

 Optically active 1,3-enyne aldehydes undergo stereoselective, Au-catalyzed conversion to bicyclo[3.3.0]octenones (up to 97% ee, good d.r.) via a domino sequence involving 1,3-acyloxy migration, Nazarov cyclization and an unprecedented aldol addition.
Optically active 1,3-enyne aldehydes undergo stereoselective, Au-catalyzed conversion to bicyclo[3.3.0]octenones (up to 97% ee, good d.r.) via a domino sequence involving 1,3-acyloxy migration, Nazarov cyclization and an unprecedented aldol addition.
Abstract
Stereoselective synthesis of bicyclo[3.3.0]octenones from chiral 1,3-enyne aldehydes bearing propargylic acetates is described. The method is based on a Au(i)-catalyzed domino sequence with concomitant transfer of chirality involving 1,3-acyloxy migration followed by Nazarov cyclization and an unprecedented aldol addition. The method furnishes densely functionalized bicyclic structures in high yields, with up to 97% ee and good diastereoselectivity.
Introduction
In recent years, Au(i) complexes have yielded powerful catalysts for the activation of alkynes, giving rise to a diversity of new and efficient catalytic transformations leading to high molecular complexity.1 Among them, Au(i)-catalyzed reactions of enynes bearing propargylic carboxylates have been found to be especially versatile.2 A particularly valuable feature of Au-catalysis is that it enables sequential formation of numerous C–C bonds via cascading cycloisomerization processes. Herein, we document a stereoselective process in which acyclic propargylic enynes with a pendant aldehyde are converted into optically active, highly functionalized diquinanes. The cascade reaction proceeds through 1,3-acyloxy migration, Nazarov cyclization, and aldol addition. Thus, in one step the method leads to the generation of functionalized, fused five-membered rings bearing three contiguous stereocenters (Fig. 1).
Fig. 1. Au(i)-catalyzed cascade reaction in this work.

In a pioneering study, Zhang and Wang reported the Au(i)-catalyzed synthesis of cyclopentenones from readily available enyne ester.3 The proposed mechanism involves 1,3-acyloxy migration followed by Nazarov cyclization, [1,2]-hydride shift, deauration, and hydrolysis of the intermediate cyclopentadienyl acetate formed (C, Fig. 2a).4 In light of the mechanistic model, we wondered whether aldehydes incorporated into the starting enynes would be trapped in situ by C and form diquinanes (2).5 The consequence of such a process is to add an unprecedented aldol addition reaction to the cascade that includes 1,3-migration and Nazarov cyclization.6 This would require that aldehydes in the starting enynes are compatible with and survive the intervening steps and thereby subsequently be engaged as electrophiles. In an ideal scenario, Au(i) would function as a catalyst in all three fundamental steps of the cascading sequence: 1,3-acyloxy migration, Nazarov cyclization, and aldol reaction. In 2009, Malacria and Fensterbank demonstrated that the Au(i)–vinylcarbene7 formed from Nazarov cyclization (5) could participate in intramolecular cyclopropanations with substrates incorporating olefin side chains (Fig. 2b).4,8 Importantly, this is the sole example of concomitant transfer of chirality from an enantioenriched propargylic acetate. The fact that optically active propargyl acetates furnish optically active cycloalkanes led to the suggestion of an intermediate “bent-allene” complex (A)4,9 that serves as a conduit for the transmission of the stereochemical information in the starting materials to enantioenriched products.10 Accordingly, starting from a readily accessible chiral linear precursor, the domino reaction would be envisioned to give rise to highly functionalized optically active bicyclo[3.3.0]octenones with the configuration of the starting propargyl acetate potentially determining the configuration of all three stereocenters in the final product.
Fig. 2. (a) Mechanistic working model for the process developed by Zhang. (b) Previous work by Fensterbank and Malacria with concomitant transfer of chirality.

