

Abstract
It is now known that the inherited prion disease is caused by over 60 different mutations in the Prion protein (PRNP) gene. Four missense mutations at codons 102, 178, 200 and 210, account for over 95% of these cases. In this study we describe, a large Indian family with familial Creutzfeldt Jakob Disease (fCJD). One affected member presented with a presenile dementia, a protracted clinical course and characateristic MRI features. Genetic analysis revealed a D178N mutation in the 2 affected individuals and 7 unaffected members. The neuropathological examination of the brain of one of the affected member was conspicuous by spongiform degeneration, neuronal loss and gliosis. This is a detailed report of a genetically and neuropathologically proven fCJD from India.
Keywords: D178N mutation, familial Creutzfeldt Jakob Disease, presenile dementia, protracted course, spongiform degeneration
INTRODUCTION
Three neurological phenotypes of inherited prion disease (IPD) are recognized clinically and neuropathologically – namely familial Creutzfeldt-Jakob disease (fCJD), Gerstmann-Straussler-Scheinker disease (GSS) and Fatal familial insomnia (FFI). It is now known that these phenotypes are caused by mutations in the prion protein gene (PRNP). Over 60 different mutations in PRNP have been found in IPD, of which four missense mutations at codons 102, 178, 200 and 210, and insertional mutations of the octapeptide repeat region account for 95% of familial cases[1,2] with the proviso that IPD has very limited ascertainment in many regions of the world which are not actively surveyed for prion diseases. In addition, a missense polymorphism at codon 129 codes for either methionine or valine in the protein has been shown to influence the phenotype of prion disease, be it of sporadic, familial or acquired aetiologies.[3,4,5]
Clinically, CJD is typically a rapidly progressive dementia associated with a combination of extra pyramidal, pyramidal and cerebellar signs with seizures and/or myoclonus. The pathological hallmarks are cerebral spongiform changes, neuronal loss, gliosis and abnormal deposits of prion protein (PrP). Prion diseases are clinically and pathologically heterogeneous, and some of this variability in IPD can be accounted for by the mutation type and the genotype at polymorphic codon 129.[6] Whether ethnicity or geography contributes to variability in phenotype is not known.
In the present article, we describe an autosomal dominant, pre-senile dementia with a prolonged clinical course in a large Indian family. Neuropathological examination of the brain of one family member was remarkable in that it showed a very severe and advanced neuronal loss, significant spongiform changes, associated with strong gliosis but exceptionally little deposition of abnormal prion protein. A D178N mutation of the PRNP gene was identified in 2 affected individuals.
Case History
The propositus (IV-15, Figure 1) was apparently asymptomatic up to April 2001. At the age of 44 years, his family initially observed memory loss – he made mistakes in receiving telephonic messages, forgot appointments and recent events. His remote memory was preserved. Subsequently, there was a gradual deterioration in behaviour and personality. He became socially withdrawn and depressed. At times, he would become irritable and even aggressive with frequent mood swings and emotional liability. In addition, there was a perceptible decline in his word output and ability to communicate. He became repetitive and would address family members with the common suffix – “aye”. Early in 2002, a fairly rapid deterioration began. His personal grooming and hygiene deteriorated. He was disoriented, became bed-bound with very poor word output, developed a hand tremor, and incontinence of urine and faeces. On neurological examination, in November 2002 he was conscious but grossly demented. Mini Mental Status Examination (MMSE) was attempted but abandoned because of a lack of comprehension. He had perseveration and compulsive manipulation of tools with forced groping and mouthing. All frontal lobe release signs were hyperactive including bilateral grasp reflex and exaggerated blepherospasm. He also had bucco-facial apraxia. The external ocular movements, face and lower cranial nerves were grossly normal. No amyotrophy or fasciculations were observed in the limbs. Gegenhalten type of paratonia at the elbow joints and polyminimyoclonus of the outstretched hands were observed. Startle myoclonus could be elicited. The deep tendon reflexes were brisk but plantar response was difficult to elicit as he would constantly withdraw.
Figure 1.

