

Abstract
Cytotoxic T lymphocyte-associated protein 4 (CTLA-4) blockade therapy is able to induce long-lasting antitumor responses in a fraction of cancer patients. Nonetheless, there is still room for improvement in the quest for new therapeutic combinations. ICOS costimulation has been underscored as a possible target to include with CTLA-4 blocking treatment. Herein, we describe an ICOS agonistic aptamer that potentiates T cell activation and induces stronger antitumor responses when locally injected at the tumor site in combination with anti-CTLA-4 antibody in different tumor models. Furthermore, ICOS agonistic aptamer was engineered as a bi-specific tumor-targeting aptamer to reach any disseminated tumor lesions after systemic injection. Treatment with the bi-specific aptamer in combination with CTLA-4 blockade showed strong antitumor immunity, even in a melanoma tumor model where CTLA-4 treatment alone did not display any significant therapeutic benefit. Thus, this work provides strong support for the development of combinatorial therapies involving anti-CTLA-4 blockade and ICOS agonist tumor-targeting agents.
Keywords: target therapeutics, cancer immunotherapy, ICOS, CTLA-4, aptamer
Graphical Abstract
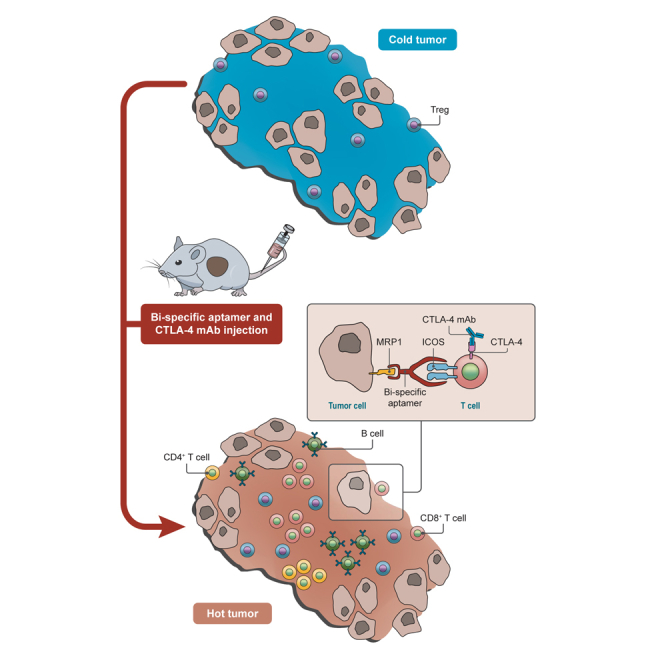
Soldevilla et al. showed a broadly applicable and clinically feasible method to enhance ICOS costimulatory capacity in disseminated tumor lesions by using a bi-specific aptamer reagent to target ICOS agonist to the tumor. This therapeutic approach is able to inflame the tumor milieu favoring the antitumor effect of CTLA-4 antibodies
Introduction
Immune-checkpoint blockade (mainly anti-cytotoxic T lymphocyte-associated protein 4 [CTLA-4] and anti-programmed cell death protein 1 [PD-1]/programmed death-ligand 1 [PD-L1]) is being introduced as a standard treatment in advanced melanoma, inducing long-term survival in a small set of cancer patients. Anti CTLA-4 antibody (ipilimumab) was approved for clinical use in 2011 for the treatment of unresectable or metastatic melanoma. The mechanisms of action of CTLA-4 and PD-1 blockade are different, which has led to the endorsement of a clinical trial combining the blockade of both immune checkpoints.1, 2, 3 The clinical trial outcomes showed therapeutic improvement, and the use of both antibodies in combination was FDA approved; the toxicities observed in combination of anti-PD-1 and anti-CTLA-4 were exacerbated (even with reduced doses of ipilimumab). The identification of other immunotherapeutic combinations with higher therapeutic efficacy and safer profile are still actively sought after. Ipilimumab monotherapy is still indicated for adults and adolescents with melanoma, and combination with anti-PD-1 is approved for adults but not for adolescent patients. Inducible T cell costimulator (ICOS) is a CD28-superfamily receptor induced in activated T lymphocytes. Providing artificial ICOS costimulation together with CTLA-4 blockade glimmers as a reasonable combination based on the observation that, first, patients treated with anti-CTLA-4 antibody induce higher expression of ICOS on tumor-infiltrating T lymphocytes and, second, that ICOS−/− or ICOS ligand (ICOSL)−/− mice have a diminished response to CTLA-4 therapy.4 Successful preclinical experiments using artificial expression of ICOS ligand in the tumor (throughout genetically engineered oncolytic virus) or irradiated tumor cells (such as IVAX vaccine) further support the pursuit of this therapeutic combination.5, 6 Conversely, there is another school of thought predicting that ICOS may be detrimental in cancer immunotherapy, as it has been associated with the expansion of immunosuppressive pathways mediated by T regulatory cells (Tregs).7 As a matter of fact, ICOS agonist and antagonist antibodies for cancer immunotherapy are currently under research in the clinical pipeline.7 To the best of our knowledge, so far there are no published data reporting the efficacy of combining agonist ICOS antibodies and CTLA-4 blockade, and no trials are foreseen as tumor-targeted delivery therapy.
Lack of accessibility to monoclonal antibodies agonistic to ICOS led us to develop an alternative agonist. In the last few years, aptamers, which are single-stranded 3D globular structured oligonucleotides, have been used as artificial agonist ligands to different costimulatory receptors with reported aptamers against 4-1BB, OX40, and CD28.8, 9 Aptamers, though still very expensive to produce at low scale for preclinical in vivo application, show some features that can make them amenable to cancer immunotherapy: They are chemically synthesized molecules and not cell-derivative products, which will likely simplify regulatory processes facing future clinical trials; they can be easily engineered to adequate their activity, for instance to generate multimers or bi-specific constructs with dual activity.10, 11 Additionally, they are poorly antigenic with reduced chances of triggering T-cell-dependent neutralizing antibodies that can hamper their usage after repetitive administrations, something that occurs frequently in chimeric recombinant protein therapeutics.8, 9 Furthermore, it is expected that as the technology is improved and expanded, prices for aptamer synthesis will decrease, making them more accessible and competitive as therapeutic tools in preclinical and clinical settings.
Herein, we describe the first agonistic aptamer against the murine ICOS receptor. ICOS agonistic aptamer was proven to elicit a higher activation of T lymphocytes in vitro. In vivo, the tumor delivery of ICOS agonistic aptamer in combination with CTLA-4 blockade antibody therapy induces stronger antitumor responses in different tumor models. Furthermore, using the oligonucleotide aptamer therapeutic platform allows us to engineer bi-specific aptamer constructs to drive ICOS costimulus at the tumor site after systemic injection. In a highly aggressive melanoma model, tumor-ICOS bi-specific aptamer in combination with CTLA-4 blockade therapy showed reduced tumor growth with higher lymphocyte infiltration.
