

Summary
Aging is an important biological phenomenon and a major contributor to human disease and disability, but no drugs have been demonstrated to delay human aging. Caenorhabditis elegans is a valuable model for studies of animal aging, and the analysis of drugs that extend the lifespan of this animal can elucidate mechanisms of aging and might lead to treatments for age‐related disease. By testing drugs that are Food and Drug Administration approved for human use, we discovered that the mood stabilizer and anticonvulsant valproic acid (VA) extended C. elegans lifespan. VA also delayed age‐related declines of body movement, indicating that VA delays aging. Valproic acid is a small carboxylic acid that is the most frequently prescribed anticonvulsant drug in humans. A structure–activity analysis demonstrated that the related compound valpromide also extends lifespan. Valproic acid treatment may modulate the insulin/IGF‐1 growth factor signaling pathway, because VA promoted dauer larvae formation and DAF‐16 nuclear localization. To investigate the mechanism of action of VA in delaying aging, we analyzed the effects of combining VA with other compounds that extend the lifespan of C. elegans. Combined treatment of animals with VA and the heterocyclic anticonvulsant trimethadione caused a lifespan extension that was significantly greater than treatment with either of these drugs alone. These data suggest that the mechanism of action of VA is distinct from that of trimethadione, and demonstrate that lifespan‐extending drugs can be combined to produce additive effects.
Keywords: aging, C. elegans, dauer, lifespan, valproic acid, valpromide
Introduction
Pharmacological compounds that extend lifespan could delay the progression of age‐related degenerative changes and age‐related illnesses such as Alzheimer's disease and cardiovascular disease. In addition, the characterization of drugs that extend lifespan can elucidate endogenous mechanisms involved in lifespan determination, because the targets of these drugs are likely to influence normal aging. The short lifespan and rapid aging of invertebrates make them powerful models for the identification of drugs that extend lifespan and for the characterization of the mechanism of action of these drugs (Lithgow et al., 2005; Collins et al., 2006; Olsen et al., 2006a). The free‐living soil nematode Caenorhabditis elegans has been a leading system for studying genetic and pharmacologic influences on lifespan. Four categories of compounds have been reported to extend C. elegans lifespan: a variety of antioxidant compounds (Harrington & Harley, 1988; Adachi & Ishii, 2000; Melov et al., 2000; Ishii et al., 2004; Keaney et al., 2004); complex mixtures derived from plants (Wu et al., 2002; Wilson et al., 2006); resveratrol, a potential modulator of Sir2 activity (Wood et al., 2004; Bass et al., 2007); and medications such as heterocyclic anticonvulsant medications that are likely to act by affecting neural activity (Evason et al., 2005; Petrascheck et al., 2007; McColl et al., 2008). Drosophila melanogaster has also been an important system for the discovery of drugs that extend lifespan, and several histone deacetylase (HDAC) inhibitors delay Drosophila aging (Kang et al., 2002; Zhao et al., 2005). Compounds that extend the lifespan of vertebrates have not been well characterized. However, a recent report showing that resveratrol can extend the lifespan of a short‐lived fish suggests that compounds that extend invertebrate lifespan may be relevant to vertebrate biology (Valenzano et al., 2006).
Current medical practice in humans often involves combining two or more drugs with different mechanisms of action to provide greater efficacy and minimize deleterious side effects. For example, HIV antiretroviral treatment can involve the use of a reverse transcriptase inhibitor and a protease inhibitor that act by distinct mechanisms to inhibit viral replication (Temesgen et al., 2006). Treatment of hypertension can involve combining a diuretic that decreases plasma volume with an ACE inhibitor that promotes vasodilation (Sica, 2002). The possibility of combining lifespan‐extending drugs to produce a longer lifespan extension than is possible with either drug alone is appealing because it might illuminate the mechanism of action of the drugs and provide therapeutic benefits. However, combinations of drugs that have additive effects on lifespan have not been reported.
Here, we report that valproic acid (VA) extends C. elegans lifespan and delays age‐related degenerative changes in body movement. Valproic acid is a small carboxylic acid that was discovered serendipitously to have anticonvulsant activity when it was used as a solvent to dissolve other potentially active compounds (Isoherranen et al., 2003). Valproic acid is now the most frequently prescribed antiepileptic drug with annual sales exceeding $1 billion (Isoherranen et al., 2003). Valproic acid is also used to treat numerous neurological and psychiatric disorders including bipolar disorder and migraine headaches (Isoherranen et al., 2003). In addition to extending C. elegans lifespan, this compound enhanced dauer formation and nuclear localization of the DAF‐16 protein. Worms treated with a combination of VA and the heterocyclic anticonvulsant trimethadione (TRI) lived significantly longer than worms treated with either drug alone. This suggests that the mechanism of action of VA is distinct from that of TRI. Furthermore, our results demonstrate that two drugs can be combined to cause an additive lifespan extension.
Results
VA extends C. elegans lifespan
To identify drugs that influence lifespan, we analyzed Food and Drug Administration‐approved compounds for the ability to extend the mean lifespan of C. elegans hermaphrodites cultured at 20 °C on agar plates with abundant Escherichia coli as a food source. We chose compounds from different structural and functional classes of drugs, and initially analyzed the effect of three drug concentrations on populations of 50 hermaphrodites. We previously described this approach and the identification of the anticonvulsant drug ethosuximide that can extend C. elegans lifespan (Evason et al., 2005). Subsequently, we analyzed VA and observed an extension of the mean lifespan at one of the initial test concentrations.
To characterize the range of effects of VA and identify the optimal concentration for lifespan extension, we conducted a dose–response analysis. Worms cultured in a medium containing 6 mm VA displayed the largest lifespan extension; lower concentrations caused smaller extensions, while higher concentrations reduced lifespan and substantially delayed development (Fig. 1A and data not shown). To accurately quantify the effects of 6 mm VA, we analyzed 530 animals in 10 separate trails. It was found that 6 mm VA extended mean adult lifespan from 16.2 days to 21.9 days (35% increase) and extended maximum adult lifespan from 23.3 days to 33.1 days (42% increase) (Table 1). These effects were temperature sensitive; VA did not extend lifespan at 15 °C or 25 °C (data not shown).
Figure 1.
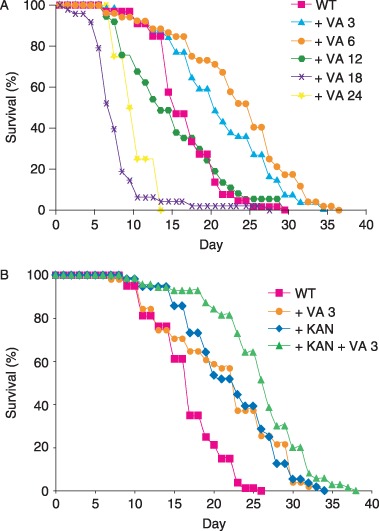
Dose–response analysis of valproic acid (VA). (A) Wild‐type (WT) hermaphrodites were cultured with no drug or VA from conception until death, and monitored for survival. Doses are in mm. The mean lifespans for 0 mm, 3 mm, 6 mm, 12 mm, 18 mm, and 24 mm VA were days [SD (n)]: 16.4 ± 4.3 (66), 20.9 ± 6.7 (56), 23.2 ± 7.3 (52), 14.7 ± 6.4 (37), 7.4 ± 3.9 (48), and 9.8 ± 2.5 (4), respectively. Treatment with 3 mm and 6 mm VA extended the mean lifespan significantly (P < 0.0001), whereas treatment with 18 mm and 24 mm VA shortened the mean lifespan significantly (P < 0.0001 and P < 0.05, respectively). Most animals cultured in the presence of 24 mm VA died or arrested development before reaching the fourth larval stage, and we analyzed the adult lifespan of the rare animals that survived to adulthood. (B) Valproic acid extends the lifespan of animals treated with kanamycin. Wild‐type hermaphrodites were cultured with no drug (WT, n = 80), with 3 mm VA alone ( + VA 3, n = 51), with kanamycin alone ( + KAN, n = 56), or with both kanamycin and VA ( + KAN + VA 3, n = 70) from conception until death. Treatment with 3 mm VA alone or with kanamycin alone significantly extended lifespan compared to animals with no drug treatment (P < 0.0002 and P < 0.0001, respectively). Treatment with kanamycin and 3 mm VA significantly extended lifespan compared to treatment with kanamycin alone (P < 0.0007). These data represent the results of a single trial; combined data for two trials are presented in Table 1.
Table 1.
