

Abstract
The Orbitrap is now a core component of several different instruments. However, evaluating the capabilities of each system is lacking in the field. Here, we compared the performance of multidimensional protein identification (MudPIT) on Velos Pro Orbitrap and Velos Orbitrap Elite mass spectrometers to reversed phase liquid chromatography (RPLC) on a Q-Exactive Plus and an Orbitrap Fusion Lumos. Using HeLa cell protein digests, we carried out triplicate analyses of 16 different chromatography conditions on four different instrumentation platforms. We first optimized RPLC conditions by varying column lengths, inner diameters, and particle sizes. We found that smaller particle sizes improve results but only with smaller inner diameter microcapillary columns. We then selected one chromatography condition on each system and varied gradients lengths. We used distributed normalized spectral abundance factor (dNSAF) values to determine quantitative reproducibility. With Pearson Product-Moment Correlation Coefficients r values routinely above 0.96, single RPLC on both the QE+ and Orbitrap Lumos outperformed MudPIT on the Orbitrap Elite mass spectrometer. In addition, when comparing dNSAF values measured for the same proteins across the different platforms, RPLC on the Orbitrap Lumos had greater sensitivity than MudPIT, as demonstrated by the detection and quantification of histone deacetylase complex components. Data are available via ProteomeXchange with identifier 10.6019/PXD009875.
Keywords: Orbitrap, Q Exactive, Orbitrap Fusion Lumos, Quantitative Proteomics, Human, Liquid Chromatography, Pearson Product-Moment Correlation Coefficient, Reproducibility, Multidimensional Protein Identification Technology, distributed Normalized Spectral Abundance Factor
Graphical Abstract
INTRODUCTION
Rapid advances in protein mass spectrometry (MS) instrumentation have taken place over the decade that has led to remarkable capture of proteomes. With each new instrumentation, optimizing its use presents a new challenge to determine the capabilities of each platform. For example, the Orbitrap was introduced more fifteen years ago1, 2 and has become the foundation of a series of mass spectrometry systems3, 4. The evolution of the Orbitrap-based systems has led to the current dichotomy of the Q Exactive (QE) platform, with the QE HF-X being one of the latest versions5 and the Orbitrap Fusion Lumos6 platform. Each time a new instrument is released, research must be conducted to evaluate the parameters of a system and determine its performance. This has occurred on the QE systems with each new QE platform5, 7–10 and the Orbitrap Fusion and Fusion Lumos systems6, 11–16.
A major challenge with the rapid advances of mass spectrometry systems is to compare the performance of distinct systems to determine optimal approaches for proteomics analysis. Sun et al compared the LTQ-Orbitrap Velos and the QE on a range of amounts of cell lysate for proteome capture17. Williamson et al compared the QE-Plus to the Orbitrap Fusion Tribrid to test quantitative accuracy of the systems12. Wei et al. pursued missing proteins from the human proteome project by comparing the LTQ-Orbitrap Velos and the QE HF where the QE-HF detected 74 missing proteins whereas the LTQ-Orbitrap Velos found 39 missing proteins18. Instrumentation comparisons are important to evaluate new systems and to determine the impacts of technological advances on the field.
Performance evaluation for proteome analysis goes beyond the capabilities of a mass spectrometer itself. Liquid chromatography (LC) is arguably as important to proteomics analysis as the mass spectrometer, and rapid advances in instrumentation have resulted in calls for improved chromatography19. Single20 and multidimensional chromatography21 play important roles in proteomics analysis. There are many aspects of chromatography that can be varied and tested. These features include the column length, gradient length, and particle size, for example20, 22–25. Therefore, it is important to analytically evaluate both mass spectrometer settings and hromatographic set-up for optimal proteomic analysis.
In this study, we compared three different Orbitrap based instrumentation platforms. We compared the performance of multidimensional protein identification (MudPIT)26 on Velos Pro Orbitrap and Velos Orbitrap Elite mass spectrometers to single dimension reversed phase (RP) liquid chromatography on a Q-Exactive Plus (QE+) or an Orbitrap Fusion Lumos (OFL) mass spectrometer. On the QE+ or OFL mass spectrometers, we tested several different combinations of microcapillary inner diameters (i.d.), column lengths, and RP particle sizes. We focused on the comparison of chromatography settings and the ability of each integrated LCMS system to capture a human HeLa cell proteome at the qualitative and quantitative levels.
EXPERIMENTAL PROCEDURES
Materials.
HeLa protein digest standard was purchased from Pierce (Rockford, IL). ReproSil-Pur C18-AQ 1.9 μm and 3 μm porous spherical silica, both with 120 Å pore diameter, were purchased from Dr. Maisch GmbH (Germany). Aqua 5 μm C18, 125 Å pore diameter, and Luna 5 μm strong cation exchange (SCX), 100 Å pore diameter, were purchased in bulk from Phenomenex (Torrance, CA). Deactivated fused silica tubing (0.100 mm internal diameter (i.d.) × 0.360 mm outer diameter (o.d.), 0.050 mm i.d. × 0.360 mm o.d., and 0.250 mm i.d. × 0.360 mm o.d.) were obtained from Polymicro (Lisle, IL). Acclaim PepMap 100 (75um × 2 cm, C18, 3 μm, 100 Å) trap column was from Thermo Scientific, (San Jose, CA). HPLC grade water and acetonitrile were from EMD Chemicals Inc. (Gibbstown, NJ). HPLC grade formic acid was purchased from Mallinckrodt Baker, Inc. (Phillipsburg, NJ).
Reverse-Phase Liquid chromatography (RPLC).
RPLC-MS/MS analyses were performed using either a Q-Exactive Plus (QE+) mass spectrometer (Thermo Scientific, San Jose, CA) connected to a Dionex UltiMate 3000 RSCLnano System and Nanospray Flex Ion Source (Thermo Scientific, San Jose, CA), or an Orbitrap Fusion Lumos (OFL) mass spectrometer (Thermo Scientific, San Jose, CA) coupled to a Dionex UltiMate 3000 RSCLnano System with Variable Wavelength Detector. Both QE+ and OFL systems were outfitted with a Nanospray Flex Ion Source (Thermo Scientific, San Jose, CA).
All analytical microcapillary columns were packed in-house with different particle sizes (ReproSil-Pur C18-AQ 1.9 μm and 3 μm porous spherical silica, or Phenomenex Aqua 5 μm C18) using custom-made stainless-steel pressurization devices. The organic solvent solutions were: water/acetonitrile/formic acid at 95:5:0.1 (v/v/v) for buffer A (pH 2.6), and at 20:80:0.1 (v/v/v) for buffer B. To saturate nonspecific binding sites, all newly packed columns were preconditioned before use by loading 2.5 μg of trypsin-digested yeast whole cell lysate, followed by elution and two wash steps consisting of three back-to-back 90 min RP gradients. The 90 min RP gradient was a 10 min column equilibration step in 2% B; a 5 min ramp to reach 10% B; 90 min from 10 to 40 % B; 5 min to reach 95% B; a 10 min wash at 95% B; 1 min to 2% B; followed by a 14 min column re-equilibration step at 2% B. During each wash step, the loading trap was washed through the loading pump at 2 μl/min, while the analytical column was washed through the nano pump at 0.3 μl/min or 0.15 μl/min for the 100 and 50 μm i.d. analytical columns, respectively.
For all 200ng HeLa digests analyses (Table S1), peptides were injected via the autosampler directly onto the analytical column at nano-flow rates set as defined above for each type of inner diameter columns. For all 3 μg HeLa digests analyses (Table S2), peptides were first loaded onto an Acclaim PepMap 100 trap column connected with the loading pump running at 0.3 μl/min. Prior to loading, all 3 μg HeLa digests were spiked with a pre-digested mixture of 6 albumin standards27, whose amounts were evenly distributed over 2.5 orders of magnitude in logarithmic scale: 0.03 pmol of human albumin; 0.095 pmol of rat albumin; 0.303 pmol of mouse albumin; 0.966 pmol of pig albumin; 3.07 pmol of rabbit albumin; and 10 pmol of bovine albumin (Table S2A).