Results
ICOS Agonist (Apt8a) Triggers T Cell Costimulation In Vitro
Anti-ICOS aptamers were selected by HT-SELEX (Supplemental Information). It has been previously shown that certain aptamers can act as agonists by multimerization.12 This happens with ligands that trigger their signaling via receptor clusterization, as in the case of costimulatory receptors that usually form part of the T cell receptor signalosome. Among these receptors are 4-1BB, OX40, and CD40, which belong to the tumor necrosis factor receptor (TNFR) family, but also some other costimulatory receptors such as CD28.9 ICOS shows high homology to CD28, making it likely that its costimulatory signal could be triggered by crosslinking of the receptor as well.13 To address this possibility, we dimerized the ICOS-binding aptamer (Apt8) (Figure S1) to convert it into an ICOS agonist (Apt8a) (Figure S2A). Apt8a were separated and purified by PAGE; the size of dimer from monomers can be easily distinguished by electrophoresis (Figure S2B).
ICOS, just like some other costimulatory receptors, is exclusively induced on effector-activated lymphocytes and not expressed on naive lymphocytes (Figures S3 and S4). We confirmed ICOS agonist aptamer (Apt8a) binding to ICOS-expressing lymphocytes by using 32P-labeled aptamers; a randomized aptamer of the same size as Apt8a was used as the control aptamer (Apt-ctrl) (Figure 1A).
Figure 1.

ICOS Agonist (Apt8a) Enhances T Lymphocyte Activation In Vitro
(A) Aptamer binding to cell detected by 32P-labeled aptamer incubated with different amounts of ICOS expressing lymphocytes or naive ICOS-null lymphocytes. (B) IFN-γ detected by ELISA in the supernatant of CD8 lymphocytes suboptimally activated with CD3 agonist antibody coated in 96-well plates and incubated with Apt8a aptamer or Apt ctrl. (Data are expressed as the mean ± SEM of triplicate cultures. The data are from one representative experiment out of two, with similar results.) (C and D) Proliferation measured by CFSE dilution of CD8 (C) and CD4 (D) isolated lymphocytes stimulated with CD3 agonist antibody and Atp8a or Apt ctrl. CFSE intensity was measured by flow cytometry on the CD8 or CD4 gated population (The data are from one representative experiment out of two with similar results.)
Importantly, costimulatory receptors such as ICOS are aimed at inducing a higher T cell activation after TCR stimulus (Figure S4), precluding a defective signal that could lead to T cell anergy. Therefore, to validate that the Apt8a was able to induce costimulation, we used two independent functional T cell assays, based on proliferation and interferon (IFN)-γ secretion. Isolated CD8 and CD4 lymphocytes were suboptimally activated with an anti-CD3 agonistic antibody. T lymphocytes incubated with 0.5 μM of ICOS Apt8a depicted a higher proliferation rate as compared to the controls, as measured by carboxyfluorescein succinimidyl ester (CFSE) dilution (Figures 1C and 1D). Another indicator of T cell activation is the secretion of IFN-γ. To that end, we stimulated CD8-purified lymphocytes with a suboptimal amount of CD3 antibody, and we provided Apt8a as costimulus. We observed that the amount of IFN-γ produced by activated T lymphocytes with ICOS aptamer was higher than that of the control groups (Figure 1B).
Intratumoral Administration of ICOS Agonist (Apt8a) Inhibits Tumor Growth in Combination with CTLA-4 Blockade
Previous work from leading groups in the field has shown that the antitumor effect of CTLA-4-blocking antibody could be enhanced by ICOSL expression.6 In preliminary experiments with B16/F10 melanoma tumor-bearing mice, intratumoral Apt8a as monotherapy did not display any antitumor effect (Figure S5) at the experimental established dose; combination with anti-CTLA-4 antibody (9H10) was required (Figure 2) to induce tumor regression. Mice subcutaneously implanted with B16/F10 cells were treated systemically with anti-CTLA-4 antibody and intratumorally with Apt8a following the schedule depicted in Figure 2A. Systemic intravenous injection of Apt8a in preliminary experiments did not show a significant therapeutic benefit under this treatment schedule (Figure S6); intratumoral injection of Apt8a was necessary for tumor regression. The group of mice that were treated with the combination (anti-CTLA-4 antibody and Apt8a intratumorally) showed slower tumor growth as compared to the controls (Figure 2B). The mice treated with the combination showed a reduced tumor volume change with tumor rejection in two mice out of 10 (Figure 2C). The overall survival of the mice treated with the combination was also enhanced (Figure 2D). To confirm these results in another tumor model and in a different mouse strain, we used a less aggressive hepatocarcinoma model (HEPA-129) in C3H mice. Hepatocarcinoma-implanted mice were treated following the schedule shown in Figure S7A but with anti-CTLA-4 monoclonal antibody 9D9 and intratumoral injection of Apt8a. We chose to use 9D9 clone in this instance as it was already calibrated and tested in this tumor model with partial antitumor effect; it is noteworthy that 9D9 has been reported to show less depleted Treg activity than 9H10.14 In this hepatocarcinoma model, the group of mice treated with the combination therapy rejected established tumors in 90% percent very quickly, while the mice treated with the Apt-ctrl with anti-CTLA-4 antibody rejected the tumor only in 40% of the cases (Figure S7B).
Figure 2.

Intratumoral ICOS Agonist (Apt8a) Potentiates CTLA-4 Blockade Therapy in a Melanoma Mouse Model
(A) B16/F10 melanoma tumors were implanted subcutaneously in C57/BL6 mice. On days 4, 7, and 10 after tumor implantation, mice were injected intraperitoneally (i.p.) with CTLA-4 blocking antibody (9H10) and intratumorally (i.t.) with Apt8a or Apt ctrl to evaluate tumor growth and survival. (B) Tumor growth kinetics was measured by calipers, and tumor volume was determined using the formula W2 × L/2; W is tumor width, and L is tumor length. The average tumor size of each group of mice treated is shown. Data are expressed as the mean ± SEM. **p < 0.01, ***p < 0.005. (C) Percentage of tumor volume change of each mouse once the treatment is initiated (isotype control in black, Apt ctrl with CTLA-4 antibody in gray, and Apt8a with CTLA-4 antibody in red). (D) Overall survival of mice treated with anti-anti-CTLA-4 antibody and Apt8a or Apt ctrl as described in the calendar. (Experiments were repeated twice with similar results.) *p < 0.05, **p < 0.01.
We next assessed whether the antitumor effect elicited by Apt8a was associated with the potentiation of an adaptive antitumor immune response. B16/F10 melanoma-bearing mice were treated with CTLA-4 blocking antibody systemically intraperitoneally and Apt8a or Apt-ctrl intratumorally as described in Figure 3A. On day 14 after treatment, mice were sacrificed, and splenocytes were cultured in vitro with B16/F10 irradiated melanoma cells. Activation of T lymphocytes was evaluated by three different independent assays: T cell proliferation by 3H thymidine incorporation, IFN-γ ELISPOT, and IFN-γ ELISA (Figure 3A). Mice that received ICOS agonist aptamer treatment, in all cases, displayed higher T cell activation, especially highlighted by a more than 2-fold increase in IFN-γ secretion compared to control groups (Figures 3B–3D). It is worth noting that CTLA-4 blocking antibody under this particular treatment regimen and in this tumor model does not reach an increased immune response that can be detected under these methods.
Figure 3.

Intratumoral ICOS Agonistic (Apt8a) Treatment in Combination with CTLA-4 Blockade Promotes Antitumor Immune Response
(A) B16/F10 melanoma tumor-bearing mice were treated with ICOS Apt8a intratumorally as well as anti-CTLA-4 antibody intraperitoneally on days 4, 7, and 10, and splenocytes were extracted at day 14 and then cocultured with B16/F10 irradiated cells. As measurement of T cell activation, proliferation was assessed by 3H incorporation (B) and IFN-γ production by ELISPOT (C) and ELISA (D). (Data are expressed as the mean ± SEM of three mice per group. Experiments were repeated twice with similar results.) *p < 0.05, ***p < 0.005.
