

Abstract
Basosquamous carcinoma (BSC) is an aggressive skin neoplasm with the features of both basal cell carcinoma (BCC) and squamous cell carcinoma (SCC). While genetic drivers of BCC and SCC development have been extensively characterized, BSC has not been well studied, and it remains unclear whether these tumors originally derive from BCC or SCC. In addition, it is unknown which molecular pathways mediate the reprogramming of tumor keratinocytes toward basaloid or squamatized phenotypes. We sought to characterize the genomic alterations underlying sporadic BSC to elucidate the derivation of these mixed tumors. We identifed frequent Hedgehog (Hh) pathway mutations in BSCs, implicating Hh deregulation as the primary driving event in BSC. Principal component analysis of BCC and SCC driver genes further demonstrate the genetic similarity between BCC and BSC. In addition, 45% of the BSCs harbor recurrent mutations in the SWI/SNF complex gene, ARID1A, and evolutionary analysis revealed that ARID1A mutations occur after PTCH1 but before SCC driver mutations, indicating that ARID1A mutations may bestow plasticity enabling squamatization. Finally, we demonstrate mitogen-activated protein kinase pathway activation and the loss of Hh signaling associated with the squamatization of BSCs. Overall, these results support the genetic derivation of BSCs from BCCs and highlight potential factors involved in modulating tumor reprogramming between basaloid and squamatized phenotypes.
INTRODUCTION
Basosquamous carcinoma (BSC) is a clinically aggressive neoplasm of the skin with the features of both basal cell carcinoma (BCC) and squamous cell carcinoma (SCC). BSC represents 1.2–2.7% of all skin carcinomas and displays an aggressive local growth pattern with high potential for recurrence and metastases, with a prevalence rate up to 7.4% for distant metastases, higher than that of SCC and BCC (Garcia et al., 2009; Volkenstein et al., 2010). While the molecular nature of BSC is unclear, most tumors contain areas of both BCC and SCC with a transitional zone of intermediate differentiation (Maloney, 2000). This entity has been referred to as basosquamous cell carcinoma, metatypical carcinoma, and BCC with squamous differentiation (Allen et al., 2014;Costantino et al., 2006;De Stefano et al., 2012;Garcia et al., 2009) and represents the phenotypic plasticity that can occur between BCC and SCC. Despite recognition of the intermediate histopathologic features of BSC (Figure 1), it remains unclear whether these tumors are initially derived from BCC or SCC, and the genetic mutations underlying the development of BSC have not been explored.
Figure 1. Histopathology of BSC.

Histopathology demonstrates a tumor with basaloid areas containing tumor cells with scant cytoplasm arranged in cords with peripheral palisading, retraction artifact, and mucinous stroma, transitioning into areas composed of squamous cells containing abundant eosinophilic cytoplasm (original magnification ×20). Transition zones between the basaloid areas (in the bottom portion of the image) and squamous areas (in the upper left portion of the image) are marked with asterisks (*). Bar = 50 μm. BSC, basosquamous carcinoma.
In contrast to BSC, the genetics of BCC and SCC are well described. Uncontrolled activation of the Hedgehog (Hh) pathway drives the development of BCCs. Loss of the tumor suppressor PTCH1 and gain of function of the G protein–coupled receptor Smoothened (SMO) are the most common mutations that inappropriately activate the Hh pathway (Atwood et al., 2015;Sharpe et al., 2015). Other genetic drivers of BCC identified through exome sequencing studies include PTEN, MYCN, PPP6C, GRIN2A, GLI1, CSMD3, DCC, PREX2, and APC (Bonilla et al., 2016; Jayaraman et al., 2014). In contrast to BCC, SCC contains greater genetic heterogeneity. Commonly mutated genes in SCC include activating mutations in HRAS and disruptions of the TGFBR1, TGFBR2, NOTCH1, and NOTCH2 genes (Cammareri et al., 2016; Rose et al., 2017; South et al., 2014). Genetic studies have also reported additional mutations in CASP8, CDKN2A, NOTCH3, KRAS, NRAS, PDK1, BAP1, AJUBA, KMT2D, MYH9, TRAF3, NSD1, CDH1, and TP63 (Pickering et al., 2014; Schwaederle et al., 2015; Yilmaz et al., 2017).
Several recent studies have demonstrated the squamatization of BCCs with SMO inhibitor vismodegib (Ransohoff et al., 2015; Zhao et al., 2015; Zhu et al., 2014). These reports describe cases of SCC arising in areas of the original BCC tumor bed, within tumors that have stopped responding clinically to vismodegib or contained areas responding differentially to treatment, suggesting that the process of the dedifferentiation of BCC into SCC may enable the BCCs to evade drug inhibition (larrobino et al., 2013; Orouji et al., 2014; Saintes et al., 2015; Zhu et al., 2014). However, it remains unclear whether these cases of SCC arose independently or resulted from squamatization of the original BCC. Recently, Ransohoff et al. (2015) described one case of a recurrent SCC-like tumor in the lymph node following the vismodegib treatment of metastatic BCC. Genetic sequencing of the original tumor and recurrent lymph node SCC identified a common PTCH1 mutation, suggesting that the SCC most likely arose from the original BCC and that selective pressure owing to treatment with vismodegib may have been responsible for this squamatization. However, no studies of the derivation of sporadic BSC have been performed to date. Further understanding of the genetics of sporadic BSC may help to elucidate the mechanisms of phenotypic plasticity that enable BCC and SCC to change fate, promote tumor progression, and enable tumors to evade therapy.
In this study, we conducted a mutational analysis of 20 histologically confirmed BSCs compared with 16 BCC and 52 SCC samples to elucidate the genetic etiology of BSC and to explore mechanisms for the phenotypic plasticity observed in BSC.