When 200 ng HeLa peptides were analyzed with 100 μm i.d. columns on the QE+, the chromatography gradient was a 20 min column equilibration step in 2% B; a 10 min ramp to reach 10% B; 120 min from 10 to 40 % B; 5 min to reach 95% B; a 14 min wash at 95% B; 1 min to 2% B; followed by a 10 min column re-equilibration step at 2% B. When 50 μm i.d. columns were used on the QE+, the same steps were implemented except equilibrating in 2% B took 50 min. When 200 ng HeLa peptides were analyzed on the OFL system with 100 μm i.d. columns, the gradient was 35 min at 2% B; 10 min to 10% B; 150 min to 40% B; 5 min to 95% B; 14 min at 95% B; 1 min to 2% B; 10 min at 2% B. When 50 μm i.d. columns were used on the OFL, the same gradient was implemented to resolve from 200 ng HeLa peptides except the column equilibration step took 50 min at 2% B.
On both QE+ and OFL systems, 3 μg HeLa peptides were loaded onto 50 μm i.d. columns and resolved with a 50 min equilibration step in 2% B; a 10 min ramp to 10% B; 120, 240, or 360 min to reach 40% B; 5 min to 95% B; a 14 min wash at 95% B; 1 min to 2% B; and a 10 min column re-equilibration at 2% B. On both systems, the flow rates were constant at 0.3 μl/min and 0.15 μl/min for the 100 μm i.d. and 50 μm i.d. analytical columns, respectively.
We defined the effective LC gradient “length” as the ramp between 10 to 40% B, which was when most peptides eluted. The LC gradients used to analyze 200 ng of HeLa digests on the QE+ and OFL systems were hence labeled as 120 and 150 min, respectively (Figure 1), while the 3 μg HeLa peptides were analyzed with 120, 240, or 360 min effective LC gradients (Figure 2), even though column equilibration and high organic wash steps added between 60 to 90 min to the actual duration of the LC-MS analyses.
Figure 1: Qualitative effects of varying column inner diameters, lengths, and particle sizes.

Average +/− standard deviation of the total spectral counts (A), unique peptides (B), and unique proteins (C) detected in different LCMS/MS settings (Table S1A). For all panels, the experimental conditions are detailed in the table under the x-axis of panel C. Three replicates of each experiment were acquired. *, **, and *** denote p-values from a homoscedastic two-tailed t-test lesser than or equal to 0.05, 0.01, and 0.002, respectively (Table S1B).
Figure 2: Qualitative effects of varying LC gradient length.

Average +/− standard deviation of the total spectral counts (A), unique peptides (B), and unique proteins (C) detected in each LC-MS/MS setting (Table S2A). For all panels, the experimental conditions are detailed in the table under the x-axis of panel C. Three replicates of each experiment were acquired. *, **, and *** denote p-values from a homoscedastic two-tailed t-test lesser than or equal to 0.05, 0.01, and 0.002, respectively (Table S2B). Lines are plotted between datapoints within each instrument series to illustrate the linearity of the response as a function of gradient length.
Multidimensional Protein Identification Technology.
MudPIT was carried out as described previously28 with the following modifications. Peptide mixtures were loaded onto a 250 μm i.d. capillary packed first with 3.5 cm of 5 μm Luna SCX, followed by 2.5 cm of 5 μm Aqua C18. The biphasic column was washed with buffer A for more than 20 column volumes. After desalting, the biphasic column was connected via a 2-μm filtered union (UpChurch Scientific) to a 100 μm i.d. column, which had been pulled to a 5 μm tip, then packed with 10 cm of 5 μm C18 RP particles (Aqua). The split three-phase column was placed in line with an Agilent 1200 quaternary HPLC pump (Palo Alto, CA) and a Velos Pro Orbitrap (VPO) mass spectrometer or a Velos Orbitrap Elite (VOE) mass spectrometer (Thermo Fisher Scientific). The 200 ng and 3 ug HeLa digests were analyzed using 10- and 12-step MudPIT, on VPO and VOE, respectively. The solvent solutions used were described above with the additional buffer C for salt bumps consisting of buffer A with 500 mM ammonium acetate.
Tandem Mass Spectrometry (MS/MS).
Instrument settings common to the three systems were as follows: MS spray voltage set at 2.5 kV; MS transfer tube temperature set at 275 °C; 50 ms MS1 injection time; 1 MS1 microscan; MS1 data acquired in profile mode; 15 MS2 dependent scans; 1 MS2 microscan; and MS2 data acquired in centroid mode. Instrument settings specific to each of the three systems were as follows for the OFL, QE+, and VPO/VOE, respectively: MS1 scans acquired in Orbitrap (OT) at 120000, 70000, and 60000 resolution; full MS1 range acquired from 375 to 1500, 375–1700, and 400–1500 m/z; MS1 AGC targets set to 4.00E+05, 1.00E+06, and 1.00E+06; MS1 charge states between 2–6, 2–5, and 2–5; MS1 repeat counts of 1, 1, and 2; MS1 dynamic exclusion durations of 30, 30, and 90 sec; ddMS2 acquired in IT, OT at 17,500 resolution, and IT; MS2 collision energy and fragmentation: 35% CID, 27 % HCD, and 35% CID; MS2 AGC targets of 1.00E+04, 1.00E+05, and 1.00E+05; MS2 max injection times of 100, 150, and 150 ms.
Data Analysis.
Collected MS/MS spectra were searched with the ProLuCID algorithm29 against a database of 73653 protein sequences combining 36636 non-redundant Homo sapiens proteins (NCBI, 2016-06-10 release), 192 common contaminants, and their corresponding 36825 randomized amino acid sequences. All cysteines were considered as fully carboxamidomethylated (+57 Da statically added), while methionine oxidation was searched as a differential modification. DTASelect v1.930 and swallow, an in-house developed software, were used to filter ProLuCID search results at given FDRs at the spectrum, peptide, and protein levels. Here, all controlled FDRs were less than 1%. All 200 ng HeLa analysis (27 data sets, Table S1A) and all 3 μg HeLa analysis results (21 data sets; Table S2A) were contrasted against their merged data set, respectively, using Contrast v1.9 and in house developed sandmartin v0.0.1. Our in-house developed software, NSAF7 v0.0.1, was used to generate spectral count-based label free quantitation results27.
Statistical and Correlation Analyses.
Significant differences between qualitative parameters (spectral, peptide, and protein counts) obtained under different LC-MS/MS conditions were assessed using a t-test performed in excel (Figure 1/Table S1B and Figure 2/Table S2B). To assess the qualitative reproducibility of protein identifications in the 3μg of HeLa digests datasets, the number of proteins shared and unique to each replicate in systematic inter-replicate pair-wise comparisons were calculated (Table S2C). To assess the reproducibility of the quantitative values measured for proteins under different LC-MS/MS conditions, Pearson product-moment correlation coefficients (r) were calculated in excel for each pair-wise comparison on the proteins detected in both analyses being compared (Table S2D). The rcorr() function from the R Hmisc package was used to perform Pearson correlation, while the chart.correlation() function from the R PerformanceAnalytics package and the corrplot() function from the R corrplot package were combined to chart the correlation matrix (Figure 3). Overlap and uniqueness in protein identifications between the MudPIT analysis and all other RPLC MS/MS datasets were qualitatively assessed (Table 1/Table S2E). Significant difference in the distributions of label-free quantitative dNSAF values measured in the MudPIT dataset were compared against the values obtained under the other RPLC-MS/MS conditions and statistically assessed using the non-parametric Mann-Whitney U test performed in OriginPro 2017 (Figure 4/Table S2F). Variability of the quantitative values in replicate measurements was assessed by calculating averages, standard deviations, and coefficients of variations in excel (Figure 5/Table S2G).
Figure 3: Reproducibility of dNSAF quantitative values obtained for the same proteins on three different mass spectrometry platforms and varying LC gradient lengths.

Pearson product-moment correlation coefficients were calculated for each of the 210 pair-wise comparisons for proteins detected in both analyses being compared (Table S2D). The 210 pairwise dNSAF correlation plots are displayed in the lower left quadrant, with the corresponding r values color-coded in the cells above the diagonal. All r values were calculated to be significant for linear correlation at the p-value ≤0.01 level (***).
