

Abstract
Alternative splicing of eukaryotic transcripts is a mechanism that significantly contributes to the generation of the vast protein diversity from a rather limited number of genes. The mechanisms and outcomes of alternative splicing of individual transcripts are relatively well understood, and recent efforts have been directed towards studying splicing networks. It has become apparent that coordinated splicing networks regulate tissue and organ development, and that alternative splicing has important physiological functions in different developmental processes in humans.
Table of content summary
Alternative splicing expands proteome complexity by generating multiple transcript (and protein) isoforms from a single gene. Numerous alternative splicing events occur during cell differentiation and tissue maturation, suggesting that alternative splicing supports proper development. Recent studies shed light on how alternative splicing and its coordination regulates organ development and tissue homeostasis.
Introduction
Alternative splicing is a ubiquitous regulatory mechanism of gene expression that allows generation of more than one unique mRNA species from a single gene. Alternative splicing can generate mRNAs that differ in untranslated regions (UTR) or the coding sequence, with mechanisms including exon skipping (removal of specific exons, referred to as cassette exons, that can be either included or excluded from the mature transcript depending on alternative splicing decisions), choice between mutually exclusive exons, the usage of alternative splice sites (that affects the boundaries between introns and exons involved in this alternative splicing event contributing to transcript diversity) and intron retention. These differences might affect mRNA stability, localization, or translation. Furthermore, some splicing mRNA isoforms could change the reading frame, resulting in the generation of different protein isoforms with diverse functions and/or localizations. Genome-wide studies estimated that 90–95% of human genes undergo some level of alternative splicing1,2. From the ~20,000 human protein-coding genes, high-resolution mass spectrometry analyses revealed that ~37% of them generates multiple protein isoforms3. This evidence certainly demonstrates that alternative splicing contributes to proteome complexity. However extensive further work is needed to identify the functional consequences for most of the identified splicing events. In addition, not all detected alternative splicing events might necessarily result in the production of functional proteins owing to several reasons: (i) the transcript is non-coding and is not translated into a protein, (ii) RNA stability is affected, (iii) mRNA localization changes precluding correct function of the transcript and/or protein. Adding another level of complexity, it is possible that alternatively spliced transcripts are non-coding, but may still have a function, for example they may modulate other RNAs by competing with them for their regulators. It also needs to be considered that nucleic acid amplification methods can generate some artifacts, including overestimation of splicing events.
The research of alternative splicing has now progressed from recording single splicing events and studying their impact on protein expression to global description of alternative splicing networks and how they are molecularly coordinated. However, whereas the progress in uncovering regulatory aspects of alternative splicing networks has been fast, the demonstration of their functional consequences has been running behind, and understanding the physiological relevance of alternative splicing events has now emerged as one of the biggest challenges of the field (Box 1).
Box 1 |. From individual splicing events to coordinated splicing networks in development.
The advances in genomics allowed the transition from cloning individual splice isoforms to global description of alternatively-spliced transcripts. The first generation of global transcriptomic analyses used microarrays to identify groups of splicing events involved in tissue formation, function and pathology as well as events controlled by certain signalling cascades. More recently, deep RNA-sequencing revealed and continues revealing in an unbiased manner how alternative splicing is coordinated during development, cell differentiation, and in diseases. Complementary studies aim at understanding roles of alternative splicing.
Studies of functions of alternatively spliced isoforms in the nervous system have been particularly insightful. Some exciting data has also been collected in the context of cardiac and skeletal muscle development as well as heart diseases and muscular dystrophies. However, much more work is still required. A very limited number of splicing variants has been associated with particular functions in vivo so far, while the number of splicing isoforms with unknown and even unexplored functions exponentially increases. The fact that in vivo splicing events are coordinated adds another level of complexity to the analysis.
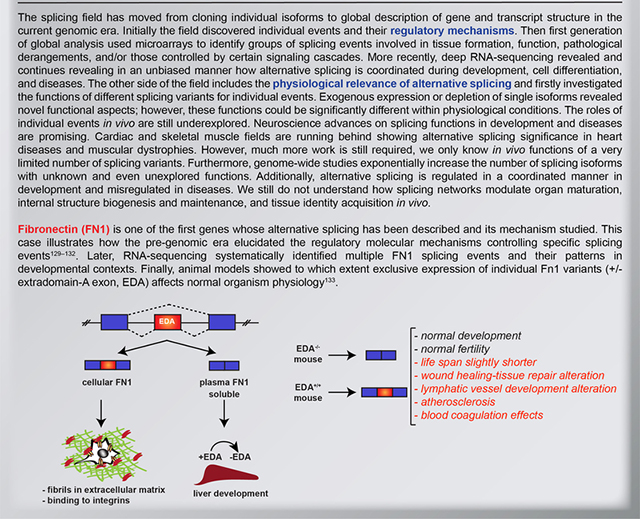
Fibronectin (FN1) was one of the first genes of which alternative splicing has been described and its mechanism studied. The Figure illustrates the early finding regarding a specific alternative splicing event in FN1 transcript, namely the retention or exclusion of the extradomain-A exon (EDA) that controls FN1 tissue localization. EDA-containing protein (referred to as cellular FN1) is an important component of extracellular matrix and is secreted (mostly by fibroblasts) to regulate cell adhesion and migration, whereas in the liver EDA is skipped generating a soluble FN1 isoform that is secreted into plasma. Plasma FN1 constitutes the main protein components of the blood and is involved in blood clotting (left)129–132. Later, RNA-sequencing systematically identified multiple other FN1 splicing events and their patterns in various developmental contexts. Finally, animal models revealed the functional impact of splice isoforms of FN1, demonstrating roles of both EDA isoforms in normal organism physiology, and at the same time revealing that these splicing transitions do not have a major impact on animal development (right)133.
It is now established that alternative splicing contributes to cell differentiation and lineage determination, tissue identity acquisition and maintenance, and organ development2. This functional impact of alternative splicing is further highlighted by the large number of human diseases caused by mutations in cis-acting pre-mRNA sequence elements (including 5’ and 3’ splice sites, exonic and intronic enhancer or silencer sequences ), trans-acting splicing factors, or other components of the spliceosome4. Studying tissue and organ development is particularly well-suited to identify functional impact and relevance of splicing networks, as in this context regulation of cell fate decisions is absolutely critical and governs the transitioning from embryonic to adult functions5.Molecular understanding of the splicing transitions that take place during development has also revealed important bases for understanding pathological mechanisms implicated in diseases where normal networks are misregulated.
Here, we comprehensively review our current knowledge about the physiological contribution of alternative splicing to tissue identity acquisition, organ development and tissue and organ physiology. First we briefly discuss the different layers of splicing regulation and coordination, moving on to outlining recent discoveries demonstrating that coordination of alternative splicing networks contributes to the development of various organs, including brain, heart, skeletal muscle, and liver. We also consider several notable examples of the role of alternative splicing in maintaining tissue homeostasis and cell function. This article will not cover the role of alternative splicing in epithelial–mesenchymal transition as well as its critical relevance to cancer and the reader is referred to other outstanding reviews on this topics6,7. Deregulation of splicing in diseases has been extensively reviewed elsewhere4,8,9 and thus we focus here on the physiological context. We will only briefly discuss throughout the manuscript some developmental disorders for which the functional impact of defective splicing has been demonstrated.
Alternative splicing regulation
Alternative splicing outcomes (that is, the relative abundance of the different alternatively-spliced isoforms) are influenced by several factors: i) splice site strength, ii) cis-regulatory sequences in pre-mRNAs that favor or impair exon recognition, and iii) expression levels of trans-acting factors (RNA-binding proteins (RBPs), splicing factors). Strong splice sites contain consensus sequences that are recognized by the spliceosome machinery and usually lead to “constitutive splicing”, that is the inclusion of the exon in question in all the mRNA molecules synthesized by the cell. However, constitutive exons containing non-consensus splice sites are also frequent and their reproducible retention is mediated by additional splicing regulatory elements. Weak splice sites differ from consensus sequences and their recognition by the spliceosome is highly dependent on the presence of cis-acting sequences and the cellular context such as expression levels of splicing factors.