We recently reported the first asymmetric total synthesis of (–)-merochlorin A (7), relying on the key Au(i)-catalyzed reaction cascade described herein to access the diquinane core (Fig. 3).11 Intrigued by the significant potential of this transformation, we were interested in investigating this novel reaction cascade because of its general applicability as a synthetic method for the synthesis of optically active bicyclo[3.3.0]octanes, which are abundant structural motifs in natural products such as anislactone A (8) and cantabrenonic acid (9).12
Fig. 3. Representative natural products incorporating a diquinane core.

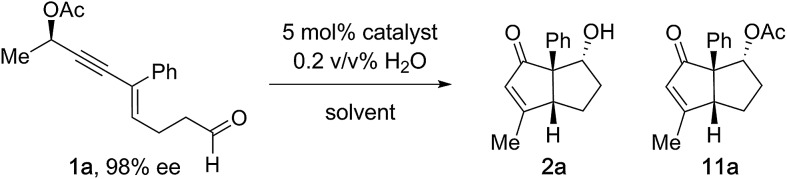
Results and discussion
Our synthetic studies commenced with the investigation of readily available chiral enyne (R)-1a (98% ee, Table 1). Thus, 1a was treated with various gold complexes. While AuCl3, AuCl(PPh3)/AgSbF6, and Ipr AuCl/AgSbF6 (10) furnished enone 2a in either poor yield or with poor chirality transfer, the treatment of 1a with 5 mol% Au(MeCN)(JohnPhos)SbF6 (Echavarren's catalyst, 3)13 in THF gave 2a in 71% yield, with 91% ee and a d.r. of 85 : 15 (entry 4). It is noteworthy that 6% of aldol acetate 11a was isolated, a product arising from acetate transfer during the terminating aldol reaction. Furthermore, initial screening revealed that the reaction benefits from the presence of 0.2 vol% H2O in the reaction mixture (see the ESI†), which is consistent with prior reports of H2O favoring the formation of A.4a Screening of reaction media (entries 4–7) revealed that solvents other than THF led to decreased d.r. and increased formation of acetate 11a. Interestingly, when the reaction was conducted in CH2Cl2 in the absence of water, aldol acetate 11a was found to be the dominant product (entry 5).
Table 1. Optimization of reaction conditions a .
| ||||||
| Entry | Catalyst | Solvent | Yield 2 b | Yield 11a b | ee c | d.r. |
| 1 | AuCl3 | THF | 42% | — | 79% | 85 : 15 |
| 2 d | AuClPPh3 | THF | 67% | — | 44% | 81 : 19 |
| 3 d | 10 e | THF | 61% | — | 67% | 81 : 19 |
| 4 | 3 | THF | 71% | 6% | 91% | 85 : 15 |
| 5 | 3 | CH2Cl2 | 19% | 74% | 92% | 74 : 26 |
| 6 | 3 | Acetone | 51% | 23% | 91% | 88 : 12 |
| 7 | 3 | Dioxane | 50% | 18% | 91% | 60 : 40 |
| 8 f | 3 | THF | 76% | — | 93% | 89 : 11 |
aReaction conditions: 5 mol% catalyst, 0.2 vol% H2O, solvent (0.05 M), 0.09–0.11 mmol 1a, and RT.
bYields of isolated products.
cEnantiomeric excess determined by supercritical fluid chromatography (SFC) on a chiral stationary phase and reported for the major diastereomer.
d5 mol% AgSbF6 was added.
eIpr = 1,3-bis(2,6-diisopropylphenyl)imidiazol-2-ylidene.
fReaction was performed at –10 °C for 24 h.
When the influence of the reaction temperature was investigated, optimal performance was found at –10 °C, leading to 76% of the desired bicyclo[3.3.0]octenone with 93% ee and 89 : 11 d.r. (entry 8).