We observed a higher infiltration of CD45 leucocytes and CD8 lymphocytes in B16/F10 tumor-bearing mice treated with CTLA-4 antibody and intratumorally Apt8a as compared to untreated mice, measured by flow cytometry (Figure S8). CTLA-4 antibody treatment alone induces a slight increase in lymphocyte infiltration, but it does not reach statistical significance, only when combined with Apt8a.
Systemic Treatment with Tumor-Targeted Bi-specific ICOS Agonistic Construct Enhances the Efficacy of CTLA-4 Blocking Therapy
In cancer patients, tumor lesions might not be accessible, precluding direct intratumoral therapeutics. With this in mind, we designed a bi-specific aptamer to target ICOS costimulation at the tumor site, as we have previously done with other costimulatory agonistic aptamers.12, 15 To that end, we used a previously described multidrug resistance protein 1 (MRP1)-targeting aptamer16 to conjugate with ICOS aptamer in order to generate a MRP1-ICOS bi-specific aptamer. MRP1 is commonly expressed in cancer stem cells and is associated with chemotherapy resistance.17 The MRP1-ICOS bi-specific aptamer was designed based on its predictive structure resolved using RNAstructure and Rosetta FARFAR software. The most thermodynamic stable MRP1-ICOS bi-specific aptamer was produced in vitro as a single molecule (Figure S10). To confirm that MRP1-ICOS bi-specific aptamer was able to recognize ICOS protein, we performed a binding blotting assay to ICOS recombinant protein (Figure 4A). MRP1-ICOS bi-specific aptamer was radiolabeled with 32P to assess binding to B16 cells overexpressing MRP1 (B16-MRP1), the aptamer bound to cells that express MRP1 but not to the parental B16/F10 cell line (Figure 4B). To ensure that the MRP1-ICOS bi-specific aptamer was able to induce costimulation on T lymphocytes, we performed T cell activation assays measuring proliferation as CFSE dilution and secretion of IFN-γ by ELISA (Figures 4C and 4D). MRP1-ICOS bi-specific aptamer was able to promote higher T cell activation by both assays as compared to the control groups. Of note is the fact that Apt8a and MRP1-ICOS bi-specific aptamer displayed similar costimulatory activity on primary lymphocytes activated with suboptimal doses of CD3 agonistic antibody (Figure S11).
Figure 4.

In Vitro Characterization of Tumor-Targeted MRP1-ICOS Bi-specific Aptamer
(A) Binding of the bi-specific aptamer to ICOS recombinant protein by 32P blotting. (B) Binding of the bi-specific aptamer labeled with 32P to B16-MRP1 cells. (C and D) T cell activation was determined as proliferation using CFSE dilution (C) or IFN-γ secretion measured by ELISA (D). (E) Assay to elucidate that both parts of the bi-specific aptamer are functional concurrently. B16-MRP1 cells were incubated with the MRP1-ICOS bi-specific aptamer and washed twice to remove the unbound aptamer fraction. Aptamer-coated tumor cells were incubated with anti-CD3-stimulated T lymphocytes. (F) Activation of T lymphocytes cocultured with tumor cells as described in (E) determined by ELISA of IFN-γ production in the cell supernatant. (All data are expressed as the mean ± SEM of experimental triplicates. The experiments were repeated twice, with similar results.) **p < 0.01, ***p < 0.005.
As a final test to validate whether MRP1-ICOS bi-specific aptamer was able to bind concurrently to MRP1-expressing cells and trigger ICOS costimulation on T lymphocytes, we performed a functional in vitro assay. Irradiated tumor B16-MRP1 cells were incubated with the MRP1-ICOS bi-specific aptamer and washed to remove any unbound aptamers. Afterward, the B16-MRP1 cells decorated with MRP1-ICOS bi-specific aptamer were cultured with suboptimal CD3-stimulated CD8 lymphocytes. As an endpoint to measure ICOS costimulation, IFN-γ secretion was determined by ELISA (Figure 4E). MRP1-ICOS bi-specific aptamer was able to significantly increase the production of IFN-γ, while MRP1/ICOS unconjugated aptamers did not (unbound aptamers were removed after tumor cell washes before being added to the T lymphocytes) (Figure 4F).
MRP1-ICOS bi-specific aptamer injected systemically intravenously preferably accumulates in B16-MRP1 as compared to B16/10 parental tumors implanted in the same mice in opposite flanks (Figures 5A and 5B, left panel). Furthermore, tumor-bearing mice (B16-MRP1 and B16/F10) injected intravenously with Apt8a displayed equal distribution in both tumors (Figure 5B, right panel). In the same tumor-bearing mice, we also studied tissue biodistribution of MRP1-ICOS bi-specific aptamer and Apt8a (Figure S12). As the bi-specific aptamer preferentially enriches in MRP1 tumors, it shows reduced accumulation in peripheral tissues; on the other hand, Apt8a was more abundant in non-tumor tissues (Figures 5B and S12).
Figure 5.

Tumor Inhibition by Systemic Injection of ICOS Agonist Targeted to the Tumor
(A) B16-MR1 and B16/F10 tumors were implanted contralaterally in the same mice, and MRP1-ICOS aptamer or Apt8a were injected intravenously to measure aptamer accumulation in each tumor by qRT-PCR using specific primers for the aptamers. (B) Accumulation of MRP1-ICOS aptamer and Apt8 in each tumor (n = 3). Data are expressed as the mean ± SEM. (C) B16-MRP1 melanoma tumors were implanted subcutaneously in C57/BL6 mice. On days 4, 9, and 11 after tumor implantation, mice were injected intraperitoneally with CTLA-4 blockade antibody (9H10) and subcutaneously with B16/F10 irradiated cells as vaccine (VAX); on days 9, 10, 11, 12, and 14, MRP1-ICOS bi-specific aptamer was injected intravenously. (D) Tumor growth kinetics represented as average of tumor volume of each group. Data are expressed as the mean ± SEM. (E) Percentage of tumor volume change after treatment initiation (PBS saline control mice in black; VAX with isotype and Apt ctrl in light gray; VAX with CTLA4 Ab and Apt Ctrl in darker gray; VAX with CTLA4 Ab and Apt MRP1-ICOS in red). (F) Overall survival of mice treated as described in (C). (The experiments were repeated twice with similar results.) **p < 0.01.
Next, we characterized the potential therapeutic effect of the MRP1-ICOS bi-specific aptamer in a melanoma model (B16-MRP1) that is actually more aggressive than B16/F10.16 As B16-MRP1 is an aggressive tumor model and the amount of MRP1-ICOS bi-specific aptamer that reaches the tumor will not be comparable with direct intratumoral injection of Apt8a, we decided to include a vaccine with irradiated cells (VAX) to improve the efficacy of the treatment. Furthermore, VAX would mimic to a certain extent the release of tumor neoantigens that can be induced in melanoma cancer patients undergoing local radiotherapy in a prominent tumor lesion. It is worthy of note that intravenous injection of Apt8a with VAX at the experimental schedule and established dose (Figure 5C) did not improve the therapeutic outcome of anti-CTLA-4 antibody therapy (Figure S9). B16-MRP1 tumor-bearing mice treated with MRP1-ICOS bi-specific aptamer combined with CTLA-4 blocking antibody and tumor-irradiated cells (VAX) (Figure 5C) reduced tumor growth significantly as compared to the control groups (Figures 5D and 5E). Anti-CTLA-4 antibody with VAX and Apt-ctrl did not show a significant drop in tumor growth, highlighting the aggressiveness of this tumor model (Figures 5D and 5E). Notwithstanding, anti-CTLA-4 antibody with VAX slightly increases mouse survival as compared to untreated mice; however, it was still required the inclusion of MRP1-ICOS aptamer to reach statistical significance (Figure 5F). These results in the survival studies indicate that the antitumor effect of CTLA-4 blockade antibody and MRP1-ICOS aptamer are additive, as the p value between these two groups is 0.052 and only when both treatments are combined does the p value drop to 0.009.
