

Abstract
Biological gels generally require polymeric chains that produce long‐lived physical entanglements. Low molecular weight colloids offer an alternative to macromolecular gels, but often require ad‐hoc synthetic procedures. Here, a short biomimetic peptide composed of eight amino acid residues derived from squid sucker ring teeth proteins is demonstrated to form hydrogel in water without any cross‐linking agent or chemical modification and exhibits a stiffness on par with the stiffest peptide hydrogels. Combining solution and solid‐state NMR, circular dichroism, infrared spectroscopy, and X‐ray scattering, the peptide is shown to form a supramolecular, semiflexible gel assembled from unusual right‐handed 310‐helices stabilized in solution by π–π stacking. During gelation, the 310‐helices undergo conformational transition into antiparallel β‐sheets with formation of new interpeptide hydrophobic interactions, and molecular dynamic simulations corroborate stabilization by cross β‐sheet oligomerization. The current study broadens the range of secondary structures available to create supramolecular hydrogels, and introduces 310‐helices as transient building blocks for gelation via a 310‐to‐β‐sheet conformational transition.
Keywords: β‐sheet transition, molecular dynamics (MD) simulations, NMR spectroscopy, peptide hydrogels, suckerin
A short biomimetic peptide self‐assembles into a hydrogel by a conformational transition from 310‐helices to antiparallel β‐sheets. The eight‐residue peptide—derived from squid sucker ring teeth proteins—assembles into hydrogels with concentration‐dependent and tunable modulus without cross‐linking agents or chemical modifications. This study discusses the gelation dynamics and introduces 310‐helices as transient building blocks for peptide hydrogels.
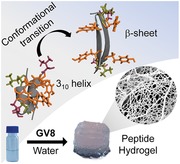
1. Introduction
Peptide hydrogels are increasingly explored for biomedical applications such as wound healing patches,1, 2 cell culture scaffolds for tissue engineering,3 drug delivery vehicles,4 or as substrates to study stem cell differentiation.5 Peptides are particularly attractive as building blocks for hydrogels because: (i) their chemical structure and polydispersity is fully controlled, (ii) they exhibit high biocompatibility,6 and (iii) their degradation products (amino acids) are readily cleared or reabsorbed by the metabolism.6 In addition, bioactive functionality can be achieved: for example RGD peptides can be incorporated into the peptide sequence7 to promote cell recognition or the peptide can be chemically modified with fluorescent probes and dye reporters8 or with functional groups to promote subsequent crosslinking reactions.9
In recent years, the ability to tune the gels' mechanical properties has become an increasingly important factor in the consideration of gel design.10 While some hydrogels employ cross‐linking to vary the elastic properties,11 others can be altered by varying the amount of salt in the gelation buffer or by adjusting the peptide concentration.12 In many cases, short peptide‐based hydrogels are assembled from β‐sheets, β‐hairpins, or coiled‐coil α‐helices.13, 14 Some peptides employ organic solvents to trigger gelation or toxic chemicals for cross‐linking, which is not ideal from a biocompatibility perspective.11, 15 Hence peptide hydrogels assembled from uncommon secondary structural constructs may expand the existing peptide hydrogel libraries and have the potential to provide new characteristics, such as a broader range of moduli and water‐based gelation.
Here, we present a short 8‐residue peptide (Ac‐GLYGGYGV‐NH2 hereafter called GV8) that gels in water. GV8 exhibits a tunable, concentration‐dependent mechanical response with ≈25‐fold variation in storage modulus (G′) and a maximum value reaching 35.5 kPa that places it among the stiffest protein‐based hydrogels. The peptide sequence originates from suckerin proteins discovered in the sucker ring teeth (SRT) of the jumbo squid.16, 17, 18 Suckerins are a protein family with a characteristic modular primary structure consisting of long Gly‐rich modules previously assumed to form mostly unordered domains, which are intervened by smaller Ala‐ and His‐rich modules that self‐assemble into stiffer β‐sheet elements.17, 18 However, a recent NMR study indicated that the Gly‐rich domain can also form β‐sheets stabilized by aromatic side‐chain interactions.19 GX8 peptides (where X = Val, Leu, and Phe) are specifically located in the Gly‐rich modules of suckerin‐19 with a high occurrence of 13 copies. Combining circular dichroism (CD), Fourier Transform Infrared Spectroscopy (FTIR), solution and solid‐state NMR, wide‐angle X‐ray scattering (WAXS), and molecular dynamics (MD) simulations, we find that GV8 peptide forms 310 helices in solution and undergoes a conformation change into antiparallel β‐sheets during gelation.
2. Results and Discussion
2.1. Peptide Gelation
We obtained the GV8 peptide hydrogel by simple incubation of the peptide in deionized (DI) water, with gelation occurring at peptide concentrations ranging from 10 to 20 × 10−3 m and a concentration‐dependent gelation time between 5 and 9 h. The minimal critical gelation concentration (C gc) in water was 10 × 10−3 m, below which we did not observe gelation. We monitored the gelation kinetics by measuring the absorbance (OD550nm) of the peptide solutions at 550 nm (Figure S1, Supporting Information), whereby OD550nm increased during the gelation process and plateaued once gelation was complete.20 We also attempted to mutate the C‐terminus Val residue with Leu (GL8), Ala (GA8), Phe (GF8), Ser (GS8), Lys (GK8), or Ile (GI8), but these peptides were not able to form gels in water, illustrating the key role of terminal Val in gelation as corroborated by NMR studies. GF8 and GI8 peptides remained in solution with some aggregates observed over time, whereas GL8 self‐assembled into large (mm‐size) and stiff beads (Figure S2, Supporting Information).
2.2. Macro‐ and Microgel Structure
We then examined the morphology and topology of GV8 hydrogel by Cryo‐Electron Microscopy (Cryo‐EM), atomic force microscopy (AFM), and scanning electron microscopy (SEM). 20 × 10−3 m GV8 peptide solution was incubated for 3 h prior to blotting and vitrification to preserve the natural nanostructure of the sample in hydrated conditions21 for Cryo‐EM imaging. Long fibers less than 10 nm wide were observed (Figure 1 a) with consistent twisted morphologies and average periods of ≈80 nm along the fibers. AFM imaging was performed on a thin layer of dried gel, revealing a surface topology of a network of fibers (Figure 1b), and the height profile (Figure S3, Supporting Information) revealed fibers of ≈5–10 nm in height thereby matching the Cryo‐EM observations. Since drying and conventional lyophilization causes the hydrogel structure to collapse, samples for SEM were prepared by snap‐freezing GV8 hydrogel in liquid N2 for at least 5 min followed by cryo‐fracture and immediate lyophilization to obtain representative cross‐sections. SEM imaging revealed a porous structure (Figure 1c, left) constructed by sheet‐like structures and closer examination indicated that the sheets were formed by a fibrous network of peptides (Figure 1c, right).
