aspartate–malate shuttle. Further, numerous data from previous studies have demonstrated that some of the TCA cycle intermediates (citrate, malate, and oxaloacetate) can act as antioxidant against a variety of pro-oxidants.45,46 Thus the reduction in free radicals could protect the membrane and increase the activities of TCA cycle enzymes.
Cytochrome-C oxidase, the terminal enzyme in the respiratory chain is located in the inner membrane of the mitochondria. Singh47 had reported that the decrease in the activities of these enzymes might be due to depletion of reducing equivalents, like NADH and NADPH, which are utilized for the formation of GSH to counter-oxidative damage of mitochondrial components. Other studies have shown that both these enzymes have an absolute requirement of cardiolipin.48 Decreased activities of cytochrome-C oxidase and NADH dehydrogenase in ISO-induced rat mitochondria could be due to the unavailability of the cardiolipin for their activity. However, morin increased the activities of these enzymes. This could be due to the increased availability of the TCA cycle intermediates such as fumarate, oxaloacetate, succinate, and malate which may protect the phospholipids from degradation.
Conclusion
In summary, the present study provides experimental evidence that morin exhibit preventive effect on ISO-induced myocardial rats due to strong antioxidant activity, and its ability to maintain cell membrane integrity, ameliorate mitochondrial respiratory dysfunction induced by high-dose ISO administration. These findings might be helpful to understand the beneficial effects of morin against myocardial injury, although further study is needed to confirm its mechanism.
Acknowledgements
This project was supported by King Saud University, Deanship of Scientific Research, College of Applied Medical Sciences Research Centre. Authors also thank National Nutrition Policy Chair, King Saud University, Riyadh, Saudi Arabia for the support.
References
- 1.Sabeena Farvin KH, Anandan R, Kumar SH, Shiny KS, Sankar TV, Thankappan TK. Effect of squalene on tissue defense system in isoproterenol-induced myocardial infarction in rats. Pharmacol Res 2004;50(3):231–6 [DOI] [PubMed] [Google Scholar]
- 2.Gilski DJ, Borkenhagen B. Risk evaluation in action for cardiovascular health, Crit. Care Nurse 2005;25(1):26–8, 26–27. [PubMed] [Google Scholar]
- 3.Rajadurai M, Stanely Mainzen Prince P. Preventive effect of naringin on cardiac markers, electrocardiographic patterns and lysosomal hydrolases in normal and isoproterenol-induced myocardial infarction in Wistar rats. Toxicology 2007;230(2–3):178–88. [DOI] [PubMed] [Google Scholar]
- 4.Chattopadhyay A, Biswas S, Bandyopadhyay D, Sarkar C, Datta AG. Effect of isoproterenolonlipid peroxidation and antioxidantenzymes of myocardial tissue of mice and protection by quinidine. Mol Cell Biochem 2003;245(1–2):43–9. [DOI] [PubMed] [Google Scholar]
- 5.Al-Numair KS, Chandramohan G, Alsaif MA. Pretreatment with morin, a flavonoid, ameliorates adenosine triphosphatases and glycoproteins in isoproterenol-induced myocardial infarction in rats. J Nat Med 2012;66(1):95–101. [DOI] [PubMed] [Google Scholar]
- 6.Echtay KS, Murphy MP, Smith RA, Talbot DA, Brand MD. Superoxide activates mitochondrial uncoupling protein 2 from the matrix side. Studies using targeted antioxidants. J Biol Chem 2002;277(49):47129–35. [DOI] [PubMed] [Google Scholar]
- 7.Murray AJ, Edwards LM, Clarke K. Mitochondria and heart failure. Curr Opin Clin Nutr Metab Care 2007;10(6):704–11. [DOI] [PubMed] [Google Scholar]
- 8.Nojiri H, Shimizu T, Funakoshi M, Zhou H, Kawakami S, Ohta Y, et al.. Oxidative stress causes heart failure with impaired mitochondrial respiration. J Biol Chem 2006;281(44):33789–801. [DOI] [PubMed] [Google Scholar]