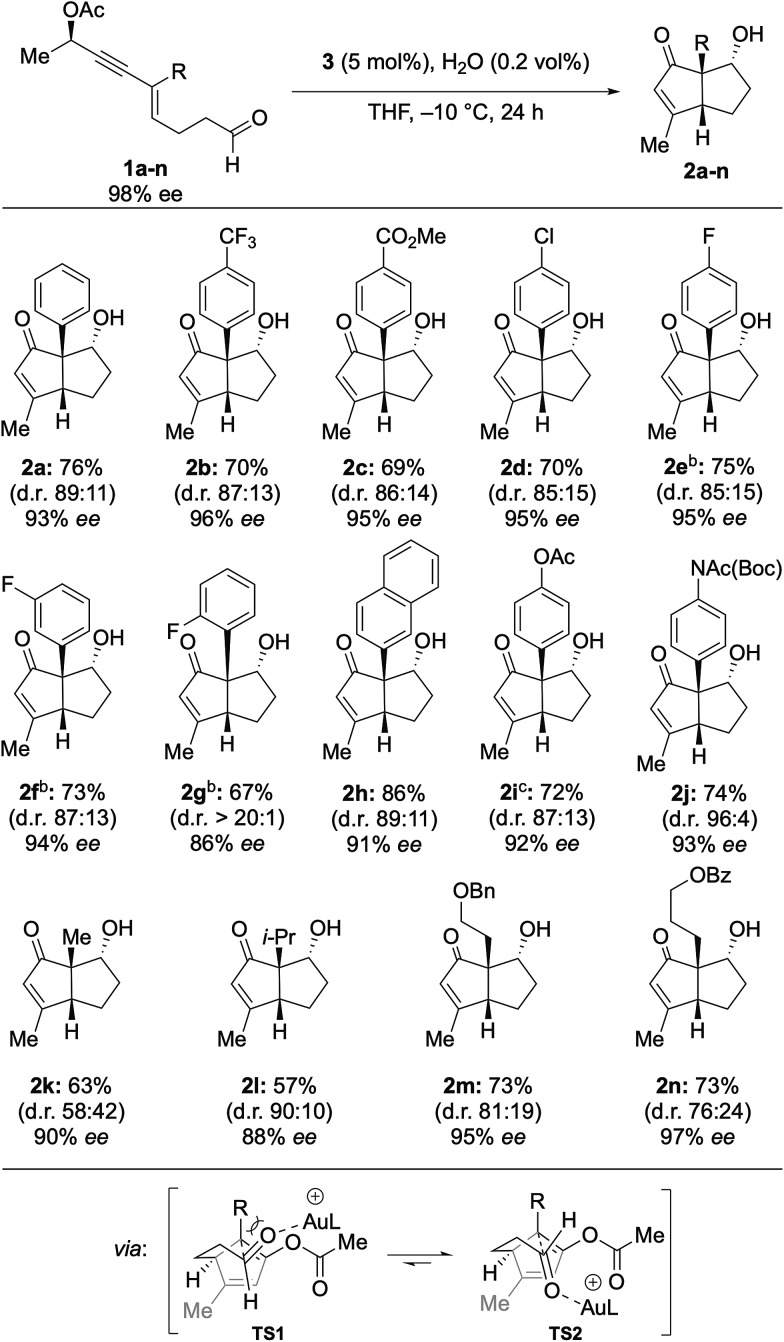
With the optimized reaction conditions in hand, the scope of this method was examined next (Table 2). To this end, arenes incorporating various substituents were examined. Electron withdrawing groups (CF3, CO2Me, Cl, and F) were well tolerated, leading to the isolation of the corresponding pentalenes in high yields with high ee (2a–2f). ortho-Fluorinated arene 2g showed some deleterious effects on chirality transfer. Strikingly, however, a d.r. of >20 : 1 was observed. A 3-naphthyl group was also compatible affording 2h in 86% yield with 91% ee. Electron donating groups were found to be problematic.14 However, the cyclization of protected aniline and phenol proceeded smoothly, furnishing the corresponding enones 2i and 2j in high yield with high ee. The observed diastereoselectivity may be explained by the consideration of a pair of possible transition states TS1 and TS2 (Table 2), in which the former suffers from destabilizing non-bonding interactions which are absent in the latter.