Figure 1.
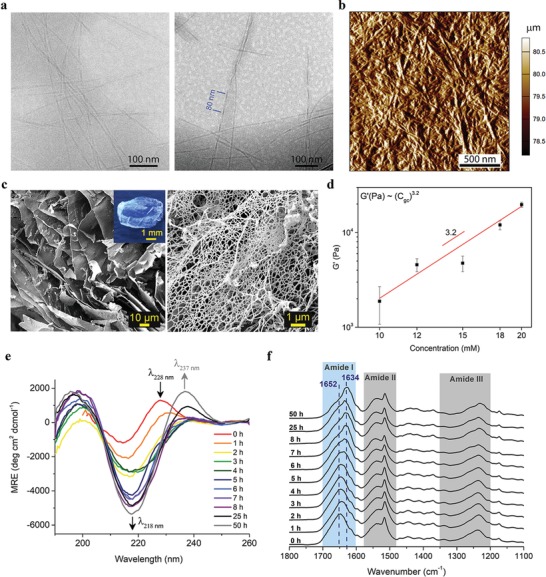
Structural features and physico‐chemical properties of GV8 peptide hydrogel observed with time‐series spectroscopy measurements during gelation. a) Cryo‐EM images of GV8 hydrogel fibrils and their twisted morphology (right image) with average periodicity of ≈80 nm. b) AFM amplitude profile of dried GV8 hydrogel with fibers of ≈6–10 nm height. c) SEM images of GV8 hydrogel cross‐section revealing sheet‐like morphology (left image) made of fibers (right image). A photograph of the hydrogel is shown in the inset. d) Scaling law plot of plateau G′ versus peptide concentration. e) CD and f) FTIR spectra recorded over 50 h indicate significant increase in β‐sheet content (intensity increase at λ218 nm and ν∼ 1634 cm −1, respectively).
2.3. Rheology Characterization
The hydrogel exhibited robust mechanical properties and could readily be manipulated and sectioned into thin slices (Movie S1, Supporting Information). In order to characterize the gel's mechanical properties, we prepared GV8 hydrogels with peptide concentrations C gc ranging from 10 to 20 × 10−3 m and conducted oscillation frequency sweeps at 0.25% shear strain (Figure S4, Supporting Information). The shear storage modulus (G′) exhibited a scaling power law as a function of peptide concentration (G′ vs C gc) with a power law index of 3.2 (Figure 1d), allowing us to tune the storage modulus ≈25‐fold over a narrow range of peptide concentration. We note that this power law index is significantly higher than the “universal” scaling law determined for protein‐based semiflexible networks22, 23, 24 where G′ scales as C 11/5 or for cross‐linked gels that exhibit a G′ ∝ C 2.5 scaling law.25 Instead, this behavior is well captured by the fractal gel model, , where d b is the fractal dimension of the connecting chain and d f is the dimensionality of the repeating fractal cluster. Taking d f = 1.6 for the fractal dimension as obtained by small angle X‐ray scattering (SAXS, Figure 2 a) yields d b = 1.5, which is a typical value of heat set protein gels.26 This behavior suggests that gelation does not proceed by entanglement of long fibrils, but rather by growth of shorter fibrillar clusters and conformational transition as evidenced by NMR and WAXS measurements described later. In line with this picture, GV8 did not strain‐stiffen, likely because the chain length is shorter than in conventional biological gels. In quantitative terms, the maximum shear modulus of GV8 of 35.5 kPa is on par with the stiffest, non‐crosslinked short peptide‐based hydrogel containing only natural amino acid residues.12 A wide range of moduli have been reported for short peptide hydrogels10, 27, 28, 29 with stiffest gels reached in gels containing modified amino acids, synthetic functional groups, or which have been crosslinked.30, 31 The ability to tune the stiffness from 1.3 to 35.5 kPa is particularly appealing for stem cell differentiation studies since gel stiffness has been well‐documented to govern cell adhesion and regulation based on the substrate's mechanical feedback,5, 32, 33, 34 with stiffness values in the range 0.1–1, 8–17, and 25–40 kPa sought after for neurogenic, myogenic, and osteogenic differentiation, respectively. The advantage of our GV8 hydrogel for such applications is that its stiffness can be modulated solely by varying the peptide concentration without any additional chemical modifications.
Figure 2.
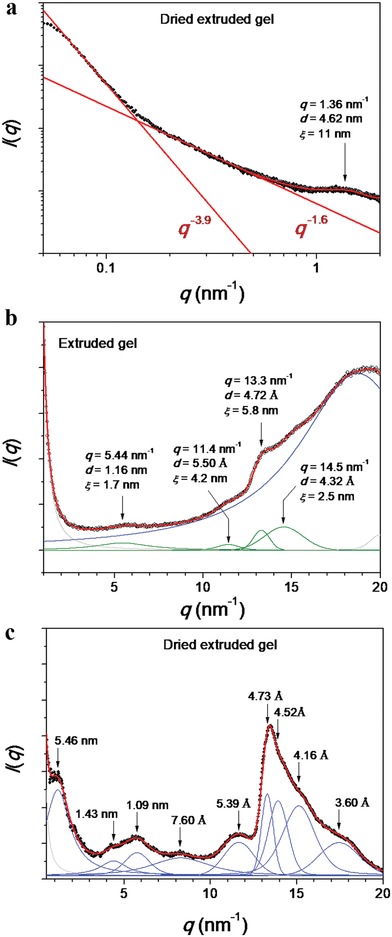
SAXS and WAXS patterns for the GV8 peptide extruded gel together with the fitting curve (red) and peaks assignment. a) SAXS profile for dried extruded GV8 gel. WAXS patterns of GV8 gel in the b) hydrated state and in the c) dried state.
2.4. Circular Dichroism and FTIR Spectroscopy
Next, we conducted time‐dependent CD and attenuated total reflection Fourier transform infrared spectroscopy (ATR‐FTIR) measurements on 20 × 10−3 m GV8 from its initial solution state until its postgelation state in order to reveal secondary structural changes during self‐assembly (Figure 1e,f). At time 0 h, the CD spectrum consisted of a minimum at 215 nm and two maxima at 200 and 228 nm (Figure 1e). The bands at 215 and 228 nm are attributed to the resultant exciton couplet of Tyr–Tyr exciton interaction of their π→π* transitions, also known as CD Cotton effect35, 36 indicating interaction between the Tyr aromatic chromophores,37, 38 as also corroborated by our 2D NMR data. Over the course of gelation (1–5 h), the minimum shifted to 218 nm with a significant increase in intensity, and the maximum at 228 nm diminished, thereby transitioning to β‐sheet secondary structure. The CD spectra remained constant after 5 h in agreement with our OD550nm measurements of 20 × 10−3 m GV8 peptide (Figure S1, Supporting Information), whereby the absorbance plateau onset at 5 h indicated that gelation was complete without any significant structural changes after 5 h. Upon further incubation postgelation (25 and 50 h), a new maximum appeared at 237 nm and can be assigned to aromatic transitions of the Tyr residues,39 which we postulate is related to the conformational transition of GV8 peptide during gelation.