- 9.Neubauer S. The failing heart – an engine out of fuel. N Engl J Med 2007;356(11):1140–51. [DOI] [PubMed] [Google Scholar]
- 10.Rice-Evans CA, Miller NJ, Paganga G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radical Biol Med 1996;20:933–56. [DOI] [PubMed] [Google Scholar]
- 11.Brown JE, Khodr H, Hider RC, Rice-Evans CA. Structural dependence of flavonoid interactions with Cu2+ ions: implications for their antioxidant properties. Biochem J 1998;330:1173–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie MX, Long M, Liu Y, Qin C, Wang YD. Characterization of the interaction between human serum albumin and morin. Biochim Biophys Acta 1760;1184–91 [DOI] [PubMed] [Google Scholar]
- 13.Wu TW, Zeng LH, Wu J, Fung KP. Morin: a wood pigment that protects three types of human cells in the cardiovascular system against oxyradical damage. Biochem Pharmacol 1994;47:1099–103. [DOI] [PubMed] [Google Scholar]
- 14.Kok LD, Wong YP, Wu TW, Chan HC, Kwok TT, Fung KP. Morin hydrate: a potential antioxidant in minimizing the free-radicals-mediated damage to cardiovascular cells by anti-tumor drugs. Life Sci 2000;67:91–9. [DOI] [PubMed] [Google Scholar]
- 15.Zeng LH, Fung KP, Wu TW. Morin hydrate protects cultured rat glomerular mesangial cells against oxyradical damage. Life Sci 1994;55:351–7. [DOI] [PubMed] [Google Scholar]
- 16.Wu TW, Zeng LH, Wu J, Fung KP. Morin hydrate is a plant-derived and antioxidant-based hepatoprotector. Life Sci 1993;53:213–8. [DOI] [PubMed] [Google Scholar]
- 17.Sivaramakrishnan V, Shilpa PNM, Kumar VRP, Niranjali Devaraj S. Attenuation of N-nitrosodiethylamine-induced hepatocellular carcinogenesis by a novel flavonol-morin. Chem Biol Interact 2008;171:79–88. [DOI] [PubMed] [Google Scholar]
- 18.Ibarretxe G, Sánchez-Gómez MV, Campos-Esparza MR, Alberdi E, Matute C. Differential oxidative stress in oligodendrocytes and neurons after excitotoxic insults and protection by natural polyphenols. Glia 2006;53:201–11. [DOI] [PubMed] [Google Scholar]
- 19.Gottlieb M, Leal-Campanario R, Campos-Esparza MR, Sánchez-Gómez MV, Alberdi E, Arranz A, et al.. Neuroprotection by two polyphenols following excitotoxicity and experimental ischemia. Neurobiol Dis 2006;23:374–86. [DOI] [PubMed] [Google Scholar]
- 20.Johnson D, Lardy H. Isolation of liver or kidney mitochondria. In: , Estabrook RW (ed.) Methods in enzymology. vol. 10 London: Academic Press; 1967. p. 94–6. [Google Scholar]
- 21.Niehaus WG, Samuelson B. Formation of malondialdehyde from phospholipid arachidonate during microsomal lipid peroxidation. Eur J Biochem 1968;6:126–30. [DOI] [PubMed] [Google Scholar]
- 22.Jiang ZY, Hunt JV, Wolff SD. Ferrous sulphate oxidation in the presence of xylenol orange for detection of lipid hydroperoxide in low density lipoprotein. Anal Biochem 1992;202:384–9. [DOI] [PubMed] [Google Scholar]
- 23.Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys 1959;82:70–7. [DOI] [PubMed] [Google Scholar]
- 24.Kakkar P, Das B, Viswanathan PN. A modified spectrophotometric assay of superoxide dismutase. Ind J Biochem Biophys 1984;21:130–2. [PubMed] [Google Scholar]
- 25.Sinha AK. Colorimetric assay of catalase. Anal Biochem 1972;47:389–94. [DOI] [PubMed] [Google Scholar]
- 26.Rotruck JT, Pope AL, Ganther HE, Swanson AB, Hafeman DG, Hoekstra WG. Selenium: biochemical role as a component of glutathione peroxidase. Science 1973;179:588–90. [DOI] [PubMed] [Google Scholar]
- 27.Habig WH, Pabst MJ, Jakoby WBC. Glutathione-S-transferases: the first enzymatic step in mercapturic acid formation. J Biol Chem 1974;249:7130–9. [PubMed] [Google Scholar]
- 28.Bell JL, Baron DN. A colorimetric method for determination of isocitrate dehydrogenase. Clin Chem Acta 1960;5:740–7. [Google Scholar]
- 29.Reed LJ, Mukherjee RB. α-Ketoglutarate dehydrogenase complex from Escherichia coli. In: , Colowick SP, Kaplon NO (eds.) Methods in enzymology. vol. 13 New York: Academic press; 1969. p. 53–61. [Google Scholar]