Table 1:
Comparison of the proteins detected in 24hr-MudPIT analyses vs all other RPLC-MS/MS pipelines
| Pair-wise comparison | Entire dNSAF Rangea | b | dNSAF >=1E-3a | c | d | 1E-3 > dNSAF >= 1E-4a | c | d | 1E-4 > dNSAF >= 1E-5a | c | d | dNSAF <1E-5a | c | d | |
|---|---|---|---|---|---|---|
|
24hr-VOE ∩ |
120-QE+ | 3023 | 79 | 133 | 98 | 4 | 1471 | 95 | 49 | 1351 | 68 | 45 | 68 | 39 | 2 |
| 240-QE+ | 3217 | 80 | 133 | 98 | 4 | 1491 | 96 | 46 | 1496 | 71 | 47 | 97 | 47 | 3 | |
| 360-QE+ | 3307 | 82 | 133 | 99 | 4 | 1493 | 96 | 45 | 1572 | 74 | 48 | 109 | 47 | 3 | |
| 120-OFL | 3279 | 74 | 133 | 97 | 4 | 1450 | 91 | 44 | 1557 | 64 | 47 | 139 | 54 | 4 | |
| 240-OFL | 3422 | 71 | 131 | 96 | 4 | 1457 | 91 | 43 | 1681 | 62 | 49 | 153 | 41 | 4 | |
| 360-OFL | 3495 | 71 | 133 | 98 | 4 | 1478 | 91 | 42 | 1728 | 64 | 49 | 156 | 31 | 4 | |
| 24hr-VOE - | 120-QE+ | 587 | 15 | 3 | 2 | 1 | 44 | 3 | 7 | 446 | 22 | 76 | 94 | 53 | 16 |
| 240-QE+ | 447 | 11 | 3 | 2 | 1 | 35 | 2 | 8 | 343 | 16 | 77 | 66 | 32 | 15 | |
| 360-QE+ | 342 | 8 | 2 | 1 | 1 | 29 | 2 | 8 | 261 | 12 | 76 | 50 | 22 | 15 | |
| 120-OFL | 418 | 9 | 3 | 2 | 1 | 67 | 4 | 16 | 312 | 13 | 75 | 36 | 14 | 9 | |
| 240-OFL | 325 | 7 | 4 | 3 | 1 | 61 | 4 | 19 | 233 | 9 | 72 | 27 | 7 | 8 | |
| 360-OFL | 232 | 5 | 2 | 1 | 1 | 49 | 3 | 21 | 165 | 6 | 72 | 16 | 3 | 7 | |
| 120-QE+ | - 24hr-VOE | 238 | 6 | 0 | 0 | 0 | 35 | 2 | 15 | 189 | 10 | 79 | 14 | 8 | 6 |
| 240-QE+ | 333 | 8 | 0 | 0 | 0 | 29 | 2 | 9 | 260 | 12 | 78 | 44 | 21 | 13 | |
| 360-QE+ | 396 | 10 | 0 | 0 | 0 | 35 | 2 | 9 | 290 | 14 | 73 | 71 | 31 | 18 | |
| 120-OFL | 728 | 16 | 1 | 1 | 0 | 82 | 5 | 11 | 564 | 23 | 77 | 81 | 32 | 11 | |
| 240-OFL | 1074 | 22 | 1 | 1 | 0 | 88 | 5 | 8 | 796 | 29 | 74 | 189 | 51 | 18 | |
| 360-OFL | 1215 | 25 | 1 | 1 | 0 | 90 | 6 | 7 | 799 | 30 | 66 | 325 | 65 | 27 | |
Number of proteins detected at least twice in each compared dataset and with averaged dNSAF values in 24-Hr VOE in the specified range (or averaged dNSAF in other RPLC-MS analyses for proteins not detected in 24hr-VOE).
% of the total number of proteins identified merging the two analyses being compared.
% of the total number of proteins in each abundance range (sum of column).
% of the total number of proteins in entire dNSAF range (sum of row).
Figure 4: Pair-wise comparisons of the label-free quantitative values.

Log-transformed dNSAF values (averaged across triplicates) measured in the 24-hr MudPIT on VOE (gray symbols in all 6 panels) were compared to the corresponding dNSAF values measured by RPLC-MS/MS analyses with varying LC gradient lengths (120, 240, and 360 min, top to bottom panels) implemented on QE+ (shades of pink symbols, left panels) and OFL (shades of blue symbols, right panels). On the right side of each panel, proteins are sorted into 4 abundance groups (gray horizontal grid lines; Table 1) and the distribution of dNSAF values within each of these groups are illustrated by box-plots (reported full size in Table S2F).
Figure 5: Label-free quantitation of HDAC1/2-containing protein complexes detected in 3μg of HeLa whole cell digests on various LC/MS platforms.

A. The average ± standard deviation of dNSAF values measured for known members of the Sin3, NuRD, and CoRest complexes are shown for each of the three LC/MS platforms. For each protein, the dNSAF values plotted are for the MudPIT analyses over 24-hr on a VOE (grey bars, left panel) and the analyses of single RPLCMS/MS with varying gradient lengths (120, 240, and 360 min) on a QE+ (shades of pink, middle panel) and OFL (shades of blue, right panel) mass spectrometers. B. An expended view of the frequency of detection (numbers above bars) and quantitation of pairs of paralogues within each HDAC1/2-containing complex (Table S2G).
Analysis of Chromatographic Peak Widths.
To extract full width at half maximum (FWHM) values for detected peptide ions, the first replicates analyses of the 3 μg HeLa analyzed on the OFL with 120, 240, and 360 min RPLC gradients were processed as follow. ProLuCID search engine sqt files were converted with an in-house script to pep.xml format, which in turn was converted to interact.pep format. Following this online tutorial for “Skyline MS1 Full-Scan Filtering”, interact.pep files were inputted into Skyline 3.631 that built a spectral library for each of the three RPLC gradient length datasets. A database containing the amino acid sequences for the detected proteins (fasta format) as well as the mass spectrometry raw files were also inputted to Skyline, from which the peak boundaries results were exported for each of the three RPLC gradient lengths (Table S3A–C). Peptide ions commonly detected in all three LC conditions were assembled in excel (Table S3D) and their respective FWHM ranges were plotted (Figure S1A/C) and statistically assessed using the non-parametric Mann-Whitney U test performed in OriginPro 2017 (Table S3D). Retention time and intensity values (Table S3E) were also exported from Skyline to plot extracted ion chromatograms for representative peptide ions (Figure S1B).
Data Accessibility.
The mass spectrometry data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository32, 33 with the identifiers 10.6019/PXD009875. All original data files underlying this manuscript may also be accessed after publication from the Stowers Original Data Repository at http://www.stowers.org/research/publications/libpb-1303.
RESULTS AND DISCUSSION
Varying Particle Size, Inner Diameter, and Column Length
To begin, we tested the performance of three Orbitrap-based mass spectrometry systems with 200 μg of HeLa cell extract analyzed using different combinations of mass spectrometer and liquid chromatography settings (Figure 1). We ran a 10-step 20-hour MudPIT28 analysis on a Velos Pro Orbitrap (VPO) mass spectrometer and carried out six different reversed phase liquid chromatography (RPLC) analyses on a Q-Exactive Plus (QE+) mass spectrometer and two different RPLC analyses on an Orbitrap Fusion Lumos (OFL) mass spectrometer (Figure 1). The mass spectrometry settings were kept constant for each instrument. Three technical replicates were acquired for each of the nine LC-MS/MS analyses. In each case, we qualitatively assessed the results based on the number of spectral counts, unique peptides, and identified proteins (Figure 1). Over the course of a 10-step 20-hour MudPIT analysis28, the data set generated on the Velos Pro Orbitrap matched 64912 ± 1,876 tandem mass spectra to 10506 ± 600 unique peptides, leading to 2150 ± 122 unique proteins/protein groups identified (Figure 1 and Supplemental Table S1AB). This served as our baseline for comparison against other chromatographic conditions and mass spectrometry systems.
We next tested six different chromatographic conditions on the QE+. Three different columns were packed in 100 μm inner diameter (i.d.) microcapillaries to better compare with the MudPIT LC condition, in which the resolutive RP column was packed in 100 μm i.d. microcapillary with 10 cm of 5 μm C18. Single phase columns were packed with 15 cm of 5 μm RP particles, 15cm of 3 μm RP particles, or 25 cm of 3 μm RP particles. Even with these varying lengths and particle sizes, there was no statistical difference in the number of spectral counts, peptide, and protein identifications between these three 100 μm i.d. columns. On the other hand, when 50 μm i.d. columns were packed with 25cm of 3 μm RP particles, peptide and protein identifications were significantly greater than the ones observed with the same column length and particle size packed in 100 μm i.d. microcapillaries. Further decreasing the particle size to 1.9 μm led to significantly greater number of peptide and spectral counts, even when using shorter column length (compare 25cm packed with 3 μm C18 vs 15 cm packed with 1.9 μm C18, Figure 1AB). However, varying column length or particle size within the same column i.d. did not seem to impact significantly the number of detected proteins (Figure 1C and Table S1B).