Table 2. Substrate scope of the stereoselective 1,3-acyloxy migration/Nazarov cyclization/aldol addition sequence a .
|
aReaction conditions: 5 mol% 3, 0.2 vol% H2O, THF (0.05 M), and –10 °C. Enantiomeric excess reported for the major diastereomer.
bReaction warmed to RT after 16 h.
cIsolated yield over two steps from the primary alcohol.
We next examined enyne aldehydes substituted with aliphatic groups. Enyne 1k bearing a methyl substituent was found to be a suitable substrate, affording 2k in 63% yield with 90% ee, albeit with poor diastereoselectivity. Nevertheless, introduction of a sterically more demanding isopropyl led to increased 90 : 10 d.r. (2l). A benzyloxy-substituted alkyl chain was tolerated to give 2m in 72% yield with 95% ee. Pentalene 2n including a benzoyl-protected alcohol is an intermediate of the total synthesis of (–)-merochlorin A (7)11 and was obtained with excellent transfer of chirality (97% ee) from the acyclic starting material (98% ee). These findings considerably expand the utility of the procedure developed, as it allows for the synthesis of highly functionalized aliphatic bicyclo[3.3.0]octenones. The absolute configuration was determined by X-ray crystallographic analysis of 2e and 2j, which confirmed the mechanistic model established by Fensterbank and Malacria for 6 (Fig. 4, see ESI†).
Fig. 4. Absolute configuration assigned by X-ray crystallographic analysis of (R,R,R)-2e and (R,R,R)-2j. For ORTEP representations the thermal ellipsoids are shown at 50% probability.

A salient feature of this transformation is the formation of a quaternary center at a ring junction. Such substitution would otherwise be cumbersome to access via conventional synthetic approaches such as α-arylation or alkylation of this sterically hindered position.
To further demonstrate the utility of this method, we were curious to explore whether the aldehyde is able to undergo the aldol reaction when placed at a different position within the starting material. For this purpose, we synthesized enyne 12 (d.r. >20 : 1) using Enders' SAMP protocol (see the ESI†).15 Remarkably, triquinane 13 was isolated as a single diastereomer in 60% yield when 12 was subjected to the standard reaction conditions (5 mol% 3, 0.2 vol% H2O, and THF) at room temperature (Fig. 5). Furthermore, propargylic enyne 14 (E/Z = 6 : 1) underwent smooth cyclization to give enone 15 (74%, with 78% ee) as a single diastereomer. The absolute configuration of 15 was determined by X-ray crystallographic analysis. Much to our delight, the Au(i)-catalyzed reaction cascade developed herein is thus useful for the synthesis of enantioenriched bicyclo[3.3.0]octane, spiro[4.4]nonane and tricyclo[6.3.0.01,5]undecane systems.
Fig. 5. Synthesis of tricyclo[6.3.0.01,5]undecane 13 and spiro[4.4]nonane 15. Reagents and conditions: 5 mol% 3, 0.2 v/v% H2O, THF (0.05 M), RT, and 24 h. For ORTEP representations the thermal ellipsoids are shown at 50% probability.

In 1974, Mukaiyama and co-workers reported the reaction of vinyl acetates with acetal- and carbonyl compounds in the presence of strong Lewis acids (TiCl4, SnCl4, and BF3·Et2O) to afford the corresponding aldol products in modest yield.8 In light of this report, the described aldol reaction of a cyclopentadienyl acetate in the presence of the unarguably poor Lewis acidic Au(i)-catalyst is remarkable.16,17 In principle, two different mechanistic pathways can be proposed (Fig. 6). Acetate 16, formed after initial 1,3-acyloxy migration/Nazarov cyclization, may first be hydrolyzed under the employed standard reaction conditions (Table 1, entry 8) to give cyclopentadienyl enolate 17, which then undergoes an intramolecular aldol reaction to afford pentalene 1. Importantly, while the initial hydrolysis may be mediated by the Au(i)-catalyst, this pathway fails to account for the formation of acetate 11. Alternatively, the aldol reaction could proceed directly from the enol acetate to give 18, which then could either undergo hydrolysis to produce enone 1 or suffer intramolecular acetate transfer to afford acetate 11.