During tissue development and cell differentiation specific RBPs are finely regulated at their expression levels, localization, their own splicing, mRNA stability, and translation efficiency. RBPs bind to cis-elements promoting or inhibiting splice site recognition, hence RBP expression coordinates alternative splicing networks during development (Supplementary Table 1). Microarray analysis and more recently RNA-sequencing studies have globally described transcriptional and posttranscriptional dynamics in development including those involving alternative splicing events (Table 1).These studies have provided several important insights: i) splicing transitions occurred during development and cell differentiation in multiple genes at the same time (splicing coordination); ii) specific RBPs contribute to this coordination; iii) genes that are regulated by alternative splicing mechanisms are not modulated at their overall expression levels (up- or down-regulation); and iv) developmental splicing networks are cell type- and/or region-specific within different tissues (for example, differences were observed between cerebral cortex, and the hippocampus, as well as between different neuronal cell types in the brain, between cardiomyocytes and cardiac fibroblasts in the heart, and between hepatocytes and non-parenchymal cells in the liver)10–15.
Table 1.
Results from recent studies investigating alternative splicing during tissue development or cell differentiation.
| Tissue / experimental conditions | Splicing targets or processes implicated in alternative splicing events | Recent review articles |
|---|---|---|
| Brain development / mouse cerebral cortex (RNA-seq), hippocampus (RNA-seq, microarrays) 13,31,139,140 | Endocytosis, vesicle mediated transport, small GTPases, cytoskeleton dynamics. | 76,141,142 |
| Heart development / mouse ventricles, cardiomyocytes, cardiac fibroblasts (RNA-seq, microarrays) 5,11,80 | Cardiomyocytes: vesicular trafficking, membrane and cytoskeleton remodelling, ion channels, chromatin modifications. Fibroblasts: cell–cell contact, cell adhesion. | 79,143,144 |
| Male germ cell development / mouse (RNA-seq)120,121 | Ralgps2, Bptf, Vapa4, Ezh2, Odf2, among others. | 145 |
| T-cell activation / human Jurkat JSL1 cells PMA activated, primary human CD4+ T-cells (RNA-seq, microarrays)10,26,128,129 | Cell cycle regulation, cell signaling (BMP, CD40, NFKB, MAPK, RhoA pathways), vesicular trafficking, immune and inflammatory responses. Focus: LEF1 (exon 6), MKK7 (exon 2) | 146,147 |
| Liver development / mouse hepatocytes and non-parenchymal cells (RNA-seq)12 | Actin-based processes, protein transport, splicing, chromatin modifications, cytoskeleton organization. | Not available to our knowledge |
| Erythropoiesis / human erythroblasts from CD34+ cord blood progenitors (RNA-seq)137,138 | Cell cycle, chromatin function, RNA processing. Subset of transitions: PTCs and/or intron retention on RNA processing and spliceosome genes, indicating that splicing–NMD coupling may contribute to gene regulation. | 148 |
| Erythropoiesis / mouse liver (RNA-seq)136 | Receptor activity, chromatin modifications, RNA processing, RNA binding, DNA repair and packaging. Focus: Ndel1. | 148 |
| Myoblast differentiation / C2C12 cells (RNA-seq, microarrays) 14,149 | Cytoskeletal properties, actin binding, integrin signalling, cell junction. | Not available to our knowledge |
| Smooth muscle cell de-differentiation / mouse primary cells from aorta and bladder (exon-junction arrays)15 | Actin cytoskeleton, non-motor actin binding protein. Differentiated cells: intron retentions lead to PTC and protein downregulation (RBPs, splicing factors, splicing regulatory kinases) | Not available to our knowledge |
Bpft: bromodomain PHD finger transcription factor, Ezh2: enhancer Of Zeste 2 Polycomb Repressive Complex 2 Subunit, LEF1: lymphoid enhancer binding factor 1, MKK7: Jun-kinase-kinase MAP-kinase-7, Ndel1: nuclear distribution protein nudE-like 1, NMD: non-sense mediated decay, Odf2: outer dense fiber of sperm tails-2, PMA: phorbol 12-myristate 13-acetate, PTC: premature termination codon, Ralpgs2: ras-specific guanine nucleotide-releasing factor-2, RBPs: RNA-binding proteins, RNA-seq: RNA-sequencing, Vapa4: vesicle-associated membrane protein-associated-4.
Regardless of the patterns of expression of RBPs in different tissues, a “splicing code” (meaning combinations of RNA features associated with the regulation of splicing) has been proposed to predict splicing outcomes in specific cell types and conditions16,17. Deciphering this splicing code is not trivial: the same regulatory sequences on different or the same transcripts can be recognized by different RBPs depending on cellular and specific sequence contexts; RBPs can be regulated by other RBPs; the same RBP can exert positive or negative regulatory effects on different splicing events depending on the location of the binding motif for this RBP relative to the alternatively spliced regions in the transcript; last but not least, we still lack a full comprehensive list of RBPs and their binding sites18. Furthermore, the goal to predict splicing patterns based solely on sequences of the pre-mRNA is even more complicate to achieve in tissues, as they are composed of different cell types, each of which may have unique splicing regulatory networks18.
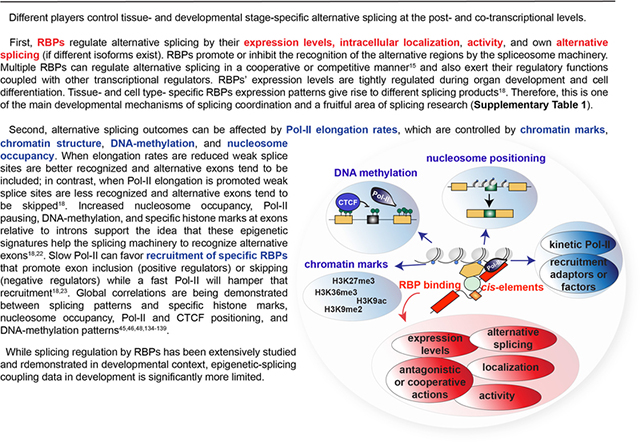
Apart from the differential expression and activity of RBPs, one more layer of regulation is provided by the fact that splicing frequently occurs co-transcriptionally19,20 allowing a mechanistic interplay between the transcriptional and splicing machineries21–24 (Box 2). Alternative splicing outcomes can be affected by RNA-polymerase II (Pol II) kinetics, which are controlled by chromatin modifications, chromatin structure, and nucleosome occupancy21. This indicates that epigenetic modifications and alternative splicing can be coupled (Box 2). Further evidence for this coupling within developmental contexts will be provided as more specific examples later in the manuscript. The transcriptional machinery has also been shown to recruit splicing factors that in turn promote or inhibit exon inclusion21,25. Combination of both scenarios is certainly possible: slow Pol II can favor recruitment of specific RBPs that promote exon inclusion or skipping while a fast Pol II will hamper that recruitment21,26.
Box 2 |. Key regulators of alternative splicing coordination.
Different players control tissue- and developmental stage-specific alternative splicing at the post- and co-transcriptional levels.
First, RNA-binding proteins (RBPs) regulate alternative splicing owing to their different expression levels, intracellular localization, activity, and in some cases their own alternative splicing. RBPs promote or inhibit the recognition of the alternative regions by the spliceosome machinery. Multiple RBPs can regulate alternative splicing in a cooperative or a competitive manner15 and also exert their regulatory functions by coupling with other splicing regulators (such as the transcriptional machinery or epigenetic readers). Expression levels of RBPs are tightly regulated during organ development and cell differentiation. Tissue- and cell type- specific RBP expression patterns give rise to different splicing products18. Therefore, it appears that modulation of splicing transitions through RBPs is one of the main mechanisms of splicing coordination, particularly during development, and the role of RBPs in regulating developmental splicing transitions has been intensively studied (Supplementary Table 1).