Lifespan analysis of wild‐type worms cultured with drugs
| Drug† | Mean lifespan (SD) (days)‡, § | % Change§ | Maximum lifespan (SD) (days)‡, §, ¶ | % Change§ | N †† |
|---|---|---|---|---|---|
| None | 16.2 ± 4.0 | 23.3 ± 1.8 | 708 (12) | ||
| VA (3) | 20.9 ± 7.0*** | +29 | 33.3 ± 2.1*** | +43 | 304 (5) |
| VA (6) | 21.9 ± 7.0*** | +35 | 33.1 ± 2.1*** | +42 | 530 (10) |
| VA (6/0) | 17.1 ± 5.0 | +6 | 26.3 ± 2.1** | +13 | 107 (2) |
| VA (0/6) | 20.8 ± 7.5*** | +28 | 33.1 ± 4.2*** | +42 | 100 (2) |
| TRI (28) | 23.5 ± 7.5*** | +45 | 36.9 ± 2.5*** | +58 | 248 (4) |
| TRI (28) + VA (6) | 26.1 ± 7.8*** | +61 | 37.9 ± 2.3*** | +63 | 222 (4) |
| Ethanol | 16.8 ± 3.8 | 22.7 ± 0.8 | 168 (3) | ||
| VPD (6) | 20.4 ± 6.7*** | +21 | 30.6 ± 2.5*** | +35 | 152 (3) |
| KAN | 20.2 ± 5.2 | 29.6 ± 2.5 | 139 (2) | ||
| KAN + VA (3) | 22.9 ± 5.6*** | +13 | 32.5 ± 2.2** | +10 | 155 (2) |
Drug concentration in mm for valproic acid (VA), trimethadione (TRI), and valpromide (VPD). VA (6/0) and VA (0/6) indicate treatment with 6 mm VA from conception until fourth larval (L4) or from L4 until death, respectively. All other drug treatments were from conception until death. Ethanol was used to solubilize VPD, and ethanol alone was the control treatment for VPD. The antibiotic kanamycin (KAN) was added to nematode growth medium at a final concentration of 50 µg mL−1. KAN alone was the control treatment for KAN + VA (3).
If the standard deviations of two groups were similar, comparisons were performed using the Student's t‐test. Otherwise, the alternate Welch test was used. P > 0.05, no stars;
P < 0.05;
P < 0.005;
P < 0.0001.
The percentage change and the statistical significance of this change were determined by comparing the experimental treatments VA (3), VA (6), VA (6/0), VA (6/0), TRI (28), and TRI (28) + VA(6) to none. The experimental treatment VPD (6) was compared to ethanol, because VPD was dissolved in ethanol. The experimental treatment KAN + VA (3) was compared to KAN.
Maximum adult lifespan is the mean lifespan of 10% of the population that had the longest lifespans.
Number of hermaphrodites analyzed, with number of independent experiments in parentheses.
Supplementation of nematode growth medium (NGM) plates with VA prevented the accumulation of E. coli, indicating that VA might inhibit bacterial proliferation. To provide an abundant food source for worms cultured with VA, we grew E. coli OP50 in standard medium, concentrated the bacteria, and dispensed the bacteria onto plates containing VA. Bacterial pathogenicity reduces the lifespan of worms cultured with E. coli, because treatment with antibiotics can extend lifespan (Garigan et al., 2002). To determine if inhibition of bacterial pathogenicity contributes to the mechanism of action of VA in lifespan extension, we compared treatment with the antibiotic kanamycin to treatment with VA and kanamycin (Fig. 1B and Table 1). In single treatments, kanamycin and 3 mm VA increased mean lifespan to 20.2 days (25%) and 20.9 days (29%), respectively (Table 1). A combined treatment of kanamycin and 3 mm VA increased the mean lifespan to 22.9 days (41%). In the presence of kanamycin, treatment with VA caused a significant extension of mean lifespan extension of 13%, indicating that VA influences lifespan by a mechanism that is independent of inhibiting bacterial proliferation. There are several possible explanations for the difference in the magnitude of the lifespan extension caused by VA alone and VA in the presence of kanamycin: (i) Kanamycin and VA have partially overlapping effects. Valproic acid might extend lifespan partly by inhibiting bacterial proliferation, or kanamycin might extend lifespan partly by directly acting on worms. Alternatively, VA might extend lifespan by increasing the ability of worms to resist bacterial pathogens, in which case the addition of kanamycin might have minimal effects (Garsin et al., 2003). (ii) Kanamycin and VA do not have overlapping effects. In this case, the failure to observe fully additive effects on lifespan might result from the toxic effects of combining these drugs, which limits the lifespan extension. Alternatively, the lifespan extension could be limited by age‐related degenerative changes that neither drug remedies.
To determine the developmental stage when VA functions to extend lifespan, we administered the drug from conception until the fourth larval (L4) stage or from the L4 stage until death. Exposure to VA only during embryonic and larval development had no significant effect on mean lifespan. In contrast, exposure to VA beginning at the L4 stage extended mean lifespan to 20.8 days (28% increase) (Fig. 2B and Table 1). These results indicate that VA does not influence lifespan by affecting development, but rather acts in adults to delay degeneration.
Figure 2.
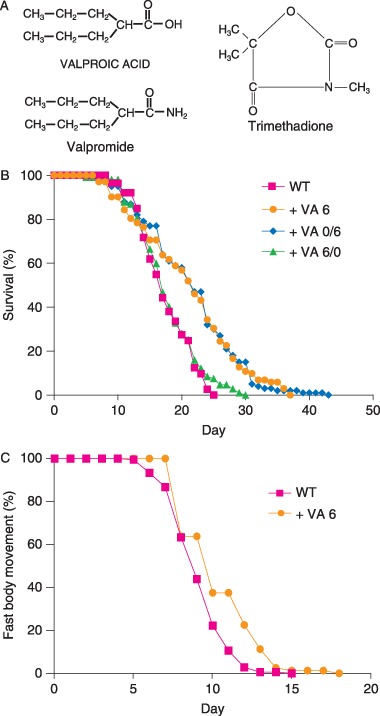
Valproic acid (VA) extends worm lifespan and delays the age‐related decline in body movement. (A) Structures of VA, valpromide, and trimethadione. (B) Wild‐type (WT) hermaphrodites were cultured without drug (WT, n = 113) or with 6 mm VA from conception until death ( + VA 6, n = 102), from fourth larval (L4) until death ( + VA 0/6, n = 100), or from conception until L4 ( + VA 6/0, n = 107). (C) Wild‐type hermaphrodites were cultured with no drug (WT, n = 180) or with 6 mm VA ( + VA 6, n = 80). Animals were classified as fast moving if they displayed continuous and well‐coordinated sinusoidal movements, and not fast moving if they displayed discontinuous or sluggish movements (Huang et al., 2004). Survival and fast body movement were monitored beginning at the L4 stage (day 0).
VA delays the age‐related decline of body movement
Caenorhabditis elegans hermaphrodites display age‐related declines in several physiological processes including a transition from rapid, coordinated body movement to sluggish, uncoordinated body movement, and a decline in pharyngeal pumping rate (Herndon et al., 2002; Huang et al., 2004; Collins et al., 2007). To determine if VA delays age‐related declines of physiological processes, we analyzed body movement and pharyngeal pumping. Valproic acid extended the span of time that animals display fast body movement from 8.2 ± 1.7 days to 9.4 ± 2.3 days (P < 0.0001) (Fig. 2C). Valproic acid did not significantly extend the span of time that animals display pharyngeal pumping (data not shown). These results indicate that VA delays age‐related degeneration of physiologic processes that mediate body movement.
It is important to monitor reproductive capacity because several manipulations that extend adult lifespan significantly affect progeny production. For example, dietary restriction and diminished mitochondrial function reduce progeny production early in life and reduce the total number of progeny produced by self‐fertile hermaphrodites (Felkai et al., 1999; Hughes et al., 2007). To analyze self‐fertile reproduction, we monitored progeny production daily. Hermaphrodites cultured in a medium containing 6 mm VA did not display an extended self‐fertile reproductive span, indicating that VA does not delay the utilization of self‐sperm (data not shown). Reproductive cessation in self‐fertile hermaphrodites occurs because animals exhaust their supply of self‐sperm. To accurately measure the full reproductive potential of hermaphrodites, we mated L4 hermaphrodites to male C. elegans for 2 days and monitored progeny production. Animals cultured with 6 mm VA produced an average of 413 ± 125 progeny, similar to animals cultured without the drug. Furthermore, culturing animals with VA neither lengthened nor shortened the mated reproductive period, because animals cultured with VA and untreated control animals produced similar numbers of progeny during the last several days of reproductive activity (Fig. 3).
Figure 3.
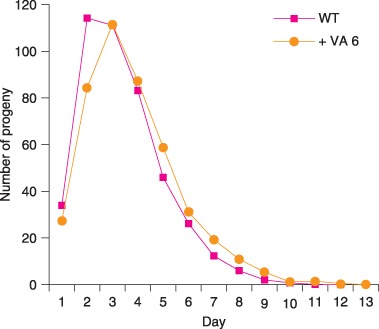
Valproic acid (VA) does not decrease reproduction of mated hermaphrodites. We mated wild‐type (WT) hermaphrodites to WT males and counted progeny of hermaphrodites cultured without drug (WT) or with 6 mm VA ( + VA 6). Hermaphrodites cultured with and without drug produced 413 ± 125 (n = 28) and 434 ± 102 (n = 65) progeny, respectively (not significantly different). Hermaphrodites cultured with and without drug produced 2.3 ± 6.9 (n = 68) and 2.7 ± 9.4 (n = 86) progeny after day 9, respectively (not significantly different).
A structurally similar compound, valpromide (VPD), extends C. elegans lifespan
Compounds that are structurally related to VA have been analyzed to characterize the mechanism of action of VA as an anticonvulsant (Bialer, 1991; Phiel et al., 2001; Isoherranen et al., 2003). Valpromide is an aliphatic amide analogue of VA and shares its potent anticonvulsant activity (Fig. 2A) (Bialer, 1991). Unlike VA, VPD is not a HDAC inhibitor (Phiel et al., 2001). We analyzed the effects of VPD on C. elegans lifespan. Valpromide was dissolved in ethanol, and control animals for VPD experiments were exposed to ethanol alone. At a concentration of 6 mm that is optimal for VA lifespan extension, VPD extended mean lifespan from 16.8 days to 20.4 days (21% increase) (P < 0.0001) and maximum lifespan from 22.7 days to 30.6 days (35% increase) (P < 0.0001) (Fig. 4, Table 1). These effects on lifespan are similar to the effects caused by VA. In contrast to VA, supplementation of NGM plates with VPD did not prevent accumulation of E. coli, indicating that VPD does not inhibit bacterial proliferation. These results suggest that inhibition of bacterial proliferation does not mediate the lifespan extension caused by these drugs.