Fig. 6. Mechanistic options for the terminal aldol reaction.

Optimization studies (Table 1) revealed that the Au(i)-catalyzed tandem 1,3-acyloxy migration/Nazarov cyclization/aldol reaction sequence occurs in the absence of water. In particular, if the reaction is conducted in dry solvents such as THF or CH2Cl2, acetate 11a is isolated as the major product (Fig. 7A). Furthermore, isolated enol acetate 19 does not participate in intramolecular aldol reactions or hydrolysis under the standard reaction conditions in the absence of the Au(i)-catalyst (Fig. 7B). However, the treatment of 19 with 5 mol% Echavarren's catalyst (3) furnished the desired products 2a and 11a in high yield. In summary, these results suggest that the Au(i)-catalyst is involved in all three reaction steps and the terminal aldol reaction indeed proceeds directly from the enol acetate. Nevertheless, the possibility of a second operative pathway via initial hydrolysis of 19 in wet solvent in the presence of the Au(i)-catalyst cannot be entirely excluded.
Fig. 7. Experiments to elucidate the mechanism of the terminal aldol reaction of cyclopentadienyl acetate 19.

In summary, we surmise that the water present under the standard reaction conditions primarily suppresses the acetate transfer via hydrolysis of oxocarbenium ion intermediate 18 (Fig. 6).
Conclusions
In conclusion, a Au(i)-catalyzed stereoselective tandem 1,3-acyloxy migration/Nazarov cyclization/aldol addition cascade has been developed. In a single step, this method allows for the generation of two rings bearing three contiguous stereocenters, one of them being quaternary. The bicyclo[3.3.0]octenones obtained are isolated in high yields and with high stereoselectivity. The ability of intermediate cyclopentadienyl acetates to undergo an intramolecular Au(i)-catalyzed aldol reaction constitutes a reactivity pattern with great potential to provide the foundation for new cascading sequences leading to highly functionalized building blocks. Studies to apply this method to complex systems and to include it in further total syntheses of natural products are currently ongoing in our laboratories and will be reported in due course.
Conflicts of interest
The authors declare no conflict of interest.
Supplementary Material
Acknowledgments
This research was funded by ETH Zurich. M. B. acknowledges a fellowship of the Stipendienfonds Schweizerische Chemische Industrie (SSCI). We are greatful to Dr Nils Trapp and Michael Solar for the X-ray crystallographic analysis.