RESULTS
BSC contains genetic alterations in PTCH1, SMO, and MYCN, similar to those of BCC
We compared gene mutations among BSC and BCC (Table 1, Figure 2, and Supplementary Table S1). Forty-five percent of the BSC tumors contained deleterious mutations in PTCH1 compared with 44% of the BCCs and 10% of the SCCs (P = 0.95 and P = 0.001, respectively). Five percent of the BSCs contained the oncogenic SMO M2 mutation, W535L, compared with 25% of the BCCs and none (0%) of the SCC samples. Fifteen percent of the BSCs had mutations in MYCN, a recently described BCC cancer driver, compared with 19% of the BCC and 6% of the SCC samples (P = 0.75 and P = 0.20, respectively). Additional mutations in the BCC drivers PPP6C, GRIN2A, CSMD3, DCC, PREX2, APC, and ARID1A were also present, totaling 100% (11 of 11) of the known BCC driver mutations accounted for in BSC. The presence of PTCH, MYCN, and SMO in the BSC samples provides genetic evidence that BSCs share cancer drivers similar to those of BCC and arise through the activation of Hh signaling.
Table 1.
Comparison of BCC and SCC Driver Gene Mutation Frequencies
| Gene | BSC Sample Mutation Frequency (N = 20) |
BSC Sample Mutation Frequency (N = 16) |
BSC Sample Mutation Frequency (N = 52) |
BCC versus BSC P- Value1 |
SCC versus BSC P- value1 |
|---|---|---|---|---|---|
| BCC Drivers | |||||
| PTCH1 | 0.45 | 0.44 | 0.10 | 0.95 | 0.001 |
| SMO2 | 0.05 | 0.25 | 0.00 | 0.08 | 0.10 |
| MYCN3 | 0.15 | 0.19 | 0.06 | 0.75 | 0.20 |
| PPP6C3 | 0.05 | 0.25 | 0.00 | 0.08 | 0.10 |
| GRIN2A | 0.35 | 0.50 | 0.27 | 0.36 | 0.50 |
| CSMD3 | 0.60 | 0.56 | 0.75 | 0.82 | 0.21 |
| DCC | 0.10 | 0.25 | 0.31 | 0.23 | 0.07 |
| PREX2 | 0.05 | 0.38 | 0.19 | 0.01 | 0.13 |
| APC | 0.10 | 0.19 | 0.27 | 0.45 | 0.12 |
| PTEN | 0.05 | 0.06 | 0.00 | 0.87 | 0.10 |
| PIK3CA3 | 0.10 | 0.13 | 0.04 | 0.78 | 0.31 |
| SCC Drivers | |||||
| CDKN2A | 0.00 | 0.00 | 0.15 | N/A | 0.02 |
| NOTCH1 | 0.15 | 0.31 | 0.46 | 0.24 | 0.01 |
| NOTCH2 | 0.20 | 0.25 | 0.37 | 0.72 | 0.18 |
| NOTCH3 | 0.15 | 0.06 | 0.19 | 0.39 | 0.68 |
| KRAS3 | 0.00 | 0.00 | 0.02 | N/A | 0.53 |
| NRAS3 | 0.00 | 0.00 | 0.02 | N/A | 0.53 |
| HRAS3 | 0.00 | 0.00 | 0.12 | N/A | 0.11 |
| RASA1 | 0.00 | 0.00 | 0.06 | N/A | 0.26 |
| TGFBR1 | 0.15 | 0.13 | 0.06 | 0.86 | 0.20 |
| TGFBR2 | 0.10 | 0.06 | 0.08 | 0.66 | 0.75 |
| CEBPA | 0.05 | 0.00 | 0.00 | 0.36 | 0.10 |
| PDK1 | 0.05 | 0.00 | 0.08 | 0.36 | 0.69 |
| BAP1 | 0.10 | 0.00 | 0.04 | 0.19 | 0.31 |
| CREBBP | 0.25 | 0.13 | 0.19 | 0.37 | 0.59 |
| BCC and SCC Drivers | |||||
| TP53 | 0.55 | 0.31 | 0.58 | 0.15 | 0.84 |
| KMT2D/MLL2 | 0.40 | 0.44 | 0.58 | 0.82 | 0.18 |
| AJUBA | 0.10 | 0.13 | 0.13 | 0.81 | 0.69 |
| MYH9 | 0.25 | 0.38 | 0.13 | 0.42 | 0.24 |
| TRAF3 | 0.10 | 0.13 | 0.06 | 0.81 | 0.53 |
| NSD1 | 0.20 | 0.13 | 0.08 | 0.55 | 0.14 |
| CDH1 | 0.05 | 0.13 | 0.10 | 0.42 | 0.53 |
| CASP8 | 0.15 | 0.06 | 0.19 | 0.41 | 0.69 |
| RAC13 | 0.05 | 0.00 | 0.04 | 0.36 | 0.85 |
| ARID1A | 0.45 | 0.19 | 0.19 | 0.10 | 0.03 |
| TP63 | 0.05 | 0.25 | 0.10 | 0.08 | 0.53 |
Abbreviations: BCC, basal cell carcinoma; BSC, basosquamous carcinoma; N/A, not applicable; SCC, squamous cell carcinoma.
Two-proportion Z-test, P-value two-tailed.
lncludes only known activating SMO mutations.
lncludes only oncogenic mutations reported in other samples identified in the Catalogue Of Somatic Mutations In Cancer (COSMIC).
Figure 2. Heat map of BCC and SCC driver gene mutations among tumor samples.