Alternative splicing outcomes can be modulated also by other means, including transcription and epigenetic changes. It has been shown that Pol II elongation rates (kinetics), which are controlled by chromatin marks, chromatin structure, DNA-methylation, and nucleosome occupancy can largely influence splicing. When elongation rates are reduced, weak splice sites are better recognized and alternative exons tend to be included; in contrast, when Pol-II elongation is promoted, recognition of weak splice sites is impaired and alternative exons tend to be skipped18. However, slow Pol II can favour recruitment of specific RBPs that promote exon inclusion (positive regulators) or skipping (negative regulators) while a fast Pol II will hamper that recruitment18,23. Increased nucleosome occupancy, Pol II pausing, DNA-methylation, and specific histone marks at exons relative to introns support the idea that these epigenetic signatures can also help the splicing machinery to recognize alternative exons18,22. Indeed, global correlations are being demonstrated between splicing patterns and specific histone marks, DNA-methylation patterns, nucleosome occupancy, Pol II positioning and RBP binding45,46,48,134–139. However, whereas splicing regulation by RBPs has been extensively studied and demonstrated in developmental context, the developmental data on the coupling between splicing events and transcription machinery and epigenetic modifications is much more limited.
Functional consequences of alternative splicing have been significantly less explored than its mechanisms and only limited examples have been studied in depth. Nevertheless, during the last decade, investigations have combined the use of transgenic animals where RBPs are induced or depleted in specific tissues, the characterization of physiological phenotypes, and high-throughput methods to identify the splicing targets of the RBP under study. The main goals of these studies are to identify the splicing targets of individual RBPs and their binding sites in developmental contexts, characterize the function of the RBPs in splicing coordination, and then infer potential roles of these splicing networks for tissue and organ development. Rescue experiments then can be used to re-introduce specific splicing isoforms in animal models in which RBP expression, and hence splicing isoform composition has been altered, confirming functions of particular splicing isoforms. Owing to these studies, we now much better understand the functional impact of specific splicing events within developmental scenarios. In the next sections we will describe some of the most notable, recent examples that demonstrate the important roles of alternative splicing for proper development and tissue homeostasis.
Splicing in the brain
Embryonic neural progenitor cells differentiate into neurons, which after migration to their final destination gradually form neuronal circuits that serve as basis for information processing and behavioral control. A transcriptome data set from mouse fetal and adult cerebral cortex has been described, including the data regarding alternative splicing isoforms13. In this study, adult cerebral cortex and embryos exhibited almost four hundred differential alternative splicing events and 31% of the genes that were found to be differentially regulated by alternative splicing during development of the brain did not change their total expression levels13. This evidence suggests that generation of alternative mRNA isoforms is the major regulatory paradigm for the expression of these genes. It is possible that alternative splicing in addition to generating different protein isoforms could change translation reading frames producing diverse translation outputs or may insert poison exons leading to nonsense-mediated mRNA decay (NMD), a well-documented mechanism of regulating mRNA levels (see also below). These transitions between splicing isoforms were shown to occur on genes involved in actin cytoskeleton organization, small GTPases, vesicle-mediated transport, synaptic connectivity, endocytosis, and membrane bending13.
RBPs and splicing networks in the brain
Different RBPs have been demonstrated to regulate splicing events during brain development (Supplementary Table 1). Among them, are PTBP1 (polypyrimidine tract-binding protein-1), PTBP2, and SRM4 (serine/arginine repetitive matrix protein-4), levels of which change during neurogenesis. Therefore, alterations of their target splicing networks occur during the transition from the neural progenitor to the fully differentiated neurons27–31. In particular PTBP1 and PTBP2 engage in a cross talk, whereby in neuronal progenitors PTBP1 represses the inclusion ofPTBP2 exon 10 leading to exon skipping and a transcript with a premature termination codon (PTC) andNMD32–34. As cells exit the cell cycle to differentiate into neurons, PTBP1 is downregulated, whereas SRRM4, which acts as a positive regulator of PTBP2 splicing is upregulated, and this promotes PTBP2 exon 10 inclusion. In result, PTBP2 is expressed promoting neuronal development and tissue maintenance29,30,32–35. Interestingly, PTBP2 has a broader role in the development of the mammalian neocortex as it is also expressed in neural progenitors, where it is in responsible for repressing the inclusion of adult-specific alternative exons in genes encoding proteins that control cell fate, proliferation, and actin cytoskeleton27,28. Another mechanism of splicing regulation by RBPs in neuronal differentiation is through their own alternative splicing changes during development, as occurs with RBFOX1 – a RBP, which has been associated with both neuronal differentiation36 and neurodevelopmental programmes that control synaptic functions37.Exon 19 of RBFOX1 is alternatively spliced by RBFOX proteins giving rise to nuclear (excluding exon 19) or cytoplasmic (including exon 19) protein isoforms. In RBFOX-depleted neurons more than 500 alternatively spliced cassette exons were misregulated, leading to significant changes in the level of exon inclusion or skipping in comparison with the control condition. Exogenous introduction of the nuclear isoform rescues these splicing changes probably by binding to the upstream GCAUG motifs within the proximal intronic region downstream of the regulated exons. In contrast, expression of the cytoplasmic variant rescues changes in mRNA levels of synaptic and autism-related genes through mRNA stabilization mechanisms (3’UTR binding, competition with microRNAs)38 (Figure 1a).
Figure 1. Alternative splicing functions in brain development.

a. Exon 19 (e.19) in Rbfox1 transcript is alternatively spliced giving rise to nuclear and cytoplasmic protein isoforms. In RBFOX-depleted neurons misregulation of splicing networks controlled by RBFOX1 leads to defects in the expression of transcription factors, other splicing factors and synaptic proteins. Introduction of exogenous nuclear RBFOX1 (RBFOX1 containing e.19) rescues changes resulting from the misregulation of splicing networks. Introduction of exogenous cytoplasmic RBFOX1 (lacking e.19) stabilizes multiple mRNAs (for example by interfering with binding of some micro-RNAs (miRNAs)), particularly of synaptic and autism-related genes. This stabilization increases protein expression of the targeted genes rescuing certain neuronal functions. b. During brain development DAB1 protein undergoes a transition from a long to a short isoform, involving skipping of exons 7b and 7c. This developmentally regulated splicing transition of DAB1 is under control of RNA-binding protein NOVA2, which governs exon skipping. DAB1 isoform containing exons 7b and 7c (DAB1–7bc) is more stable and when misexpressed in adults it antagonizes the short variant inducing neuronal migration defects. Consistent with this, Nova2 knock out mice as well as mice in which the DAB1–7bc has been ectopically introduced show defects in neuronal migration, and these defects are rescued by expression of the short DAB1 isoform. c. Alternative splicing of the chromatin remodeler EHMT2/G9a in neuronal differentiation regulates neuronal differentiation. EHMT2 exon 10 inclusion increases during neuronal differentiation and favors EHMT2 nuclear localization. This leads to the increase in histone H3 Lys9 dimethylation (H3K9me2), a histone mark generally associated with transcriptional silencing. In this case, it is plausible to hypothesis that the increase in H3K9me2 might contribute to repression of progenitor-specific genes. Isoform containing exon 10 is thus required for neuronal differentiation through the promotion of exon 10 inclusion on its own pre-mRNA. In consequence, this positive feedback loop reinforces commitment to differentiation24. d. Honeybee larvae development into a queen or a worker bee is regulated by DNA-methylation (green pins represent methylation sites), which is influenced by nutrition (larvae developing into queens have lower DNA methylation levels and differentiation into queens can be promoted by silencing DNA-methyl-transferase-3 (DNMT3)). Alternative splicing is more prevalent in methylated genes than in non-methylated ones. In particular, intron between exons 25 and 26 in pre-mRNA of Alk (a metabolic regulator with the capacity to enable growth in a nutrient-independent manner)is differentially methylated between queens and workers. Low methylation correlates with exon 25 inclusion and leads to the development of the larvae as queens.