Figure 4.
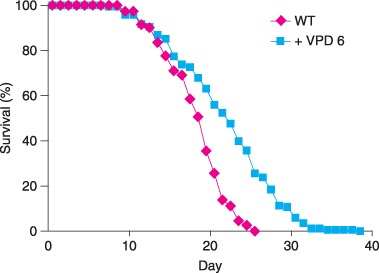
Valpromide (VPD) extends Caenorhabditis elegans lifespan. Wild‐type (WT) hermaphrodites were cultured with ethanol alone (WT, n = 168) or with 6 mm VPD dissolved in ethanol ( + VPD 6, n = 152) from conception until death.
VA and VPD enhance dauer formation, and VA promotes DAF‐16::GFP nuclear localization
Pioneering work by many investigators studying dauer formation in C. elegans has established that there is a significant overlap between genes that mediate entry into the dauer larvae stage of development and genes that influence adult lifespan (Friedman & Johnson, 1988; Kenyon et al., 1993; Larsen et al., 1995; Kimura et al., 1997; Gems et al., 1998). Mutations in genes such as daf‐2, which encodes an insulin‐like receptor, and age‐1, which encodes a PI3 kinase, both promote dauer formation and extend adult lifespan (Morris et al., 1996; Kimura et al., 1997). The lifespan extension caused by these mutations requires the activity of the daf‐16 gene which encodes a forkhead family transcription factor that is regulated by the insulin/IGF signaling pathway (Lin et al., 1997; Ogg et al., 1997). Because VA and VPD extend adult lifespan, we investigated the ability of these drugs to promote dauer formation. Neither VA nor VPD caused wild‐type (WT) animals to form dauer larvae at 20 °C (data not shown). To analyze the ability of these drugs to promote dauer formation in a more sensitive background, we analyzed daf‐7 mutations that cause a partially penetrant dauer constitutive (Daf‐c) phenotype at 20 °C (Table 2). Valproic acid treatment increased dauer formation of daf‐7(m62) animals from 49% of untreated animals to 91% (Table 2A). Similarly, VPD treatment increased dauer formation of daf‐7(m62) animals from 71% on control plates with ethanol alone to 93% on plates with VPD dissolved in ethanol (Table 2B). Thus, VA and VPD enhanced dauer formation of daf‐7(m62) animals. It is notable that these effects of VA and VPD are different from the results observed with ethosuximide and TRI, which caused slight decreases in dauer formation of daf‐7(m62) animals (Table 2A).
Table 2.
Drugs influence dauer formation of daf‐7(m62) at 20 °C
| A | ||
|---|---|---|
| Drug treatment | % Dauer (95% CI) | N (number of trials) |
| None | 49 (44–55) | 332 (3) |
| Ethosuximide (14 mm) | 28 (23–34) | 238 (2) |
| Trimethadione (28 mm) | 21 (15–29) | 150 (2) |
| Valproic acid (6 mm) | 91 (87–94) | 242 (3) |
| B | ||
|---|---|---|
| Drug treatment | % Dauer (95% CI) | N (number of trials) |
| Vehicle | 71 (67–75) | 526 (2) |
| Valpromide (6 mm) + vehicle | 93 (90–95) | 588 (2) |
CI, confidence interval.
The DAF‐16 forkhead transcription factor has been demonstrated to localize to the nucleus when the activity of the daf‐2 signaling pathway is reduced (Henderson & Johnson, 2001; Lin et al., 2001). Therefore, the extent of DAF‐16 nuclear localization has been used to monitor the activity of the signaling pathway. Our studies of dauer formation indicate that VA might influence the insulin‐signaling pathway. To test this hypothesis, we monitored the effect of VA on the nuclear localization of DAF‐16 using a DAF‐16::GFP reporter construct that is integrated in the genome (Berdichevsky et al., 2006). In the absence of VA, less than 1% of animals displayed DAF‐16::GFP nuclear localization (Table 3). Treatment with VA caused 11.5% of animals to display prominent DAF‐16 nuclear localization (Table 3). Because we used stringent criteria for scoring animals as positives in this assay, it is possible that VA promoted lower levels of DAF‐16 nuclear localization in additional animals. These results suggest that VA functions upstream of daf‐16 and promotes nuclear localization.
Table 3.
Valproic acid (VA) enhances DAF‐16 nuclear localization
| Drug treatment | % Nuclear localization (95% CI) | N (number of trials) |
|---|---|---|
| None | 0.44 (0–2) | 459 (4) |
| VA (6 mm) | 11.5 (9–15) | 428 (4) |
CI, confidence interval.
VA functions differently from ethosuximide and TRI, and the lifespan extension of VA and TRI is additive
Ethosuximide and TRI extend lifespan of daf‐16 mutants, indicating that at least part of the activity of these drugs does not require daf‐16 (Evason et al., 2005). To define the relationship between the activity of VA and daf‐16, we tested the effect of a variety of VA doses on the lifespan of daf‐16(m26) and daf‐16(mu86) mutants. Valproic acid did not extend mean lifespan of either of these daf‐16 mutants (Table 4).
Table 4.
Mean and maximum lifespans of mutants treated with valproic acid
| Genotype | Dose (mm) | Mean lifespan (SD) (days)†, ‡ | % Change‡ | Maximum lifespan (SD) (days)†, ‡, § | % Change‡ | N ¶ |
|---|---|---|---|---|---|---|
| Wild type | None | 16.2 ± 4.0 | 23.3 ± 1.8 | 708 (12) | ||
| 3 | 20.9 ± 7.0*** | +29 | 33.3 ± 2.1*** | +43 | 304 (5) | |
| 6 | 21.9 ± 7.0*** | +35 | 33.1 ± 2.1*** | +42 | 530 (10) | |
| daf‐16 (m26) | None | 16.1 ± 3.0 | 20.6 ± 1.0 | 65 (1) | ||
| 0.6 | 14.9 ± 4.3 | –7 | 22.6 ± 2.5 | +10 | 70 (1) | |
| 3 | 16.6 ± 4.6 | +3 | 25.6 ± 2.2** | +24 | 51 (1) | |
| 6 | 13.0 ± 3.1*** | –19 | 18.6 ± 3.2 | –10 | 53 (1) | |
| daf‐16 (mu86) | None | 13.5 ± 3.5 | 20.0 ± 1.8 | 116 (2) | ||
| 0.6 | 12.0 ± 4.0** | –11 | 20.4 ± 3.1 | +2 | 118 (2) | |
| 3 | 11.8 ± 4.1** | –13 | 19.7 ± 1.7 | –2 | 95 (2) | |
| 6 | 12.4 ± 2.9* | –8 | 17.6 ± 1.2** | –12 | 110 (2) | |
| aex‐3 (ad418) | None | 21.6 ± 5.8 | 30.3 ± 1.0 | 43 (1) | ||
| 6 | 21.0 ± 6.2 | –3 | 31.8 ± 2.6 | +5 | 64 (1) | |
| age‐1 (hx546) | None | 23.1 ± 8.0 | 36.9 ± 1.2 | 64 (1) | ||
| 6 | 25.8 ± 14.6 | +12 | 46.7 ± 1.3*** | +28 | 29 (1) | |
| daf‐2 (e1370) | None | 34.7 ± 12.9 | 56.1 ± 3.7 | 78 (2) | ||
| 6 | 36.2 ± 13.2 | +4 | 52.3 ± 1.8* | –7 | 79 (2) | |
| eat‐2 (ad465) | None | 21.9 ± 9.7 | 38.4 ± 2.9 | 51 (1) | ||
| 0.6 | 21.2 ± 8.7 | –3 | 36.8 ± 5.2 | –4 | 28 (1) | |
| 3 | 20.8 ± 10.8 | –5 | 39.0 ± 6.9 | +2 | 34 (1) | |
| 6 | 17.6 ± 8.8* | –20 | 33.8 ± 1.6* | –12 | 31 (1) | |
| isp‐1 (qm150) | None | 17.9 ± 8.9 | 35.8 ± 1.4 | 70 (1) | ||
| 0.6 | 19.7 ± 10.9 | +10 | 42.0 ± 4.0* | +17 | 62 (1) | |
| 3 | 15.2 ± 7.2 | –15 | 28.0 ± 1.3*** | –22 | 31 (1) | |
| 6 | 15.5 ± 5.6 | –12 | 28.1 ± 5.0 | –22 | 41 (1) |
If the standard deviations of two groups were similar, comparisons were performed using the Student's t‐test. Otherwise, the alternate Welch test was used. P > 0.05, no stars;
P < 0.05;
P < 0.005;
P < 0.0001.
The percent change and the statistical significance of this change were determined by comparison to the same genotype with no drug treatment.
Maximum adult lifespan is the mean lifespan of 10% of the population that had the longest lifespans.
Number of hermaphrodites analyzed, with number of independent experiments in parentheses.