The analysis of signature genes revealed a greater similarity of mutation frequencies between BCCs and BSCs as compared with those of SCCs. includes only known activating SMO mutations. *Includes only oncogenic mutations reported in other samples identified in the Catalogue Of Somatic Mutations In Cancer (COSMIC). FS, Frameshift insertion or deletion; IF, In-frame insertion or deletion; MS, Missense mutation; NS, Nonsense mutation,
BSC lacks commonly found SCC driver mutations
We next compared genetic mutations between BSC and SCC (Table 1, Figure 2, and Supplementary Table S1). Fifteen percent and 20% of the BSC tumors were found to have NOTCH1 and NOTCH2 mutations, respectively, lower than the 46% and 37% of the SCC samples (P = 0.01 and P = 0.18) and more comparable to the frequencies observed in the BCCs, 31% and 25%, respectively (P = 0.24 and P = 0.72), suggesting that the BSCs do not harbor elevated NOTCH mutations at the frequency reported in SCCs. In addition, BSC also lacked oncogenic HRAS and KRAS mutations, which have been reported to be mutated in SCC (Que et al., 2018). Finally, there were no CDKN2A mutations present in BSC compared with 15% of SCC (P = 0.02). The differences in mutation frequencies suggest that BSC has a mutational landscape differing from SCC and lacks classic SCC driver mutations.
Principal component analysis
Principal component analysis demonstrated overlap between all three tumor groups, indicating some amount of similarity between all three. However, there was a statistically significant difference in the genetic landscape between the BSC and SCC tumors (P = 0.02) compared with the similar genetic distribution of the BSC and BCC tumor samples (P = 0.56) (Figure 3).
Figure 3. Principal component analysis of the BSC, BCC, and SCC tumor types.

Analysis of the BCC and SCC driver genes listed in Table 1 demonstrated similarity in the genetic distribution of the BSC and BCC tumor types (P = 0.56) and statistically significant difference in the genetic landscape of the BSC and SCC tumor types (P = 0.02). BCC, basal cell carcinoma; BSC, basosquamous carcinoma; PC, principal component; SCC, squamous cell carcinoma.
ARID1A is highly mutated in BSC
We identified the top 20 cancer genes mutated in BSC (Supplementary Tables S2 and S3). ARID1A was highly mutated in 45% of the BSCs compared with 19% of the BCC and 19% of the SCC tumors (P = 0.10, P = 0.03, respectively). ARID1A encodes a component of the SWI/SNF chromatin remodeling complex and plays a role in differentiation in a number of cancer types. ARID1A is frequently mutated across many cancers, and its mutations are associated with increased cell proliferation (Nagl et al., 2007; Zang et al., 2012), loss of differentiation (Gao et al., 2008; Nagl et al., 2007), and poor prognosis in multiple cancer types (Mamo et al., 2012; Wu and Roberts, 2013). Recent studies have shown that the loss of ARID1A attenuates the lineage-enforcing transcriptional activities of C/ebpα, thereby promoting cellular plasticity (Sun et al., 2016). In the setting of BSC, mutations in ARID1A may enable keratinocytes to acquire plasticity to squamatize. Other frequent cancer gene alterations among all tumor types included CSMD3, TP53, NAV3, and KMT2D (Bolshakov et al., 2003; Hertzler-Schaefer et al., 2014; Sharpe et al., 2015). These shared mutated genes are likely to be cancer drivers that contribute to cancer progression but do not contribute to the differentiation of specific tumor types.
ERK1/2 activation and loss of Hh signaling associated with squamatization in BSC
A recent study demonstrated the activation of ERK1/2 signaling in a single BCC that squamatized while undergoing SMO inhibition (Zhao et al., 2015). We hypothesized that activation of the mitogen-activated protein kinase (MAPK) pathway may also contribute to the squamatization of sporadic BSC. The Gli1 transcription factor is shuttled between the nucleus and cytoplasm by chaperone protein systems. Nuclear Gli1 is required for the Hh signaling pathway, while Gli1 is inactive in the cytoplasm (Mirza et al., 2019). Staining of the BSC samples with Gli1 and p-ERK demonstrated that typical of BCC, basaloid keratinocytes displayed higher nuclear Gli1 and minimal p-MEK staining (Figure 4, Supplementary Figures S1 and S2). In comparison, squamatized keratinocytes had higher p-MEK staining and loss of Gli1 expression. We also detected higher expression of p-ERK in the basaloid cells adjacent to the squamatized areas, indicating that activation of the RAS-MAPK pathway may drive squamatization with the subsequent loss of Gli1 expression as a secondary event. These results implicate the MAPK pathway in modulating the tumor plasticity observed in BSC.
Figure 4. RAS-MAPK pathway is activated in keratinocytes during squamatization.