ATR‐FTIR was performed on dried 20 × 10−3 m GV8 hydrogels incubated over the same time points as in CD studies. The samples were snap‐freezed in liquid N2 to arrest the structural assembly of the peptides at stipulated time points. Amide I bands were deconvoluted and peaks assigned to β‐sheets, unordered regions, helices and turns, or 310‐helices (Figure S5, Supporting Information).40, 41 β‐turns and 310‐helices were grouped together in our assignments as they are structurally alike42, 43 with similar hydrogen bond strengths, and hence close frequency positions within the Amide I band. Over the course of gelation, the Amide I band maximum at ν˜ max = 1652 cm−1 shifted to 1634 cm−1 (Figure 1f), confirming secondary structural change towards β‐structures. Semiquantitative analysis by deconvolution of Amide I band (Table S1, Supporting Information) indicated that the initial dominating secondary structures of GV8 peptide were turns and/or 310, whereas in the gel state β‐sheet structures were the most abundant (≈65 % at 50 h).
2.5. Solution NMR
In order to obtain the molecular level structure of GV8, we analyzed the 3D structure of the peptide in solution using NMR. To maintain the peptide in soluble form, its concentration was kept at 0.5 × 10−3 m. 2D 1H–1H TOCSY (TOtal Correlation SpectroscopY) and 1H–1H NOESY (Nuclear Overhauser Effect SpectroscopY) spectra showed well‐resolved cross‐peaks assigned to the individual amino acid residues of GV8 (Figure 3 a,c). The 1Hα chemical shift deviations (CSD)44 exhibited negative chemical shifts suggestive of a predominantly helical structure (Figure S6a, Supporting Information). However, precise examination of the 1H–1H NOESY spectrum revealed the absence of (i, i+4) medium range (Hα–HN) NOE connectivities typically observed in α‐helix (Figure 3b). Instead, we only detected (i, i+3) Hα–HN NOEs in addition to the strong (i, i+1) HN–HN NOEs, suggesting the presence of 310 helix.45 Further analysis also revealed the presence of ring proton NOEs between Y3 and Y6 stabilizing the 310 helical structure (Figure 3c). Collectively, these data indicated that the aromatic side chain interactions between Y3 and Y6 may lead the GV8 peptide to adopt a well‐defined 310 helix. This was also supported by NOEs between aliphatic side chains of L2 and V8 along the helical axis (Figure 3c). The 3D structure of the GV8 monomeric 310 helix calculated using a total of 39 NOE constraints (Table 1 ) is shown in Figure 3d,e. When Val8 was mutated to Leu (GL8) and Ala (GA8), the aromatic interactions between Y3 and Y6 disappeared and both mutated peptides remained in extended conformations (Figure S6c,d, Supporting Information).
Figure 3.
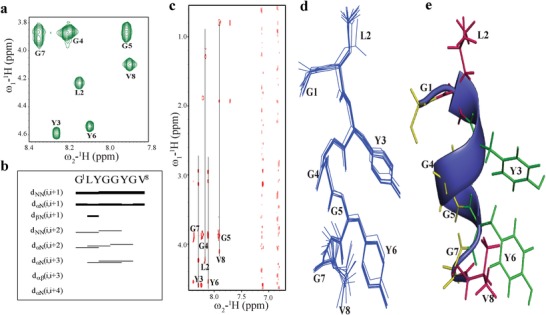
Solution NMR characterization of GV8 at monomeric concentration (0.5 × 10−3 m). a) 2D 1H–1H TOCSY spectrum of 0.5 × 10−3 m GV8 peptide delineating the individual spins of GV8 amino acid residues. b) Bar diagram representation of NOE connectivities detected for GV8 peptide. c) 2D 1H–1H NOESY spectra displaying weak ring interaction of Y3 and Y6. d) Superimposition of ten lowest energy structures of GV8 peptide. e) Representative structure of monomeric 310‐helix showing side chain stacking of Y3 and Y6.
Table 1.
Structural statistics summary of ten lowest energy structures of GV8 monomer and oligomer obtained from solution state NMR, and GV8 hydrogel by ssNMR
| GV8 monomer (solution NMR) | GV8 oligomer (solution NMR) | GV8 hydrogel (ssNMR) | |
|---|---|---|---|
| Distance restraints | |||
| Intraresidue (|i−j| = 0) | 9 | 18 | 22 |
| Sequential (|i−j| = 1) | 14 | 32 | 27 |
| Medium range (2≤ |i−j|≤4) | 16 | 56 | 0 |
| Long range (|i−j|≤ 5) | 0 | 32 | 10 |
| Total NOE constraints (solution NMR)/dipolar contacts (ssNMR) | 39 | 138 | 59 |
| Distance restraints violations | |||
| Number of violations | 9 | 51 | 33 |
| Maximum violation | ≤0.5 | ≤0.5 | ≤0.5 |
| Average target function value | 4.49 | 34.36 | 17.37 |
| Deviation from mean structure | |||
| Backbone atoms [Å] | 0.52 | 0.65 | 1.49 |
| Heavy atoms [Å] | 0.92 | 0.70 | 1.75 |
| Ramachandran plot for the mean structure | |||
| % residues in the most favorable and additionally allowed regions | 100 | 100 | 85 |
| % residues in the generously allowed region | 0 | 0 | 15 |
| % residues in the disallowed region | 0 | 0 | 0 |
To monitor gel formation, both 1D proton and 2D 1H–1H NOESY spectra of 20 × 10−3 m GV8 were recorded during a 4 h period. The peak intensities of the amide protons arising from residual peptides in solution decreased with time (Figure S6b, Supporting Information), implying that an increasing amount of peptide underwent structural rearrangement and were incorporated in the hydrogel.
Analysis of 2D 1H–1H TOCSY spectra acquired after 20 h demonstrated well resolved cross‐peaks corresponding to individual spins of GV8 peptide (Figure 4 a). 2D 1H–1H NOESY spectra displayed the (i, i+3) NOEs that are fingerprints of a 310‐helix45 (Figure 4c). Strikingly, residues at the C‐terminal (G7 and V8) were involved in displaying long range NOEs with residues at N‐terminal (Y3 and L2) (Figure 4c). The Hα of G7 interacted with L2 and Y3 residues (blue arrows and dotted lines, Figure 4c) and side chain β protons of V8 were also found to interact with L2 protons. These long‐range NOEs are attributed to cross‐strand NOEs resulting from oligomerization of the GV8 peptide after 20 h (Figure 4b). The aromatic packing interactions between Y3 and Y6 were also clearly detected owing to the high peptide concentration (Figure 4c).