Ethosuximide and TRI further extend the lifespan of several long‐lived mutants, including eat‐2(ad465), isp‐1(qm150), daf‐2(e1370), aex‐3(ad418), and age‐1(hx546) (Evason et al., 2005). To define the relationship between VA activity and these mutations, we analyzed the lifespan of these mutants treated with 6 mm VA. Valproic acid did not significantly extend mean lifespan in any of these genetic backgrounds, although in some cases the drug caused a small extension of maximum lifespan (Table 4). There are two possible interpretations for the lack of an additive lifespan extension in long‐lived mutants treated with VA. First, VA and a particular mutation may extend lifespan by similar mechanisms. Second, VA and a particular mutation may extend lifespan by different mechanisms, but also cause toxicities, and the combination of the toxicities limits the potential lifespan extension. Because eat‐2(ad465), isp‐1(qm150), daf‐2(e1370), and aex‐3(ad418) are likely to extend lifespan by different mechanisms, we favor the interpretation that the combination of VA and these mutations results in toxicity. Consistent with this possibility, VA caused a significant decrease in the mean or maximum lifespan of several mutants, including daf‐16, daf‐2, eat‐2, and isp‐1 (Table 4). Therefore, these experiments do not clearly establish the relationship between the mechanism of action of VA and the mechanism of action of eat‐2, isp‐1, aex‐3, age‐1, and daf‐2 mutations. However, these studies do show that VA interacts with longevity mutations in a manner that is strikingly different from the interactions previously observed with ethosuximide and TRI (Evason et al., 2005).
Ethosuximide and TRI affect several neuromuscular behaviors. These drugs increase egg laying, a process mediated by HSN neurons and vulval muscles (Trent et al., 1983; Evason et al., 2005). They also cause hypersensitivity to aldicarb, an acetylcholinesterase inhibitor that causes paralysis because of accumulation of acetylcholine at the neuromuscular junction (Miller et al., 1996; Evason et al., 2005). By contrast, VA did not stimulate egg laying or cause hypersensitivity to aldicarb (data not shown).
Our experiments demonstrate several differences between the effects of VA and TRI, suggesting the two drugs might act differently in nematodes to extend lifespan. One approach to testing this possibility is analyzing combinations of drugs. The interpretation of these experiments requires well‐established dose–responses for each of the drugs being tested. For example, if two drugs extend lifespan by a similar mechanism, then suboptimal doses of the drugs could be combined to produce an additive lifespan extension, but optimal doses should not combine to produce an additive lifespan extension. By contrast, if two drugs work by different mechanisms of action, then combinations of the optimal doses of each drug might cause an additive lifespan extension. In this case, the combination of drugs would produce a longer lifespan extension than can be achieved with either drug individually at any concentration. To investigate this issue, we first analyzed combinations of ethosuximide and TRI, two structurally related compounds that are likely to function by the same mechanism (Levy et al., 2002; Evason et al., 2005). The optimal dosage for lifespan extension is approximately 14–28 mm for ethosuximide and approximately 28 mm for TRI. Combination of 14 mm ethosuximide and 14 mm TRI caused a larger lifespan extension than either drug alone, indicating that these two drugs can display an additive effect when used at suboptimal dosages (Fig. 5A). However, combination of 28 mm ethosuximide and 28 mm TRI did not cause a lifespan extension that was longer than either drug alone and actually shortened the lifespan (Fig. 5B). These results document that two lifespan‐extending drugs that act by a similar mechanism of action cannot be combined to produce a longer lifespan extension than either drug used alone.
Figure 5.
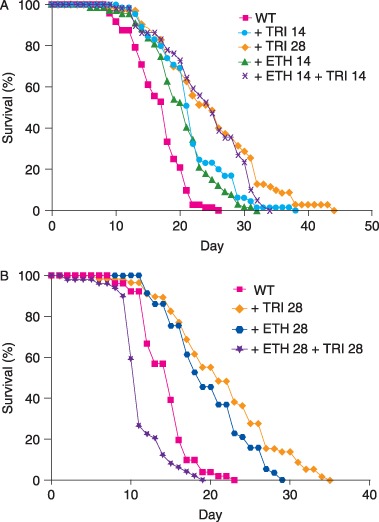
Lifespan‐extending effects of ethosuximide and trimethadione (TRI) can be additive at suboptimal dosages, but are not additive at optimal individual dosages. (A) Wild‐type worms were cultured with only 14 mm TRI ( + TRI 14, mean lifespan 21.1 ± 5.3, n = 65), only 28 mm TRI ( + TRI 28, mean lifespan 24.2 ± 7.8, n = 70), only 14 mm ethosuximide ( + ETH 14, mean lifespan 19.6 ± 5.1, n = 67), or with a combination of 14 mm ethosuximide and 14 mm TRI ( + ETH 14 + TRI 14, mean lifespan 23.3 ± 6.4, n = 59). Treatment with a combination of ethosuximide and TRI caused a greater lifespan extension than treatment with 14 mm ethosuximide alone (P = 0.0005) or 14 mm TRI alone (P < 0.05), but treatment with the combination did not cause a greater lifespan extension than treatment with the optimal dose of 28 mm TRI alone (P = 0.51). (B) Worms cultured with a combination of 28 mm ethosuximide and 28 mm TRI ( + ETH 28 + TRI 28, mean lifespan 11.4 ± 2.9, n = 49) displayed a significantly shorter lifespan than worms cultured with only 28 mm ethosuximide ( + ETH 28, mean lifespan 20.0 ± 5.0, n = 57) (P < 0.0001) or only 28 mm TRI ( + TRI 28, mean lifespan 21.9 ± 6.6, n = 58) (P < 0.0001).
To analyze the interaction of VA with TRI, we combined the optimal lifespan‐extending dose of 6 mm VA with the optimal lifespan‐extending dose of 28 mm TRI. Worms cultured with 6 mm VA had a mean lifespan of 20.9 ± 7.1 days (N = 229), and worms cultured with 28 mm TRI had a mean lifespan of 23.5 ± 7.5 days (N = 248). Worms cultured with a combination of 28 mm TRI and 6 mm VA had a mean lifespan of 26.1 ± 7.8 days (N = 222), a 61% increase compared to control worms cultured without drugs (Fig. 6, Table 1). Importantly, this mean lifespan was significantly greater than the mean lifespan of worms treated with 28 mm TRI (P < 0.0005) or with 6 mm VA (P < 0.0001).
Figure 6.
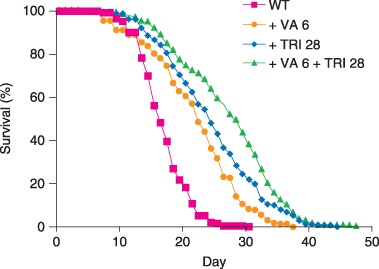
Lifespan‐extending effects of valproic acid (VA) and trimethadione (TRI) are additive. Worms were cultured with only VA ( + VA 6), only TRI ( + TRI 28), or a combination of VA and TRI ( + VA 6 + TRI 28).
Discussion
Specific models for the mechanism of action of VA in lifespan extension
Our results demonstrate that VA extends C. elegans lifespan and delays age‐related diminishment of vigorous body movement. In humans VA has multiple effects, including inhibiting convulsions, reducing migraine headaches, mood stabilization, and teratogenic effects during pregnancy. Critical questions that are raised by these findings include: What is the mechanism of action of VA in extending lifespan? How does this mechanism relate to the activities in humans? The mechanism of action is likely to involve direct binding of VA to one or more targets, which triggers a series of events that result in lifespan extension in worms or neural effects in humans. Because VA was discovered serendipitously to affect seizure activity in vertebrates (Isoherranen et al., 2003), a major challenge has been defining the molecular mechanism of action of this drug. The investigations of candidate processes and proteins have implicated several targets, including effects on γ‐amino butyric acid levels, sodium channels, HDACs, and the endoplasmic reticulum stress response pathway (Wang et al., 1999; Phiel et al., 2001; Levy et al., 2002). Valproic acid might have multiple therapeutic targets in humans, which would help explain its diverse clinical effects (Levy et al., 2002).
Structure–activity studies have been an effective approach used to determine how the molecular actions of VA relate to the physiological effects of the drug. Elegant studies have been performed with the structurally related anticonvulsant VPD (Phiel et al., 2001). Valproic acid is a potent HDAC inhibitor; by contrast, VPD does not inhibit HDACs. Therefore, activities that are displayed by both drugs are not likely to be caused by inhibiting HDACs, and activities displayed by VA but not VPD may be caused by inhibiting HDACs. Valproic acid is a teratogen, whereas VPD is not a terotogen, indicating that HDAC inhibition causes teratogenicity (Phiel et al., 2001). By contrast, VPD and VA are both anticonvulsants, indicating that HDAC inhibition does not cause anticonvulsant activity.
Histone deacetylase activity has been implicated in the control of lifespan, because drugs that inhibit HDAC activity and mutations that reduce HDAC activity can extend the lifespan of Drosophila (Kang et al., 2002; Rogina et al., 2002; Zhao et al., 2005). To address the role of HDAC inhibition in the mechanism of action of VA in C. elegans, we conducted a structure–activity study. We observed that VPD extended C. elegans lifespan similar to VA, indicating that lifespan extension is not caused by inhibiting HDAC activity, but rather is caused by an activity that is shared by VA and VPD.