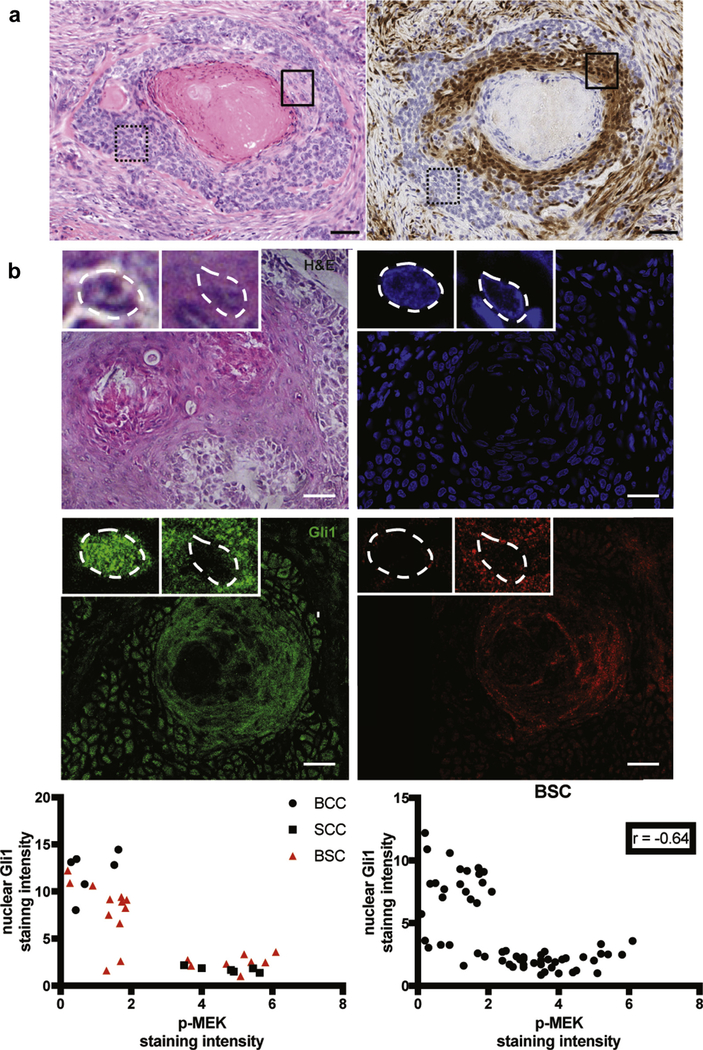
(a) Immunohistochemical staining demonstrates that basaloid keratinocytic cells typical of BCC, such as those contained in the dashed box, have minimal expression of p-ERK, whereas squamatized keratinocytes and adjacent basaloid cells, such as those contained in the solid box, show high expression of p-ERK. Left panel, H&E staining original magnification ×20. Right panel, p-ERK staining, original magnification ×20. Bar = 50 μm. (b) Representativepicturesofthe H&E, DAPI, nuclear Gli1 (as readoutfor Hh pathwayactivation) and p-MEK (as readout for Ras/MAPK pathway activation) immunostainings in human BSC samples. Higher magnifications of the basal and squamous areas of the tumor are shown in the top left and top midline framed pictures, respectively, with nuclei highlighted by white dashed lines. Basaloid keratinocytic cells typical of BCC have a higher expression of nuclear Gli1 and low p-MEK, whereas squamatized keratinocytes have high p-MEK and low Gli1. BCC, basal cell carcinoma; BSC, basosquamous carcinoma; H&E, hematoxylin and eosin; Hh, Hedgehog; MAPK, mitogen-activated protein kinase; SCC, squamous cell carcinoma. Bar = 25 μm.
Evolutionary analysis
Of the 20 BSC samples, 10 (50%) had mutations in PTCH1 or SMO, drivers of BCC, with nine mutations in PTCH1 and one mutation in SMO. All 10 of these mutations were clonal (Figure 5, Supplementary Figure S3). Nine of the 20 BSC samples had mutations in ARID1A, with five of these mutations being clonal and four being subclonal. Of these nine mutations, six occurred directly after mutations in PTCH1, and five occurred directly before mutations in TP53, indicating a possible sequence of squamous transformation (Supplementary Figure S4). Analysis of the individual allele frequencies for the driver genes in the 20 BSC samples demonstrated that the ARID1A allele frequency is less than that of PTCH1 in six of seven samples in which they co-occur, supporting the hypothesis that the PTCH1 mutation occurred before the ARID1A mutation occurring as a secondary event (Supplementary Table S4).
Figure 5. Evolutionary trajectories of select BSC samples.

Analysis of the evolutionary trajectories for BSC samples demonstrated mutations in PTCH1 before mutations in ARID1A, and then followed by TP53 and SCC driver genes in several BSC samples including the two displayed. Shaded regions represent clones and subclones. BSC, basosquamous carcinoma; GL, germline; SCC, squamous cell carcinoma.
DISCUSSION
Although BSC has histologic features of both BCC and SCC, the genetic etiology is poorly understood with controversy surrounding whether these tumors originate from SCC or BCC (Garcia et al., 2009). This study identified underlying PTCH1 and SMO mutations in most BSCs, supporting a role for the Hh signaling pathway as the initial driver of BSC. In addition, mutations in other known BCC drivers were identified, including MYCN, PPP6C, GRIN2A, CSMD3, DCC, PREX2, APC, PTEN, and PIK3CA. Consistent with this, principal component analysis of the mutations revealed that BSC has much closer genetic similarity to BCC than SCC. Evolutionary analysis demonstrated early PTCH1 mutations, followed by ARID1A, and then subsequent mutations suggesting a sequence of squamous transformation. Our data support the hypothesis that the BSCs originally derive from the BCCs and subsequently acquire mutations leading to squamatization.
Recently, there have been reports of BCCs evading Hh pathway suppression by SMO inhibitors through squamatization (Ransohoff et al., 2015; Zhao et al., 2015;Zhu et al., 2014). The plasticity of cancer cells may enable the BCCs to acquire changes in cell fate and become more “SCC-like” to escape reliance on the Hh signaling pathway (Jia et al., 2017). Based on our data, we hypothesize that mutations in the SWI/SNF complex may bestow a permissive environment allowing BCC cells to gain cellular plasticity. This suggests that the use of PARP inhibitors may be a potential therapeutic approach to BSC tumors, as recent studies have demonstrated that ARID1A deficiency sensitizes human cancer cells to PARP inhibitors (Shen et al., 2015). ARID1A mutations are also associated with defects in DNA mismatch repair and increased PD-L1 expression, allowing for the increased sensitivity of tumors to immunotherapy with PD-1 inhibitors (Shen et al., 2018). Additional molecular signals, such as activation of the MAPK pathway in BCC, in a permissive environment, may bestow squamatization. Furthermore, BSCs demonstrate decreased Gli1 and increased p-ERK in squamatized keratinocytes and adjacent basaloid cells but retain Gli1 expression in classic BCC-like portions of tumors. This has implications for the treatment of BSC, as they are likely to be resistant to SMO inhibitor therapeutics but may respond to combination therapies targeting both the Hh and MAPK pathways. Overall, the genetic landscape of BSC support the derivation of BSCs from BCCs and implicate the interplay of the MAPK and Hh pathways in modulating tumor plasticity between basaloid and squamatized phenotypes. Further studies are needed to elucidate the full spectrum of sequential events involved in the progression and squamatization of BSC.