Figure 4.
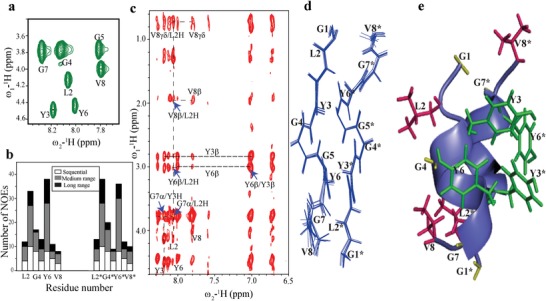
Solution NMR characterization of GV8 at oligomeric concentration (20 × 10−3 m). a) 2D 1H–1H TOCSY spectrum of 20 × 10−3 m GV8 peptide delineating the individual spins of GV8 amino acid residues. b) Bar diagram representation of residues that display sequential, medium range, and long‐range NOEs detected at 20 × 10−3 m concentration. c) 2D 1H–1H NOESY spectra displaying long range cross‐strand NOEs and ring proton interactions of Y3 and Y6 (marked with blue arrows and dotted lines). d) Superimposition of ten lowest energy structures of GV8 peptide at oligomeric 20 × 10−3 m concentration. e) Representative structure of dimeric 310‐helix of GV8 oligomer showing side chain arrangement. Aliphatic residues (L2, V8, L2*, and V8*) are shown in pink color and aromatic residues (Y3, Y6, Y3*, and Y6*) in green.
Using a total of 138 NOE constraints (Table 1), we calculated an ensemble of ten structures for GV8 composed of dimeric 310‐helical building blocks (adding 5 Gly as a linker between the two monomers). The aromatic residues Y3 and Y6 delineated a higher number of medium and long‐range NOEs (Figure 4b). Superposition of ten lower energy conformers led to root mean square deviations (RMSD) of backbone and heavy chains of 0.65 and 0.70 Å, respectively (Table 1 and Figure 4d). 3D structure calculation revealed that the hydrophobic face of the dimeric 310‐helix is composed of π‐stacking interactions between Y3 and Y6, while the exposed side of the dimeric helix is made up of aliphatic side chains L2 and V8 (Figure 4d,e). Procheck analysis of the 3D structure revealed that all residues resided in the sterically allowed regions (Table 1).
2.6. Amide Temperature Coefficient and H/D Exchange NMR Studies
The role of hydrogen bonds in stabilizing the 310‐helix was studied by calculating the protection factors from H/D exchange as well as the amide proton temperature coefficients (ΔδNH/ΔT) at various temperatures. A series of 2D 1H–1H TOCSY spectra were recorded every 30 min for the 0.5 and 20 × 10−3 m GV8 peptide dissolved in D2O. All Gly residues for both the monomer (0.5 × 10−3 m) and the oligomer (20 × 10−3 m) concentrations displayed protection factor of 60–80, supporting a significant H/D exchange protection inside the core of the 310‐helical structure (Figure S7a, Supporting Information). The protection factor of Y3 and Y6 increased with the peptide concentration, indicating enhanced aromatic interactions for the oligomeric form (Figure S7a, Supporting Information). Comparably, the amide proton temperature coefficients of all GV8 residues at both monomer and oligomer concentrations exhibited values more positive than −4.6 ppb/K.46 The Gly residues also exhibited more positive values in line with their higher protection factor values (Figure S7b, Supporting Information).
2.7. Solid State NMR
NMR characterizations of the gel state were conducted by ssNMR under magic angle spinning (MAS) conditions. All amino acids of GV8 hydrogel prepared with uniformly labeled 13C and 15N peptide were unambiguously assigned using the sequential walking method of 3D NCACX, NCOCX, and CANcoCX spectra (Figure 5 a). Analysis of the 2D 13C–13C DARR spectra acquired at 50 ms contact time revealed long range dipolar contacts between L2 and V8 side chains (Figure 5a,b). The Y3/Y6 ring packing interactions that were detected in oligomeric solutions of GV8 were no longer present in the hydrogel state.
Figure 5.
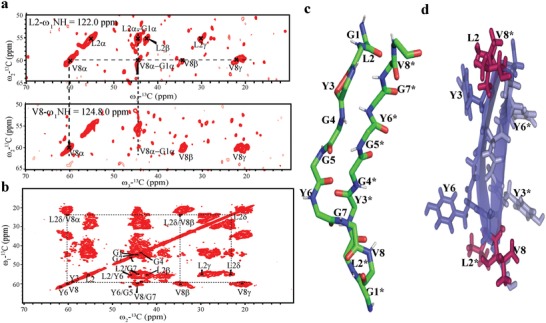
Solid NMR characterization of 13C–15N labeled GV8 hydrogel. a) Strip plots of L2 and V8, 3D NCACX spectra of 13C–15N labeled GV8 peptide hydrogel displaying long‐range contact of residues. b) 2D 13C–13C DARR spectra with contact time of 50 ms showing long‐range dipolar contacts between L2 and V8 side chains (β, δ, γ). c) Representative structure of dimeric extended conformation of GV8 hydrogel. d) Side chain disposition representation of antiparallel β‐sheets of GV8 hydrogel displaying interchain connectivity between L2 and V8 residues (L2/V8* and L2*/V8).
We then calculated the dimeric conformation of GV8 in the hydrogel state using intraresidue and sequential dipolar constraints (Experimental Section). An overview of ten lowest energy structures resulted in RMSD value of 1.49 Å for backbone atoms and 1.75 Å for heavy side chain atoms (Table 1). The lowest energy structure revealed that the GV8 hydrogel comprised of extended antiparallel β‐sheets (Figure 5c). The absence of Tyr ring packing interactions strongly suggests that during gelation, Tyr side chains (Y3 and Y6) rearranged to be exposed to solvent, and that at the same time stronger hydrophobic interactions between L2, V8, L2*, and V8* stabilized the antiparallel β‐strand conformation of the GV8 hydrogel (Figure 5d). Extended 310‐helices have been shown to act as intermediate seeds for the formation of amyloid (β‐sheet rich) aggregates,47 but to the best of our knowledge it has previously not been reported to induce hydrogel formation. Furthermore, none of the Val8‐mutated peptides (Figure S1, Supporting Information) gelled under the same conditions and neither control peptides GL8 nor GA8 formed 310‐helices in solution (Figure S6c,d, Supporting Information) hence corroborating that 310‐helix is a critical transient conformation leading to gelation of GV8. Val8 at the C‐terminus therefore plays a crucial role in stabilizing the 310‐helix structural intermediate through an intrachain hydrophobic interaction, which is not achieved with Ala or Leu residues.