PTBP and RBFOX proteins can exert antagonistic splicing actions controlling neurogenesis. In neuronal progenitor cells PTBP1 maintains the lamination of neural progenitor cells by promoting the skipping of the poison exon, exon-N (57-nt in length in Filamin A transcript. Exon-N inclusion introduces a PTC originating a truncated protein and/or NMD31. On the other hand, RBFOX proteins promote neuronal differentiation through the splicing switch of Ninein exons 18 (2,000-nt) and 29 (61-nt) from a centrosomal non-neuronal isoform to a non-centrosomal neuronal variant associated with microtubules31. In summary, auto-regulatory loops of RBPs are another mechanisms of how they control splicing during development.
The functional consequences of alternative splicing have been convincingly documented in neuronal differentiation. NOVA2 is a splicing regulator implicated in brain but also vascular development (where it controls cell polarity)39–41. In the brain NOVA2 exerts this function by controlling DAB1 (disabled-1) splicing. DAB1 is an adaptor protein involved in Reelin signalling, an important pathway in development of the mammalian cortex42,43. When exons 7b and 7c are included in DAB1, the protein becomes more stable. This longer isoform also interferes with the function of its shorter form that is important for regulating neuronal migration. Expression of this long isoform of DAB1 in neurons (which express the short, alternatively spliced isoform) induces neuronal migration defects similar to those observed in Nova2 knocked out (Nova2−/−) mice40. Furthermore, short isoform re-introduction in Nova2−/− animals rescues these migration defects40 (Figure 1b). RBFOX3 controls another splicing event that has been functionally implicated in neuronal differentiation. Via binding to an upstream intronic UGCAUG element, RBFOX3 promotes the skipping of one alternative exon in Numb, a signalling adaptor protein. When RBFOX3 expression is silenced in the developing chicken spinal cord this Numb exon is included blocking the progression of neuronal differentiation44. Another axis connecting one RBP with a specific splicing event and a clear functional consequence in brain development has been recently demonstrated in a very elegant manner. The RBP SLM2 (also known as KHDRBS3) has been found to control functional specification of glutamatergic synapses in mouse hippocampus through alternative splicing. Slm2−/− mice exhibit impaired synaptic plasticity and behavioral defects that are rescued by genetic correction of one single Slm2-dependent alternative spliced region in the neurexin-1 (Nrxn1) gene45. This segment is known in the field as the “alternative spliced segment-4” (AS4), which is spatio-temporally controlled in murine brains and responds to neuronal activity 46.
Alternative splicing and epigenetic landscapes
As mentioned above, there is evidence that alternative splicing and epigenetic regulation of chromatin are coupled. This appears to be the case in the developing brain, where alternative splicing of precise chromatin remodelers has been shown to impact neuronal differentiation and neurite morphogenesis24,47. It has also been recently demonstrated that neuronal differentiation responds to alternative splicing of EHMT2 (G9a/KMT1C), an enzyme controlling histone-H3 lysine-9 mono- and di-methylation (H3K9me1, H3K9me2). The generation of EHMT2 isoform harbouring alternatively-spliced exon 10 increases during the differentiation of mouse neuroblastoma (N2A) cell line, and also during mouse brain development. This alternative splicing event enhances H3K9me2 levels without altering enzyme catalytic activity, likely through favouring EHMT2 nuclear localization. Presence of exon 10 does not alter EHMT2 protein domains but is adjacent to a nuclear localization signal (NLS)48 and so its inclusion might favour the exposure of the NLS24. This exon 10-containig isoform is required for neuronal differentiation, most likely by increasing H3K9 methylation levels. It also regulates its own splicing promoting inclusion of exon 10, generating a positive feedback loop that further reinforces commitment to differentiation24 (Figure 1c). On the other hand, the histone lysine specific demethylase-1 (LSD1/KDM1) is also alternatively spliced, and an isoform containing exon 8a (encoding four amino acids) is specific to the nervous system. Expression of neural LSD1 isoform containing exon 8a in cultured cortical neurons reduced LSD1 activity as a transcriptional repressor. Depletion of this neuro-specific isoform inhibited neurite maturation whereas overexpression enhanced it. In contrast, experimental changes of the expression of LSD1 isoforms lacking exon 8a had no effect on neurite morphogenesis47. More recently, LSD1 isoform containing exon 8a has been shown to mediate H3K9me2 demethylation by interacting with supervillin protein. Depletion of LSD1+8a isoform led to an increase in H3K9me2 and thus changes in specific gene expression programmes that in turn alter neuronal differentiation49. This collectively demonstrates that alternative splicing can have an impact on the epigenetic landscape of the cell, thereby modulating gene expression programmes.
The opposite scenario, whereby epigenetic changes influence splicing outcomes has also been documented in the brain. Global genomic studies have shown that DNA-methylation is enriched within exons50,51, but interestingly lower levels of DNA-methylation were detected within skipped exons in comparison with those included52,53. In addition, GC-content of exon–intron boundaries has been shown to impact splicing outcomes54–56, suggesting that DNA methylation regulates splicing events. One interesting example of this regulatory paradigm has been recently described in brain development in honeybees (Apis mellifera). Larva development into a queen or worker honeybee depends on nutrition, which in turn impacts DNA-methylation levels57 (Figure 1d). DNMT3 (DNA-methyl-transferase-3) silencing in embryos generates adult queen bees demonstrating that DNA methylation controls caste determination (with demethylation promoting queen development)57. Adult queen and worker honeybees have different brain methylomes. Interestingly, spliceosome components and splicing factors are enriched within the group of differentially methylated genes and alternative splicing has been shown to be more prevalent in methylated than in non-methylated spliced genes58,59, indicating that in these animal DNA methylation and alternative splicing are closely associated. Among the differentially methylated genes was the anaplastic lymphoma kinase (Alk) gene that encodes a metabolic regulator and undergoes extensive alternative splicing59. Alk gene is differentially methylated between queens and workers mostly within intron 25 and exon 959. Low intron 25 methylation correlates with exon 25 inclusion59. Similar mechanism has been also described in mammalian B-cells, where differential methylation affects isoform expression of a surface molecule CD45. In more detail, CCCTC-binding factor (CTCF) promotes exon 5 inclusion when bound to chromatin by inducing Pol II pausing at the intron-exon boundary; DNA-methylation inhibits CTCF binding, thereby preventing exon 5 inclusion60. CTCF promotes inclusion of weak upstream exons also on a more global scale, also through mediating Pol II pausing60 and CTCF-binding sites are highly conserved between tissues61. Therefore, developmental DNA-methylation might contribute to tissue-specific splicing60. This mechanism is relevant in diseases such as cancer where global DNA-methylation and CTCF-binding changes occur together with misregulation of splicing networks60.
Perturbation of alternative splicing networks in neurological disorders
Aggregation of the RBP TDP43 (transactive-response DNA-binding protein-43) is the pathological hallmark of amyotrophic lateral sclerosis (ALS) and fronto-temporal lobar degeneration (FTD) 62–68. TDP43 regulates the levels of its own mRNA and protein by a self-regulatory loop. Excess TDP43 results in increased binding to the 3’UTR of its mRNA and the splicing of a normally silent 3’UTR intron. A second, rarely used polyadenylation site is located downstream of this 3’ splice site. The proximity between the spliceosome assembled in that splice site and the polyadenylation complex induces disruptive interactions, the splicing process is thus aborted, and the partially processed pre-mRNA is degraded, allowing regulation of TDP43 expression69.In pathological state TDP43 accumulates in neurons, depleting the nucleus of this protein. TDP43 self-regulation loop detects low TDP43 nuclear levels and increases mRNA production. In consequence, aggregates become larger and entrap further newly synthesized TDP43. Consequently, the splicing of TDP43 targets is altered64,70. In particular, mis-splicing and expression of genes involved in neuromuscular junction maintenance are controlled by TDP43.
Global misregulation of splicing networks has been shown in autism spectrum disorder (ASD). Mutations in RBFOX proteins are present in patients suffering from autism altering splicing of transcripts involved in ASD such as SHANK3, CACNA1C, TSC271–73. Furthermore, a splicing network in functionally related genes has been described to affect “microexons” (3–15nt). These microexons modulate interaction domains of proteins involved in neurogenesis and are mainly regulated by nSR100/SRRM4. Neural microexon inclusion is misregulated in patients with ASD through SRRM4 upregulation. As a result, in developing autistic brains protein-interaction networks might be remodelled in comparison with normal neurogenesis programmes74.