- 30.Slater EC, Bonner WD. The effect of fluoride on succinic oxidase system. Biochem J 1952;52:185–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mehler AH, Kornberg A, Grisolia S, Ochoa S. The enzymatic mechanism of oxidation-reductions between malate or isocitrate and pyruvate. J Biol Chem 1948;174:961–77. [PubMed] [Google Scholar]
- 32.Pearl W, Cascarano J, Zweifach BW. Microdetermination of cytochrome oxidase in rat tissues by the oxidation of N-phenyl-p-phenylene diamine or ascorbic acid. J Histochem Cytochem 1963;2:102–4. [Google Scholar]
- 33.Minakami S, Ringler RL, Singer TP. Studies on the respiratory chain-linked dihydrodiphosphopyridine nucleotide dehydrogenase I. Assay of the enzyme in particulate and in soluble preparation. J Biol Chem 1962;237:569–76. [PubMed] [Google Scholar]
- 34.Mukesh Nandave, Ipseeta Mohanty, Nag1 TC, Shreesh Kumar Ojha, Rajan Mittal, Santosh Kumari, et al.. Cardioprotective response to chronic administration of vitamin E in isoproterenol induced myocardial necrosis: hemodynamic, biochemical and ultrastructural studies. Indian J Clin Biochem 2007;22(1):22–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bakan E, Taysi S, Polat MF. Nitric oxide levels and lipid peroxidation in plasma of patients with gastric cancer. Jpn J Clin Oncol 2002;32:162–6. [DOI] [PubMed] [Google Scholar]
- 36.Raghavendran HRB, Sathivel A, Devaki T. Antioxidant effect of Sargassum polycystum against acetaminophen induced changes in hepatic mitochondrial enzymes during toxic hepatitis. Chemosphere 2005;61:276–81. [DOI] [PubMed] [Google Scholar]
- 37.Sathish V, Vimal V, Ebenezar KK, Devaki T. Synergistic effect of nicorandil and amlodipine on mitochondrial function during isoproterenol-induced myocardial infarction in rats. J Pharm Pharmacol 2001;54:133–7. [DOI] [PubMed] [Google Scholar]
- 38.Tappel AL. Lipid peroxidation damages to cell components. Fed Proc 1973;32:1870–4. [PubMed] [Google Scholar]
- 39.Yogeeta SK, Gnanapragasam A, Kumar SS, Subhashini R, Sathivel A, Devaki T. Synergistic interaction of ferulic acid with ascorbic acid: its cardio protective role during isoproterenol induced myocardial infarction in rats. Mol Cell Biochem 2006;283:139–46. [DOI] [PubMed] [Google Scholar]
- 40.Bloom S, Davis DL. Calcium as mediator of isoproterenol-induced myocardial necrosis. Am J Pathol 1972;69:459–70. [PMC free article] [PubMed] [Google Scholar]
- 41.Li J, Gao X, Qian M, Eaton JW. Mitochondrial metabolism underlies hyperoxic cell damage. Free Radic Biol Med 2004;36:1460–70. [DOI] [PubMed] [Google Scholar]
- 42.Chambers DE, Parks DA, Patterson G, Roy R, McCord JM, Yoshida S, et al.. Xanthine oxidase as a source of free radical damage in myocardial ischemia. J Mol Cell Cardiol 1985;17(2):145–52. [DOI] [PubMed] [Google Scholar]
- 43.Maxwell SR, Lip GY. Reperfusion injury: a review of the pathophysiology, clinical manifestations and therapeutic options. Int J Cardiol 1997;58(2):95–117. [DOI] [PubMed] [Google Scholar]
- 44.Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest 2005;115(3):500–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mallet RT, Sun J. Antioxidant properties of myocardial fuels. Mol Cell Biochem 2003;253(1–2):103–11. [DOI] [PubMed] [Google Scholar]
- 46.Yamamoto HA, Mohanan PV. Effect of alpha-ketoglutarate and oxaloacetate on brain mitochondrial DNA damage and seizures induced by kainic acid in mice. Toxicol Lett 2003;143(2):115–22. [DOI] [PubMed] [Google Scholar]
- 47.Singh RJ. Glutathione: a marker and antioxidant for aging. J Lab Clin Med 2002;140:380–1. [DOI] [PubMed] [Google Scholar]
- 48.Nicolay K, Van Der Neut R, Fok JJ, de Kruijff B. Effects of adriamycin on lipid polymorphism in cardiolipin-containing model and mitochondrial membranes. Biochim et Biophys Acta 1985;819:55–65. [DOI] [PubMed] [Google Scholar]