Footnotes
†Electronic supplementary information (ESI) available. CCDC 1517154–1517155 and 1913669. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02828e
References
- For recent reviews on homogenous gold catalysis, see: ; (a) Fürstner A., Davies P. W. Angew. Chem., Int. Ed. 2007;46:3410–3449. doi: 10.1002/anie.200604335. [DOI] [PubMed] [Google Scholar]; (b) Hashmi A. S. K. Chem. Rev. 2007;107:3180–3211. doi: 10.1021/cr000436x. [DOI] [PubMed] [Google Scholar]; (c) Gorin D. J., Sherry B. D., Toste F. D. Chem. Rev. 2008;108:3351–3378. doi: 10.1021/cr068430g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Jiménez-Núñez E., Echavarren A. M. Chem. Rev. 2008;108:3326–3350. doi: 10.1021/cr0684319. [DOI] [PubMed] [Google Scholar]; (e) Krause N., Winter C. Chem. Rev. 2011;111:1994–2009. doi: 10.1021/cr1004088. [DOI] [PubMed] [Google Scholar]; (f) Dorel R., Echavarren A. M. Chem. Rev. 2015;115:9028–9072. doi: 10.1021/cr500691k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected examples of gold(i)-catalyzed reactions of enynes bearing propargylic carboxylates, see: ; (a) Zhang L. J. Am. Chem. Soc. 2005;127:16804–16805. doi: 10.1021/ja056419c. [DOI] [PubMed] [Google Scholar]; (b) Gorin D. J., Dubé P., Toste F. D. J. Am. Chem. Soc. 2006;128:14480–14481. doi: 10.1021/ja066694e. [DOI] [PubMed] [Google Scholar]; (c) Buzas A., Gagosz F. J. Am. Chem. Soc. 2006;128:12614–12615. doi: 10.1021/ja064223m. [DOI] [PubMed] [Google Scholar]; (d) Lemière G., Gandon V., Cariou K., Fukuyama T., Dhimane A.-L., Fensterbank L., Malacria M. Org. Lett. 2007;9:2207–2209. doi: 10.1021/ol070788r. [DOI] [PubMed] [Google Scholar]; (e) Schwier T., Sromek A. W., Yap D. M. L., Chernyak D., Gevorgyan V. J. Am. Chem. Soc. 2007;129:9868–9878. doi: 10.1021/ja072446m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Dudnik A. S., Schwier T., Gevorgyan V. Tetrahedron. 2009;65:1859–1870. [Google Scholar]; (g) Shapiro N. D., Shi Y., Toste F. D. J. Am. Chem. Soc. 2009;131:11654–11655. doi: 10.1021/ja903863b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Teng T.-M., Liu R.-S. J. Am. Chem. Soc. 2010;132:9298–9300. doi: 10.1021/ja1043837. [DOI] [PubMed] [Google Scholar]; (i) Rao W., Susanti D., Chan P. W. H. J. Am. Chem. Soc. 2011;133:15248–15251. doi: 10.1021/ja2052304. [DOI] [PubMed] [Google Scholar]; (j) Rao W., Sally, Berry S. N., Chan P. W. H. Chem.–Eur. J. 2014;20:13174–13180. doi: 10.1002/chem.201402500. [DOI] [PubMed] [Google Scholar]; (k) Zi W., Wu H., Toste F. D. J. Am. Chem. Soc. 2015;137:3225–3228. doi: 10.1021/jacs.5b00613. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Bürki C., Whyte A., Arndt S., Hashmi A. S. K., Lautens M. Org. Lett. 2016;18:5058–5061. doi: 10.1021/acs.orglett.6b02505. [DOI] [PubMed] [Google Scholar]
- Zhang L., Wang S. J. Am. Chem. Soc. 2006;128:1442–1443. doi: 10.1021/ja057327q. [DOI] [PubMed] [Google Scholar]