2.8. WAXS of Extruded GV8 Hydrogel and MD Simulations
Confirmation of β‐sheet presence in the gel was gained by performing WAXS measurements in both the wet and dry states. In the wet state (Figure 2b) a peak at q = 5.44 nm−1 and a shoulder at 13.3 nm−1 were observed, corresponding to distances of 1.16 nm and 4.7 Å, respectively. These features are the hallmark of β‐sheet rich amyloid fibrils, with 1.16 nm corresponding to the inter‐β‐sheet spacing and 4.7 Å to the interstrand distance of β‐sheets. Interestingly, both features were greatly enhanced upon drying of the gel: the peak at 1.16 nm shifted to 1.09 nm due to dehydration, whereas the 4.7 Å peak now became the dominant scattering peak of the dried gel. The emergence of additional correlations is highlighted by a deconvolution of the intensity profile with Laurentian curves as shown in Figure 2c.
To further assess the conformation propensity of GV8, we conducted MD simulations on both oligomeric and 40‐mer constructs. These simulations predicted that oligomers of GV8 prefer the antiparallel β‐sheet conformation, especially the Leu2, Tyr3, and Tyr6 residues (Figure 6 a), whereas 310‐helices were not stable thus resulting in a very low concentration of 310‐helices after 200 ns simulations. These results indicate that antiparallel β‐sheets constituted the most stable structure at equilibrium. It is important to emphasize that the peptide concentration during MD simulations is much higher than in the soluble form and more representative of the gel state. Therefore, the very low concentration of 310‐helices at equilibrium is in line with the conformational transition detected in the gel by NMR. Based on solid‐state NMR and WAXS data in the gel state, we then conducted simulations on a 40‐mer antiparallel β‐sheet construct. The 100 ns simulation indicated a very high stability of antiparallel β‐sheets, in agreement with the WAXS measurements. Furthermore, the simulations indicated that β‐strands were stabilized by intersheet π–π stacking of Tyr residues (Figure 6b). This result corroborates the molecular‐level structure of GV8 by solid‐state NMR, which indicated that Tyr side‐chains in the gel state pointed out perpendicular to the strand direction, making them available to engage in intersheet interactions as predicted by the simulations. In addition, in‐register antiparallel β‐sheets were observed for both ssNMR and MD simulations, which we postulate is due to the arrangement of the Tyr side‐chains that leads to the least sterically‐hindered conformation.
Figure 6.
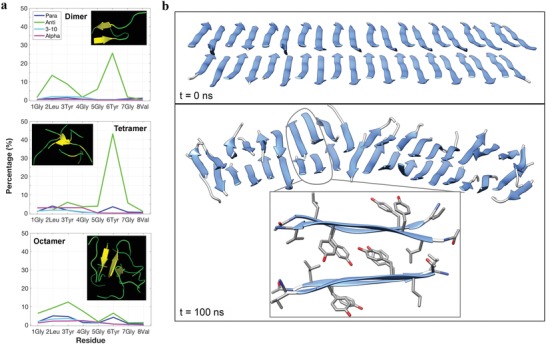
MD simulations of GV8 conformation and oligomeric self‐assembly. a) Secondary structure distribution of each residue in dimer, tetramer, and octamer of GV8 at 300K for 200 ns. b) Initial and final structures of a 40‐mer model of the GV8 peptide with β‐sheets structures shown in blue. The inset in the bottom panel shows the representative structure in the model, whereby the π–π stacking and hydrophobic interactions mainly contribute to intersheet association.
3. Conclusions
GV8 is an eight amino acid long peptide repeat from suckerin‐19—the most abundant protein forming the load‐bearing squid sucker ring teeth—that forms stiff hydrogels in water with tunable elastic modulus. Using CD, FTIR, and solution NMR spectroscopy, we have determined that GV8 self‐assembles into unusual 310 monomeric helices at low peptide concentration, which are intramolecularly stabilized by π–π stacking aromatic interactions between Y3 and Y6 residues, as well by the aliphatic side chains L2 and V8. As the concentration increases, GV8 dimerizes into antiparallel 310 helices driven by π‐stacking interactions between Tyr residues Y3, Y6, Y3*, and Y6*. In the gel state, ssNMR and WAXS measurements indicate that GV8 is made of antiparallel β‐sheets, inferring that gelation proceeds by a 310‐helix to β‐sheet conformational rearrangement. This mechanism is starkly different from previous reports on fibrous peptide‐based hydrogels. During this conformational transition, Tyr side‐chains reorient perpendicular to the chain direction according to both ssNMR and MD simulations, allowing to mediate intersheet interactions. Peptide‐based hydrogels with water gelation and the ability to tune the stiffness 25‐fold simply by increasing the peptide concentration may find notable opportunities for biomedical applications, such as tissue engineering, encapsulation of therapeutics, soft tissue adhesives, or matrix for stem cell differentiation.
4. Experimental Section
Materials: Ac‐GLYGGYGV‐NH2 peptide and Ac‐GLYGGYGX‐NH2 peptides (where X = V, L, A, F, S, K, and I) were purchased from GL Biochem (Shanghai) Ltd. Peptides were checked to be >98% purity via trace HPLC and LC/MS prior to use. All of the peptides were acetylated at the N‐terminal and amidated at the C‐terminal to prevent end‐to‐end charge interactions. 13C–15N uniformly labeled GV8 crude peptide purchased from Cambridge Isotopes was purified to >95% purity via HPLC and checked with LC/MS prior to use.
UV–vis Spectroscopy: The peptides were dissolved in DI water at the respective concentrations and 100 µL was aliquoted into each well of a 96‐well microtiter plate, with a minimum of three wells per condition. UV–vis absorbance measurements at 550 nm were recorded on a Tecan infinite M200 Pro microplate reader at intervals of 30 min for the first 16 h and subsequently at increased time intervals.
Peptide Hydrogel: GV8 peptide was dissolved in DI water at the desired concentration (between 10 and 20 × 10−3 m) and incubated at ambient temperature for at least 12 h.
CD Spectroscopy: GV8 peptide was dissolved at 20 × 10−3 m concentration in DI water and spectra were collected using a 0.2 mm path length quartz cuvette. Data acquisition was performed using AVIV 420 Circular Dichroism (New Jersey, USA) spectrometer. A quartz sandwich cuvette with optical path length of 0.2 mm was used for all data collection and the edges of the cuvette were sealed with parafilm to prevent loss of liquid. Data were acquired over a wavelength range of 190–260 nm and acquisition parameters were 0.5 nm wavelength steps with an averaging time of 0.1 s, 1.00 nm bandwidth, and readings were averaged over three scans. Obtained spectra were smoothed at 12 pts via adjacent‐averaging method (ensuring that no visible existing peaks were removed or artefacts introduced) and plotted via OriginPro 9.1.