As evidence of immune-mediated tumor response associated with the treatment, we measured the infiltration of lymphocytes in the tumor. We analyzed B16-MRP1 tumors from mice treated with CTLA-4 blocking antibody, VAX, and MRP1-ICOS bi-specific aptamer injected intravenously (Figure 6A). Tumors treated with the combination showed a significant increase in the percentage of CD3+ tumor-infiltrating T lymphocytes as compared to CTLA-4 blockade and VAX-treated groups, measured by immunofluorescence (Figures 6B and 6C). Importantly, we detected a significant enrichment of CD8+ and CD4+ infiltrating cells (Figures 6B and 6C); CD8+ lymphocyte infiltration was also corroborated by flow cytometry (Figure 6D). No significant changes in the FOXP3+ infiltrating cells were observed (Figures S13 and S14F). ICOS has been considered important in the activation of CD4 follicular lymphocytes that are involved in the activation and expansion of B lymphocytes. Furthermore, tumors with higher infiltration of B lymphocyte areas are associated with better prognosis in many tumor types.18, 19 Based on these observations, we decided to evaluate whether the treatment could also promote higher infiltration of B lymphocytes in the tumor. Combination therapy in mice displayed higher infiltration of B220+ cells in the tumor than the control group, as measured by immunofluorescence (Figures 6B and 6C). It is to be noted that CTLA-4 blockade in combination with VAX on its own does not induce a significant increase in T or B lymphocyte infiltration under this treatment schedule in B16-MRP1 tumors.
Figure 6.

ICOS Agonist Targeted to the Tumor Enhances Lymphocyte Tumor Infiltration
(A) Treatment schedule of B16-MRP1 tumor-bearing mice. (B) Immunofluorescence imaging of tumors stained with CD3, CD8, CD4, and B220 antibodies. (C) CD3+, CD8+, CD4+, and B220 infiltration in treated B16-MRP1 tumors. It is represented as the relative area of stained cell over the whole tumor slide. Data are expressed as the mean ± SEM. (D) Percentage of CD8+ cells of the whole CD45+ fraction of cells that infiltrates each tumor, quantified by flow cytometry (five mice per group of control, VAX, and VAX with CTLA-4 Ab treatment; eight mice per group of MRP1-ICOS aptamer, anti-CTLA-4 antibody, and VAX combination treatment). Data are expressed as the mean ± SEM. *p < 0.05, **p < 0.01.
To better evaluate the stage of immunosuppression that is taking place in the tumor milieu after each treatment, we performed gene expression analysis of the most relevant immune genes by qRT-PCR (Figure S14). As expected, CD8 gene was highly expressed in the group of mice treated with MRP1-ICOS bi-specific aptamer (Figure S14A). Interestingly, in tumors of mice treated with MRP1-ICOS bi-specific aptamer, the T cell exhaustion markers PD-1 and LAG-3 were upregulated, but not TIM-3 (Figures S14B–S14D). No changes were observed in the immunosuppressive markers expressed on Tregs (Foxp3, CTLA-4, transforming growth factor β [TGF-β], CD73, interleukin-10 [IL-10], and IL-6), indicating that Treg activity is not enhanced by MRP1-ICOS bi-specific aptamer (Figure S14).
Discussion
CTLA-4 blocking antibody therapy has shown striking long-term responses in melanoma patients, yet the clinical benefit of this therapy is limited to a small fraction of patients.20 Thus, the quest to improve response rates by combining other immunomodulatory compounds with CTLA-4 blockade therapy is highly pursued in cancer immunotherapy.21, 22 A compelling set of publications have highlighted that anti-CTLA-4 antibody activity might depend on the induction of a T cell population that expresses ICOS.4, 5, 6 ICOS is expressed by T lymphocytes, and upon engagement with its ligand (CD275) on antigen-presenting cells (APC), T lymphocytes are further reinvigorated. However, its role in tumor immunology has been under debate, as ICOS is also expressed on Treg lymphocytes, leading to the possibility that artificial ICOS costimulus may promote Treg expansion.7, 23, 24
The primary mechanism by which CTLA-4 blocking antibody may function is by competing with CD28 for binding to CD80 and CD86. Other findings indicate that anti-CTLA-4 antibodies with immunoglobulin G2a (IgG2a) murine isotope or IgG1 human isotope could trigger depletion of tumor-infiltrating Tregs in some instances.6, 14, 25 A recent publication claims that ipilimumab actually does not deplete tumor Tregs in spite of being humanized with IgG1 isotype.26 Under our experimental conditions, we have not observed a significant reduction of Foxp3 expression in the treated tumors (Figures S13 and S14F). This is probably due to the lower dose of anti-CTLA-4 antibody used as compared to previous reports.14 Despite the presence of Treg lymphocytes in the tumor, ICOS agonistic aptamer improves anti-CTLA-4 antibody therapy. It is also possible that ICOS costimulation at the tumor site in combination with anti-CTLA-4 and Treg depletion might exhibit an even higher antitumor effect. Experiments using other modalities of Treg depletion (independent of anti-CTLA-4 antibody) together with ICOS costimulus will likely address this possibility in further studies.27, 28, 29
Using the B16-MRP1 tumor model16 and calibrated doses of anti-CTLA-4 antibody, we successfully generated a setting in which CTLA-4 blockade therapy did not display a profound effect on tumor kinetics and T cell infiltration (Figures 5 and 6). These settings mimic to a certain extent the conditions observed in non-responding ipilimumab-treated melanoma cancer patients.30 Our results suggest that ICOS-targeted costimulation at the tumor site induced a significant reduction in tumor growth and an outburst of T and B lymphocyte infiltration, turning an immunologically cold tumor microenvironment into a hot one.
ICOS is involved in the development and maintenance of T follicular helper cells (Tfh), Th1, Th2, Th17, Tc, and even memory effector T cells. ICOS is highly expressed in tonsillar T cells, which are usually associated with B lymphocytes in germinal centers facilitating the formation and maturation of B lymphocytes, and lack of ICOS triggers disruption of germinal center formation and reduction of lymphoid tissue.31 A tantalizing possibility is that persistent ICOS costimulus at the tumor site favors long-term formation of ectopic tertiary-like lymphoid tissue with enriched organized B and T lymphocyte areas in the tumor. CTLA-4 antibody blockade might reshape the tumor antigen-specific CD8 lymphocyte repertoire,32 leading to the activation of new tumor-specific CD8 lymphocytes within the tumor. It is possible that the new wave of tumor-reactive lymphocytes homing to the tumor triggered by CTLA-4 blockade would be optimally primed and expanded in situ thanks to ICOS costimulus at the tumor site, triggering the potential formation of ectopic lymphoid tissue. We also observed a significant enrichment of B lymphocytes in the tumors of mice treated with ICOS agonist aptamer. Activated B lymphocytes in the tumor can provide further activation signals to T lymphocytes potentiating antitumor immunity.33 Longer follow-up times will be required to further investigate whether these tumor-infiltrating B cells triggered by the ICOS agonist aptamer will be able to stimulate formation of organized, ectopic lymphoid tissue-like structures.