- The mechanistic model is deduced from the following work: ; (a) Shi F.-Q., Li X., Xia Y., Zhang L., Yu Z.-X. J. Am. Chem. Soc. 2007;129:15503–15512. doi: 10.1021/ja071070+. [DOI] [PubMed] [Google Scholar]; (b) Lemiére G., Gandon V., Cariou K., Hours A., Fukuyama T., Dhimane A.-L., Fensterbank L., Malacria M. J. Am. Chem. Soc. 2009;131:2993–3006. doi: 10.1021/ja808872u. [DOI] [PubMed] [Google Scholar]
- Mukaiyama T., Izawa T., Saigo K. Chem. Lett. 1974;3:323–326. [Google Scholar]
- For a comprehensive overview on Au(i)-catalyzed aldol reactions, see: Hubbert C. and Hashmi S. K., Gold-Catalyzed Aldol and Related Reactions in Modern Gold Catalyzed Synthesis, ed. A. S. K. Hashmi and D. F. Toste, Wiley-VCH, Weinheim, 2012, pp. 237–261. [Google Scholar]
- (a) Comprido L. N. D., Klein J. E. M. N., Knizia G., Kastner J., Hashmi A. S. K. Angew. Chem., Int. Ed. 2015;54:10336–10340. doi: 10.1002/anie.201412401. [DOI] [PubMed] [Google Scholar]; (b) Pflästerer D., Hashmi S. A. K. Chem. Soc. Rev. 2016;45:1331–1367. doi: 10.1039/c5cs00721f. [DOI] [PubMed] [Google Scholar]; (c) Rudolph M., Hashmi A. S. K. Chem. Soc. Rev. 2012;41:2448–2462. doi: 10.1039/c1cs15279c. [DOI] [PubMed] [Google Scholar]; (d) Hashmi A. S. K., Rudolph M. Chem. Soc. Rev. 2008;37:1766–1775. doi: 10.1039/b615629k. [DOI] [PubMed] [Google Scholar]
- Lemière G., Gandon V., Cariou K., Fukuyama T., Dhimane A.-L., Fensterbank L., Malacria M. Org. Lett. 2007;9:2207–2209. doi: 10.1021/ol070788r. [DOI] [PubMed] [Google Scholar]
- For selected reports on “bent-allenes” and their transition metal complexes, see: ; (a) Tonner R., Frenking G. Angew. Chem., Int. Ed. 2007;46:8695–8696. doi: 10.1002/anie.200701632. [DOI] [PubMed] [Google Scholar]; (b) Kaufhold O., Hahn F. E. Angew. Chem., Int. Ed. 2008;47:4057–4061. doi: 10.1002/anie.200800846. [DOI] [PubMed] [Google Scholar]; (c) Fürstner A., Alcarazo M., Goddard R., Lehmann C. W. Angew. Chem., Int. Ed. 2008;47:3210–3214. doi: 10.1002/anie.200705798. [DOI] [PubMed] [Google Scholar]; (d) Dyker C. A., Lavallo V., Donnadieu B., Bertrand G. Angew. Chem., Int. Ed. 2008;47:3206–3209. doi: 10.1002/anie.200705620. [DOI] [PubMed] [Google Scholar]; (e) Hsu Y.-C., Shen J.-S., Lin B.-C., Chen W.-C., Chan Y.-T., Ching W.-M., Yap G. P. A., Hsu C.-P., Ong T.-G. Angew. Chem., Int. Ed. 2015;54:2420–2424. doi: 10.1002/anie.201406481. [DOI] [PubMed] [Google Scholar]; (f) en W.-C., Shen J.-S., Jurca T., Peng C.-J., Lin Y.-H., Wang Y.-P., Shih W.-C., Yap G. P. A., Ong T.-G. Angew. Chem., Int. Ed. 2015;54:15207–15212. doi: 10.1002/anie.201507921. [DOI] [PubMed] [Google Scholar]; (g) Pranckevicius C., Fan L., Stephan D. W. J. Am. Chem. Soc. 2015;137:5582–5589. doi: 10.1021/jacs.5b02203. [DOI] [PubMed] [Google Scholar]; (h) Pranckevicius C., Liu L., Bertrand G., Stephan D. W. Angew. Chem., Int. Ed. 2016;55:5536–5540. doi: 10.1002/anie.201600765. [DOI] [PubMed] [Google Scholar]