Propositus - IV:15 denoted by an arrow; • below family member denotes asymptomatic carriers
The blood counts, serology and serum biochemistry were unremarkable. Cerebrospinal fluid (CSF) analysis could not be performed as consent for lumbar puncture was not given. Magnetic Resonance Imaging (MRI) done in November 2002 showed hyperintensities on the diffusion-weighted images (DW1) in the basal ganglia (BG), frontal and temporal lobe cortices [Figure 2]. The electro-encephalography (EEG) showed non-specific slowing. Periodic slow wave complexes (PSWC) were not seen. The patient was re-evaluated by one of us (AP) in December 2003, 21 months after the onset of his disease. He was bedridden and in an akinetic mute state. Minimyoclonus-like twitches of the hands and startle myoclonus were more marked. Thereafter, he rapidly deteriorated and died in June 2004, 38 months after the onset of the disease. Genealogical assessment was done by interviewing non-affected members of this pedigree. The pedigree over 5 generations is shown in Figure 1.
Figure 2.
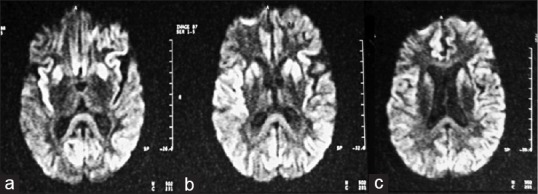
Diffusion weighted axial MRI shows hyperintensity in: (a) the insular cortex and splenium of the corpus callosum; (b) the bilateral caudate and putamen; (c) the bilateral parietal and para-falcine frontal cortices
Subsequently, another member of this pedigree (IV-2) with a similar dementia and a protracted course was looked after by one of us (AP). He rapidly deteriorated and died in June 2003. A post-mortem examination limited to the brain was performed.
METHODS
Histopathological studies
The brain from patient IV-2 was removed and fixed in 10% buffered formalin. Sections for histological examination were taken bilaterally from the frontal, temporal, parietal and occipital lobes, basal ganglia, thalamus, cerebellum, midbrain, pons and medulla. Sections of 5 μm nominal thickness were prepared and stained with Haematoxylin and Eosin (H and E), Periodic Acid Shiff, Alcian blue, Congo red and Luxol fast blue. On 2 brain regions, basal ganglia, adjacent to the lateral ventricle and from the frontal cortex and white matter, immunohistochemical preparations for abnormal PrP (ICSM35) and glial fibrillary acidic protein (GFAP) were performed. The brain homogenates were exposed to Anti-PrP ICSM35 (D-Gen Ltd, London, UK), raised in Prnpo/o mice against recombinant human PrP as previously described.[7] To detect abnormal PrP deposition, mounted sections were treated with 98% formic acid for 5 min, placed on an automated Ventana Discovery staining machine, heated to 95°C in 2.1 mM Tris-HCl, 1.3 mM EDTA, 1.1 mM sodium citrate, pH 7.8, for 30 minutes, digested for 16 minutes with a low concentration of protease. Gliosis was detected with antibody directed against GFAP (DAKO Z0334). Detection was accomplished by a biotinylated secondary antibody and visualised using the iView detection kit. Haematoxylin was used as counterstain.
Genetic studies: Analysis of the PRNP gene
The genetic studies were performed on 27 members of this pedigree. Genomic DNA was extracted from peripheral blood leucocytes. All the individuals were above 18 years of age. The blood for DNA extraction was obtained after appropriate genetic counselling following protocols for those established for Huntington Disease[8] and informed consent in their mother tongue. Sanger sequencing of PRNP was done according to established protocols[9] in the two probands, which identified the D178N mutation of PRNP in the heterozygous state. Genotypes were confirmed using PflF1 restriction endonuclease digestion of PCR amplicons and size fractionation by gel electrophoresis as previously described.[9] This genotype is typically associated with fCJD.
RESULTS
Figure 1 shows the pedigree of this large family over five generations. According to the information given by the relatives, 22 individuals spanning four generations were affected. The propositus (IV-15) was clinically examined by us. The others had died and no medical records were available for scrutiny. One individual (IV-2) was confirmed to be affected by neuropathological examination of the brain.