A very recent study has demonstrated the functional consequences of the unbalanced generation of different Tau splicing isoforms. Tau is a microtubule-associated protein involved in axonal transport and neurite growth. Exon 10 is alternatively spliced generating two isoforms with three (3R) or four (4R) microtubule-binding repeats. Imbalanced 3R-4R ratios are associated with neurodegeneration by impairing the intracellular transport of the Alzheimer’s disease-related amyloid precursor protein (APP)75.
Overall, brain development and neuronal differentiation are highly suitable models for studying the regulatory and functional aspects of alternative splicing in vivo. In this tissue, extensive alterations of splicing events are translated into changes in specific protein isoforms over development and these alterations then impact key physiological processes such as neuronal differentiation, cell migration, synaptogenesis, synapse functions, motor neuron development, and neuronal excitability76. These splicing networks are often controlled by RBPs that developmentally change their expression levels, but can also be modulated by (and engage in the modulation of) epigenetic factors (see also Box 2).
Splicing in striated muscle
Striated muscle was one of the first tissues where it has been demonstrated that alternative splicing regulation increases protein complexity. Together with the brain, heart and skeletal muscle are where most tissue-specific alternative splicing takes place77,78.
Cardiac muscle
Extensive alternative splicing transitions during cardiac development
The heart is the first organ to form and function during embryogenesis and thus embryonic heart development has been very well studied at the transcriptional and posttranscriptional levels. However, the postnatal period was not so deeply investigated until recent years. The changes that occur in the heart postnatally are very interesting, as it is after birth that the heart needs to adjust to largely different oxygen and metabolic conditions, and these adaptations are synchronized with changes in cardiomyocyte intracellular structure (Figure 2a). These physiological and morphological changes are mainly driven by transcriptional and posttranscriptional transitions. During heart development there are dramatic changes in the expression levels of multiple RBPs including CELF1 (CUGBP Elav-like family member-1), MBNL1 (muscleblind-like protein-1), RBFOX1, RBFOX2, and RBM24, among others79. The splicing targets of these RBPs have been identified using animal models in combination with genome-wide approaches (Supplementary Table 1)5,11,80–82. Similar to the situation in the brain, also during heart development RBPs can engage in cooperative and antagonistic actions (for example, CELF1 and MBNL1 were shown to antagonistically regulate alternative splicing in heart development80).
Figure 2. Alternative splicing in heart development.

a. The heart is the first organ to form and function during embryogenesis and prenatal heart development has been very well studied at the transcriptional and posttranscriptional levels. Many changes in the heart also occur postnatally, and are associated with the switch between extremely different oxygen and metabolic conditions as well as changes in cardiomyocyte intracellular structure. These physiological and morphological changes are mainly driven by transcriptional and posttranscriptional transitions, including changes in the expression of RNA-binding proteins (RBPs) and splice isoforms. b. During heart development RBM20 regulates splicing of titin – a giant protein able to act as a molecular spring – with exons 50–219 being developmentally regulated87–89. In neonates primarily long isoforms (N2BA) are expressed, whereas a shorter isoform (N2B) dominates in adults. The alternatively spliced region of titin is known as the I-band, and controls titin elasticity (with longer regions promoting elasticity). The ratio between N2BA and N2B isoforms thus determines sarcomere length, cardiomyocyte passive tension, and myocardium wallstiffness87. RBM20 mutations were identified in humans with cardiomyopathies, linking changes in titin alternative splicing to human pathology89–93. FN3: fibronectin type-3 domain, Ig: immunoglobulin-like, PVEK: region rich in proline, glutamate, valine, and lysine.
Microarray experiments revealed splicing transitions during differentiation of human embryonic stem cells into cardiac precursors83 and in postnatal heart development5. More recently, transcriptome dynamics in mouse cardiac ventricles, cardiomyocytes, and cardiac fibroblast at different postnatal stages have been described11. These analyses revealed that alternative splicing transitions in cardiomyocytes are particularly concentrated within the first four weeks after birth, suggesting that cardiomyocytes utilize splicing mechanisms for maturation11. In contrast, splicing transitions are not restricted to the first four postnatal weeks in cardiac fibroblasts and occur within the entire postnatal period. In cardiomyocytes the genes most significantly regulated by alternative splicing are involved in vesicular trafficking and membrane remodelling11. Four of these splicing events were further shown to impact skeletal muscle functions and structure in vivo. Antisense oligonucleotides targeting splice sites in these transcripts induced the reversion of splicing pattern from adult to fetal ones when delivered into the foot muscles of adult mice. After splicing reversion, muscle force generation was reduced, calcium signalling was misregulated, and internal cell architecture alterations were observed84.
Titin splicing contributes to the regulation of mechanical properties
In the heart, probably the best-studied splicing events are those occurring in titin (TTN). In mammals, TTN gene contains the largest number of exons. Exons 50–219 are developmentally regulated: a 3.7 MDa protein (N2BA-isoform) is primarily expressed in neonates, whereas in adults a shorter, 2.97 MDa isoform (N2B) is the most abundant one85,86. Within cardiomyocytes the amino-terminus of TTN is located at the Z-line and the carboxy-terminus at the M-line. Alternative exons encode part of the immunoglobulin-like region, the N2A portion, and the PVEK region (rich in proline, glutamate, valine, and lysine)85. This alternative region is known as the I-band. Sequence variability in the I-band region determines the elastic properties of the protein, thereby modulating the elasticity of the muscle (the longer the region the more elastic the protein). And so, the ration of N2BA (long isoform) to N2B (short isoform) isoforms determines sarcomere length, cardiomyocyte passive tension, and thus myocardium wall-stiffness during ventricular filling87. TTN splicing in the I-band is regulated by RBM2087–89 and mutations in RBM20 gene were identified in humans with dilated cardiomyopathies89–93 (Figure 2b). RBM20 regulates TTN splicing by a two-step sequential mechanism: i) before splicing reaction starts RBM20 binds the newly transcribed pre-mRNA blocking intron removal within bound regions, while the rest of the pre-mRNA is spliced87, ii) the partially spliced pre-mRNA is retained in the nucleus and the 5’ and 3’ splice sites on the exons flanking the RBM20-bound region are spliced together leading to that region removal87. Alternative splicing also occurs in the M-line associated portion of TTN. Here, splicing regulates the inclusion of Mex5 exon, which encodes for the specific is7 domain located between the immunoglobulin-like domains. Mex5 exon inclusion is muscle type-, developmental stage-, and specie-specific and its regulation is altered in Myotonic Dystrophy Type-1. Recently a mouse model was generated using CRISPR-Cas9 editing to constitutively prevent the expression of isoforms containing the is7 domain. These animals exhibited a dystrophic phenotype in striated muscles (i.e. soleus and heart), tissues that exclusively express TTN isoforms containing Mex5 exon demonstrating that alternative splicing of this region has tissue-specific functions94.
Perturbation of alternative splicing networks in heart diseases
In cardiac diseases, there is a global reversion to neonatal transcriptional programmes, accompanied by adult heart reverting to neonatal oxygen consumption and metabolic programmes, and by severe alterations in the internal architecture and functionalities of cardiomyocytes (Figure 2a, right).This overall reversion from adult to neonatal programmes is also associated with changes in the expression of multiple RBPs (including RBFOX, RBM20, and RBM24 among many others) and thus, leads to the perturbation of their targets79,81,95,96. In particular, a global hallmark of heart failure and cardiomyopathies is the reversion from adult to fetal alternative splicing patterns, particularly affecting the expression of proteins involved in RNA-processing, cytoskeletal organization, development, and metabolism97. Diverse examples were reported in the literature and one of the most recent has been described in cardiac hypertrophy (Table 2). Hypoxia, a hallmark of cardiac hypertrophy, induces the expression of the splicing factor-3b subunit-1 (SF3B1) that in turn deregulates splicing of two mutually exclusive exons of ketohexokinase biasing the generation of an isoform primarily produced in the liver. Since the ketohexokinase is the central fructose-metabolizing enzyme, and is involved in glucose metabolism mis-splicing promotes anabolic metabolism, leading to heart muscle overgrowth and severe cardiac contractile problems98.