Therapeutic approaches combining ICOS costimulus and CTLA-4 blockade have been previously explored: IVAX, an irradiated-cell-based vaccine generated from tumor cells genetically modified to express ICOSL (IVAX) as well as an oncolytic virus that encodes ICOSL have been shown to synergize with CTLA-4 blockade therapy.5, 6 Nevertheless, the clinical translation of such therapeutic approaches (IVAX or oncolytic virus encoding ICOSL) might be more technically cumbersome in a clinical setting than a single tumor-targeted ICOS agonist therapeutic agent. There are some ICOS agonist antibodies under development and in clinical trials for cancer immunotherapy, but to the best of our knowledge, there are no published data so far reporting the combination of anti CTLA-4 checkpoint blockade and anti-ICOS therapy concurrently (0.1158/2326-6074.CRICIMTEATIAACR15-A059) (JTX-2011, GSK3359609). Furthermore, none of the described approaches have explored the intratumoral delivery of this treatment.
Herein, we described an alternative ICOS agonist, an oligonucleotide-based aptamer that can be engineered as a bi-specific target molecule to deliver ICOS costimulus at the tumor in situ. We show that ICOS costimulation at the tumor site after treatment with CTLA-4 blocking antibody significantly amplifies the existing antitumor response and perhaps even potentiates it de novo. Furthermore, treatment with the ICOS agonist favors the generation of a systemic, T-cell-based immune response against the tumor cell that can be detected ex vivo.
Tumor targeting bi-specific therapeutic aptamers have the potential to reach any disseminated tumor lesions, mobilizing an immune response in situ and thus precluding the escape of variants that express differentially immunodominant neoantigens in different tumor lesions.34, 35 Moreover, the costimulatory tumor-targeting bi-specific aptamer elicits strong tumor inflammation with a high T cell infiltration (Figure 6), stronger than with intratumoral injection of Apt8a (Figure S8). This might be explained as the bi-specific aptamer being able to persist in the tumor milieu for a longer time attached to the tumor cell membrane, favoring also the immobilization of the costimulatory aptamer, which renders a more efficient crosslink of ICOS receptor. Tumor-targeted therapeutics usually displays better therapeutic indices, increasing efficacy and reducing undesirable side effects.11, 36 CTLA-4 blocking antibody has previously been shown to trigger severe autoinflammatory-like reactions in some instances that can force the interruption of the treatment.37, 38 With the use of ICOS costimulation targeted to the tumor, it might allow the reduction of anti-CTLA-4 antibody dosage, minimizing toxicity significantly.37 Furthermore, by customizing the specificity of the targeting tumor aptamers, this approach could be easily adapted for broad use in most types of cancers.10, 11, 36, 39
Materials and Methods
Binding to Cells with 32P-Labeled Aptamer
Aptamers were transcribed in vitro using the DuraScribe T7 transcription kit, following manufacturer’s instructions in the presence of ATP32 (PerkinElmer, Boston, MA, USA; ref. BLU003H250UC).
CD4+ lymphocytes were isolated from the spleen of a C57/BL6 mouse using Myltenyi negative selection kit (Miltenyi Biotec, Bergisch Gladbach, Germany; ref. 130-104-454). Afterward, 2.5 × 105 CD4+ purified cells were activated with anti-mouse 1 μ/mL CD3e on a coated plate, washed with lymphocyte medium, and seeded by serial dilution up to blank. Naive lymphocytes and activated CD4+ lymphocytes were seeded in 96-well plate in serial dilutions, and 1 × 105 cpm of 32P-labeled Apt8a dimer aptamer and Apt-ctrl were added to cells and incubated for 30 min at 37°C. Afterward, cells were washed three times with 37°C pre-warmed lymphocyte medium (Supplemental Information). Binding to cells was measured by scintillation.
B16/F10 or B16-MRP1 melanoma cells were seeded by serial dilution up to blank, and 105 cpm of 32P-labeled MRP1-ICOS bi-specific aptamer or control aptamer were added to the cells and incubated for 30 min at 37°C. Afterward, cells were washed three times with 37°C pre-warmed medium. Binding to cells was measured by scintillation.
In Vitro Measurement of T Cell Activation
To evaluate the costimulatory capacity of each aptamer construct, CD4 and CD8 polyclonal proliferation assays by CFSE dilution were performed. CD4+ and CD8+ lymphocytes were isolated from the spleen using the Miltenyi negative selection kit (Miltenyi Biotec, Bergisch Gladbach, Germany; ref. 130-104-454 and 130-104-075, respectively). 105 purified CD4+ and CD8+ lymphocytes were stained with CFSE at the final concentration of 3 μM. CFSE-labeled cells were incubated with 1 μg/mL of anti-mouse CD3e previously coated to wells of a U-bottom 96-well plate (BD Biosciences, San Jose, CA, USA; ref. 353077). 0.5 μM of ICOS agonist aptamer (Apt8a or MRP1-ICOS aptamer) was added and incubated for 72 h. Proliferation rate was measured using BD FACSCantoTM II and analyzed using FlowJo vX.0.7.
A similar experiment was performed to measure IFN-γ secretion in purified CD8 lymphocytes stimulated with a suboptimal dose or CD3e agonistic antibody and 0.5 μM of Apt8a. INF-γ was measured by ELISA in the supernatant after 72 h (BD Biosciences, San Diego, CA, USA; ref. 555138). ELISA was quantified by TECAN Sunrise (TECAN, Männedorf, Switzerland) and analyzed with Magellan (TECAN, Männedorf, Switzerland).
To assess the dual function of MRP1-ICOS bi-specific aptamer, we performed an IFN-γ ELISA. 104 B16-MRP1-irradiated tumor cells were incubated with 100 pmol of MRP-ICOS bi-specific aptamer or MRP1/ICOS unconjugated aptamer and a control aptamer. After 30 min of incubation at 37°C in PBS (Gibco, Life Technologies, Waltham, MA, USA; ref. 14190-094), cells were washed twice and added to 105 isolated CD8+ T cells activated in 96-well anti-mouse CD3e (BD Biosciences, San Jose, CA, USA; ref. 145-2C11)-coated U-bottom 96-well plates (BD Biosciences, San Jose, CA, USA; ref. 353077) at 1 μg/mL. IFN-γ was measured by ELISA in the supernatant after 72 h (BD Biosciences, San Diego, CA, USA; ref. 555138). ELISA was quantified by TECAN Sunrise (TECAN, Männedorf, Switzerland) and analyzed with Magellan (TECAN, Männedorf, Switzerland).
In Vivo Tumor Model Studies
Ethical approval for animal studies was granted by the Animal Ethical Committee of the University of Navarra. Animal studies were performed in the veterinary facilities of the Center for Applied Medical Research (CIMA) following the institutional, regional, and national laws and ethical guidelines for experimental animal care. Animal wellness was monitored on a daily basis. All the mice used in the study were received from Harlan Laboratories.