Of the chromatography conditions tested on the QE+ mass spectrometer, the optimal setup consisted of 1.9 μm RP particles packed to 15 cm length in a 50 μm i.d. column (Figure 1 and Supplemental Table S1B). The HPLC system coupled to the QE+ was able to tolerate the back pressure this configuration generated across all replicates. This chromatographic configuration on the QE+ captured 23997 ± 763 spectral counts, identified 17863 ± 342 unique peptides, and identified 3394 ± 76 unique proteins (Figure 1 and Table S1B). When compared to the MudPIT results, this chromatographic configuration on the QE+ captured 2.7-fold fewer spectral counts but 70% more unique peptides and 58% more unique proteins on average in substantially shorter instrumentation time. This trend was consistent with all chromatography configurations on the QE+ where MudPIT captured many more spectral counts but identified fewer unique peptides and fewer unique proteins than any given analysis on the QE+ (Figure 1 and Supplemental Table S1AB). When using 100 μm i.d. microcapillary columns, no significant gains were achieved with varying particles sizes or packed length. The general trend across these six conditions was smaller particle sizes in smaller i.d. columns gave superior results. Increasing column length when holding other features constant also yielded better results. These results are consistent with general liquid chromatography knowledge.
Using the QE+ results as a guide, we next tested three LC conditions on the OFL mass spectrometer using 200ng of HeLa cell extract digests. The chosen LC conditions were identical to three of the conditions tested on the QE+. Two of these three conditions yielded tolerable back pressures and reproducible results, however, when we directly adopted the optimal LC conditions observed on the QE+ (50 μm i.d. column packed with 15cm of 1.9 μm RP particles), the acquisition failed 2 of 3 times (data not shown) due to high back pressure on the LC system coupled to the OFL. Slightly different configurations in the valves plumbing could be responsible to the HPLC systems coupled to our QE+ and OFL tolerating back-pressure differently.
For both LC conditions that were identical on the QE+ and the OFL (5 and 3 μm RP particles packed in 100 μm i.d. and 50 μm i.d. columns, respectively), the OFL significantly outperformed the QE+ runs. Furthermore, both LC conditions tested on the OFL outperformed the top performing LC condition on the QE+ (Figure 1). The top performing OFL chromatographic configuration (50 μm i.d. column packed with 25 cm of 3 μm RP particles) yielded the second most spectral counts (49362 ± 2977), the most unique peptides (26099 ± 964), and the most unique proteins (4738 ± 101) across all conditions (Figure 1 and Supplemental Table S1AB). With LC conditions being otherwise identical, the main performance difference between the QE+ and OFL instruments likely derives from their MS2 settings: while the MS2 scans were acquired in the ion trap on the OFL (“FTIT” mode), the MS2 scans must be acquired in the Orbitrap on the QE+, increasing the instrument duty cycle. Acquiring MS data in “FTFT” mode (MS2 scans in the OT) on an OFL should result in protein counts similar to those obtained on a QE+16.
Varying Chromatography Gradient Length
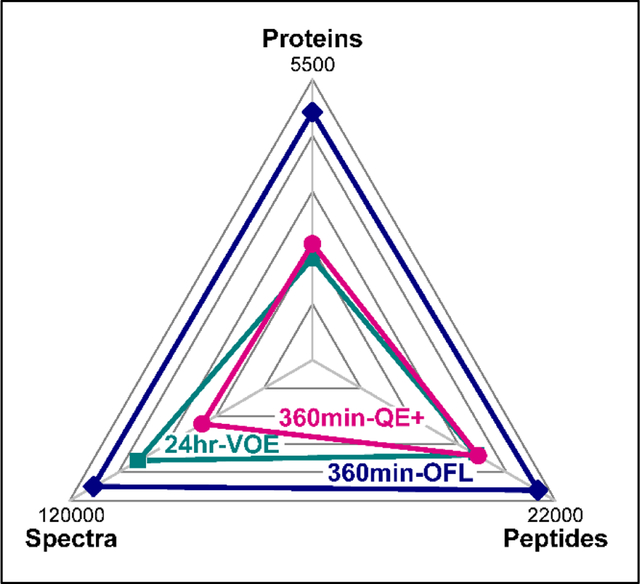
Next, in a distinct set of experiments, we used the top performing chromatography setups for the QE+ and OFL LCMS systems to analyze 3 μg of HeLa whole cell digests and compared these results to a 12-step 24-hour MudPIT analysis on a Velos Orbitrap Elite mass spectrometer. As defined above, the peptides mixtures were analyzed on 50 μm i.d. columns packed with 15cm of 1.9 μm RP particles and 25cm of 3 μm RP particles for the QE+ and OFL systems, respectively (Figure 2). We then tested these column configurations at 120 min, 240 min, and 360 min of effective organic gradient time (Figure 2). The effective LC gradient “length” was defined as the ramp between 10 to 40% B, which was when most peptides eluted. MudPIT was again used as a baseline and captured 91756 ± 16698 spectral counts, 19402 ± 1388 unique peptides, and 3908 ± 36 unique proteins (Supplemental Table S2A).
As expected on the QE+ and OFL, as gradient time increased, the numbers of spectral counts, unique peptides, and unique proteins increased with the largest numbers obtained at 360 minutes of gradient time. With a 360-min gradient, the QE+ captured 65471 ± 1926 spectral counts, 19464 ± 394 unique peptides, and 4034 ± 39 unique proteins. Compared to MudPIT, under these conditions the QE+ captured fewer spectral counts, essentially the same number of unique peptides, and slightly more unique proteins (Table S2B). The OFL operated in “FTIT” mode again outperformed both systems. While a comparable number of unique peptides were identified between the QE+ and the OFL with a 120-min gradient, the OFL captured more spectral counts and detected more unique proteins (Figure 2). With the 360-min gradient, the OFL captured 110211 ± 1894 spectral counts, 21425 ± 420 unique peptides, and 5209 ± 72 unique proteins, which again outperforms MudPIT and the QE+ in all qualitative categories.
The shorter 120-min gradient appeared to under sample the complexity of the HeLa cell extract given the near equal capture of peptide numbers on both QE+ and OFL (Table S2B). For both QE+ and OFL, significant gains were hence made when doubling the effective organic gradient length from 120 to 240 min. For both QE+ and OFL systems, the spectral counts showed a linear relationship with the length of the organic gradient over the three time points tested (Figure 2A), while peptide and protein identifications started to plateau at 360 min (Figure 2BC). The spectral and peptide counts obtained with a 360-min RPLC-MS/MS analysis on the QE+ or 240/360-min RPLC-MS/MS analyses on the OFL were not statistically different from the ones obtained with a 12-step MudPIT (Table S2B), while the number of identified proteins was slightly or very significantly increased on the QE+ and OFL systems, respectively. Even considering that column equilibration and high organic wash steps added between 60 to 90 min to the actual duration of the LC-MS analyses using single phase RP columns, the overall duration of each experiment on the QE+ and OFL systems was still significantly shorter than a 24-hour MudPIT run on the Orbitrap Elite. RPLC-MS/MS on the newest Orbitrap instruments hence leads to equivalent or better qualitative results than MudPIT in a shorter amount of time.
Assessment of Quantitative Reproducibility on Different LC-MS/MS Platforms
In terms of protein identification, the LC-MS/MS analyses of the proteins detected from 3μg of HeLa whole cell digests had inter-replicate reproducibility of 77% commonly detected proteins within the MudPIT dataset, while inter-replicate overlaps of 83% and 85% were measured for the RPLC-MS/MS datasets acquired on the QE+ and OFL, respectively (Table S2C). Increasing the length of the LC gradient had no effect on inter-replicate reproducibility of detection. However, detection and identification of peptides and proteins is not the primary objective of modern proteomics. Detection, identification, and quantitation of peptides and proteins is the primary goal. In addition, the reproducibility of quantitative results is critical for extracting meaningful biological knowledge. Here, we used the distributed normalized spectral abundance factor (dNSAF)34 to quantitatively analyze the results of the 21 LC-MS/MS analyses performed on 3 μg of HeLa cell extract (Supplemental Table S2D).
To assess reproducibility both between technical replicates and between various data acquisition schemes, 210 systematic pair-wise comparisons were performed and Pearson product-moment correlation coefficients (r) were calculated (Figure 3). The quantitative reproducibility between the three technical replicates acquired under the same LC-MS/MS conditions was high for the QE+ and OFL instruments (Figure 3), reaching r values of 0.99, while the three replicate MudPIT analyses showed Pearson r values in the 0.9 range. This low variation is not surprising considering that the RPLC analyses were performed back to back with loading of the peptide mixtures via autosampler, while the MudPIT columns had to be loaded offline before each analysis, hence increasing chromatographic variability.