Table 2.
Examples of developmental diseases associated with deregulation of splicing networks.
| Human disease | Mechanism | Mis-splicing events | Features associated with splicing defects |
|---|---|---|---|
| Amyotrophic lateral sclerosis and fronto-temporal lobar degeneration62–68 | TDP43 and FUS mutations lead to cytoplasmic aggregation (nuclear depletion). Aggregation then leads to TDP43 loss of function and FUS gain or loss of function. | TDP43 and FUS targets. TDP43 loss induces splicing alterations that alter neuronal function | Motor neuron degeneration |
| Autism spectrum disorder71–74 | RBFOX1 mutations | Transcripts involved in autism disease (e.g. SHANK3, CACNA1C, TSC2) | Neurogenesis and neuronal functions |
| SRRM4 upregulation | Neural microexons within protein–proteininteraction domains important for neurogenesis | ||
| Cardiac hypertrophy98 | SF3B1 upregulation through hypoxia pathways | Ketohexokinase mutually exclusive exons | Contractility alterations through metabolic switch |
| Dilated cardiomyopathy87,89,95,96,150 | RBM20 mutations | TTN, RYR2 (exon 24-nt), LMO7 (exons 9–10), RTN4 (exons 3–4), PDLIM3 (exons 4–6), CAMK2D, LDB3, among other sarcomeric genes | Arrhythmias, sudden death |
| Myotonic dystrophy type-1 and type-2140,151–153 | CTG) or (CCTG) repeats in the 3’UTR of DMPK gene or intron 1 of CNBP gene lead to MBNL1/2 loss of function and CELF1 gain of function | CELF1 and MBNL1 targets: INSR (exon 11), BIN1 (exon 11), RYR1, ATP2A1, DMD, CACNA1S (exon 29), CLCN1, SCN5A (exon 6a/b), among many others | Insulin resistance, myotonia, dystrophia, muscle weakness, T-tubule disorganization, cardiac conduction defects, arrhythmias, cardiomyopathies, learning impairment, synaptic defects |
For exceptional articles revising the literature about splicing and diseases the reader may consult the suggested references4,8,9. ATP2A1: ATPase sarcoplasmic/endoplasmic reticulum calcium transporting-1, BIN1: bridging integrator-1, CACNA1C: calcium voltage-gated channel subunit alpha1c, CACNA1S: calcium voltage-gated channel subunit alpha1s, CAMK2D: calcium/calmodulin dependent protein kinase II delta, CLCN1: chloride voltage-gated channel-1, DMD: dystrophin, DMPK: dystrophia myotonica protein kinase, FUS: FUS RNA binding protein, INSR: insulin receptor, LDB3: LIM domain binding-3, LMO7: LIM domain-7, NMD: non-sense mediated decay, PDLIM3: PDZ and LIM domain-3, RYR1: ryanodine receptor 1, RYR2: ryanodine receptor 2, SCN5A: sodium voltage-gated channel alpha subunit 5, SHANK3: SH3 and multiple ankyrin repeat domains protein, TTN: titin,.
Skeletal muscle
Alternative splicing in skeletal muscle differentiation
Although, more than 1,000 muscle-specific splicing events have been mapped99, little is known about the regulatory mechanisms that drive skeletal muscle-specific developmental splicing programmes in vivo. No genome-wide data is yet available about splicing transitions during skeletal muscle development in vivo but splicing networks operating during muscle cell differentiation have been identified in the murine myoblast cell line C2C1214. Specific RBPs coordinate these splicing networks including PTB, Qk (quaking), and RBFOX2 proteins14,100,101. In particular, RBFOX2 controls numerous splicing transitions and its depletion leads to myoblast fusion defects. These abnormalities are partially rescued when differentiated cell-specific splicing isoforms of the transcriptional regulator MEF2D (myocyte-specific enhancer factor-2D) and the cytoskeleton regulator ROCK2 (rho-associated protein kinase-2) are re-introduced14. Furthermore, it has been demonstrated that the ubiquitous (MEF2Da1) and the muscle-specific (MEF2Da2) splicing isoforms antagonistically regulate muscle cell differentiation102. Only MEF2Da2 activates late muscle gene transcription programmes owing to its decreased susceptibility to phosphorylation. Protein kinase-A phosphorylates MEF2Da1 leading to its association with corepressors and thus preventing its activating role on transcription. In contrast, MEF2Da2 variant remains unphosphorylated and recruits the coactivator Ash2L, involved in activation of the muscle-specific genes102.
Alternative splicing and myotonic dystrophy
Myotonic dystrophy is the most common muscular dystrophy in adults. Patients with myotonic dystrophy exhibit symptoms affecting different organs: muscle weakness, myopathy, insulin resistance, cardiac arrhythmias, hypogonadism, and cataracts, among others103. Two main forms of this pathology exist and are caused by expansion of repeat sequences in the 3’UTR of the DMPK gene (Myotonic dystrophy type 1 or the first intron of ZFN9 gene (Myotonic dystrophy type 2). These expansions lead to the nuclear sequestration and/or deregulation of specific splicing factors. In Myotonic dystrophy type 1, the RBP MBNL binds to the expanded repeats in the DMPK 3’UTR in the nucleus and thus its functionality as a transcriptional and posttranscriptional regulator is reduced. On the other hand, the repeats in the 3’UTR of the DMPK gene lead to CELF1 activation and its gain of functions. One of the consequences of this molecular mechanism is that CELF and MBNL splicing targets are misregulated and adult tissues (mainly brain, heart, and skeletal muscle) express the fetal protein isoforms. Embryonic protein variants are functionally efficient in fetal and neonatal organs and cells but in adult tissues they do not fulfill the functional requirements. Therefore, misregulation of alternative splicing in myotonic dystrophy directly contributes to the pathological features observed in patients (Table 2). For example, mis-splicing of insulin receptor exon 11 reduces metabolic insulin response104,105. Muscle weakness is proposed to be a consequence of misregulated calcium signalling as a result of mis-splicing of genes encoding the ryanodine receptor-1 (RYR1, exon 70), ATPase sarcoplasmic/endoplasmic reticulum calcium transporting-1 (ATP2A1, exon 22), the calcium voltage-gated channel subunit alpha1-S (CACNA1S, exon 29), and the bridging integrator-1 (BIN1, exon 11)106,107. Myotonia (prolonged muscle contraction) is attributed to abnormal inclusion of exon 7a in the muscle chloride channel CLCN1 that produces a frame-shift and a non-functional protein. Approximately 80% of patients with myotonic dystrophy also develop cardiac dysfunction. Analysis of samples from hearts of patients with myotonic dystrophy type 1 revealed global deregulation of splicing networks that affects genes encoding for ion channels, intracellular transport, and cell architecture108. In particular, mis-splicing of the cardiac voltage-gated sodium channel α-subunit (SCN5A) causes cardiac conduction defects and arrhythmias108.
In summary, alternative splicing plays an important functional role in striated muscle (heart and skeletal muscle) development and physiology. Alternative splicing regulates the properties of proteins that exert precise actions in these tissues: ion transport, structural functions, excitation-contraction coupling, and metabolism. As a result, misregulation of splicing networks in these tissues can lead to major diseases.
Splicing in other tissues and organs
The brain and striated muscle are best studied with regards to the impact of alternative splicing on physiology. Nevertheless, several interesting examples of functional roles of alternative transcripts and splicing networks have been provided for other systems.