FTIR Spectroscopy: ATR‐FTIR spectroscopy of lyophilized GV8 samples were performed on a Bruker Vertex 70 (Massachusetts, USA) equipped with a PIKE Technologies MIRacle attenuated total reflection (ATR) ZnSe‐Diamond 3‐reflection accessory and a LN2 cooled MCT detector. Scans were obtained at ambient temperature over the range of 4000–750 cm−1 with a resolution of 2 cm−1, averaged over 128 scans. GV8 peptide solutions were prepared at 20 × 10−3 m concentration in separate vials of 20 µL and snap freezed by dipping the vials in liquid N2 for 5 min at the stipulated time points and lyophilized immediately. All spectra processing were performed on OPUS 6.5, and processed in the sequence of water vapor subtraction, baseline correction, then normalized using amide I band. Amide I band was deconvoluted by secondary derivation, with peak fitting performed using 100% Gaussian curves with individual FWHM kept relatively consistent. The deconvoluted peaks were assigned to β‐sheet, unordered, helix and turns or 310 structures.40, 41, 48, 49
Cryo‐EM: GV8 peptide was dissolved at a concentration of 20 × 10−3 m and incubated for 3 h. Copper grids with Ultrathin C Film on Lacey Carbon support film was plasma‐treated with JEOL DATUM HDT‐400 for 300 s to increase hydrophilicity of grid surfaces to allow aqueous samples to adhere and spread. Vitrified samples were prepared using Gatan CryoplungeTM3 (Cp3). 4 µL of sample was pipetted onto each plasma‐treated copper grid and blotted for 5 s followed by vitrification in liquid ethane at −180 °C. All images were taken in bright‐field mode with objective aperture inserted. Imaging was carried out with energy filtered Carl Zeiss TEM, LIBRA 120 with in‐column Omega spectrometer and operated an acceleration voltage of 120 kV and the sample temperature was maintained below −180 °C during imaging.
SEM: Peptide hydrogel was snap freezed by dipping into liquid N2 for at least 5 min. The frozen hydrogel was then cryo‐fractured with tweezers to expose the porous cross‐section and the fractured surfaces were placed face‐up on carbon tape and lyophilized immediately. Samples were Platinum‐coated below 5 Pa, at 20 mA for 30 s and imaging was performed using JEOL JSM‐FESEM 7600F (Massachusetts, USA), at SEI‐mode, 5 kV, and 92 µA emission current.
AFM: 10 µL of GV8 peptide at 20 × 10−3 m concentration was deposited onto freshly cleaved mica and air‐dried overnight. AFM images were obtained on Asylum Cypher S AFM (Oxfordshire, UK) in tapping mode using Nanoworld NCSTR silicon nitride soft‐tip cantilevers (R f = 160 kHz, k = 7.4 N m−1). All images were flattened to remove background curvature using Igor Pro software and no further image processing was carried out.
Rheology: Rheological measurements were performed at ambient temperature on Anton Paar MCR501 rheometer with a parallel plate PP10 geometry. Five different concentrations (10, 12, 15, 18, and 20 × 10−3 m) of GV8 hydrogels were prepared in DI water, each pipetted into 1 mL syringes with nozzles removed, then left overnight to gelate. The hydrogel was extruded from the syringes and cut to 1–2 mm thick slices with a sterile blade and placed on the rheometer plate for measurements. Strain sweeps were first conducted at constant frequency of 1 Hz, from 0.1% to 10 % strain to identify the linear viscoelastic region and 0.25% strain was selected for subsequent frequency sweeps that were conducted from 0.01 to 100 Hz. All measurements were triplicated with repeated measurements performed on fresh samples.
NMR: All solution state NMR experiments were carried out on a Bruker 700 MHz spectrometer equipped with a cryoprobe. NMR data were processed with TOPSPIN (Bruker), then analyzed using Sparky50 programs. 0.5 or 20 × 10−3 m GV8 peptide was dissolved in water, pH 6.8 with 10% D2O for deuterium lock and DSS for signal reference. 2D 1H–1H TOCSY and NOESY spectra were acquired with 80 and 200 ms mixing times, respectively. In order to monitor hydrogel formation, 1D 1H spectrum and a series of 2D 1H–1H NOESY spectra were recorded every 4 h for 20 h using the 20 × 10−3 m peptide solution. For H/D exchange experiments, 0.5 and 20 × 10−3 m peptide samples were dissolved in 100% D2O and 2D 1H–1H TOCSY spectra were recorded at 30 min intervals. The extrinsic exchange rates were obtained by fitting the peak intensity versus time to a single‐exponential decay equation. The protection factor were calculated as the ratio of intrinsic exchange rates (calculated from SPHERE51) to the extrinsic exchange rates. A protection factor above 30 is indicative of stable hydrogen bonds, while values between 10 and 30 indicate an intermediate range of hydrogen bond strength.52 Amide temperature coefficients were also determined by recording 1D 1H spectra of 0.5 and 20 × 10−3 m GV8 peptide at 298, 303, 308, and 313 K. Amide proton chemical shift deviations were fitted linearly against temperature and the temperature coefficients were calculated as σδHN/ΔT (ppb K−1).
Solid state NMR experiments were carried out on a Bruker 600 MHz spectrometer equipped with a 1.7 mm MAS probe. The MAS spinning frequency was 13333 Hz. 20 × 10−3 m of 13C–15N labeled GV8 peptide was dissolved in water, pH 6.8 and allowed to incubate overnight for hydrogel formation. Sample was loaded in a 1.7 mm thin wall zirconia rotor (Bruker) manually and the rotor was spun at 70 000 rpm for 30 min by ultracentrifugation (Beckman Proteomelab XL, IN, USA). 2D 13C–13C DARR spectra were recorded over contact times ranging from 50 to 400 ms. 3D NCACX, NCOCX, and CANcoCX experiments were also recorded with 50 ms contact time.
NMR Structure Calculation: The structure calculations were carried out using the CYANA 2.1 program. The monomeric conformation of GV8 was calculated using the intensities of 1H–1H NOE cross peaks that were classified as strong, medium, and weak and translated to upper bound distance limits of 2.5, 3.5, and 5.0 Å. The dihedral Φ and Ψ angles were constrained between −120° to −30° and −120° to 120° as suggested in the CYANA program files. Out of the 100 structures generated, the ten lowest energy structures were used for more analysis. The dimer structures of GV8 were also calculated using the same constraints as monomeric structure calculation. The two monomeric units were linked by five glycine linkers. The structure of hydrogel was calculated using 13C–13C dipolar contacts derived from solid state NMR spectra. All of the conformations were validated using PROCHECK.53 For structure calculation from ssNMR, a total of 47 intraresidue and sequential dipolar constraints were used. The long‐range dipolar contacts included in the structure calculation were cross‐strand contacts used to generate a dimeric conformation.