B16/F10 Mouse Tumor Model
1.5 × 105 B16/F10 melanoma cells were subcutaneously injected in the right flank of 6- to 8-week-old female C57BL6/J mice. At days 4, 7, and 10 after tumor inoculation, 50 μg of anti-mouse CTLA-4 clone 9H10 (BioXCell, West Lebanon, NH, USA; ref. BE0121) or isotype antibody (BioXCell, West Lebanon, NH, USA; ref. BE0094) was systemically administered via peritoneum. At days 4, 7, and 10 after tumor injection, 250 pmol of Apt8a or Apt-ctrl were administered intratumorally. Tumor volume was measured using a caliper and calculated using the following formula: tumor volume = [length × (width)2]/2. Log rank test was used to evaluate mouse survival. Mice were euthanized when the tumor reached larger diameter of 15 mm or they showed signs of discomfort with significant reduction of weight and mobility.
B16-MRP1 Mouse Tumor Model
1.5 × 105 B16-MRP1 cells were injected in C57BL6/J mice in the right flank. At days 4, 9, and 11 post-tumor-inoculation, mice were injected with irradiated B16/F10 cells (VAX) subcutaneously in the inguinal area of both rear legs, and 50 μg of anti-mouse CTLA-4 clone 9H10 intraperitonially (BioXCell, West Lebanon, NH, USA; ref. BE0121) or isotype antibody (BioXcell, West Lebanon, NH, USA; ref. BE0094). VAX consists of B16/F10 cells irradiated at 5,000 rad, and 5 × 105 irradiated cells per leg (1 × 106 total cells) were subcutaneously injected in the inguinal area. At days 9, 10, 11, 12, and 14 post-tumor-inoculation, 250 pmol of MRP1-ICOS bi-specific aptamer or the Apt-ctrl were intravenously administered. Assessment of antitumor effect was evaluated by calculating the tumor size and mouse survival. Tumor size was calculated as follows: tumor volume = [length × (width)2]/2 .
Aptamer Biodistribution
200 pmol of Apt8a or MRP-ICOS bi-specific aptamer were intravenously administered via tail vein in 6- to 8-week-old female mice implanted with B16/F10 and B16-MRP1 tumors contralaterally. Mice were allocated into two groups of three mice each. 12 h later, cells isolated from each tumor, brain, lungs, spleen, heart, kidney, liver, and bone marrow were processed, and total RNA was extracted with TRIzol (Invitrogen, Life Technologies, Carlsbad, CA, USA; ref. 15596018). 4 μg of RNA was retro-transcribed (Invitrogen, Thermo Fisher Scientific, Carlsbad, CA, USA; ref. 28205013). qPCR was performed with Fast SYBR green (Applied Biosystems, Vilnius, Lithuania; ref. 4385612) using the Fast RT-PCR system (7900 HT, Applied Biosystems, Foster City, CA, USA); the RNA fluorinated aptamers were retrotranscribed and amplified by qRT-PCR using the Sel_Fwd and Sel_Rev primers indicated in the Supplemental Information and normalized to an endogenous gene HPRT as previously described.16 The aptamer amplicon was confirmed by Sanger sequencing.
Monitoring Immune Responses
ELISpot IFN-γ
4 × 105 splenocytes of C57BL/6J mice previously treated as mentioned in Figure 3A were incubated in the presence of 104 irradiated B16/F10 melanoma cells. The experiment was performed following manufacturer’s instructions (BD Biosciences, San Diego, CA, USA; ref. 551083). Assessment of IFN-γ-expressing cells was measured and analyzed with CTL ImmunoSpot analyzer (CTL-Europe, Bonn, Germany).
3H Thymidine Proliferation Assay
105 splenocytes of C57BL/6J mice previously treated as described in Figure 3A (BD Biosciences, San Jose, CA, USA; ref. 145-2C11) in the presence of 1 × 104 B16 or B16-MRP1-irradiated melanoma cells. The coculture was incubated for 72 h in U-bottom 96-well plates (BD Biosciences, San Jose, CA, USA; ref. 353077). 0.5 μCi of H3 thymidine (PerkinElmer, Boston, MA, USA; ref. NET027E005MC) was added overnight and measured by scintillation.
ELISA IFN-γ
105 splenocytes of C57BL6 mice were treated as in Figure 3A (BD Biosciences, San Jose, CA, USA; ref. 145-2C11) for 72 h in U-bottom 96-well plates in presence of irradiated B16/F10 melanoma cells. The supernatant was collected to perform ELISA experiments. IFN-γ was measured by ELISA (BD Biosciences, San Diego, CA, USA; ref. 555138) following manufacturer’s instructions. ELISA was quantified by TECAN Sunrise (TECAN, Männedorf, Switzerland) and analyzed with Magellan (TECAN, Männedorf, Switzerland).
Tumor-Infiltrating Lymphocytes by Immunofluorescence Labeling
For immunofluorescence studies, paraffin sections (3 μm thick) were cut, dewaxed, and hydrated. Antigen retrieval was performed for 30 min at 95°C in 0.01 M Tris-1 mM EDTA buffer (pH 9) in a Pascal pressure chamber (S2800, Dako). Tumor-embedded paraffin slices were incubated overnight at 4°C with primary antibodies. Rabbit monoclonal α-CD8a antibody clone D4W2Z was used as a primary antibody at dilution 1:300 (Cell Signaling, Danvers, MA, USA; ref. 98941). Rabbit monoclonal α-CD3 antibody clone SP7 was used as a primary antibody at dilution 1:300 (Thermo Fisher, Freemont, CA, USA; ref. RM9107). Rabbit monoclonal α-CD4 antibody clone EPR19514 was used as a primary antibody at dilution 1:1,000 (Abcam, Cambridge, UK; ref. ab183685). Slices underwent a second incubation with Alexa Fluor 555 goat anti-rabbit IgG (H+L) (Thermo Fisher Scientific, Freemont, CA, USA; ref. A31572) as secondary antibody at 1:200 dilution. Anti-mouse α-B220 antibody clone RA3-6B2 (BD PharMingen, San Diego, CA, USA) was used as primary antibody at 1:20,000 dilution and donkey anti-rabbit Alexa Fluor 555 (Thermo Fisher Scientific, Freemont, CA, USA; ref. A31572) was used as a secondary antibody at 1:200 dilution. For FoxP3 staining, anti-rat monoclonal antibody at dilution 1:800, clone FJK-16 (Thermo Fisher, Freemont, CA, USA; ref. 14-5773) was used as described above. Biotinylated rabbit anti-rat (Vector Labs, Burlingame, CA, USA; ref. 416490) was added and followed by incubation with Alexa Fluor 555 streptavidin (Thermo Fisher, Freemont, CA, USA; ref. S32355). All samples were counterstained with DAPI. Fluorescent images were recorded at 40× with AxioImager M1 microscope with Zen software and AxioCam MRm camera (Zeiss, Oberkochen, Germany).
Fluorescent images were taken as described above, and relative area was quantified using Fiji (ImageJ). Five randomized pictures were recorded from each slide. The relative area was defined as the summatory of positive areas for the specific marker divided by the tumor area of each picture. For the calculation, “positive staining area” was divided by “tumor area” to obtain the “relative area” and multiplied by 100 to obtain the percentage. “Relative area” is represented as the percentage of “positive staining area” out of the “tumor area.” Relative area (%) = [positive staining area/tumor area] × 100.