The number of proteins detected in both analyses used for the pair-wise comparisons was on average 3397 for the inter-replicate comparisons within the MudPIT pipeline, while similar numbers of 3165, 3491, and 3626 were derived from the QE+ platform using 120-, 240- and 360-min LC gradients, respectively (Supplemental Table S2D). With 240- and 360-min LC gradients, the QE+ platform essentially matched the protein output of MudPIT but with substantially improved quantitative reproducibility. At over 4000 for the shortest LC gradient and up to 4700 for the longest, the number of proteins quantified between any two replicates from the OFL platform outperformed both VOE and QE+ at any gradient time, with excellent inter-replicate quantitative reproducibility. Doubling the LC gradient to 240-min on the OFL led to a large increase in the number of reproducibly quantified proteins but did not affect the Pearson r correlation (above 0.99 for all three LC gradients). While, not unexpectedly, the largest r values (>0.99) were observed for pair-wise comparisons of the technical replicates acquired on the QE+ and OFL instruments, comparisons of dNSAF values across platforms were still clearly linear, indicating that this relative quantitative parameter is stable across the different LC-MS/MS platforms.
Qualitative assessment of the proteins detected in 24hr-MudPIT analyses compared to other RPLC-MS/MS pipelines
To further explore the similarities and differences in proteome coverage between a multidimensional chromatography approach on an older generation orbitrap and the other two mass spectrometry orbitrap platforms coupled with single RPLC, we performed systematic pair wise comparisons of the proteins confidently detected (in at least two replicates analyses) in the MudPIT dataset and each of the other six RPLC-MS/MS datasets.
The overlap in identifications (Table S2E) between the 24-hr MudPIT analyses on VOE and the QE datasets at all three LC gradients was large with between 79 and 82% of the total proteins identified combining both datasets (over 3000 proteins, Table 1a). The overlap between 24-hr MudPIT VOE and the OFL datasets was lower at 71–74% (Table 1b), yet still over 3200 to 3495 proteins were detected in both for the shortest and longest LC gradients, respectively. In all sets of comparisons, the number of commonly detected proteins increased as the LC gradient length increased. Concomitantly, as the LC gradient length increased, the number of proteins uniquely detected in the MudPIT analyses decreased, while the number of proteins uniquely detected in the RPLC datasets increased. The number of proteins unique to the 24-hr VOE dataset or QE+ datasets was low, on average around 390 and 320 proteins respectively, corresponding to between 6 to 15% of the proteins detected when merging both analyses being compared (Table 1b). The overlap and uniqueness in protein identifications between 24-hr VOE and QE+ datasets are hence in line with inter-replicate reproducibility calculations (Table S2C) and could be explained by the overall stochasticity in peptide identification by LC/MS, especially in the multidimensional chromatography settings. On the other hand, the OFL datasets contained 728 to 1215 unique proteins, that is, contributed between 16 to 25% of the total proteins identified when combined with the 24-hr VOE dataset (Table 1b). This illustrates a clear technological advantage of this instrument platform over the MudPIT implementation on the older orbitrap instrument.
To further characterize the type of proteins commonly or uniquely detected in each dataset, proteins were sorted into four groups of abundance based on their dNSAF values measured in MudPIT (or measured in the other LC/MS dataset for the proteins not detected in MudPIT): dNSAF >= 1E-3 (high abundance); 1E-3 > dNSAF >=1E-4 (medium-high abundance); 1E-4 > dNSAF >=1E-5 (medium-low abundance); dNSAF <1E-5 (low abundance). These cut-off values roughly corresponded to the inflexion points in log-transformed dNSAF values ranked in decreasing order (Figure 4). Not surprisingly, the large majority (over 91%) of proteins falling in the high and medium-high abundance groups were detected in both analyses being compared (Table 1c). For all six pair-wise comparisons with the MudPIT dataset, the number of proteins that were either commonly or uniquely detected and belonged to the two higher abundance groups was essentially constant and independent on the length of the LC gradient used in the RPLC-MS/MS analyses. Conversely, for proteins in the two lowest abundance ranges, the overlap between MudPIT and the QE+ datasets slightly increased with the longer LC gradient durations. Most (between 66 and 79%) of the proteins uniquely detected in one dataset and not the other fell into the medium-low abundance range (Table 1d). With 51 and 65% (Table 1c) of the proteins in the lowest abundance group, respectively, the 240 and 360 min OFL datasets essentially doubled the recovery of such proteins, which demonstrates once again the greater sensitivity of the Lumos instrument.
In the high abundance group, the MudPIT experiments identified seven proteins not detected in at least one of the six RPLC-MS/MS datasets, with one protein not detected in any of these datasets (Table S2E). Five of these seven proteins were isoforms that shared peptides with other proteins. The non-reproducible detection of peptides unique to each isoform resulted in the differential protein detection across the LC-MS analyses. Similarly, the three OFL datasets consistently and uniquely identified one protein with dNSAF value above 1E-3. This protein, PRKCSH isoform 2, also shared peptides with glucosidase 2 subunit beta isoform 3 and no unique peptide for isoform 2 were detected in the MudPIT or QE+ datasets. Interestingly, a peptide level comparison of the PSMs detected across all LC-MS analyses (Table S2E) revealed that the two peptides unique to PRKCSH isoform 2 and only identified in the OFL datasets were very long and carried a +4 or +5 charge. Although all three instruments were set up to acquire MS1 ions at least up to a +5 charge, no spectra matching precursor ions above +3 were positively identified in the VOE and QE+ datasets. This led to the lack of PRKCSH isoform 2 identification on these instruments. The total number of +4 and +5 peptides spectrum matches (PSMs) contributed by the six OFL datasets only represented 2.5% to the total PSMs combining all datasets. Nevertheless, as illustrated by the differential detection of the PRKCSH isoforms, such PSMs of higher charge states may yet be an additional technical feature unique to the Lumos that provides an advantage in protein detection and quantitation.
Effect of LC/MS Pipeline on Label-Free dNSAF Quantitation
As discussed above, the lowest Pearson product-moment correlation coefficients values amongst the 210 pair-wise comparisons performed to evaluate quantitative reproducibility were measured when comparing the 24hr-VOE MudPIT dataset against the other LC/MS datasets (Figure 3 and Table S2D). To dig deeper into these differences, we performed an analysis of the variability in the quantitative dNSAF values obtained on the different LC-MS/MS platforms. As defined for the qualitative analysis of protein overlap described above (Table 1), the proteins considered in these pair-wise comparisons had to be detected at least twice in the 24hr-MudPIT replicates acquired on Velos Orbitrap Elite (24hr-VOE) and at least twice in the replicate RPLC-MS/MS analyses acquired on the Q-Exactive (120/240/360-QE+) and Orbitrap Fusion Lumos (120/240/360-OFL). Calculating Pearson correlation coefficients on the averaged dNSAF values for these stringently selected groups of proteins increased the r values to above 0.86 and 0.8 for the pair-wise comparison with the QE+ and OFL datasets, respectively (Table S2F).
Proteins were next ranked by decreasing log(dNSAF) in the 24-hr VOE experiment and dNSAF values measured in both experiments were plotted (Figure 4). In agreement with the higher r values measured when comparing MudPIT with the QE+ datasets, their paired dNSAF values appeared less scattered than the comparisons with the OFL data (Figure 4). In particular, the dNSAF values measured in the OFL datasets for the proteins falling in the two lower abundance groups deviated from the MudPIT dNSAF values and most appeared larger. To better assess differences in dNSAF distributions, the dNSAF values were also displayed as box-plots over the entire dNSAF range (Table S2F) or for each of the four groups of protein abundance (Figure 4). While the differences in mean, median, and box boundaries were subtle when plotting the entire dNSAF range (Table S2F), splitting the proteins into abundance groups revealed drastic changes in distribution profiles for high and low abundance protein groups (Figure 4 and Table S2F).
To determine whether such differences were statistically significant, the Mann-Whitney U Test was used to compare the distribution of dNSAF values (Table S2F). Based on the calculated asymptotic probabilities, the dNSAF values measured in the MudPIT dataset for the highly abundant proteins tended to be higher than the corresponding dNSAF values measured in the other six LC/MS datasets. The MudPIT experiments with their longer chromatography, longer acquisition time, and longer dynamic exclusion duration hence tend to oversample the most abundant proteins, which is a technical advantage when such proteins are of biological interest like in the analysis of affinity purified or immunoprecipitated protein complexes.