MATERIALS & METHODS Sample collection
This study was approved by Stanford University’s Institutional Review Board. BSC samples were collected through a database search of patients from the Stanford Dermatopathology Service, and consent was waived given the institutional approval for the use of previously collected deidentified tissue (IRB #29381) (Supplementary Table S5). Cases of BSC since 2000 were identified using the following search terms: “basosquamous and carcinoma“; “basal and carcinoma and squamous and features”; and “metatypical.” The hematoxylin and eosin–stained slides of 32 skin biopsy samples matching the search terms were independently reviewed by a Stanford dermatopathologist. Twenty cases of BSC were found to meet the histologic criteria and are included in the study. All the BSC tumors included contained areas with more classic BCC features and areas with more classic SCC features, with areas of transition between the two. Tumor cellularity ranged from approximately 30% to 80%. The proportion of squamatized areas ranged from 20% to 80%, with a median of 50% and a mean of 46.8%. The proportion of basaloid areas ranged from 20% to 80%, with a median of 50% and a mean of 53.2%. A total of 52 cases of SCC with matched normal samples were included. Whole-genome sequence data of 13 SCC samples were obtained from UCSF. Whole-exome sequence data of 39 SCC samples were obtained from a study by Pickering et al. (2014). Sixteen cases of BCC with matched normal samples were obtained from Stanford University Clinics with written informed patient consent.
Immunofluorescence
Paraffin-embedded samples of three BSC samples, three BCC samples, and three SCC samples were immunostained for Gli1 and p-Mek. Gli1 (NBP1–78259, 1:200, Novus Biologicals, Littleton, CO), p-MEK (EPR3338, 1:250, Abcam, Cambridge, MA), and nuclei (Hoechst 33342, 1:2000, Invitrogen, Waltham, MA) were stained using a standard immunofluorescence protocol for formalin-fixed paraffin-embedded tissues. Antigen retrieval was performed in pH 6.0 citrate buffer (Vector Laboratories, Burlingame, CA). Pictures were taken on a Leica SP8 confocal microscope equipped with an adjustable white light laser and hybrid detectors (Leica Camera, Allendale, NJ). Nuclear Gli1 and cytoplasmic p-MEK quantifications were performed using ImageJ software (NIH, Bethesda, MD). Nuclear Gli1 and p-MEK were measured in similar fields of adjacent tumor slides. BCC and SCC tumors were used as positive controls for both nuclear Gli1 and cytoplasmic p-MEK stainings, respectively.
Immunohistochemistry
Immunohistochemistry for p-ERK was performed by HistoWiz Inc. (Brooklyn, NY) using standard operating procedures and fully automated workflow. The samples were processed, embedded in paraffin, and sectioned at 4 mm. Immunohistochemistry was performed on a Bond Rx autostainer (Leica Biosystems) with enzyme treatment (1:1000) using standard protocols. The antibodies used were rabbit p-ERK (Cell Signaling, 4307S, 1:100). Bond Polymer Refine anti-rabbit HRP Detection (Leica Biosystems, Wetzlar, Germany) was used according to manufacturer’s instructions. Sections were then counterstained with hematoxylin, dehydrated and film coverslipped using a TissueTek-Prisma and Coverslipper (Sakura, Torrance, CA). Whole slide scanning (original magnification ×40) was performed on an Aperio AT2 (Leica Biosystems). Images were quantified using Halo image analysis software (Indica Labs, Corrales, NM) using CytoNuclear module.
Targeted sequencing and analysis
DNA was isolated from the 20 BSC formalin-fixed paraffin-embeddedsections using the Qiagen DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. The 260:280 absorbance ratio of each sample was measured to confirm the purity of the isolated DNA.
Capture libraries were constructed using the ACE Extended Cancer Panel spanning the coding regions of 1,641 cancer genes (Personalis, Menlo Park, CA). Sequencing was performed on the HiSeq 2500 sequencing system (Illumina, San Diego, CA) at 2 × 100-bp sequencing of paired-ends to an average 134-fold coverage. Sequence reads were aligned via Burrows-Wheeler Aligner version 0.7.15 to human reference genome hg19, and Picard tools, version 1.130 was used for sorting and marking duplicates (Li and Durbin, 2010). We applied the Genome Analysis Toolkit (GATK), version 3.6, for base quality score recalibration, indel realignment, and duplicate removal (McKenna et al., 2010). A panel of normals was created via GATK MuTect2 using all BCC and SCC matched normal samples, which was used for variant calling via MuTect2 according to the GATK best practices (Cibulskis et al., 2013; DePristo et al., 2011; Van der Auwera et al., 2013). Identified variants were annotated with AnnoL, version 2.8 (Personalis, Menlo Park, CA) for presence in the Single Nucleotide Polymorphism database (dbSNP138), Polymorphism Phenotyping v2 (PolyPhen-2), Sorting Intolerant From Tolerant (SIFT), and MutationTaster as provided in the GATK resource bundle (Adzhubei et al., 2010; Schwarz et al., 2014).