Splicing responses in the pancreas
To our knowledge there is scant developmental transcriptome data for the pancreas. Only, a few splicing networks have been described to date and these are regulated by inflammatory cytokines109,110 and fatty acids111. One of these networks is coordinated by NOVA1 and affects genes involved in exocytosis, apoptosis, insulin signalling, and transcription regulation110. Another splicing program is coordinated by the RNA-binding motif protein-4 (RBM4). RBM4 depletion in mice induces metabolic alterations and mis-splicing of factors required for pancreatic cell differentiation and function. In particular, RBM4 regulates alternative splicing of the transcription factors ISL1 (ISL1 transcription factor, LIM/homeodomain) and PAX4 (paired box-4) that in turn modulate insulin gene expression112.
Alternative splicing transitions in liver development and maturation
Liver develops from an embryonic haematopoietic tissue to a metabolic adult organ. The major hepatic parenchymal cell type (>75% volume), the hepatocytes, switches from highly proliferative to quiescent stages and misregulation of this quiescence can lead to hypertrophic growth during postnatal life. Early studies demonstrated the molecular mechanisms by which different splicing factors regulate liver development, homeostasis, and metabolism in health and disease. For example, the serine/arginine-rich splicing factor-3 (SRSF3) is required for hepatocyte differentiation113, SRSF10 has been implicated in obesity through its role in regulating lipogenesis114, and SLU7 (splicing factor homolog) is essential for liver homeostasis115. More recently, coordinated, cell-type specific (hepatocytes versus non-parenchymal cells) transcriptional and posttranscriptional transitions were shown to drive postnatal liver remodelling and maturation12. In liver postnatal development RBPs are mostly downregulated12. ESRP2 (epithelial splicing regulatory protein-2) is one of the few RBPs postnatally induced and regulates a conserved set of splicing transitions specifically in hepatocytes, governing hepatocyte terminal differentiation and maturation12.
Splicing and epigenetics in germline development
During development, the cells that will become gametes need to differentiate into primordial germ cells and migrate to the proper location to form the animal germline. Not much is known about alternative splicing during ovarian germline development. What is known is that during ovary development alternative splicing regulates RBFOX1 localization and thus modulates its cellular functions. Recent rescue experiments in Drosophila melanogaster showed that two specific cytoplasmic Rbfox1 splicing isoforms are responsible for germ cell differentiation during early germline cyst development116. Cytoplasmic Rbfox1 isoforms repress translation of mRNAs containing (U)GCAUG elements within their 3’UTRs, including mRNA of Pumilio – a protein that drives germline stem cell maintenance by repressing the translation of specific mRNAs117. In consequence, Pumilio repression promotes germ cell differentiation. Therefore, cytoplasmic Rbfox1 acts as a genetic barrier avoiding reversion of germline cysts back to earlier developmental stages116.
Regulation of splicing is also important for spermatogenesis, and in this scenario epigenetic–splicing as well as transcription–splicing interplay has been proposed (see also Box 2).Spermatogenesis occurs within the seminiferous tubules where postnatal male germ cells develop. Mammalian testis are among the tissues with the highest transcriptome complexity, including alternative splicing regulation118,119. Extensive splicing transitions occur during progression from mitotic spermatogonia to meiotic spermatocytes120. The majority of these changes affect conserved protein-coding exons120, suggesting large impact of these transition on physiological functions of the alternatively spliced protein isoforms. Numerous RBPs are differentially expressed during spermatogenesis and flanking sequences of the regulated alternative exons are enriched for binding sites for PTBP1, PTBP2, TRA2B (transformer-2 protein homolog beta), and STAR (signal transduction activator of RNA metabolism) family proteins120. Whereas PTBP1 is mainly expressed in spermatogonia (mitotic cells), PTBP2 predominates in meiotic spermatocytes and postmeiotic spermatids120,121. PTBP2 is essential for mouse spermatogenesis and for alternative splicing coordination in testis (and as discussed above for neuronal differentiation in the brain)121. During spermatogenesis, chromatin structure transitions from a closed to a more open state enabling the majority of the genome to be transcribed in spermatocytes and spermatids118.
Male germ cell development is one of the physiological contexts where transcription-splicing coupling has been elegantly demonstrated. In vivo, SAM68 (Khdrbs1), which binds to transcriptionally active chromatin in spermatocytes, modulates splicing of exon 8 in SGCE (epsilon-sarcoglycan). Molecularly, this regulation occurs through SAM68 binding to the intron–exon junction in SGCE exon 8, which results in the recruitment of phosphorylated Pol II, and at the same time interferes with the recruitment of the splicing factor U2AF65, leading to exon 8 skipping122.
A pioneering study has shown that in cell culture the adaptor protein MRG15 binds to H3K36me3 and recruits PTBP1 that in turns promotes skipping of exon IIIb in fibroblast growth factor receptor 2 transcript123. Recently, a similar splicing–epigenetics interplay has been demonstrated during spermatogenesis124. Knockout mice lacking MRG15 specifically in postnatal male germ cells exhibit spermatogenic arrest at the round spermatid stage (just before terminal differentiation into mature spermatozoa) and are thus sterile124. MRG15 depletion triggers removal of specific mRNA sequences in transcripts and at the same time results in the retention of specific intronic sequences in mRNAs of four genes124. One of these intronic retentions occurs in transition nuclear protein 2 (TNP2) that together with other transition nuclear proteins replaces histones during chromatin condensation of maturing sperm. TNPs are later replaced by protamines, and this allows optimal chromatin packaging that enables proper elongation of the sperm head. In normal round spermatids MRG15 colocalizes with PTBP1 and PTBP2 at H3K36me3 sites between the two TNP2 exons and the intron is correctly removed generating a functional TNP2 protein. In contrast, when MRG15 is absent PTBPs are not recruited to the splicing machinery on Tnp2 gene and intron is retained leading to a drastic reduction in TNP2 protein levels124 (Figure 3a).
Figure 3. Interplay between splicing and epigenetic modifications in spermatogenesis and regulatory feedback loops involving splicing in T-cell activation.

a. Interplay between splicing and epigenetics during spermatogenesis has been demonstrated using a conditional knock out mice lacking MRG15 specifically in postnatal male germ cells. These animals exhibit spermatogenic arrest and sterility. MRG15 depletion triggers intronic segment retentions and one of them occurs in transition nuclear proteins 2 (TNP2). In normal round spermatids MRG15 binds to histone H3 Lys36 trimethylated (H3K36me3) sites between the two TNP2 exons (e.1 and e.2) and it recruits RNA-binding proteins PTBP1 and PTBP2 to these sites, enabling the correct removal of the intron, which generates a functional TNP2 protein. TNP2 and other related proteins then replace histones in chromatin of maturing sperm, and are eventually replaced by protamines, which allow optimal chromatin packing for sperm head elongation observed in mature sperm. When MRG15 is absent PTPB2 is not recruited and the intron is retained leading to a drastic reduction in TNP2 protein levels, and in result arrest of spermatogenesis at a round spermatid stage. Panel is adapted from124. b. A positive regulatory feedback loop between JNK signalling and splicing in T-cell activation. This feedback loop comprises the CELF2 splicing factor, which is responsible for alternative splicing of Jun-kinase-kinase MAP-kinase-7 (MKK7). T-cell activation induces CELF2-mediated MKK7 exon 2 (e.2) skipping. This generates a shorter MKK7 isoform that harbours an additional JNK-docking site, which enhances JNK activity and JNK signalling. Enhanced JNK signalling induces CELF2 mRNA stabilization and protein up-regulation, and so CELF2 levels increase with sustained T-cell activation, establishing a positive regulatory feedback loop128,129,131.