A number of factors have been demonstrated to extend C. elegans lifespan, and the experiments described here have clarified the relationship between VA and these factors. Caloric restriction can extend the lifespan of C. elegans and many other animals (Klass, 1977; Koubova & Guarente, 2003; Partridge et al., 2005). Characteristics of calorically restricted worms include significant reductions in progeny production and reduced body size (Klass, 1977). Valproic acid did not significantly reduce progeny production or body size, suggesting that it does not function primarily by causing caloric restriction.
Bacterial pathogenicity affects C. elegans lifespan (Gems & Riddle, 2000; Garigan et al., 2002; Garsin et al., 2003). The observation that VA prevents bacterial accumulation raises the possibility that one mechanism of VA lifespan extension is by inhibiting bacterial pathogenicity. However, the following observations indicate that VA acts directly on C. elegans to influence lifespan. First, the structurally related compound VPD extends lifespan and does not affect bacterial proliferation substantially. Second, VA extended lifespan of worms cultured with an antibiotic that inhibits bacterial proliferation, indicating that VA acts, at least in part, by a separate mechanism. Third, VA treatment caused lethality, promoted dauer formation, and promoted DAF‐16 nuclear localization, and these phenotypes are unlikely to be related to bacterial proliferation.
The well‐characterized insulin/IGF signaling pathway affects DAF‐16 nuclear localization, the probability of forming dauer larvae, and the adult lifespan (Friedman & Johnson, 1988; Kenyon et al., 1993; Larsen et al., 1995; Kimura et al., 1997; Gems et al., 1998). Valproic acid treatment promoted DAF‐16 nuclear localization, increased the formation of dauer larvae, and extended the adult lifespan. Because these phenotypes are caused by mutations that partially reduce the activity of daf‐2 and age‐1, these results suggest that VA might reduce the activity of the insulin/IGF‐1 signaling pathway. However, a significant number of genes can be mutated to promote dauer larvae formation, including genes that are not directly involved in the insulin/IGF‐1 signaling pathway (Riddle et al., 1997). Furthermore, pathways in addition to the insulin/IGF‐1‐signaling pathway have been demonstrated to influence DAF‐16 nuclear localization (Oh et al., 2005). Thus, our results are consistent with the model that VA influences the insulin‐signaling pathway, but also consistent with other models. We observed that VA did not extend the lifespan of daf‐16(lf) mutants and did not further extend the lifespan of daf‐2(lf) and age‐1(lf) mutants. These results are consistent with a model that VA acts on the insulin/IGF‐1 signaling pathway to extend lifespan. However, the lack of an effect of VA in these mutants is a negative result that should be interpreted cautiously. Furthermore, this finding was not specific to mutants in the insulin/IGF‐1 signaling pathway, because VA did not further extend the lifespan of long‐lived mutants that are defective in other pathways, such as eat‐2 and isp‐1.
Our results demonstrate that VA promotes DAF‐16 nuclear localization, and here we consider three models to explain this finding. The first model is that VA directly binds DAF‐16 and thereby influences DAF‐16 nuclear localization. The second model is that VA directly binds one of the proteins that functions upstream of DAF‐16 in a linear signaling pathway, such as DAF‐2 or AGE‐1. The third model is that VA affects DAF‐16 nuclear localization very indirectly by acting on proteins that function far upstream of the insulin/IGF‐1 signaling pathway or on proteins that act in parallel to the insulin/IGF‐1 signaling pathway to regulate DAF‐16. In one version of model 3, VA binding to a target protein induces a stress response in the worm that promotes DAF‐16 nuclear localization. Further experiments are necessary to distinguish between these three models. Valproic acid treatment caused DAF‐16 nuclear localization and lifespan extension, and this correlation suggests that DAF‐16 nuclear localization might cause the lifespan extension. Consistent with this model, VA did not extend the lifespan of daf‐16(lf) mutants. However, it is also possible that the effect of VA on lifespan is independent of DAF‐16 nuclear localization, and the finding that VA did not extend the lifespan of daf‐16(lf) mutants reflects the toxicity of combining the mutation and the drug treatment.
General models for the mechanism of action of VA in lifespan extension
We have discussed specific mechanisms of action for VA in lifespan extension including HDAC inhibition, caloric restriction, effects on bacterial pathogenicity, and effects on the DAF‐2/AGE‐1/DAF‐16 signaling pathway. Next, we discuss general models for the mechanism of action of VA in lifespan extension. These general models are important to consider because they apply to a wide variety of genetic, environmental, and pharmacological treatments that can extend lifespan.
If a treatment extends adult lifespan, then by definition the treatment increases the function of animals at the end of adult life; treated animals that are alive have a higher level of function than untreated animals that are dead. A critical issue is how the treatment affects the function of the animals during the beginning and middle of adult life compared to untreated animals to achieve the lifespan extension. Here, we consider three models. The first model is that the treatment directly increases the function of a process that promotes longevity. The second model is that the treatment directly acts on a system that does not control longevity, thereby inducing a response that increases longevity by increasing the function of processes that promote longevity. The third model is that the treatment directly or indirectly decreases the function of a process that inhibits longevity. We consider each model in turn, and relate these models to our results with VA.
The first model postulates that there are processes that promote longevity, and the activity of these processes can be increased to extend lifespan. For example, if aging is caused by damage accumulation, then increasing the activity of processes that limit damage accumulation is predicted to extend longevity. Specific processes that have been proposed to promote longevity and have the capacity to be increased and extend lifespan include pathways that increase resistance to oxidative stress and increase repair of macromolecules like DNA (Balaban et al., 2005; Lombard et al., 2005). Although these models are conceptually appealing, there is limited experimental support for the proposal that increasing the function of one or a small number of damage repair systems can extend lifespan (Melov et al., 2000; Phillips et al., 2000; Keaney & Gems, 2003; Keaney et al., 2004; Landis & Tower, 2005). In cases where ectopic or over‐expression of a gene causes a lifespan extension, it is possible that the lifespan extension results from the deleterious consequences of misexpression rather than a positive effect of increased gene activity. Our results with VA are not suggestive of model 1. In particular, VA displayed dose‐dependent toxicity. If the mechanism of toxicity is related to the mechanism of lifespan extension, then it is unlikely that VA directly increases the activity of a process that promotes longevity, because these processes are not predicted to cause toxicity.
The second model postulates that lifespan‐extending treatments act directly on systems that do not control longevity, thereby inducing a complex response that increases longevity. This complex response may involve changes in gene expression and metabolism, and includes increasing processes that promote longevity. This logic is the basis for the hormesis hypothesis that proposes that low‐level stresses early in life induce a complex stress response that causes a longer lifespan primarily by enhancing damage repair systems (Rattan, 2007). In particular, heat stress early in life causes a lifespan extension and induces gene expression changes described as the stress response (Verbeke et al., 2001). This has been observed in multiple animals including C. elegans (Cypser & Johnson, 2002; Olsen et al., 2006b). The response is characterized by the induction of heat shock proteins that function as molecular chaperones. The finding that lifespan extension and induction of the stress response are correlated changes caused by heat stress suggests two possible models. The first model, termed hormesis, is it that the heat stress causes a stress response, and the stress response is characterized by increased activity of processes that promote longevity, thereby causing a lifespan extension. The second model is that the heat stress causes the lifespan extension by damaging and thereby decreasing the activity of processes that inhibit longevity, and the stress response is not the cause of the lifespan extension. Although the first model is frequently discussed and the second model is rarely discussed, both models should be considered seriously because definitive experimental evidence that distinguishes between the two models is lacking. The effect of the stress in the absence of a response has not been analyzed, nor has the effect of the response in the absence of the stress. Because low levels of VA cause a lifespan extension and high levels cause lethality, VA treatment conforms to definitions proposed for hormesis. Thus, it is possible that VA toxicity induces a stress response that increases the activity of processes that promote longevity, and this stress response causes the lifespan extension.
The third model postulates that there are processes that promote a short lifespan, and inhibiting these processes can result in a lifespan extension. What processes promote a short lifespan, and why do animals have such processes? Here, we consider three categories of processes that might promote a short lifespan. The first category includes processes that generate deleterious waste products, such as lipofuscin, or products that are capable of causing damage, such as reactive oxygen species. According to the model that these products cause aging, reducing the activity of processes that generate these products is predicted to extend lifespan. For example, mutations that decrease mitochondrial function can extend C. elegans lifespan (Felkai et al., 1999). Because mitochondria are the site of oxygen metabolism and generate reactive oxygen species, one interpretation of these findings is that decreasing mitochondrial function results in decreased reactive oxygen species production, decreased oxidative damage, and an extended lifespan. The second category of processes is based on the model that there is a trade‐off between reproduction and somatic maintenance (Kirkwood & Rose, 1991). According to this model, processes that promote reproduction divert resources from somatic maintenance and thereby shorten lifespan. Therefore, reducing such processes is predicted to extend lifespan. This explanation is unlikely to apply to VA, because drug treatment did not reduce reproduction. We propose that there is a third category of processes that can be inhibited to result in a lifespan extension, based on the model that life is based on a limited set of instructions. The instructions for the animal to develop, grow, and mature are encoded in the DNA. Because animals have a finite amount of DNA, then the instructions are necessarily finite. When an animal has fully executed the instructions to form a mature adult, and instructions for further maintenance do not exist, then age‐related degenerative changes will begin to accumulate. It is important to note that the finite instructions encoded in the DNA are adequate to encode an indefinite life cycle that proceeds from egg to adult to egg. At the same time, these instructions do not encode for an immortal adult that can indefinitely meet the new challenges of living an additional period of time. According to the model that the life program encoded by the DNA is limited, then the lifespan will be shortened by processes that accelerate the execution of the life program. Inhibiting these processes is predicted to result in a lifespan extension. There are likely to be a broad spectrum of gene products and processes that accelerate the execution of the life program, and thereby promote a rapid lifespan. For example, genes that are involved in acquiring nutrients and energy from the environment and converting that energy to ATP are expected to promote the execution of the life program. Because mitochondria generate ATP, the observation that mutations that decrease mitochondrial function can extend lifespan could indicate that a decreased rate of ATP synthesis delays the execution of the life program, and thereby extends lifespan. Furthermore, the life program requires the execution of gene expression programs, and inhibiting gene expression might extend lifespan. Consistent with this possibility, modifying the activity of specific HDACs that regulate gene expression causes lifespan extensions (Kang et al., 2002; Rogina et al., 2002; Zhao et al., 2005). Our results with VA are consistent with the possibility that VA inhibits a process that promotes the execution of the life program. The process inhibited by VA may be essential, which would explain the lethality caused by high doses of VA.