The propositus was 44 years old at the onset of his illness. Recent memory loss and personality changes were the initial symptoms followed by speech disturbance, and then a rapidly progressive dementia. On clinical examination extrapyramidal Gagenhalten type of paratonia, hyperorality and frontal lobe release reflex signs were prominent features. Minimyoclonus of the fingers and startle myoclonus were subtle in the beginning but quite marked when he was in an akinetic mute stage. Absence of seizures was noted, not only in the propositus but also in the other affected members of this pedigree. The duration of the illness was more protracted when compared to sCJD, and lasted 38 months. The EEG showed only non-specific slowing. The classical EEG finding of PSWC were conspicuous by their absence. The MRI showed DW1 hyperintensity in the frontal and temporal lobe cortices as well as the basal ganglia (BG), findings which are suggestive of CJD[10,11] [Figure 2].
Neuropathological study
Histological examination of the brain showed widespread cortical neuronal loss with shrinkage of the cortex, as well as severe spongiform degeneration and proliferation of glial cells [Figure 3a–c] whilst the white matter was relatively well preserved with only occasional vacuoles. In some areas, the neuropil was focally wiped out leaving areas of vacuolated residual tissue. These changes were most severe in the frontal, and to a lesser extent in the temporal lobes. In the caudate nucleus, the grey matter was severely vacuolated, leaving only the white matter tracts - “pencil fibres”- intact. Focal spongiform changes were also seen in the region of the corpus callosum, thalamus and mid brain. GFAP immunostaining [Figure 3b] confirmed a very substantial gliosis. No foci of necrosis, amyloid plaques or vascular changes were seen. PrP immunostaining showed very rare small plaques [Figure 3c] and virtually no synaptic staining. There were 10-15 microplaques in the cortex area and 4-6 small plaques in the BG.
Figure 3.
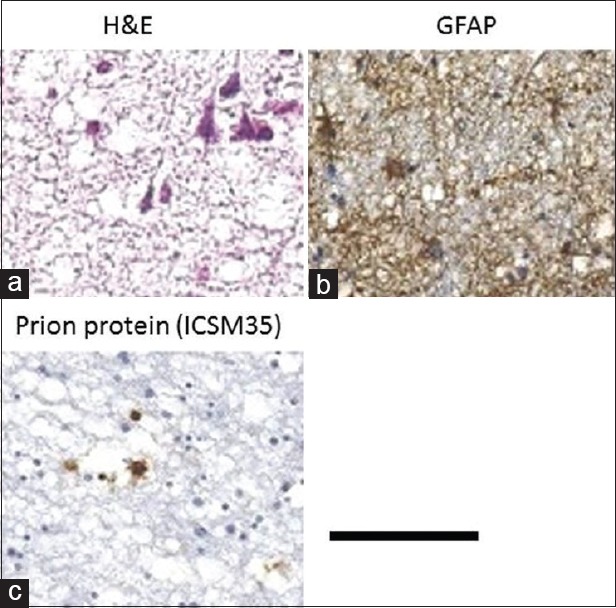
Neuropathology: (a) Grey matter (H and E): Severe spongiform changes and neuronal loss; (b) Grey matter: (GFAP stain) gliosis along the cortex; (c) Caudate nucleus: Immunostaining showing the position of prion protein. Scale bar: 50 μm – A, 200 μm – B and C
Genetic studies
We found a D178N mutation in PRNP by direct sequencing in 11 of the 28 samples. This missense mutation involves a single base-pair transition in codon 178 from GAC to AAC (CCDS 13080.1 c. 532G > A). The presence of the D178N mutation was confirmed by PflF1 restriction enzyme digestion. At codon 129, the genotype was methionine-valine (MV) heterozygous in all but 2 individuals who were homozygous for valine (VV). Analysis of the co-segregation of codons 178 and 129 in the pedigree showed that the 178N allele is linked to 129V, a haplotype typically associated with fCJD.
DISCUSSION
The diagnosis of IPD requires a combination of characteristic clinical and pathological features, and the identification of a disease-causing mutation. In this article, we report a large Indian family with fCJD in which the pathological mutation was at codon 178 of PRNP. The D178N mutation is one of the many known mutations in fCJD and involves a point mutation (GAC to AAC) at the first nucleotide of codon 178. This mutation was first reported in 1991 in a Finnish family.[12] Subsequently the D178N mutation has been reported in apparently unrelated families from America, England, Germany, Hungary, Israel, Korea, the Netherlands and Spain.[13] Recently, Sawal et al.[14] have reported fCJD from India in which 3 family members had a D178N mutation of the PrP gene. However, no pathological studies were done. This is a detailed report of a genetically and pathologically proven fCJD in a large Indian family.