SAXS and WAXS: SAXS and WAXS experiments were performed using Rigaku MicroMax‐002+ equipped with a microfocused beam (40 W, 45 kV, 0.88 mA) with the λCu Kα = 0.15418 nm radiation collimated by three pinhole collimators (0.4, 0.3, and 0.8 mm). The SAXS and WAXS intensities were collected by a two‐dimensional Triton‐200 gas‐filled X‐ray detector (20 cm diameter, 200 µm resolution) and a 2D Fujifilm BAS‐MS 2025 imaging plate system (15.2 × 15.2 cm2, 50 µm resolution), respectively. An effective scattering vector range of 0.05 nm−1 < q < 25 nm−1 was obtained, where q is the scattering wave vector defined as q = 4π sin θ/λCu Kα with a scattering angle of 2θ.
Hamiltonian‐Replica Exchange Molecular Dynamics (H‐REMD) Simulations: H‐REMD simulations54 were performed for the dimer, tetramer, and octamer of the Ac‐GLYGGYGV‐NH2 peptide for 200 ns each. The CHARMM 36 mm force field parameters55 were applied to peptides, and the dimer, tetramer, and octamer were put in a cubic box with TIP3P waters56 and 0.15 m NaCl. The minimum distance between the peptides and the box edge was larger than 1.5 nm. The dimer, tetramer, and octamer systems have 8, 12, and 16 replicas from 300 to 600 K, respectively, and each was simulated for 200 ns. The trajectories were saved every 2 ps.
Conventional MD Simulations: Conventional MD simulations were performed for the 40‐mer two‐layer antiparallel β‐sheet model for 100 ns using the AMBER 16 software57 together with the AMBER14SB force field. SHAKE algorithm58 was used to constrain all bonds involving hydrogens and electrostatic interactions were treated by the particle mesh Ewald sum method59 with a 8 Å cutoff for nonbonded interactions in direct space. The model was solvated in a rectangular box filled with TIP3P waters,56 with an at least 1.0 nm distance between the peptides and the box edge. The whole system was first energy‐minimized, with a series of position restraints on the solute (all heavy atoms, backbone atoms, and Cα atoms). The simulation was continued for 100 ns at 1 bar and 298.15 K.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supplementary
Supplementary
Acknowledgements
S.H.H. and H.M. contributed equally to this work. This research was funded by research grants from the Singapore Ministry of Education (grant #MOE 2018‐T2‐1‐043) and by the Skin Research Institute of Singapore (SIG18023). The authors also acknowledge financial support from the Strategic Initiative on Biomimetic and Sustainable Materials (IBSM) at NTU. M.Y. acknowledges support from MOE Tier 1 Grant RG 146/17 and the computing resources of the National Supercomputing Centre, Singapore (NSCC).
Hiew S. H., Mohanram H., Ning L., Guo J., Sánchez‐Ferrer A., Shi X., Pervushin K., Mu Y., Mezzenga R., Miserez A., A Short Peptide Hydrogel with High Stiffness Induced by 310‐Helices to β‐Sheet Transition in Water. Adv. Sci. 2019, 6, 1901173 10.1002/advs.201901173
References
- 1. Loo Y., Wong Y. C., Cai E. Z., Ang C. H., Raju A., Lakshmanan A., Koh A. G., Zhou H. J., Lim T. C., Moochhala S. M., Hauser C. A. E., Biomaterials 2014, 35, 4805. [DOI] [PubMed] [Google Scholar]
- 2. Carrejo N. C., Moore A. N., Silva T. L. L., Leach D. G., Li I. C., Walker D. R., Hartgerink J. D., ACS Biomater. Sci. Eng. 2018, 4, 1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nagai Y., Yokoi H., Kaihara K., Naruse K., Biomaterials 2012, 33, 1044. [DOI] [PubMed] [Google Scholar]
- 4. Li J. Y., Mooney D. J., Nat. Rev. Mater. 2016, 1, 16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Trappmann B., Gautrot J. E., Connelly J. T., Strange D. G. T., Li Y., Oyen M. L., Stuart M. A. C., Boehm H., Li B. J., Vogel V., Spatz J. P., Watt F. M., Huck W. T. S., Nat. Mater. 2012, 11, 642. [DOI] [PubMed] [Google Scholar]
- 6. Stephanopoulos N., Ortony J. H., Stupp S. I., Acta Mater. 2013, 61, 912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cui H. G., Webber M. J., Stupp S. I., Biopolymers 2010, 94, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hedegaard S. F., Derbas M. S., Lind T. K., Kasimova M. R., Christensen M. V., Michaelsen M. H., Campbell R. A., Jorgensen L., Franzyk H., Cardenas M., Nielsen H. M., Sci. Rep. 2018, 8, 6327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gonzalez M. A., Simon J. R., Ghoorchian A., Scholl Z., Lin S. T., Rubinstein M., Marszalek P., Chilkoti A., Lopez G. P., Zhao X. H., Adv. Mater. 2017, 29, 1604743. [DOI] [PubMed] [Google Scholar]
- 10. Yan C. Q., Pochan D. J., Chem. Soc. Rev. 2010, 39, 3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Khalily M. A., Goktas M., Guler M. O., Org. Biomol. Chem. 2015, 13, 1983. [DOI] [PubMed] [Google Scholar]
- 12. Loo Y. H., Lakshmanan A., Ni M., Toh L. L., Wang S., Hauser C. A. E., Nano Lett. 2015, 15, 6919. [DOI] [PubMed] [Google Scholar]
- 13. Banwell E. F., Abelardo E. S., Adams D. J., Birchall M. A., Corrigan A., Donald A. M., Kirkland M., Serpell L. C., Butler M. F., Woolfson D. N., Nat. Mater. 2009, 8, 596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sahoo J. K., VandenBerg M. A., Webber M. J., Adv. Drug Delivery Rev. 2018, 127, 185. [DOI] [PubMed] [Google Scholar]
- 15. Souza S. F., Kogikoski S., Silva E. R., Alves W. A., J. Braz. Chem. Soc. 2017, 28, 1619. [Google Scholar]
- 16. Guerette P. A., Hoon S., Ding D. W., Amini S., Masic A., Ravi V., Venkatesh B., Weaver J. C., Miserez A., ACS Nano 2014, 8, 7170. [DOI] [PubMed] [Google Scholar]