To summarize, there are three general models for lifespan extension by VA. First, VA might directly increase a process that promotes longevity. Second, VA might act by a hormesis mechanism and indirectly increase a process that promotes longevity. Third, VA might directly and/or indirectly decrease a process that inhibits longevity. We think the first model is unlikely, because VA caused dose‐dependent toxicity. Our results are consistent with the second and third models, but we favor model 3. Genetic analyses in C. elegans have identified hundreds of genes that can be modified by loss‐of‐function mutations or RNAi treatment to cause a lifespan extension, and many of these genes are involved in basic metabolic processes such as mitochondrial function and protein translation (Lee et al., 2003; Hamilton et al., 2005; Hansen et al., 2005, 2007; Curran & Ruvkun, 2007; Pan et al., 2007; Syntichaki et al., 2007). We think that the simplest interpretation of these findings is that lifespan extension is typically caused by decreasing processes that inhibit longevity. The alternative explanation is that these diverse genetic changes induce a stress response, which seems unlikely. We think that most genetic changes and pharmacological treatments that extend lifespan, including VA, decrease the function of processes that inhibit longevity and increase the time required to execute gene expression and metabolic programs that constitute the life program.
Additive lifespan extension can be achieved by combining drugs from different structural families – TRI and VA
We previously described that the heterocyclic anticonvulsants ethosuximide and TRI extend C. elegans lifespan and are likely to function by affecting neural activity (Evason et al., 2005). Valproic acid also affects neural activity in vertebrates and is used as an anticonvulsant, raising the possibility that VA and heterocyclic ring anticonvulsants have a similar mechanism of action in extending C. elegans lifespan. However, our results indicate that VA and heterocyclic ring anticonvulsants act very differently in C. elegans and are likely to have different mechanisms of lifespan extension. Valproic acid promotes dauer formation in daf‐7(m62) mutants, whereas ethosuximide and TRI do not cause this effect. Ethosuximide and TRI affect egg laying and hypersensitivity to aldicarb‐mediated paralysis, whereas VA did not cause these effects. Ethosuximide and TRI caused a lifespan extension in a variety of mutant backgrounds, whereas VA did not extend lifespan in those genetic backgrounds. These differences suggest that VA and heterocyclic ring anticonvulsants extend lifespan by distinct mechanisms, and therefore it might be possible to combine the drugs to obtain an additive lifespan extension. Consistent with this prediction, the combination of VA and TRI extended the mean lifespan by 61%, which is significantly greater than the lifespan extension caused by either compound alone. This is an important result because it demonstrates the feasibility of achieving additive lifespan extensions with a combination of pharmacologic agents.
Combining drugs is common in clinical practice. Anticonvulsants are often combined to achieve greater seizure control. Interestingly, ethosuximide and VA can be combined to achieve greater seizure control than either drug alone, and this combination is currently used in clinical practice (Rowan et al., 1983; Bourgeois, 1988). This result is consistent with our data suggesting that these two drugs act differently and form an effective combination. The possibility of combining drugs to extend lifespan is a promising area, and future studies may reveal many different combinations of drugs that have additive effects and can profoundly delay aging.
Experimental procedures
General methods and strains
Caenorhabditis elegans strains were cultured at 20 °C on 6 cm petri dishes containing NGM agar and a lawn of E. coli strain OP50 unless stated otherwise (Brenner, 1974). The WT N2 strain and most mutant strains were obtained from the Caenorhabditis Genetics Center. Strain GR1352 was used for DAF‐16::GFP nuclear localization experiments and contains the integrated array xrIs87[daf‐16alpha::GFP::DAF‐16B + rol‐6(su1006)] that rescues the daf‐16(mgDf47) mutant phenotype (Lee et al., 2001; Berdichevsky et al., 2006). The following well‐characterized mutations that affect lifespan and/or dauer formation were used: daf‐2(e1370 P1465S) is a partial loss‐of‐function mutation that affects the kinase domain of the DAF‐2 receptor tyrosine kinase (Kimura et al., 1997); age‐1 (hx546 P2416S) is a partial loss‐of‐function mutation that affects the AGE‐1 PI3 kinase (Morris et al., 1996; Ayyadevara et al., 2007); eat‐2(ad465) is a strong loss‐of‐function or null mutation that would result in the production of only the first 45 amino acids of the EAT‐2 non‐alpha nicotinic receptor subunit (McKay et al., 2004); daf‐16(m26) is a partial loss‐of‐function mutation that disrupts mRNA splicing of the DAF‐16 forkhead transcription factor (Ogg et al., 1997); daf‐16(mu86) is a probable null mutation that results in deletion of most of the DAF‐16 coding sequence including all of the forkhead domain (Lin et al., 1997); isp‐1(qm150 P225S) is a partial loss‐of‐function mutation that affects the ISP‐1 iron sulfur protein of mitochondrial complex III (Feng et al., 2001); and daf‐7(m62 Q246Stop) is a probable null mutation of the TGF‐β ligand DAF‐7 (Ren et al., 1996). daf‐7(m62) mutants display a temperature‐sensitive dauer constitutive (Daf‐c) phenotype and were propagated at 15 °C.
Measurement of lifespan and age‐related changes in physiological processes
For measurements of lifespan, hermaphrodites were chosen for analysis at the L4 stage (defined as day 0) and analyzed every 1–2 days from day 3 until death. Approximately 15 hermaphrodites were cultured on each petri dish. Hermaphrodites were transferred to fresh petri dishes about every 2 days until the cessation of progeny production and about every week thereafter. Animals were scored as dead if they displayed no spontaneous movement or response when prodded. Dead worms that displayed internally hatched progeny, an extruded gonad, or desiccation caused by crawling off the agar were excluded from the data. Lifespan is the number of days from the L4 stage to the average of the last day a worm was observed to be alive and the first day a worm was observed to be dead.
Self‐fertile reproductive span, fast pharyngeal pumping span, pharyngeal pumping span, and fast body movement span were determined as described previously (Huang et al., 2004). Reproductive aging of mated hermaphrodites was analyzed as described previously (Hughes et al., 2007). To determine total progeny production, L4 hermaphrodites were placed on individual petri dishes (time zero) and transferred to fresh dishes every day until death or at least 4 days without progeny production. Progeny were counted after about 2 days. To allow mating, three young, WT male C. elegans that had not been exposed to drug were added to the dish and removed after 2 days. Hermaphrodites that did not mate to the male C. elegans were recognized by a lack of male progeny and excluded from the data. Sterile hermaphrodites were also excluded from the data. Fertile animals that died during the experiment were included in the data until the day of death, and N values are the number of animals at the start of the experiments.
Dauer formation and DAF‐16::GFP nuclear localization
Dauer formation assays were performed as described previously (Gunther et al., 2000). Briefly, five to ten young adult hermaphrodites were allowed to lay eggs on one petri dish at 20 °C for 4–10 h. An observer that was blind to the drug treatment status counted the number of eggs that had formed adults or dauer larvae after 4 days and 5 days.
For DAF‐16 nuclear localization experiments, animals were cultured from conception to the L3 larval stage in the presence or absence of 6 mm VA, and representative L3 animals were scored for DAF‐16::GFP localization using an Olympus SZX12 dissecting microscope (Tokyo, Japan) equipped for fluorescence microscopy at 144× magnification. Animals were scored as positive if they displayed multiple, intensely fluorescing puncta across the length of the animal. Animals were scored as negative if there was diffuse fluorescence and few or no intensely fluorescing puncta.
Pharmacological compounds
Valproic acid sodium salt, ethosuximide, TRI, and kanamycin sulfate were obtained from Sigma Chemical Co. (St. Louis, MO, USA). Concentrated solutions of each drug were prepared by dissolving compounds in water at 50–100 mg mL−1. Valpromide was obtained as a gift from Katwijk chemie bv (Katwijk, ZH, The Netherlands). A concentrated solution of VPD was prepared by dissolving VPD in ethanol at 250 mg mL−1. Solutions of concentrated compounds were added to liquid NGM that had been autoclaved and cooled to 50–60 °C, the mixture was agitated to ensure the compounds were dissolved, and then the medium was dispensed into petri dishes. For experiments with VPD, we prepared control petri dishes containing 3.4 mL ethanol L−1 NGM. Petri dishes were stored in the dark at 4 °C until 1–2 days before use and then moved to room temperature and seeded with E. coli OP50. Escherichia coli did not form a thick lawn on NGM containing VA, indicating that VA might inhibit bacterial proliferation. By contrast, E. coli did form a thick lawn on NGM containing VPD. To provide an abundant source of E. coli food to worms cultured on plates containing VA, we cultured bacteria in a medium not containing VA, and aliquoted the concentrated bacteria onto dishes containing VA. By contrast, experiments with VPD were conducted using the standard approach of plating a thin lawn of E. coli OP50 on the petri dish and allowing the bacteria to proliferate 1–2 days at room temperature. Experiments with kanamycin were conducted by aliquoting concentrated bacteria onto dishes.