Tumor-Infiltrating Lymphocytes by Flow Cytometry
For the assessment of tumor-infiltrating lymphocytes (TILs), mice were euthanized and tumors were excised. Tumors were placed each in 100/15 mm Petri dishes (Greiner Bio-One, Monroe, NC, USA; ref. 663102) with 5 mL of collagenase D (Roche, Basel, Switzerland; ref. 11088866001)-DNase I (Roche, Basel, Switzerland; ref. 284932001)-containing medium and digested for 30 min at 37°C. After incubation, 60 μL of EDTA (Invitrogen, Life Technologies, Waltham, MA, USA; ref. 15575-038) were added to the tumors in order to stop the reaction. Tumor samples were smashed and filtered through a 40-μm nylon cell strainer (Falcon, Corning, NY, USA; ref. 352340) to a 50-mL centrifuge conical tube (Corning, Corning, NY, USA; ref. 430291). Cells were spun down at 1,500 rpm for 5 min. Supernatants were discarded, and 1 mL of ACK lysis buffer (Gibco, Life Technologies, Waltham, MA, USA; ref. 15575-038) was added to each pellet for 1 min on agitation. Free medium was added up to 50 mL to neutralize the lysis, and cells were spun down again at 1,500 rpm for 5 min. The pellet was re-suspended in PBS buffer for counting, and 3 × 106 cells were washed with PBS buffer for staining. Cells were incubated with Zombie Aqua fixable viability kit at dilution 1:500; CD45 APC/Cy7 clone 30-F11 at dilution 1:400, CD8 APC clone 53.67 at dilution 1:400 were all purchased from BioLegend (San Diego, CA, USA; ref. 423102, 103115, 100712, and 107705, respectively). The expression level of these markers was measured using BD FACSCanto II (BD Biosciences, San Diego, CA, USA) and analyzed using FlowJo vX.0.7.
Statistics
One-way ANOVA was used to assess the best treatment followed by post-hoc Tukey tests. Long rank test on Kaplan-Meier curves was conducted for survival and tumor-rejection experiments. Data are presented as mean ± SEM using GraphPad Prism 5.
Author Contributions
M.M.S. performed in vivo studies and characterized the immunological responses. H.V. selected the ICOS aptamers and assisted in the in vivo studies. D.M.-C., M.M.S., A.P.M., and M.V. contributed to the performance and analysis of the immune responses and the lymphocyte infiltration study. T.L., M.M.-S., A.P., and H.V. characterized the ICOS aptamer in vitro. M.R., D.L., and P.S. performed the studies on the hepatocarcinoma model. J.C. and B.M. were responsible for the production ICOS agonist aptamer and bi-specific aptamer used in the study. F.P. designed and supervised the whole study and wrote the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by a Worldwide Cancer Research grant under grant 15-1208, Melanoma Research Alliance (509510), and Fundación Ramón Areces (CIVP18A3916). Instituto de Salud Carlos III cofunded Fondos FEDER (PI17/00372). F.P. was supported by Ramón y Cajal (10699). This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 721358.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2019.07.013.
Supplemental Information
References
- 1.Larkin J., Chiarion-Sileni V., Gonzalez R., Grob J.J., Cowey C.L., Lao C.D., Schadendorf D., Dummer R., Smylie M., Rutkowski P. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antonia S.J., López-Martin J.A., Bendell J., Ott P.A., Taylor M., Eder J.P., Jäger D., Pietanza M.C., Le D.T., de Braud F. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016;17:883–895. doi: 10.1016/S1470-2045(16)30098-5. [DOI] [PubMed] [Google Scholar]
- 3.Motzer R.J., Tannir N.M., McDermott D.F., Arén Frontera O., Melichar B., Choueiri T.K., Plimack E.R., Barthélémy P., Porta C., George S., CheckMate 214 Investigators Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018;378:1277–1290. doi: 10.1056/NEJMoa1712126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fu T., He Q., Sharma P. The ICOS/ICOSL pathway is required for optimal antitumor responses mediated by anti-CTLA-4 therapy. Cancer Res. 2011;71:5445–5454. doi: 10.1158/0008-5472.CAN-11-1138. [DOI] [PubMed] [Google Scholar]
- 5.Zamarin D., Holmgaard R.B., Ricca J., Plitt T., Palese P., Sharma P., Merghoub T., Wolchok J.D., Allison J.P. Intratumoral modulation of the inducible co-stimulator ICOS by recombinant oncolytic virus promotes systemic anti-tumour immunity. Nat. Commun. 2017;8:14340. doi: 10.1038/ncomms14340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fan X., Quezada S.A., Sepulveda M.A., Sharma P., Allison J.P. Engagement of the ICOS pathway markedly enhances efficacy of CTLA-4 blockade in cancer immunotherapy. J. Exp. Med. 2014;211:715–725. doi: 10.1084/jem.20130590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amatore F., Gorvel L., Olive D. Inducible Co-Stimulator (ICOS) as a potential therapeutic target for anti-cancer therapy. Expert Opin. Ther. Targets. 2018;22:343–351. doi: 10.1080/14728222.2018.1444753. [DOI] [PubMed] [Google Scholar]
- 8.Pastor F., Berraondo P., Etxeberria I., Frederick J., Sahin U., Gilboa E., Melero I. An RNA toolbox for cancer immunotherapy. Nat. Rev. Drug Discov. 2018;17:751–767. doi: 10.1038/nrd.2018.132. [DOI] [PubMed] [Google Scholar]
- 9.Zhou J., Rossi J. Aptamers as targeted therapeutics: current potential and challenges. Nat. Rev. Drug Discov. 2016;16:181–202. doi: 10.1038/nrd.2016.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schrand B., Verma B., Levay A., Patel S., Castro I., Benaduce A.P., Brenneman R., Umland O., Yagita H., Gilboa E., Ishkanian A. Radiation-Induced Enhancement of Antitumor T-cell Immunity by VEGF-Targeted 4-1BB Costimulation. Cancer Res. 2017;77:1310–1321. doi: 10.1158/0008-5472.CAN-16-2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pastor F., Kolonias D., McNamara J.O., 2nd, Gilboa E. Targeting 4-1BB costimulation to disseminated tumor lesions with bi-specific oligonucleotide aptamers. Mol. Ther. 2011;19:1878–1886. doi: 10.1038/mt.2011.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McNamara J.O., Kolonias D., Pastor F., Mittler R.S., Chen L., Giangrande P.H., Sullenger B., Gilboa E. Multivalent 4-1BB binding aptamers costimulate CD8+ T cells and inhibit tumor growth in mice. J. Clin. Invest. 2008;118:376–386. doi: 10.1172/JCI33365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hutloff A., Dittrich A.M., Beier K.C., Eljaschewitsch B., Kraft R., Anagnostopoulos I., Kroczek R.A. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature. 1999;397:263–266. doi: 10.1038/16717. [DOI] [PubMed] [Google Scholar]