Whole-exome sequencing and analysis
Fresh tissue samples of 16 BCCs along with paired-matched blood samples were obtained and the DNA isolated using the DNeasy Blood & Tissue kit according to the manufacturer’s instructions (Qiagen). Capture libraries were constructed using the Agilent SureSelect XT Human All Exon V4 Kit (Agilent Technologies, Palo Alto, CA) according to the manufacturer’s instructions, and enriched exome libraries were multiplexed and sequenced on the Illumina HiSeq 2500 platform to generate 100-bp paired-end reads to an average 72-fold coverage.
Whole-exome sequencing data for 39 SCC samples were obtained from the supplementary materials of a study by Pickering et al. (2014). Exomes were sequenced on an Illumina HiSeq 2000 platform to an average 115-fold coverage.
Sequence reads were aligned via Burrows-Wheeler Aligner, version 0.7.15, to human reference genome hg19, and Picard tools, version 1.130, was used for sorting and marking duplicates (Li and Durbin, 2010). We applied GATK, version 3.6 for base quality score recalibration, indel realignment, and duplicate removal (McKenna et al., 2010). MuTect2 was used for variant calling according to the GATK best practices (Cibulskis et al., 2013; DePristo et al., 2011; Van der Auwera et al., 2013). Variants were annotated with AnnoL, version 2.8, for their presence in dbSNP138, PolyPhen-2, SIFT, and MutationTaster as provided in the GATK resource bundle (Adzhubei et al., 2010; Schwarz et al., 2014).
Whole-genome sequencing and analysis
Whole-genome sequencing data for 13 SCC samples at 40-fold coverage were obtained from Dr Raymond Cho at UCSF (dbGaP accession phs000830.v1.p1) (Zheng et al., 2014). Sequence reads were aligned via Burrows-Wheeler Aligner, version 0.7.15, to human reference genome hg19, and Picard tools, version 1.130, was used for sorting and marking duplicates (Li and Durbin, 2010). We applied GATK, version 3.6 for base quality score recalibration, indel realignment, and duplicate removal (McKenna et al., 2010). MuTect2 was used for variant calling according to GATK best practices (Cibulskis et al., 2013; DePristo et al., 2011; Van der Auwera et al., 2013). Variants were annotated with AnnoL, version 2.8, for presence in dbSNP138, PolyPhen-2, SIFT, and MutationTaster as provided in the GATK resource bundle (Adzhubei et al., 2010; Schwarz et al., 2014).
Mutational analysis
Mutations were identified among the 1,641 genes used for BSC targeted sequencing using the following criteria. Candidate mutations were selected on the basis that they were found exclusively in tumor sample and not identified in normal controls. Synonymous mutations, noncoding mutations, allele frequency less than 5%, and mutations with ExAC Non-Finnish European > 0.01 were filtered from the analysis. Frequently mutated genes commonly found in exome sequencing data were also excluded as identified in previous reports (Shyr et al., 2014). SMO mutations were filtered to only include known activating mutations. The oncogenes MYCN, PPP6C, PIK3CA, KRAS, NRAS, HRAS, and RAC1 were filtered to only include variants reported in other samples identified in the Catalogue Of Somatic Mutations In Cancer. Tumor suppressor genes were filtered to exclude missense mutations annotated as benign in dbSNP138. Inclusion criteria required that tumor suppressor gene missense mutations be predicted pathogenic by at least two out of three of prediction scores SIFT, PolyPhen2, and MutationTaster (Dong et al., 2015). The number of nonsynonymous mutations and in-frame indels were calculated for each sample. Candidate cancer driver gene mutations were filtered for expression in the skin according to the HPA data set for RNA expression based on RNA-Seq on The Human Protein Atlas (http://www.proteinatlas.org) with a threshold value of 1.0 transcripts per million or above (Uhlén et al., (2015). The calculated mutation rates were 19.875 nonsynonymous mutations/Mb for BCCs, 19.596 nonsynonymous mutations/Mb for SCCs, and 21.9 nonsynonymous mutations/Mb for BSCs.
A comprehensive search of the literature was performed to determine the gene sets associated with BCC and SCC. The final gene sets consisted of 11 BCC-associated genes (PTCH1, SMO, MYCN, PPP6C, GRIN2A, CSMD3, DCC, PREX2, APC, PIK3CA, and PTEN), 14 SCC-associated genes (CDKN2A, NOTCH1, NOTCH2, NOTCH3, KRAS, NRAS, HRAS, RASA1, TGFBR1, TGFBR2, CEBPA, PDK1, BAP1, and CREBBP), and 11 genes found to be commonly mutated in both BCC and SCC (TP53, KTM2D, AJUBA, MYH9, TRAF3, NSD1, CDH1, CASP8, RAC1, ARID1A, and TP63).
Statistical analysis
A two proportion z-test was used to compare the gene mutation frequencies between tumor types. All the P-values were two-sided with P < 0.05 considered as a gene having statistically significant different mutation frequencies between compared tumor types. Fisher exact test was used for comparison of mutational signatures among tumor types. All P-values were two-sided with P < 0.05 considered as compared tumor types having a statistically significant different distribution of mutational signatures. Principal component analysis was used to compare the presence of BCC and SCC driver genes between different tumor types, and ellipses were drawn assuming a normal distribution at a 75% confidence level. Mahalanobis Distance was used to measure the distance between different gene points and central distribution of the tumor types calculated from the principal component analysis (Goodpaster and Kennedy 2011; Team, 2016). Hotelling’s two-sample t-test was used to compare the principal components of different pairs of tumor groups (Nordhausen et al., 2012), with P < 0.05 considered as a statistically significant difference.