A polymorphism at PRNP codon 129 leads to the protein containing either methionine or valine. This polymorphism may affect the phenotype of many PRNP gene mutation in a cis-acting effect (the variant alleles are on the same DNA strand) or a trans-acting effect (the variant alleles are on different DNA strands).[15,16] More than 20 years ago, it was reported that in patients with a D178N mutation and valine at Codon 129 typically produces a phenotype consistent with CJD. By contrast, when codon 129 codes for methionine, the phenotype is often that of FFI.[15] In the members of our family, codon 129 coded for valine, which is consistent with the phenotype of CJD in the proband.
The genotype of codon 129 on the normal allele may modify the age at onset and duration of the disease in some IPDs. In one study, the mean age at onset was 39 + 8 years (range 26-47) for those who are homozygous (VV), and 49 + 4 years (range 45-56) for those who are heterozygous (VM).[17] Likewise, the mean duration was shorter for those who were VV homozygous (range 9-18 months) compared to MV heterozygous patients (range 7-51 months). In our family, the propositus was heterozygous at Codon 129 and had a more protracted illness (38 months). The 2 individuals who are homozygous for valine are now in their mid-thirties, and still unaffected. Thus, any effect of homozygosity at codon 129 on the age at onset and severity of the disease in this family is yet to be established.
The clinical phenotype of fCJD generally has an earlier age at onset and a more protracted course compared to sCJD. The propositus was the only member of this family whose clinical phenotype was well documented. His illness began at the age of 44 years, which is much earlier than that of sCJD. Judging from his clinical course and the age at death of the other affected family members, it appears to be earlier than sCJD. The initial symptoms were those of memory loss, followed by a combination of abnormal behaviour, speech impairment, extrapyramidal signs and myoclonus. In the advanced stage of the disease, he was in an akinetic mute stage with incontinence. The clinical phenotype and duration of his illness are consistent with D178N mutation in fCJD.[17] The EEG in this illness usually shows generalized slowing without PSWCs, a pattern noted in our patient. In the MRI, DWI and FLAIR images are more sensitive than T2W images in demonstrating hyperintensities in the cortical ribbon of the frontal and temporal lobe insular cortex and basal ganglia. This pattern has been reported in sporadic and familial CJD with a high degree of specificity compared to other forms of rapidly progressive dementia.[10,11] In the propositus, the MRI showed a similar pattern and was the first investigation which suggested the possibility of fCJD and lead to the final diagnosis [Figure 2].
The salient features of the neuropathology were remarkable spongiform degeneration with neuronal loss and gliosis of the grey matter of the frontal and temporal lobes. These features are consistent with the diagnosis human prion disease. The PrP immunostaining pattern is unusual in that there is virtually no deposition of abnormal PrP, with only focal presence of microplaques. A possible explanation of this striking finding in D178N prion disease could be a widespread presence of toxic PrP species that have resulted in severe neuronal toxicity with spongiosis and neuronal death.