For a typical lifespan experiment, parental worms were cultured in the presence of the drug, and progeny were selected at the L4 stage for lifespan analysis. Thus, these progenies were exposed to drug from the time of conception until death. Lifespan experiments involving pharmacological compounds were always done in parallel with a control group.
Statistical methods
For each experimental group, comparisons were made to a control group maintained in the same incubator and analyzed at the same time points. Mean, standard deviation, P values, 95% confidence intervals, and other statistical parameters were calculated using InStat 2.03 software. If the standard deviations of two groups were similar, comparisons were performed using the Student's t‐test. Otherwise, the alternate Welch test was used.
Acknowledgments
We thank Amanda Fisher for assistance with the graphics. Some strains were provided by the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health (NIH) National Center for Research Resources. Support was provided by the Washington University Alzheimer's Disease Research Center (Grant No. P50 AG05681), the National Science Foundation (NSF) (IOB0446240), the NIH (R01AG02656101A1), and the Longer Life Foundation, an RGA/Washington University Partnership (Grant No. 1999‐001). Kerry Kornfeld was a scholar of the Leukemia and Lymphoma Society, and is a senior scholar of the Ellison Medical Foundation. Cheng Huang and Stacie Hughes were supported by predoctoral fellowships from the Howard Hughes Medical Institute and the NSF, respectively.
References
- Adachi H, Ishii N (2000) Effects of tocotrienols on life span and protein carbonylation in Caenorhabditis elegans . J Gerontol A Biol Sci Med Sci. 55, B280–285. [DOI] [PubMed] [Google Scholar]
- Ayyadevara S, Alla R, Thaden JJ, Shmookler Reis RJ (2008) Remarkable longevity and stress resistance of nematode PI3K‐null mutants. Aging Cell. 7, 13–22. [DOI] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T (2005) Mitochondria, oxidants, and aging. Cell. 120, 483–495. [DOI] [PubMed] [Google Scholar]
- Bass TM, Weinkove D, Houthoofd K, Gems D, Partridge L (2007) Effects of resveratrol on lifespan in Drosophila melanogaster and Caenorhabditis elegans . Mech Ageing Dev. 128, 546–552. [DOI] [PubMed] [Google Scholar]
- Berdichevsky A, Viswanathan M, Horvitz HR, Guarente L (2006) C. elegans SIR‐2.1 interacts with 14‐3‐3 proteins to activate DAF‐16 and extend life span. Cell. 125, 1165–1177. [DOI] [PubMed] [Google Scholar]
- Bialer M (1991) Clinical pharmacology of valpromide. Clin Pharmacokinet. 20, 114–122. [DOI] [PubMed] [Google Scholar]
- Bourgeois BF (1988) Combination of valproate and ethosuximide: antiepileptic and neurotoxic interaction. J Pharmacol Exp Ther. 247, 1128–1132. [PubMed] [Google Scholar]
- Brenner S (1974) The genetics of Caenorhabditis elegans . Genetics. 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins JJ, Evason K, Kornfeld K (2006) Pharmacology of delayed aging and extended lifespan of Caenorhabditis elegans . Exp Gerontol. 41, 1032–1039. [DOI] [PubMed] [Google Scholar]
- Collins JJ, Huang C, Hughes S, Kornfeld K (2007) The measurement and analysis of age‐related changes in Caenorhabditis elegans (January 24, 2008), WormBook, ed. The C. elegans Research Community, WormBook, doi/DOI: 10.1895/wormbook.1.137.1,, http://www.wormbook.org. [DOI] [PMC free article] [PubMed]
- Curran SP, Ruvkun G (2007) Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 3, e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cypser JR, Johnson TE (2002) Multiple stressors in Caenorhabditis elegans induce stress hormesis and extended longevity. J Gerontol A Biol Sci Med Sci. 57, B109–114. [DOI] [PubMed] [Google Scholar]
- Evason K, Huang C, Yamben I, Covey DF, Kornfeld K (2005) Anticonvulsant medications extend worm life‐span. Science. 307, 258–262. [DOI] [PubMed] [Google Scholar]
- Felkai S, Ewbank JJ, Lemieux J, Labbe JC, Brown GG, Hekimi S (1999) CLK‐1 controls respiration, behavior and aging in the nematode Caenorhabditis elegans . Embo J. 18, 1783–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Bussiere F, Hekimi S (2001) Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans . Dev Cell. 1, 633–644. [DOI] [PubMed] [Google Scholar]
- Friedman DB, Johnson TE (1988) A mutation in the age‐1 gene in Caenorhabditis elegans lengthens life and reduces hermaphrodite fertility. Genetics. 118, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garigan D, Hsu AL, Fraser AG, Kamath RS, Ahringer J, Kenyon C (2002) Genetic analysis of tissue aging in Caenorhabditis elegans: a role for heat‐shock factor and bacterial proliferation. Genetics. 161, 1101–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garsin DA, Villanueva JM, Begun J, Kim DH, Sifri CD, Calderwood SB, Ruvkun G, Ausubel FM (2003) Long‐lived C. elegans daf‐2 mutants are resistant to bacterial pathogens. Science. 300, 1921. [DOI] [PubMed] [Google Scholar]
- Gems D, Riddle DL (2000) Genetic, behavioral and environmental determinants of male longevity in Caenorhabditis elegans . Genetics. 154, 1597–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gems D, Sutton AJ, Sundermeyer ML, Albert PS, King KV, Edgley ML, Larsen PL, Riddle DL (1998) Two pleiotropic classes of daf‐2 mutation affect larval arrest, adult behavior, reproduction and longevity in Caenorhabditis elegans . Genetics. 150, 129–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther CV, Georgi LL, Riddle DL (2000) A Caenorhabditis elegans type I TGF beta receptor can function in the absence of type II kinase to promote larval development. Development. 127, 3337–3347. [DOI] [PubMed] [Google Scholar]
- Hamilton B, Dong Y, Shindo M, Liu W, Odell I, Ruvkun G, Lee SS (2005) A systematic RNAi screen for longevity genes in C. elegans . Genes Dev. 19, 1544–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M, Hsu AL, Dillin A, Kenyon C (2005) New genes tied to endocrine, metabolic, and dietary regulation of lifespan from a Caenorhabditis elegans genomic RNAi screen. PLoS Genet. 1, 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M, Taubert S, Crawford D, Libina N, Lee SJ, Kenyon C (2007) Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans . Aging Cell. 6, 95–110. [DOI] [PubMed] [Google Scholar]
- Harrington LA, Harley CB (1988) Effect of vitamin E on lifespan and reproduction in Caenorhabditis elegans . Mech Ageing Dev. 43, 71–78. [DOI] [PubMed] [Google Scholar]
- Henderson ST, Johnson TE (2001) daf‐16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans . Curr Biol. 11, 1975–1980. [DOI] [PubMed] [Google Scholar]
- Herndon LA, Schmeissner PJ, Dudaronek JM, Brown PA, Listner KM, Sakano Y, Paupard MC, Hall DH, Driscoll M (2002) Stochastic and genetic factors influence tissue‐specific decline in ageing C. elegans . Nature. 419, 808–814. [DOI] [PubMed] [Google Scholar]
- Huang C, Xiong C, Kornfeld K (2004) Measurements of age‐related changes of physiological processes that predict lifespan of Caenorhabditis elegans . Proc Natl Acad Sci U S A. 101, 8084–8089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes SE, Evason K, Xiong C, Kornfeld K (2007) Genetic and Pharmacological Factors That Influence Reproductive Aging in Nematodes. PLoS Genet. 3, e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii N, Senoo‐Matsuda N, Miyake K, Yasuda K, Ishii T, Hartman PS, Furukawa S (2004) Coenzyme Q10 can prolong C. elegans lifespan by lowering oxidative stress. Mech Ageing Dev. 125, 41–46. [DOI] [PubMed] [Google Scholar]
- Isoherranen N, Yagen B, Bialer M (2003) New CNS‐active drugs which are second‐generation valproic acid: can they lead to the development of a magic bullet? Curr Opin Neurol. 16, 203–211. [DOI] [PubMed] [Google Scholar]
- Kang HL, Benzer S, Min KT (2002) Life extension in Drosophila by feeding a drug. Proc Natl Acad Sci U S A. 99, 838–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keaney M, Gems D (2003) No increase in lifespan in Caenorhabditis elegans upon treatment with the superoxide dismutase mimetic EUK‐8. Free Radic Biol Med. 34, 277–282. [DOI] [PubMed] [Google Scholar]
- Keaney M, Matthijssens F, Sharpe M, Vanfleteren J, Gems D (2004) Superoxide dismutase mimetics elevate superoxide dismutase activity in vivo but do not retard aging in the nematode Caenorhabditis elegans. Free Radic Biol Med. 37, 239–250. [DOI] [PubMed] [Google Scholar]
- Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R (1993) A C. elegans mutant that lives twice as long as wild type. Nature. 366, 461–464. [DOI] [PubMed] [Google Scholar]
- Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G (1997) daf‐2, an insulin receptor‐like gene that regulates longevity and diapause in Caenorhabditis elegans . Science. 277, 942–946. [DOI] [PubMed] [Google Scholar]
- Kirkwood TB, Rose MR (1991) Evolution of senescence: late survival sacrificed for reproduction. Philos Trans R Soc Lond B Biol Sci. 332, 15–24. [DOI] [PubMed] [Google Scholar]
- Klass MR (1977) Aging in the nematode Caenorhabditis elegans: major biological and environmental factors influencing life span. Mech Ageing Dev. 6, 413–429. [DOI] [PubMed] [Google Scholar]
- Koubova J, Guarente L (2003) How does calorie restriction work? Genes Dev. 17, 313–321. [DOI] [PubMed] [Google Scholar]
- Landis GN, Tower J (2005) Superoxide dismutase evolution and life span regulation. Mech Ageing Dev. 126, 365–379. [DOI] [PubMed] [Google Scholar]
- Larsen PL, Albert PS, Riddle DL (1995) Genes that regulate both development and longevity in Caenorhabditis elegans . Genetics. 139, 1567–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RY, Hench J, Ruvkun G (2001) Regulation of C. elegans DAF‐16 and its human ortholog FKHRL1 by the daf‐2 insulin‐like signaling pathway. Curr Biol. 11, 1950–1957. [DOI] [PubMed] [Google Scholar]
- Lee SS, Lee RY, Fraser AG, Kamath RS, Ahringer J, Ruvkun G (2003) A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet. 33, 40–48. [DOI] [PubMed] [Google Scholar]
- Levy RH, Mattson RH, Meldrum BS, Perucca E (2002) Antiepileptic drugs. Philadelphia: Lippincott, Williams and Wilkins. [Google Scholar]
- Lin K, Dorman JB, Rodan A, Kenyon C (1997) daf‐16: An HNF‐3/forkhead family member that can function to double the life‐span of Caenorhabditis elegans . Science. 278, 1319–1322. [DOI] [PubMed] [Google Scholar]
- Lin K, Hsin H, Libina N, Kenyon C (2001) Regulation of the Caenorhabditis elegans longevity protein DAF‐16 by insulin/IGF‐1 and germline signaling. Nat Genet. 28, 139–145. [DOI] [PubMed] [Google Scholar]
- Lithgow GJ, Gill MS, Olsen A, Sampayo JN (2005) Pharmacological intervention in invertebrate aging. AGE. 27, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW (2005) DNA repair, genome stability, and aging. Cell. 120, 497–512. [DOI] [PubMed] [Google Scholar]
- McColl G, Killilea DW, Hubbard AE, Vantipalli MC, Melov S, Lithgow GJ (2008) Pharmacogenetic Analysis of Lithium‐induced Delayed Aging in Caenorhabditis elegans . J Biol Chem. 283, 350–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay JP, Raizen DM, Gottschalk A, Schafer WR, Avery L (2004) eat‐2 and eat‐18 are required for nicotinic neurotransmission in the Caenorhabditis elegans pharynx. Genetics. 166, 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melov S, Ravenscroft J, Malik S, Gill MS, Walker DW, Clayton PE, Wallace DC, Malfroy B, Doctrow SR, Lithgow GJ (2000) Extension of life‐span with superoxide dismutase/catalase mimetics. Science. 289, 1567–1569. [DOI] [PubMed] [Google Scholar]
- Miller KG, Alfonso A, Nguyen M, Crowell JA, Johnson CD, Rand JB (1996) A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc Natl Acad Sci U S A. 93, 12593–12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JZ, Tissenbaum HA, Ruvkun G (1996) A phosphatidylinositol‐3‐OH kinase family member regulating longevity and diapause in Caenorhabditis elegans . Nature. 382, 536–539. [DOI] [PubMed] [Google Scholar]
- Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G (1997) The Fork head transcription factor DAF‐16 transduces insulin‐like metabolic and longevity signals in C. elegans . Nature. 389, 994–999. [DOI] [PubMed] [Google Scholar]
- Oh SW, Mukhopadhyay A, Svrzikapa N, Jiang F, Davis RJ, Tissenbaum HA (2005) JNK regulates lifespan in Caenorhabditis elegans by modulating nuclear translocation of forkhead transcription factor/DAF‐16. Proc Natl Acad Sci U S A. 102, 4494–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen A, Vantipalli MC, Lithgow GJ (2006a) Lifespan extension of Caenorhabditis elegans following repeated mild hormetic heat treatments. Biogerontology. 7, 221–230. [DOI] [PubMed] [Google Scholar]
- Olsen A, Vantipalli MC, Lithgow GJ (2006b) Using Caenorhabditis elegans as a model for aging and age‐related diseases. Ann N Y Acad Sci. 1067, 120–128. [DOI] [PubMed] [Google Scholar]
- Pan KZ, Palter JE, Rogers AN, Olsen A, Chen D, Lithgow GJ, Kapahi P (2007) Inhibition of mRNA translation extends lifespan in Caenorhabditis elegans . Aging Cell. 6, 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge L, Piper MD, Mair W (2005) Dietary restriction in Drosophila. Mech Ageing Dev. 126, 938–950. [DOI] [PubMed] [Google Scholar]
- Petrascheck M, Ye X, Buck LB (2007) An antidepressant that extends lifespan in adult Caenorhabditis elegans . Nature. 450, 553–556. [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS (2001) Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 276, 36734–36741. [DOI] [PubMed] [Google Scholar]
- Phillips JP, Parkes TL, Hilliker AJ (2000) Targeted neuronal gene expression and longevity in Drosophila. Exp Gerontol. 35, 1157–1164. [DOI] [PubMed] [Google Scholar]
- Rattan SI (2008) Hormesis in aging. Ageing Res Rev. 7, 63–78. [DOI] [PubMed] [Google Scholar]
- Ren P, Lim CS, Johnsen R, Albert PS, Pilgrim D, Riddle DL (1996) Control of C. elegans larval development by neuronal expression of a TGF‐beta homolog. Science. 274, 1389–1391. [DOI] [PubMed] [Google Scholar]
- Riddle D, Blumenthal T, Meyer B, Priess J (1997) C. elegans II. Plainview, New York: Cold Spring Harbor Laboratory Press. [PubMed] [Google Scholar]
- Rogina B, Helfand SL, Frankel S (2002) Longevity regulation by Drosophila Rpd3 deacetylase and caloric restriction. Science. 298, 1745. [DOI] [PubMed] [Google Scholar]
- Rowan AJ, Meijer JW, De Beer‐Pawlikowski N, Van Der Geest P, Meinardi H (1983) Valproate‐ethosuximide combination therapy for refractory absence seizures. Arch Neurol. 40, 797–802. [DOI] [PubMed] [Google Scholar]
- Sica DA (2002) Rationale for fixed‐dose combinations in the treatment of hypertension: the cycle repeats. Drugs. 62, 443–462. [DOI] [PubMed] [Google Scholar]
- Syntichaki P, Troulinaki K, Tavernarakis N (2007) eIF4E function in somatic cells modulates ageing in Caenorhabditis elegans . Nature. 445, 922–926. [DOI] [PubMed] [Google Scholar]
- Temesgen Z, Warnke D, Kasten MJ (2006) Current status of antiretroviral therapy. Expert Opin Pharmacother. 7, 1541–1554. [DOI] [PubMed] [Google Scholar]
- Trent C, Tsuing N, Horvitz HR (1983) Egg‐laying defective mutants of the nematode Caenorhabditis elegans . Genetics. 104, 619–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzano DR, Terzibasi E, Genade T, Cattaneo A, Domenici L, Cellerino A (2006) Resveratrol prolongs lifespan and retards the onset of age‐related markers in a short‐lived vertebrate. Curr Biol. 16, 296–300. [DOI] [PubMed] [Google Scholar]
- Verbeke P, Fonager J, Clark BF, Rattan SI (2001) Heat shock response and ageing: mechanisms and applications. Cell Biol Int. 25, 845–857. [DOI] [PubMed] [Google Scholar]
- Wang JF, Bown C, Young LT (1999) Differential display PCR reveals novel targets for the mood‐stabilizing drug valproate including the molecular chaperone GRP78. Mol Pharmacol. 55, 521–527. [PubMed] [Google Scholar]
- Wilson MA, Shukitt‐Hale B, Kalt W, Ingram DK, Joseph JA, Wolkow CA (2006) Blueberry polyphenols increase lifespan and thermotolerance in Caenorhabditis elegans . Aging Cell. 5, 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, Sinclair D (2004) Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 430, 686–689. [DOI] [PubMed] [Google Scholar]
- Wu Z, Smith JV, Paramasivam V, Butko P, Khan I, Cypser JR, Luo Y (2002) Ginkgo biloba extract EGb 761 increases stress resistance and extends life span of Caenorhabditis elegans . Cell Mol Biol (Noisy-le-grand). 48, 725–731. [PubMed] [Google Scholar]
- Zhao Y, Sun H, Lu J, Li X, Chen X, Tao D, Huang W, Huang B (2005) Lifespan extension and elevated hsp gene expression in Drosophila caused by histone deacetylase inhibitors. J Exp Biol. 208, 697–705. [DOI] [PubMed] [Google Scholar]