- (a) Gandon V., Lemière G., Hours A., Fensterbank L., Malacria M. Angew. Chem., Int. Ed. 2008;47:7534–7538. doi: 10.1002/anie.200802332. [DOI] [PubMed] [Google Scholar]; (b) Brown T. J., Sugie A., Leed M. G. D., Wiedenhoefer R. A. Chem.–Eur. J. 2012;18:6959–6971. doi: 10.1002/chem.201103289. [DOI] [PubMed] [Google Scholar]; (c) Harris R. J., Nakafuku K., Widenhoefer R. A. Chem.–Eur. J. 2014;20:12245–12254. doi: 10.1002/chem.201402729. [DOI] [PubMed] [Google Scholar]
- Brandstätter M., Freis M., Huwyler N., Carreira E. M. Angew. Chem., Int. Ed. 2019;58:2490–2494. doi: 10.1002/anie.201813090. [DOI] [PubMed] [Google Scholar]
- (a) Paquette L. A., Doherty A. M., Synthesis of Nonpolycyclopentanoid Natural Products by Way of Diquinane Intermediates, in Polyquinane Chemistry. Reactivity and Structure Concepts in Organic Chemistry, Springer, Berlin, Heidelberg, 1987, vol. 26, pp. 112–126. [Google Scholar]; (b) Paquette L. A., Doherty A. M., Synthesis of Diquinane Natural Products, in Polyquinane Chemistry. Reactivity and Structure Concepts in Organic Chemistry, Springer, Berlin, Heidelberg, 1987, vol. 26, pp. 127–168. [Google Scholar]
- Herrero-Gomez E., Nieto-Oberhuber C., López S., Benet-Buchholz J., Echavarren A. M. Angew. Chem., Int. Ed. 2006;45:5455–5459. doi: 10.1002/anie.200601688. [DOI] [PubMed] [Google Scholar]
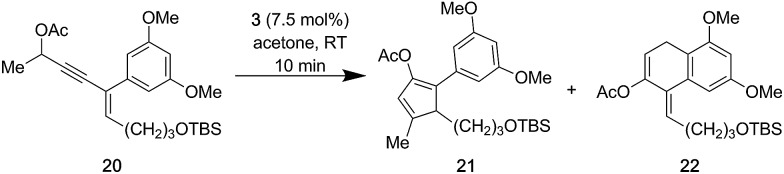
- During our studies, electron rich enynesDuring our studies, electron rich enynes
such as 20 furnished substantial amounts of such as 20 furnished substantial amounts of 22, which is derived from a 6-endo-trig cyclization of the intermediate Au(i)–allene complex and the electron rich arene
- Bockstiegel B., Enders D. Synthesis. 1989;7:493–496. [Google Scholar]
- For the pioneering and first asymmetric Au(i)-catalyzed aldol reaction, see: Ito Y., Sawamura M., Hayashi T., J. Am. Chem. Soc., 1986, 108 , 6405 –6406 , , For a comprehensive overview on Au(i)-catalyzed aldol reactions, see: C. Hubbert and S. K.Hashmi, Gold-Catalyzed Aldol and Related Reactions, in Modern Gold Catalyzed Synthesis, ed. A. S. K. Hashmi and D. F. Toste, Wiley-VCH, Weinheim, Germany, 2012, pp. 237–261 . [Google Scholar]
- For Au(i)-catalyzed reaction cascades containing aldol reactions, see: ; (a) Jin H. M., Tian B., Song X. L., Xie J., Rudolph M., Rominger F., Hashmi A. S. K. Angew. Chem., Int. Ed. 2016;55:12688–12692. doi: 10.1002/anie.201606043. [DOI] [PubMed] [Google Scholar]; (b) Wang G., Liu X. H., Chen Y. S., Yang J., Li J., Lin L. L., Feng X. M. ACS Catal. 2016;6:2482–2486. [Google Scholar]; (c) Yu Z. Z., Qiu H. L., Liu L., Zhang J. L. Chem. Commun. 2016;52:2257–2260. doi: 10.1039/c5cc08880a. [DOI] [PubMed] [Google Scholar]; (d) Kang D., Park S., Ryu T., Lee P. H. Org. Lett. 2012;14:3912–3915. doi: 10.1021/ol301660f. [DOI] [PubMed] [Google Scholar]; (e) Korner C., Starkov P., Sheppard T. D. J. Am. Chem. Soc. 2010;132:5968–5969. doi: 10.1021/ja102129c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.