In conclusion, we describe a large Indian family with a familial prion disease presenting as a presenile dementia, a protracted clinical course and characteristic MRI features. The classical pathological features of spongiform degeneration, neuronal loss and gliosis were evident in the brain.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Collinge J. Prion diseases of humans and animals: Their causes and molecular basis. Annu Rev Neurosci. 2001;24:519–50. doi: 10.1146/annurev.neuro.24.1.519. [DOI] [PubMed] [Google Scholar]
- 2.Mead S, Poulter M, Beck J, Webb TE, Campbell TA, Linehan JM, et al. Inherited prion disease with six octapeptide repeat insertional mutation--molecular analysis of phenotypic heterogeneity. Brain. 2006;129:2297–317. doi: 10.1093/brain/awl226. [DOI] [PubMed] [Google Scholar]
- 3.Palmer MS, Dryden AJ, Hughes JT, Collinge J. Homozygous prion protein genotype predisposes to sporadic Creutzfeldt-Jakob disease. Nature. 1991;352:340–42. doi: 10.1038/352340a0. [DOI] [PubMed] [Google Scholar]
- 4.Poulter M, Baker HF, Frith CD, Leach M, Lofthouse R, Ridley RM, et al. Inherited prion disease with 144 base pair gene insertion. 1. Genealogical and molecular studies. Brain. 1992;115:675–85. doi: 10.1093/brain/115.3.675. [DOI] [PubMed] [Google Scholar]
- 5.Monari L, Chen SG, Brown P, Parchi P, Petersen RB, Mikol J, et al. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: Different prion proteins determined by a DNA polymorphism. Proc Natl Acad Sci U S A. 1994;91:2839–42. doi: 10.1073/pnas.91.7.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mead S. Prion disease genetics. Eur J Hum Genet. 2006;14:273–81. doi: 10.1038/sj.ejhg.5201544. [DOI] [PubMed] [Google Scholar]
- 7.Khalili-Shirazi A, Summers L, Linehan J, Mallinson G, Anstee D, Hawke S, et al. PrP glycoforms are associated in a strain-specific ratio in native PrPSc. J Gen Virol. 2005;86:2635–44. doi: 10.1099/vir.0.80375-0. [DOI] [PubMed] [Google Scholar]
- 8.Guidelines for the molecular genetics predictive test in Huntington's Disease. International huntington association (IHA) and the World federation of neurology (WFN) research group on huntington's chorea. Neurology. 1994;44:1533–6. [PubMed] [Google Scholar]
- 9.Wadsworth JD, Powell C, Beck JA, Joiner S, Linehan JM, Brandner S, et al. Molecular diagnosis of human prion disease. Methods Mol Biol. 2008;459:197–227. doi: 10.1007/978-1-59745-234-2_14. [DOI] [PubMed] [Google Scholar]
- 10.Demaerel P, Sciot R, Robberecht W, Dom R, Vandermeulen D, Maes F, et al. Accuracy of diffusion weighted MR imaging in the diagnosis of sporadic Creutzfeldt-Jacob disease. J Neurol. 2003;250:222–5. doi: 10.1007/s00415-003-0983-6. [DOI] [PubMed] [Google Scholar]
- 11.Vitali P, Maccagnano E, Caverzasi E, Henry RG, Haman A, Torres-Chae C, et al. Diffusion-weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias. Neurology. 2011;76:1711–9. doi: 10.1212/WNL.0b013e31821a4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldfarb LG, Haltia M, Brown P, Nieto A, Kovanen J, McCombie WR, et al. New mutation in scrapie amyloid precursor gene (at codon 178) in finnish Creutzfeldt-Jakob kindred. Lancet. 1991;337:425. doi: 10.1016/0140-6736(91)91198-4. [DOI] [PubMed] [Google Scholar]
- 13.Montagna P, Gambetti P, Cortelli P, Lugaresi E. Familial and sporadic fatal insomnia. Lancet Neurol. 2003;2:167–76. doi: 10.1016/s1474-4422(03)00323-5. [DOI] [PubMed] [Google Scholar]
- 14.Sawal N, Chakravarty K, Puri I, Goyal V, Garg A, Shi Q, et al. Familial Creutzfeldt-Jakob diseases: The first reported kindred from South-East Asia.Ann. Indian Acad Neurol. 2019;22:225–7. doi: 10.4103/aian.AIAN_441_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldfarb LG, Petersen RB, Tabaton M, Brown P, LeBlanc AC, Montagna P, et al. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: Disease phenotype determined by a DNA polymorphism. Science. 1992;258:806–8. doi: 10.1126/science.1439789. [DOI] [PubMed] [Google Scholar]
- 16.Kaski DN, Pennington C, Beck J, Poulter M, Uphill J, Bishop MT, et al. Inherited prion disease with 4-octapeptide repeat insertion: Disease requires the interaction of multiple genetic risk factors. Brain. 2011;134:1829–38. doi: 10.1093/brain/awr079. [DOI] [PubMed] [Google Scholar]
- 17.Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial CJD: Classification and characterisation. Br Med Bull. 2003;66:213–39. doi: 10.1093/bmb/66.1.213. [DOI] [PubMed] [Google Scholar]