The dNSAF values for proteins in the medium-high abundance group were either slightly higher in the QE+ datasets compared to MudPIT or not statistically different in the OFL data. On the other hand, the dNSAF values for the proteins in the two groups of lower abundance tended to be greater in the RPLC-MS/MS datasets than the ones measured in MudPIT. Especially, more than 75% of the proteins in the lowest abundance group (dNSAF <1E-5) in the MudPIT dataset had dNSAF values falling in the second lowest abundance group in all three OFL datasets (dNSAF ≥ 1E-5). In the OFL datasets, an entirely different group of proteins constitute the lowest abundance range, proteins that were not detected in either the MudPIT or QE+ analyses (Table 1). These results illustrate once again the greater depth of proteome coverage observed in the OFL datasets.
Effect of RPLC gradient length on peak widths
Longer RPLC gradients are known to broaden chromatographic peak widths, which has consequences on label-free quantitation based on peak areas or ion intensities. MS1 chromatographic features were extracted from the first replicate analyses of the data acquired using 120, 240, and 360 min RPLC gradients on the OFL (Table S3A–C). Full width at half maximum (FWHM) and maximum height values were compared for over 2900 peptide ions detected in all three LC conditions (Table S3D). Doubling gradient length from 120 to 240 min predictably increased the median and range of FWHM values when comparing all 2938 peptides (Figure S1A), however the FWHM range for the 360 min dataset had a lower median. To investigate this surprising trend further, peptide ions were binned into three intensity groups based on their maximum height measured in the 360-min dataset (Table S3D). Regardless of ion intensity, the FWHM values measured at 120 min always tended to be lower than the ones measured at 240 min (see results of non-parametric statistic U test in Table S3D). On the other hand, in the 360-min dataset, only the peptide ions of higher intensity (with maximum height greater or equal to 1E8) showed a statistically significant peak broadening when compared to the shorter 120-min gradient, but there was no significant difference in the FWHM range compared to the 240-min dataset. Plotting the data points next to the notched boxes revealed that a large number of FWHM values were measured at less than 0.1 min (6 sec), especially for the peptides of lower abundance in the 360-min dataset (red symbols in Figure S1A).
As illustrated by the extracted ion chromatograms (Table S3E) for representative peptides falling in the three intensity bins (Figure S1B), the issue with peptide ions of lower intensity was that peak boundaries could not be measured accurately due to noisy signal, resulting in FWHM values being inaccurately calculated in the chromatographic profiles. In the 360-min dataset, over 68% of the peptides of low abundance were assigned such incorrect FWHM values of less than 0.1 min (Table S3D). After removing peptides with FWHM values lower than 6 sec in the 360-min dataset, the distribution of peak widths observed for the remaining 1553 peptides now followed the expected trend of peak broadening as the LC gradient increased (Figure S1C). The differences in median and range of peak widths observed between the shorter 120-min gradient and the other two chromatographic conditions were statistically supported as assessed by U-test (Table S3D).
Overall, for the 360-min RPLC dataset, a large portion of the chromatographic features calculated using standard Skyline data processing settings could not be trusted, especially for peptides of lower abundance. In other words, if label-free quantitation is to be performed using chromatographic peak areas or ion intensities, RPLC gradients longer than four hours should be avoided.
Depth of Proteome Coverage
As previously shown, the Orbitrap Fusion Lumos operated in FT/IT mode is more sensitive than the QE+ and hence provides superior results in terms of depth of coverage of the HeLa cell proteome16. To illustrate this point further, we queried the proteome detected in the current analyses of 3μg of HeLa whole cell digests for known members of the histone deacetylase (HDAC) 1 and 2 containing complexes (Table S2G). The Sin3, NuRD, and CoRest complexes all contain HDAC1 and HDAC235 and affinity purifications using HDAC1 and HDAC2 as baits effectively recover components from all three complexes36, 37.
In all, 35 known members of these three complexes were detected in at least one of the 21 HeLa whole cell extracts analyzed on the VOE, QE+, or OFL (Figure 5A). The 360-min OFL dataset was the most comprehensive with all 35 proteins recovered in at least one of the three replicates analyses (Table S2G). Not surprisingly, the four core subunits shared by multiple complexes (HDAC1 and 2; RBBP4 and 7) were reproducibly detected in all 21 independent analyses and at higher levels (in the medium-high abundance range defined in Figure 4) than subunits unique to each complex. NuRD components were detected more comprehensively than the other two complexes with at least 8 out of 10 components detected by all three LC/MS platforms (with all 10 detected by the OFL). On the other hand, SIN3 components were more sporadically detected and only the longer LC gradient on the OFL was able to recover 15 SIN3 subunits. On all three platforms, NuRD subunits were detected at overall higher levels than SIN3 components, suggesting that the NuRD complex may be present in higher abundance than the Sin3 complex in vivo (Figure 5A).
Analyzing the same protein samples on the OFL enabled the more robust detection of subunits also detected on the other two platforms, such as the BBX, FOXK1 and FOXK2 transcription factors associated with the SIN3A complex. In addition, the OFL dataset contained some uniquely detected subunits. For example, paralogues of different abundance exist within each of the three complexes, such as Sin3A/Sin3B, ZMYM3/ZMYM2, and CHD4/CHD3, which were differently recovered at both the quantitative and frequency levels. Sin3B and ZMYM2 were only found in the Lumos analyses (Figure 5B) and fell in the lowest dNSAF abundance range. The case of the MAX transcription factor is also worth noting. MAX is a short 97 amino acid protein with only one tryptic peptide detectable by positive ion mass spectrometry. The doubly- and triply-charged ions of this peptides were detected in all three analyses on the OFL. For all three LC gradient lengths, MAX’s peptide reproducibly eluted early in the organic gradient (at 19.5, 16.5, and 15.5 % B in the 120, 240, and 360 min gradients, respectively; data not shown), which is likely the reason why this peptide was “missed” in the multidimensional MudPIT analysis. The more controlled LC settings of the single RPLC (such loading via autosampler) compared to the “leakier” chromatography implemented in the MudPIT set-up (offline loading) may hence be another contributing factor to the better performance of the OFL LC/MS system. These results further demonstrate the powerful depth of analysis that is achievable on the Lumos system using a longer organic LC gradient on a single RP column.
CONCLUSIONS
Rapid advances in mass spectrometry present the challenge of determining the optimal setup of a platform where a mass spectrometer and the liquid chromatographic system must work in tandem for maximum proteome coverage. Multidimensional chromatography approaches like MudPIT28 can occupy an instrument for up to 24 hrs, a significant amount of time to dedicate to one sample. However, multidimensional LC can yield very deep analyses of proteomes and post translational modifications38. On the other hand, trying to comprehensively capture a proteome in a short period of time is also of great interest39. Different studies have different objectives and practical considerations hence play a role. Essentially, there will be a tradeoff between depth of proteome analysis and the amount of time an instrument can be used. Therefore, it is important to analytically evaluate both MS settings and LC set-up for optimal proteomic analysis.
In this work, we compared chromatographic and mass spectrometry systems for the analysis of the HeLa cell proteome, the end goal being to evaluate the reproducibility of the number of proteins detected, identified, and quantified. Not surprisingly and consistent with general liquid chromatography theory, smaller RP particle size packed over longer lengths in smaller i.d. columns led to improved performance. In addition, as a major goal of modern proteomics is the quantitative analysis of proteomes, we used spectral counts and the dNSAF34 as the basis for our quantitative comparisons and assessed the Pearson r values of replicate analyses on three mass spectrometers under several different chromatographic conditions. Single phase RP chromatography on both the QE+ and Lumos outperformed MudPIT on the Orbitrap Elite mass spectrometer, which led to maximum Pearson’s correlation coefficient r of 0.92 between two replicates. On the QE+, we obtained r values greater than 0.99 for more than 3100 proteins at 120 min of gradient time and r values greater than 0.98 for more than 3600 proteins with a 360 min gradient. On the Lumos, we obtained r values greater than 0.99 for all LC gradients tested with over 4700 proteins reproducibly quantified at 360 minutes of gradient time, demonstrating the remarkable performance of this system. The Pearson’s r values were impressive across replicates on the QE+ and the Lumos, and more proteins can be quantified as gradient lengths increased. However, increasing gradient time by 240 min led to an increase of about 500 and 700 proteins on the QE+ and the Lumos, respectively, yet the identification of these additional proteins of lower abundance may not be useful for all studies. Therefore, LC conditions should hence be tailored to each experimental goal to mitigate the tradeoff between spending chromatographic time and gaining deeper proteome coverage. Especially on the Lumos system at 360 minutes of gradient time, highly reproducible spectral count-based quantitative proteomic analysis of the HeLa cell proteome was achieved.