Evolutionary trajectories
Mutations in the BSC samples were analyzed with REVOLVER (Caravagna et al., 2018) using transfer learning to detect repeated patterns of cancer evolution in multiple patients and to generate evolutionary trajectories. Clonal architecture was inferred with SciClone (Miller et al., 2014) using a mixture of beta distributions on variant allele frequencies. Clones and subclones were ordered usingClonEvol (Dang et al., 2017) to generate consensus trees that satisfied all subclonal relationships among the mutation frequencies in the clones.
Data availability statement
Datasets related to this article, (BSC) targeted sequencing and (BCC) whole-exome sequencing data, have been deposited to the NIH Sequence Read Archive (SRP139077) and GEO (GSE58377), respectively.
Supplementary Material
ACKNOWLEDGMENTS
The research reported in this publication was supported by funds from the American Medical Association Foundation Seed Grant (AC), the Swiss National Science Foundation, Switzerland, under award number P300PB_171576 (FK), the American Skin Association (KYS), the National
Cancer Institute of the National Institutes of Health under award number K23 CA21 1793 (KYS), and a philanthropic gift by Mr Robert McIntyre (KYS). This work was supported in part by NIH P30 CA124435 using the Stanford Cancer Institute Shared Resource, Genetics Bioinformatics Service Center.
Abbreviations:
- BCC
basal cell carcinoma
- BSC
basosquamous carcinoma
- Hh
Hedgehog
- MAPK
mitogen-activated protein kinase
- SCC
squamous cell carcinoma
Footnotes
SUPPLEMENTARY MATERIAL
Supplementary material is linked to the online version of the paper at www.jidonline.org, and at https://doi.org/10.10167j.jid.2019.03.1163.
CONFLICT OF INTEREST
ALSC is a clinical investigator for studies funded by Regenero, Novartis and Merck which are not related to this study. The other authors state no conflicts of interest.
REFERENCES
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen KJ, Cappel MA, Killian JM, Brewer JD. Basosquamous carcinoma and metatypical basal cell carcinoma: a review of treatment with Mohs micrographic surgery. Int J Dermatol 2014;53:1395–403. [DOI] [PubMed] [Google Scholar]
- Atwood SX, Sarin KY, Whitson RJ, Li JR, Kim G, Rezaee M, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell 2015;27:342–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolshakov S, Walker CM, Strom SS, Selvan MS, Clayman GL, El-Naggar A, et al. p53 mutations in human aggressive and nonaggressive basal and squamous cell carcinomas. Clin Cancer Res 2003;9:228–34. [PubMed] [Google Scholar]
- Bonilla X, Parmentier L, King B, Bezrukov F, Kaya G, Zoete V, et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat Genet 2016;48:398–406. [DOI] [PubMed] [Google Scholar]
- Cammareri P, Rose AM, Vincent DF, Wang J, Nagano A, Libertini S, et al. Inactivation of TGFbeta receptors in stem cells drives cutaneous squamous cell carcinoma. Nat Commun 2016;7:12493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caravagna G, Giarratano Y, Ramazzotti D, Tomlinson I, Graham TA, Sanguinetti G, et al. Detecting repeated cancer evolution from multi-region tumor sequencing data. Nat Methods 2018;15:707–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino D, Lowe L, Brown DL. Basosquamous carcinoma-an underrecognized, high-risk cutaneous neoplasm: case study and review of the literature. J Plast Reconstr Aesthet Surg 2006;59:424–8. [DOI] [PubMed] [Google Scholar]
- Dang HX, White BS, Foltz SM, Miller CA, Luo J, Fields RC, et al. ClonEvol: clonal ordering and visualization in cancer sequencing. Ann Oncol 2017;28:3076–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefano A, Dispenza F, Petrucci AG, Citraro L, Croce A. Features of biopsy in diagnosis of metatypical basal cell carcinoma (basosquamous Carcinoma) of head and neck. Otolaryngol Pol 2012;66:419–23. [DOI] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet 2015;24: 2125–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Tate P, Hu P, Tjian R, Skarnes WC, Wang Z. ES cell pluripotency and germ-layer formation require the SWI/SNF chromatin remodeling component BAF250a. Proc Natl Acad Sci USA 2008;105:6656–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia C, Poletti E, Crowson AN. Basosquamous carcinoma. J Am Acad Dermatol 2009;60:137–43. [DOI] [PubMed] [Google Scholar]
- Goodpaster AM, Kennedy MA. Quantification and statistical significance analysis of group separation in NMR-based metabonomics studies. Chemometr Intell Lab Syst 2011;109:162–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertzler-Schaefer K, Mathew G, Somani AK, Tholpady S, Kadakia MP, Chen Y, et al. Pten loss induces autocrine FGF signaling to promote skin tumorigenesis. Cell Rep 2014;6:818–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- larrobino A, Messina JL, Kudchadkar R, Sondak VK. Emergence of a squamous cell carcinoma phenotype following treatment of metastatic basal cell carcinoma with vismodegib. J Am Acad Dermatol 2013;69:e33–4. [DOI] [PubMed] [Google Scholar]
- Jayaraman SS, Rayhan DJ, Hazany S, Kolodney MS. Mutational landscape of basal cell carcinomas by whole-exome sequencing. J Invest Dermatol 2014;134:213–20. [DOI] [PubMed] [Google Scholar]
- Jia D, Jolly MK, Kulkarni P, Levine H. Phenotypic plasticity and cell fate decisions in cancer: insights from dynamical systems theory. Cancers (Basel) 2017;9(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010;26:589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney ML. What is basosquamous carcinoma? Dermatol Surg 2000;26: 505–6. [DOI] [PubMed] [Google Scholar]
- Mamo A, Cavallone L, Tuzmen S, Chabot C, Ferrario C, Hassan S, et al. An integrated genomic approach identifies ARID1A as a candidate tumorsuppressor gene in breast cancer. Oncogene 2012;31:2090–100. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20: 1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CA, White BS, Dees ND, Griffith M, Welch JS, Griffith OL, et al. Sci-Clone: inferring clonal architecture and tracking the spatial and temporal patterns of tumor evolution. PLOS Comput Biol 2014;10:e1003665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza AN, McKellar SA, Urman NM, Brown AS, Hollmig T, Aasi SZ, et al. LAP2 proteins chaperone GLI1 movement between the lamina and chromatin to regulate transcription. Cell 2019;176:198–212.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagl NG Jr, Wang X, Patsialou A, Van Scoy M, Moran E. Distinct mammalian SWI/SNF chromatin remodeling complexes with opposing roles in cell-cycle control. EMBO J 2007;26:752–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordhausen K, Sirkia S, Oja H, Tyler DE. ICSNP: tools for Multivariate Nonparametrics. R package version 1.0–9 https://cran.r-project.org/package=ICSNP; 2012. (accessed 1 May 2018).