- 14.Simpson T.R., Li F., Montalvo-Ortiz W., Sepulveda M.A., Bergerhoff K., Arce F., Roddie C., Henry J.Y., Yagita H., Wolchok J.D. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013;210:1695–1710. doi: 10.1084/jem.20130579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pastor F., Soldevilla M.M., Villanueva H., Kolonias D., Inoges S., de Cerio A.L., Kandzia R., Klimyuk V., Gleba Y., Gilboa E., Bendandi M. CD28 aptamers as powerful immune response modulators. Mol. Ther. Nucleic Acids. 2013;2:e98. doi: 10.1038/mtna.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soldevilla M.M., Villanueva H., Casares N., Lasarte J.J., Bendandi M., Inoges S., López-Díaz de Cerio A., Pastor F. MRP1-CD28 bi-specific oligonucleotide aptamers: target costimulation to drug-resistant melanoma cancer stem cells. Oncotarget. 2016;7:23182–23196. doi: 10.18632/oncotarget.8095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He S.M., Li R., Kanwar J.R., Zhou S.F. Structural and functional properties of human multidrug resistance protein 1 (MRP1/ABCC1) Curr. Med. Chem. 2011;18:439–481. doi: 10.2174/092986711794839197. [DOI] [PubMed] [Google Scholar]
- 18.Garnelo M., Tan A., Her Z., Yeong J., Lim C.J., Chen J., Lim K.H., Weber A., Chow P., Chung A. Interaction between tumour-infiltrating B cells and T cells controls the progression of hepatocellular carcinoma. Gut. 2017;66:342–351. doi: 10.1136/gutjnl-2015-310814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nielsen J.S., Sahota R.A., Milne K., Kost S.E., Nesslinger N.J., Watson P.H., Nelson B.H. CD20+ tumor-infiltrating lymphocytes have an atypical CD27- memory phenotype and together with CD8+ T cells promote favorable prognosis in ovarian cancer. Clin. Cancer Res. 2012;18:3281–3292. doi: 10.1158/1078-0432.CCR-12-0234. [DOI] [PubMed] [Google Scholar]
- 20.Wei S.C., Duffy C.R., Allison J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018;8:1069–1086. doi: 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- 21.Mahoney K.M., Rennert P.D., Freeman G.J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 2015;14:561–584. doi: 10.1038/nrd4591. [DOI] [PubMed] [Google Scholar]
- 22.Melero I., Berman D.M., Aznar M.A., Korman A.J., Pérez Gracia J.L., Haanen J. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat. Rev. Cancer. 2015;15:457–472. doi: 10.1038/nrc3973. [DOI] [PubMed] [Google Scholar]
- 23.Chen Q., Mo L., Cai X., Wei L., Xie Z., Li H., Li J., Hu Z. ICOS signal facilitates Foxp3 transcription to favor suppressive function of regulatory T cells. Int. J. Med. Sci. 2018;15:666–673. doi: 10.7150/ijms.23940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burmeister Y., Lischke T., Dahler A.C., Mages H.W., Lam K.P., Coyle A.J., Kroczek R.A., Hutloff A. ICOS controls the pool size of effector-memory and regulatory T cells. J. Immunol. 2008;180:774–782. doi: 10.4049/jimmunol.180.2.774. [DOI] [PubMed] [Google Scholar]
- 25.Arce Vargas F., Furness A.J.S., Litchfield K., Joshi K., Rosenthal R., Ghorani E., Solomon I., Lesko M.H., Ruef N., Roddie C. Fc Effector Function Contributes to the Activity of Human Anti-CTLA-4 Antibodies. Cancer Cell. 2018;33:649–663.e4. doi: 10.1016/j.ccell.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharma A., Subudhi S.K., Blando J., Scutti J., Vence L., Wargo J., Allison J.P., Ribas A., Sharma P. Anti-CTLA-4 immunotherapy does not deplete FOXP3+ regulatory T cells (Tregs) in human cancers. Clin. Cancer Res. 2018;25:1233–1238. doi: 10.1158/1078-0432.CCR-18-0762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lahl K., Loddenkemper C., Drouin C., Freyer J., Arnason J., Eberl G., Hamann A., Wagner H., Huehn J., Sparwasser T. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J. Exp. Med. 2007;204:57–63. doi: 10.1084/jem.20061852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lozano T., Soldevilla M.M., Casares N., Villanueva H., Bendandi M., Lasarte J.J., Pastor F. Targeting inhibition of Foxp3 by a CD28 2′-Fluro oligonucleotide aptamer conjugated to P60-peptide enhances active cancer immunotherapy. Biomaterials. 2016;91:73–80. doi: 10.1016/j.biomaterials.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 29.Taylor N.A., Vick S.C., Iglesia M.D., Brickey W.J., Midkiff B.R., McKinnon K.P., Reisdorf S., Anders C.K., Carey L.A., Parker J.S. Treg depletion potentiates checkpoint inhibition in claudin-low breast cancer. J. Clin. Invest. 2017;127:3472–3483. doi: 10.1172/JCI90499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sharma P., Hu-Lieskovan S., Wargo J.A., Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell. 2017;168:707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tafuri A., Shahinian A., Bladt F., Yoshinaga S.K., Jordana M., Wakeham A., Boucher L.M., Bouchard D., Chan V.S., Duncan G. ICOS is essential for effective T-helper-cell responses. Nature. 2001;409:105–109. doi: 10.1038/35051113. [DOI] [PubMed] [Google Scholar]
- 32.Fehlings M., Simoni Y., Penny H.L., Becht E., Loh C.Y., Gubin M.M., Ward J.P., Wong S.C., Schreiber R.D., Newell E.W. Checkpoint blockade immunotherapy reshapes the high-dimensional phenotypic heterogeneity of murine intratumoural neoantigen-specific CD8+ T cells. Nat. Commun. 2017;8:562. doi: 10.1038/s41467-017-00627-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Germain C., Gnjatic S., Dieu-Nosjean M.C. Tertiary Lymphoid Structure-Associated B Cells are Key Players in Anti-Tumor Immunity. Front. Immunol. 2015;6:67. doi: 10.3389/fimmu.2015.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McGranahan N., Furness A.J.S., Rosenthal R., Ramskov S., Lyngaa R., Saini S.K., Jamal-Hanjani M., Wilson G.A., Birkbak N.J., Hiley C.T. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–1469. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McGranahan N., Swanton C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell. 2017;168:613–628. doi: 10.1016/j.cell.2017.01.018. [DOI] [PubMed] [Google Scholar]
- 36.Schrand B., Berezhnoy A., Brenneman R., Williams A., Levay A., Kong L.Y., Rao G., Zhou S., Heimberger A.B., Gilboa E. Targeting 4-1BB costimulation to the tumor stroma with bispecific aptamer conjugates enhances the therapeutic index of tumor immunotherapy. Cancer Immunol. Res. 2014;2:867–877. doi: 10.1158/2326-6066.CIR-14-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ascierto P.A., Del Vecchio M., Robert C., Mackiewicz A., Chiarion-Sileni V., Arance A., Lebbé C., Bastholt L., Hamid O., Rutkowski P. Ipilimumab 10 mg/kg versus ipilimumab 3 mg/kg in patients with unresectable or metastatic melanoma: a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2017;18:611–622. doi: 10.1016/S1470-2045(17)30231-0. [DOI] [PubMed] [Google Scholar]
- 38.Blank C.U., Rozeman E.A., Fanchi L.F., Sikorska K., van de Wiel B., Kvistborg P., Krijgsman O., van den Braber M., Philips D., Broeks A. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nat. Med. 2018;24:1655–1661. doi: 10.1038/s41591-018-0198-0. [DOI] [PubMed] [Google Scholar]
- 39.Wei J., Marisetty A., Schrand B., Gabrusiewicz K., Hashimoto Y., Ott M., Grami Z., Kong L.Y., Ling X., Caruso H. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a therapeutic target. J. Clin. Invest. 2019;129:137–149. doi: 10.1172/JCI121266. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.