Supplementary Material
Supporting Information (excel workbook): Table S1: LC-MS/MS analyses of 200ng of HeLa whole cell digests on three different mass spectrometry platforms and varying particle sizes, inner diameters, and column lengths. (A) Label-free quantitative features of the detected proteins; (B) Descriptive statistics of the numbers of spectra, peptides, and proteins. Table S2: LC-MS/MS analyses of 3ug of HeLa whole cell digests on three different mass spectrometry platforms and varying liquid chromatography gradient lengths. (A) Label-free quantitative features of the detected proteins; (B) Descriptive statistics of the numbers of spectra, peptides, and proteins; (C) Inter-replicate reproducibility of protein identification; (D) Reproducibility of the label-free quantitative values measured for the detected proteins; (E) Qualitative comparison of the proteins detected in the 24hr-MudPIT analyses vs all other RPLC-MS/MS pipelines; (F) Pair-wise comparisons of the label-free quantitative dNSAF values measured for proteins detected in both 24hr-MudPIT analyses and the other RPLC-MS/MS analyses; (G) Label-free quantitation of HDAC1/2-containing complexes. Table S3: Analysis of peptide ions peak widths as a function of varying RPLC gradient lengths. (A) MS1 chromatographic features of peptide ions detected in the first replicate analysis of 3ug HeLa digests analyzed with a 120-mim RPLC gradient coupled to an Orbitrap Fusion Lumos mass spectrometer; (B) MS1 chromatographic features of peptide ions detected in the first replicate analysis of 3ug HeLa digests analyzed with a 240-mim RPLC gradient coupled to an Orbitrap Fusion Lumos mass spectrometer; (C) MS1 chromatographic features of peptide ions detected in the first replicate analysis of 3ug HeLa digests analyzed with a 360-mim RPLC gradient coupled to an Orbitrap Fusion Lumos mass spectrometer; (D) MS1 chromatographic features of peptide ions detected in all three RPLC gradient lengths; (E) Extracted ion chromatograms of representative peptide ions of varying abundance levels. Supporting Figure S1: Effect of RPLC gradient length on MS1 chromatography. (A) Distribution of FWHM values. (B) Extracted ion chromatograms for representative peptide ions. (C) Distribution of FWHM values after filtering out inaccurate peak boundaries assignments.
Funding Sources
This work was supported by the Stowers Institute for Medical Research and the National Institute of General Medical Sciences of the National Institutes of Health under Award Number RO1GM112639 to MPW. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATIONS
- WCL
whole cell lysate
- VPO
Velos Pro Orbitrap
- VOE
Velos Orbitrap Elite
- QE+
Q-Exactive Plus
- OFL
Orbitrap Fusion Lumos
- FT
Fourier-Transform
- IT
Ion Trap
- MudPIT
Multidimensional Protein Identification Technology
- MS/MS
tandem Mass Spectrometry
- RP
reverse phase
- LC
Liquid Chromatography
- i.d.
internal diameter
- o.d.
outer diameter
- dNSAF
distributed Normalized Spectrum Abundance Factor
- PSM
peptide spectrum match
Footnotes
Supporting Information.
The following files are available free of charge at http://pubs.acs.org.
REFERENCES
- 1.Hardman M; Makarov AA, Interfacing the orbitrap mass analyzer to an electrospray ion source. Anal Chem 2003, 75, (7), 1699–705. [DOI] [PubMed] [Google Scholar]
- 2.Hu Q; Noll RJ; Li H; Makarov A; Hardman M; Graham Cooks R, The Orbitrap: a new mass spectrometer. J Mass Spectrom 2005, 40, (4), 430–43. [DOI] [PubMed] [Google Scholar]
- 3.Perry RH; Cooks RG; Noll RJ, Orbitrap mass spectrometry: instrumentation, ion motion and applications. Mass Spectrom Rev 2008, 27, (6), 661–99. [DOI] [PubMed] [Google Scholar]
- 4.Eliuk S; Makarov A, Evolution of Orbitrap Mass Spectrometry Instrumentation. Annu Rev Anal Chem (Palo Alto Calif) 2015, 8, 61–80. [DOI] [PubMed] [Google Scholar]
- 5.Kelstrup CD; Bekker-Jensen DB; Arrey TN; Hogrebe A; Harder A; Olsen JV, Performance Evaluation of the Q Exactive HF-X for Shotgun Proteomics. J Proteome Res 2018, 17, (1), 727–738. [DOI] [PubMed] [Google Scholar]
- 6.Riley NM; Mullen C; Weisbrod CR; Sharma S; Senko MW; Zabrouskov V; Westphall MS; Syka JE; Coon JJ, Enhanced Dissociation of Intact Proteins with High Capacity Electron Transfer Dissociation. J Am Soc Mass Spectrom 2016, 27, (3), 520–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Michalski A; Damoc E; Hauschild JP; Lange O; Wieghaus A; Makarov A; Nagaraj N; Cox J; Mann M; Horning S, Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole Orbitrap mass spectrometer. Mol Cell Proteomics 2011, 10, (9), M111 011015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelstrup CD; Young C; Lavallee R; Nielsen ML; Olsen JV, Optimized fast and sensitive acquisition methods for shotgun proteomics on a quadrupole orbitrap mass spectrometer. J Proteome Res 2012, 11, (6), 3487–97. [DOI] [PubMed] [Google Scholar]
- 9.Nagaraj N; Kulak NA; Cox J; Neuhauser N; Mayr K; Hoerning O; Vorm O; Mann M, System-wide perturbation analysis with nearly complete coverage of the yeast proteome by single-shot ultra HPLC runs on a bench top Orbitrap. Mol Cell Proteomics 2012, 11, (3), M111 013722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scheltema RA; Hauschild JP; Lange O; Hornburg D; Denisov E; Damoc E; Kuehn A; Makarov A; Mann M, The Q Exactive HF, a Benchtop mass spectrometer with a pre-filter, high-performance quadrupole and an ultra-high-field Orbitrap analyzer. Mol Cell Proteomics 2014, 13, (12), 3698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brunner AM; Lossl P; Liu F; Huguet R; Mullen C; Yamashita M; Zabrouskov V; Makarov A; Altelaar AF; Heck AJ, Benchmarking multiple fragmentation methods on an orbitrap fusion for top-down phospho-proteoform characterization. Anal Chem 2015, 87, (8), 4152–8. [DOI] [PubMed] [Google Scholar]
- 12.Williamson JC; Edwards AV; Verano-Braga T; Schwammle V; Kjeldsen F; Jensen ON; Larsen MR, High-performance hybrid Orbitrap mass spectrometers for quantitative proteome analysis: Observations and implications. Proteomics 2016, 16, (6), 907–14. [DOI] [PubMed] [Google Scholar]
- 13.Espadas G; Borras E; Chiva C; Sabido E, Evaluation of different peptide fragmentation types and mass analyzers in data-dependent methods using an Orbitrap Fusion Lumos Tribrid mass spectrometer. Proteomics 2017, 17, (9), 1600416. [DOI] [PubMed] [Google Scholar]
- 14.Ferries S; Perkins S; Brownridge PJ; Campbell A; Eyers PA; Jones AR; Eyers CE, Evaluation of Parameters for Confident Phosphorylation Site Localization Using an Orbitrap Fusion Tribrid Mass Spectrometer. J Proteome Res 2017, 16, (9), 3448–3459. [DOI] [PubMed] [Google Scholar]
- 15.Barbier Saint Hilaire P; Hohenester UM; Colsch B; Tabet JC; Junot C; Fenaille F, Evaluation of the High-Field Orbitrap Fusion for Compound Annotation in Metabolomics. Anal Chem 2018, 90, (5), 3030–3035. [DOI] [PubMed] [Google Scholar]
- 16.Levy MJ; Washburn MP; Florens L, Probing the Sensitivity of the Orbitrap Lumos Mass Spectrometer Using a Standard Reference Protein in a Complex Background. J Proteome Res 2018, 17, (10), 3586–3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun L; Zhu G; Dovichi NJ, Comparison of the LTQ-Orbitrap Velos and the Q-Exactive for proteomic analysis of 1–1000 ng RAW 264.7 cell lysate digests. Rapid Commun Mass Spectrom 2013, 27, (1), 157–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei W; Luo W; Wu F; Peng X; Zhang Y; Zhang M; Zhao Y; Su N; Qi Y; Chen L; Zhang Y; Wen B; He F; Xu P, Deep Coverage Proteomics Identifies More Low-Abundance Missing Proteins in Human Testis Tissue with Q-Exactive HF Mass Spectrometer. J Proteome Res 2016, 15, (11), 3988–3997. [DOI] [PubMed] [Google Scholar]