- Orouji A, Goerdt S, Utikal J, Leverkus M. Multiple highly and moderately differentiated squamous cell carcinomas of the skin during vismodegib treatment of inoperable basal cell carcinoma. Br J Dermatol 2014;1 71: 431–3. [DOI] [PubMed] [Google Scholar]
- Pickering CR, Zhou JH, Lee JJ, Drummond JA, Peng SA, Saade RE, et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin Cancer Res 2014;20:6582–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que SKT, Zwald FO, Schmults CD. Cutaneous squamous cell carcinoma: incidence, risk factors, diagnosis, and staging. J Am Acad Dermatol 2018;78:237–47. [DOI] [PubMed] [Google Scholar]
- Ransohoff KJ, Tang JY, Sarin KY. Squamous change in basal-cell carcinoma with drug resistance. N Engl J Med 2015;373:1079–82. [DOI] [PubMed] [Google Scholar]
- Rose AM, Sansom OJ, Inman GJ. Loss of TGF-beta signaling drives cscC from skin stem cells - More evidence. Cell Cycle 2017;16:386–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saintes C, Saint-Jean M, Brocard A, Peuvrel L, Renaut JJ, Khammari A, et al. Development of squamous cell carcinoma into basal cell carcinoma under treatment with vismodegib. J Eur Acad Dermatol Venereol 2015;29: 1006–9. [DOI] [PubMed] [Google Scholar]
- Schwaederle M, Elkin SK, Tomson BN, Carter JL, Kurzrock R. Squamousness: next-generation sequencing reveals shared molecular features across squamous tumor types. Cell Cycle 2015;14:2355–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 2014;11: 361–2. [DOI] [PubMed] [Google Scholar]
- Sharpe HJ, Pau G, Dijkgraaf GJ, Basset-Seguin N, Modrusan Z, Januario T, et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 2015;27:327–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Ju Z, Zhao W, Wang L, Peng Y, Ge Z, et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med 2018;24: 556–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Peng Y, Wei L, Zhang W, Yang L, Lan L, et al. ARID1A deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors. Cancer Discov 2015;5:752–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyr C, Tarailo-Graovac M, Gottlieb M, Lee JJ, van Karnebeek C, Wasserman WW. FLAGS, frequently mutated genes in public exomes. BMC Med Genomics 2014;7:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- South AP, Purdie KJ, Watt SA, Haldenby S, den Breems NY, Dimon M, et al. NOTCH1 mutations occur early during cutaneous squamous cell carcinogenesis. J Invest Dermatol 2014;134:2630–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Chuang JC, Kanchwala M, Wu L, Celen C, Li L, et al. Suppression of the SWI/SNF component Arid1a promotes mammalian regeneration. Cell Stem Cell 2016;18:456–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team RC. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2016. [Google Scholar]
- Uhlén M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science 2015;347:1260419. [DOI] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy- Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 2013;43:11.10.1–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkenstein S, Wohlschlaeger J, Liebau J, Arens A, Lehnerdt G, Jahnke K, et al. Basosquamous carcinoma-a rare but aggressive skin malignancy. J Plast Reconstr Aesthet Surg 2010;63:e304–6. [DOI] [PubMed] [Google Scholar]
- Wu JN, Roberts CW. ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov 2013;3:35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz AS, Ozer HG, Gillespie JL, Allain DC, Bernhardt MN, Furlan KC, et al. Differential mutation frequencies in metastatic cutaneous squamous cell carcinomas versus primary tumors. Cancer 2017;123: 1184–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang ZJ, Cutcutache I, Poon SL, Zhang SL, McPherson JR, Tao J, et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat Genet 2012;44:570–4. [DOI] [PubMed] [Google Scholar]
- Zhao X, Ponomaryov T, Ornell KJ, Zhou P, Dabral SK, Pak E, et al. RAS/ MAPK activation drives resistance to smo inhibition, metastasis, and tumor evolution in shh pathway-dependent tumors. Cancer Res 2015;75: 3623–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng CL, Wang NJ, Chung J, Moslehi H, Sanborn JZ, Hur JS, et al. Transcription restores DNA repair to heterochromatin, determining regional mutation rates in cancer genomes. Cell Rep 2014;9:1228–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu GA, Sundram U, Chang AL. Two different scenarios of squamous cell carcinoma within advanced basal cell carcinomas: cases illustrating the importance of serial biopsy during vismodegib usage. JAMA Dermatol 2014;150:970–3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets related to this article, (BSC) targeted sequencing and (BCC) whole-exome sequencing data, have been deposited to the NIH Sequence Read Archive (SRP139077) and GEO (GSE58377), respectively.