- 17. Hiew S. H., Guerette P. A., Zvarec O. J., Phillips M., Zhou F., Su H., Pervushin K., Orner B. P., Miserez A., Acta Biomater. 2016, 46, 41. [DOI] [PubMed] [Google Scholar]
- 18. Hiew S. H., Miserez A., ACS Biomater. Sci. Eng. 2016, 3, 680. [DOI] [PubMed] [Google Scholar]
- 19. Kumar A., Mohanram H., Kong K. W., Goh R., Hoon S., Lescar J., Miserez A., Biomater. Sci. 2018, 21, 401. [DOI] [PubMed] [Google Scholar]
- 20. Zhao H. G., Ma L., Zhou J., Mao Z. W., Gao C. Y., Shen J. C., Biomed. Mater. 2008, 3, 015001. [DOI] [PubMed] [Google Scholar]
- 21. Newcomb C. J., Moyer T. J., Lee S. S., Stupp S. I., Curr. Opin. Colloid Interface Sci. 2012, 17, 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mackintosh F. C., Kas J., Janmey P. A., Phys. Rev. Lett. 1995, 75, 4425. [DOI] [PubMed] [Google Scholar]
- 23. Storm C., Pastore J. J., MacKintosh F. C., Lubensky T. C., Janmey P. A., Nature 2005, 435, 191. [DOI] [PubMed] [Google Scholar]
- 24. Cao Y. P., Bolisetty S., Adamcik J., Mezzenga R., Phys. Rev. Lett. 2018, 120, 158103. [DOI] [PubMed] [Google Scholar]
- 25. Gardel M. L., Shin J. H., MacKintosh F. C., Mahadevan L., Matsudaira P., Weitz D. A., Science 2004, 304, 1301. [DOI] [PubMed] [Google Scholar]
- 26. Mezzenga R., Fischer P., Rep. Prog. Phys. 2013, 76, 046601. [DOI] [PubMed] [Google Scholar]
- 27. Greenfield M. A., Hoffman J. R., de la Cruz M. O., Stupp S. I., Langmuir 2010, 26, 3641. [DOI] [PubMed] [Google Scholar]
- 28. Ozbas B., Kretsinger J., Rajagopal K., Schneider J. P., Pochan D. J., Macromolecules 2004, 37, 7331. [Google Scholar]
- 29. Bertolani A., Pirrie L., Stefan L., Houbenov N., Haataja J. S., Catalano L., Terraneo G., Giancane G., Valli L., Milani R., Ikkala O., Resnati G., Metrangolo P., Nat. Commun. 2015, 6, 7574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Adams D. J., Butler M. F., Frith W. J., Kirkland M., Mullen L., Sanderson P., Soft Matter 2009, 5, 1856. [Google Scholar]
- 31. Seow W. Y., Salgado G., Lane E. B., Hauser C. A. E., Sci. Rep. 2016, 6, 32670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Discher D. E., Janmey P., Wang Y. L., Science 2005, 310, 1139. [DOI] [PubMed] [Google Scholar]
- 33. Engler A. J., Sen S., Sweeney H. L., Discher D. E., Cell 2006, 126, 677. [DOI] [PubMed] [Google Scholar]
- 34. Ahearne M., Interface Focus 2014, 4, 20130038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grishina I. B., Woody R. W., Faraday Discuss. 1994, 99, 245. [DOI] [PubMed] [Google Scholar]
- 36. Khan M. A., Neale C., Michaux C., Pomes R., Prive G. G., Woody R. W., Bishop R. E., Biochemistry 2007, 46, 4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cochran A. G., Skelton N. J., Starovasnik M. A., Proc. Natl. Acad. Sci. USA 2001, 98, 5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu L., McElheny D., Takekiyo T., Keiderling T. A., Biochemistry 2010, 49, 4705. [DOI] [PubMed] [Google Scholar]
- 39. Woody R. W., Biopolymers 1978, 17, 1451. [Google Scholar]
- 40. Kong J., Yu S., Acta Biochim. Biophys. Sin. 2007, 39, 549. [DOI] [PubMed] [Google Scholar]
- 41. Yang H. Y., Yang S. N., Kong J. L., Dong A. C., Yu S. N., Nat. Protoc. 2015, 10, 382. [DOI] [PubMed] [Google Scholar]
- 42. Armen R., Alonso D. O. V., Daggett V., Protein Sci. 2003, 12, 1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. de Brevern A. G., Sci. Rep. 2016, 6, 33191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wishart D. S., Sykes B. D., Richards F. M., Biochemistry 1992, 31, 1647. [DOI] [PubMed] [Google Scholar]
- 45. Millhauser G. L., Stenland C. J., Hanson P., Bolin K. A., van de Ven F. J. M., J. Mol. Biol. 1997, 267, 963. [DOI] [PubMed] [Google Scholar]
- 46. Tornasz Cierpicki J. O., J. Biomol. NMR 2001, 21, 249. [DOI] [PubMed] [Google Scholar]
- 47. Singh Y., Sharpe P. C., Hoang H. N., Lucke A. J., McDowall A. W., Bottomley S. P., Fairlie D. P., Chem. ‐ Eur. J. 2011, 17, 151. [DOI] [PubMed] [Google Scholar]
- 48. Wilson D., Valluzzi R., Kaplan D., Biophys. J. 2000, 78, 2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vass E., Hollosi M., Besson F., Buchet R., Chem. Rev. 2003, 103, 1917. [DOI] [PubMed] [Google Scholar]
- 50. Lee W., Tonelli M., Markley J. L., Bioinformatics 2015, 31, 1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bai Y., Milne J. S., Mayne L., Englander S. W., Proteins: Struct., Funct., Genet. 1993, 17, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kuwata T. M. K., Cheng H., Nagayama K., James T. L., Roder H., Proc. Natl. Acad. Sci. USA 2003, 100, 14790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M., J. Appl. Crystallogr. 1993, 26, 283. [Google Scholar]
- 54. Bussi G., Mol. Phys. 2014, 112, 379. [Google Scholar]
- 55. Huang J., MacKerell A. D. Jr., J. Comput. Chem. 2013, 34, 2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jorgensen W., Chandrasekhar J., Madura J., Impey R., Klein M., J. Chem. Phys. 1983, 79, 926. [Google Scholar]
- 57. Case D. A., Cheatham T. E., Darden T., Gohlke H., Luo R., Merz K. M., Onufriev A., Simmerling C., Wang B., Woods R. J., J. Comput. Chem. 2005, 26, 1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ryckaert J. P., Ciccotti G., Berendsen H. J. C., J. Comput. Phys. 1977, 23, 327. [Google Scholar]
- 59. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., Pedersen L. G., J. Chem. Phys. 1995, 103, 8577. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary
Supplementary