- 19.Shishkova E; Hebert AS; Coon JJ, Now, More Than Ever, Proteomics Needs Better Chromatography. Cell Syst 2016, 3, (4), 321–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sandra K; Moshir M; D’Hondt F; Verleysen K; Kas K; Sandra P, Highly efficient peptide separations in proteomics Part 1. Unidimensional high performance liquid chromatography. J Chromatogr B Analyt Technol Biomed Life Sci 2008, 866, (1–2), 48–63. [DOI] [PubMed] [Google Scholar]
- 21.Sandra K; Moshir M; D’Hondt F; Tuytten R; Verleysen K; Kas K; Francois I; Sandra P, Highly efficient peptide separations in proteomics. Part 2: bi- and multidimensional liquid-based separation techniques. J Chromatogr B Analyt Technol Biomed Life Sci 2009, 877, (11–12), 1019–39. [DOI] [PubMed] [Google Scholar]
- 22.Hsieh EJ; Bereman MS; Durand S; Valaskovic GA; MacCoss MJ, Effects of column and gradient lengths on peak capacity and peptide identification in nanoflow LC-MS/MS of complex proteomic samples. J Am Soc Mass Spectrom 2013, 24, (1), 148–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu H; Finch JW; Lavallee MJ; Collamati RA; Benevides CC; Gebler JC, Effects of column length, particle size, gradient length and flow rate on peak capacity of nano-scale liquid chromatography for peptide separations. J Chromatogr A 2007, 1147, (1), 30–6. [DOI] [PubMed] [Google Scholar]
- 24.Wohlbrand L; Rabus R; Blasius B; Feenders C, Influence of NanoLC Column and Gradient Length as well as MS/MS Frequency and Sample Complexity on Shotgun Protein Identification of Marine Bacteria. J Mol Microbiol Biotechnol 2017, 27, (3), 199–212. [DOI] [PubMed] [Google Scholar]
- 25.Wang H; Yang Y; Li Y; Bai B; Wang X; Tan H; Liu T; Beach TG; Peng J; Wu Z, Systematic optimization of long gradient chromatography mass spectrometry for deep analysis of brain proteome. J Proteome Res 2015, 14, (2), 829–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Washburn MP; Wolters D; Yates JR 3rd, Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol 2001, 19, (3), 242–7. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y; Wen Z; Washburn MP; Florens L, Refinements to label free proteome quantitation: how to deal with peptides shared by multiple proteins. Anal Chem 2010, 82, (6), 2272–81. [DOI] [PubMed] [Google Scholar]
- 28.Florens L; Washburn MP, Proteomic analysis by multidimensional protein identification technology. Methods Mol Biol 2006, 328, 159–75. [DOI] [PubMed] [Google Scholar]
- 29.Xu T; Park SK; Venable JD; Wohlschlegel JA; Diedrich JK; Cociorva D; Lu B; Liao L; Hewel J; Han X; Wong CCL; Fonslow B; Delahunty C; Gao Y; Shah H; Yates JR 3rd, ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J Proteomics 2015, 129, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tabb DL; McDonald WH; Yates JR 3rd, DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res 2002, 1, (1), 21–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ, Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, (7), 966–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deutsch EW; Csordas A; Sun Z; Jarnuczak A; Perez-Riverol Y; Ternent T; Campbell DS; Bernal-Llinares M; Okuda S; Kawano S; Moritz RL; Carver JJ; Wang M; Ishihama Y; Bandeira N; Hermjakob H; Vizcaino JA, The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res 2017, 45, (D1), D1100–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vizcaino JA; Deutsch EW; Wang R; Csordas A; Reisinger F; Rios D; Dianes JA; Sun Z; Farrah T; Bandeira N; Binz PA; Xenarios I; Eisenacher M; Mayer G; Gatto L; Campos A; Chalkley RJ; Kraus HJ; Albar JP; Martinez-Bartolome S; Apweiler R; Omenn GS; Martens L; Jones AR; Hermjakob H, ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol 2014, 32, (3), 223–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y; Wen Z; Washburn MP; Florens L, Improving label-free quantitative proteomics strategies by distributing shared peptides and stabilizing variance. Anal Chem 2015, 87, (9), 4749–56. [DOI] [PubMed] [Google Scholar]
- 35.Kelly RD; Cowley SM, The physiological roles of histone deacetylase (HDAC) 1 and 2: complex co-stars with multiple leading parts. Biochem Soc Trans 2013, 41, (3), 741–9. [DOI] [PubMed] [Google Scholar]
- 36.Banks CAS; Thornton JL; Eubanks CG; Adams MK; Miah S; Boanca G; Liu X; Katt ML; Parmely TJ; Florens L; Washburn MP, A Structured Workflow for Mapping Human Sin3 Histone Deacetylase Complex Interactions Using Halo-MudPIT Affinity-Purification Mass Spectrometry. Mol Cell Proteomics 2018, 17, (7), 1432–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Banks CAS; Miah S; Adams MK; Eubanks CG; Thornton JL; Florens L; Washburn MP, Differential HDAC1/2 network analysis reveals a role for prefoldin/CCT in HDAC1/2 complex assembly. Sci Rep 2018, 8, (1), 13712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bekker-Jensen DB; Kelstrup CD; Batth TS; Larsen SC; Haldrup C; Bramsen JB; Sorensen KD; Hoyer S; Orntoft TF; Andersen CL; Nielsen ML; Olsen JV, An Optimized Shotgun Strategy for the Rapid Generation of Comprehensive Human Proteomes. Cell Syst 2017, 4, (6), 587–599 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hebert AS; Richards AL; Bailey DJ; Ulbrich A; Coughlin EE; Westphall MS; Coon JJ, The one hour yeast proteome. Mol Cell Proteomics 2014, 13, (1), 339–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information (excel workbook): Table S1: LC-MS/MS analyses of 200ng of HeLa whole cell digests on three different mass spectrometry platforms and varying particle sizes, inner diameters, and column lengths. (A) Label-free quantitative features of the detected proteins; (B) Descriptive statistics of the numbers of spectra, peptides, and proteins. Table S2: LC-MS/MS analyses of 3ug of HeLa whole cell digests on three different mass spectrometry platforms and varying liquid chromatography gradient lengths. (A) Label-free quantitative features of the detected proteins; (B) Descriptive statistics of the numbers of spectra, peptides, and proteins; (C) Inter-replicate reproducibility of protein identification; (D) Reproducibility of the label-free quantitative values measured for the detected proteins; (E) Qualitative comparison of the proteins detected in the 24hr-MudPIT analyses vs all other RPLC-MS/MS pipelines; (F) Pair-wise comparisons of the label-free quantitative dNSAF values measured for proteins detected in both 24hr-MudPIT analyses and the other RPLC-MS/MS analyses; (G) Label-free quantitation of HDAC1/2-containing complexes. Table S3: Analysis of peptide ions peak widths as a function of varying RPLC gradient lengths. (A) MS1 chromatographic features of peptide ions detected in the first replicate analysis of 3ug HeLa digests analyzed with a 120-mim RPLC gradient coupled to an Orbitrap Fusion Lumos mass spectrometer; (B) MS1 chromatographic features of peptide ions detected in the first replicate analysis of 3ug HeLa digests analyzed with a 240-mim RPLC gradient coupled to an Orbitrap Fusion Lumos mass spectrometer; (C) MS1 chromatographic features of peptide ions detected in the first replicate analysis of 3ug HeLa digests analyzed with a 360-mim RPLC gradient coupled to an Orbitrap Fusion Lumos mass spectrometer; (D) MS1 chromatographic features of peptide ions detected in all three RPLC gradient lengths; (E) Extracted ion chromatograms of representative peptide ions of varying abundance levels. Supporting Figure S1: Effect of RPLC gradient length on MS1 chromatography. (A) Distribution of FWHM values. (B) Extracted ion chromatograms for representative peptide ions. (C) Distribution of FWHM values after filtering out inaccurate peak boundaries assignments.
Data Availability Statement
The mass spectrometry data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository32, 33 with the identifiers 10.6019/PXD009875. All original data files underlying this manuscript may also be accessed after publication from the Stowers Original Data Repository at http://www.stowers.org/research/publications/libpb-1303.