Crosstalk between splicing, signalling and T-cell activation
Human immune system relies on the ability of T-cells to respond to antigen on antigen presenting cells, which triggers extensive changes in protein expression and cell activation. These transitions occur through transcriptional and posttranscriptional gene expression changes. Global splicing networks are coordinated during T-cell activation through specific signalling pathways, nucleosome occupancy, and RBPs, including HNRNPL125,126, PSF,127 and CELF210,128,129. Recent RNA-sequencing experiments have revealed that global intron retention programmes might regulate T-cell activation. During T-cell activation intron retention is globally reduced and this correlates with an increase in mRNA steady-state levels. As, the majority of genes upregulated during T-cell activation show a reduction in intron retention, it has been proposed that quick responses of T-cells to acute extracellular signals are regulated posttranscriptionally, with an important role of alternative splicing130. Regulation of T-cell activation by splicing involves a feedback loop between CELF2 and JNK signalling cascade. T-cell activation induces skipping of exon 2 in MKK7 (Jun-kinase-kinase MAP-kinase-7), which introduces an additional site for JNK docking that is absent in the larger isoform. In consequence, protein–protein interactions are promoted and JNK pathway is enhanced. This then induces CELF2 mRNA stabilization and upregulates CELF2 protein expression. CELF2 binds upstream of MKK7 exon 2 and further promotes its skipping resulting in a positive regulatory feedback loop that amplifies JNK activity, which in turn promotes T-cell differentiation and cytokine production128,129,131 (Figure 3b).
HNRNPL mostly causes exon skipping, and this occurs when it is bound within cassette exons or to their flanking regions, owing to the presence of specific exon silencing sequences132,133One of HNRNPL targets in T-cells is CD45, an important surface molecule in T-cell differentiation and activation. CD45 undergoes HNRNPL-dependent alternative splicing leading to exon 4 skipping following T-cell activation133. Furthermore, exon skipping in CD45 pre-mRNA is simultaneously promoted by PSF127. Interestingly, HNRNPL can also promote the inclusion of some exons in T-cells. These exons, retention of which is positively regulated by HNRNPL, share sequence and context features: they are flanked by short GC-rich introns but themselves are not GC-rich. Consequently, the typical high GC-differential between exons and introns (high GC in exons but low in introns) is absent in these exons 132. These ‘low GC-differential exons’ exhibit poor nucleosome occupancy and are more likely to be skipped54. Evidence suggests that HNRNPL depletion results in reduced nucleosome occupancy, and so HNRNPL might prevent skipping of low GC-differential exons through an epigenetic mechanism, involving regulation of nucleosome occupancy. It is also likely that HNRNPL promotes exon retention by enhancing the recognition of low GC-differential exons for example by transcription machinery132.
Splicing and cell differentiation
Studies of the mechanisms of cell differentiation and de-differentiation (transition from differentiated state to the proliferative one) have also provided novel knowledge of the functional impact of splicing regulation. In particular, it has been shown that NMD is an important regulator of cell fate transitions.
Concerted splicing programmes leading to PTC and NMD regulate smooth muscle cells
Smooth muscle cells contribute to cardiovascular physiology due to plasticity properties from contractile (blood vessels) to more synthetic and secretory phenotypes (atherosclerosis, arterial injury). During de-differentiation of primary aorta cells extensive splicing transitions occur with very little changes in RBPs’ mRNA levels. This evidence suggests that splicing programmes might be orchestrated at an additional level15. An important piece of the splicing programme involves exons that are skipped during de-differentiation. This repression is mediated by PTBP1, which is two-fold upregulated during de-differentiation15. PTBP1 is thus responsible for repressing multiple smooth muscle specific exons in proliferative stages and its downregulation during differentiation can be expected to be a key driver of the expression of smooth muscle specific genes15. In addition, differentiated cells exhibit more frequent intron retention, inclusion of poison-exons that introduce PTC, and alternative polyadenylation usages. These concerted events lead to protein downregulation due to NMD, protein truncation, or RNA nuclear retention and mostly affect genes encoding splicing factors, splicing regulatory kinases, and other transcriptional regulators15.
Alternative splicing and PTCs in erythropoiesis
During erythropoiesis, erythroid progenitors mature into red blood cells (erythrocytes). Starting at the haematopoietic stem cell differentiation, a finely orchestrated transcriptional and posttranscriptional program drives the synthesis of the appropriate stage-specific proteome to fulfill the functions of progressively more specialized cells134. In erythroid cells, the best-studied splicing event is the stage-specific switch of exon 16 (inclusion versus skipping) in the gene encoding 4.1R protein that mechanically stabilizes erythrocyte membrane. A decrease in heterogeneous nuclear ribonucleoprotein A (HNRNPA) and HNRNPB expression during erythropoiesis mediates this splicing switch135. Microarrays identified several alternatively spliced cassette exons and alternative first exons during erythropoiesis134. This studies also revealed that various RBPs, including SNRP70, HNRPLL, and MBNL2 are also regulated by splicing mechanisms during erythropoiesis demonstrating a regulatory feedback loop134. More recently, coordinated splicing networks operating during terminal erythropoiesis have been described using RNA-sequencing, and MBNL1 was shown to act as a regulator of splicing transitions in this network136. Genes regulated by alternative splicing during erythropoiesis were shown to be involved in transmembrane receptor activity, chromatin modification, RNA-binding and processing, and DNA repair packaging136. In particular, during mouse erythroid terminal development MBNL1 promoted the inclusion of a 35-nt cassette exon in Ndel1 (nuclear distribution protein nudE-like-1) gene. The retention of this alternative exon changes the protein C-terminus and can influence NDEL1 interactions with other proteins. Only NDEL1 isoform containing this alternative exon could partially rescue the differentiation defects observed after NDEL1 depletion, which, importantly, are similar to those observed when MBNL1 is absent136. Another study employed RNA-sequencing to investigate the transcriptional landscape of distinct human erythroblasts representing the last four cell divisions before terminal differentiation (demarcated as cell enucleation)137. Interestingly, it was revealed that switches between splicing isoforms in these differentiating erythroblasts introduce PTCs into a group of transcripts and that this occurs in a differentiation stage-specific manner137. Recently, these discoveries have been expanded by showing that a dynamic intron retention programme that affects RNA-processing genes is responsible for regulating gene expression during terminal erythropoiesis138.
In conclusion, the coupling between alternative splicing and PTC-NMD emerges as a regulator of the genes expressed during cell differentiation. This concerted programme mainly impacts genes encoding RNA-processing regulators, thereby amplifying the functional consequences of alternative splicing.
Conclusion and perspectives
Global transcriptomic studies in recent years have extended the identification and description of coordinated transitions between splicing isoforms in different tissue contexts. The field is currently moving towards understanding the physiological roles of specific splicing isoforms, particularly during development, when changes between alternatively spliced isoforms are especially common. However, significant more effort is still required to fully understand functional roles of developmental splicing networks. Thousands of protein isoforms derived from splicing changes associated with development still need to be functionally characterized. A combined effort of numerous labs will be necessary to achieve the complete characterization of the roles of specific splicing isoforms and networks in developmental processes. Another challenge is that in multiple cases, dramatic splicing changes lead to only a slight physiological impact. Therefore, a great care should be taken in making statements and conclusions and it would be important to combine genome-wide approaches with molecular studies of different systems to fully determine the functional impact of developmental alternative splicing transitions.
We envision that future directions in this exciting field will be centered on three main aspects: further understanding of the regulation and coordination of splicing events, for example by mechanisms such as epigenetic modifications and transcription-related events; uncovering the physiological functions of individual splicing isoforms (which is still an underexplored area in comparison with the regulatory mechanisms); and finally, understanding how the entire splicing networks govern development and tissue homeostasis.
Supplementary Material
Online summary.
Alternative splicing explains how a single gene can generate more than one mRNA transcript expanding proteome complexity
During normal development a large number of alternative splicing changes occurs and it is now apparent that these transitions between alternatively-spliced isoforms contribute to the acquisition of adult tissue functions and identity
Individual splicing changes are coordinated during development, establishing splicing networks
Owing to recent progress we now better understand the mechanisms coordinating alternative splicing networks and the roles of these networks in cell differentiation, organ development and tissue homeostasis
Acknowledgments
J.G. is supported by start-up funds, a Junior Faculty Development Award and a Nutrition Obesity Research Center (NORC) Pilot and Feasibility Grant (P30DK056350) from The University of North Carolina at Chapel Hill.
Glossary terms
- Cis-acting factors
DNA sequences that are bound by proteins (trans-acting factors) that control gene expression
- Spliceosome
Large macromolecular complex composed by small nuclear RNAs and protein factors that removes regions (mostly introns) from transcribed pre-mRNA
- RNA-binding proteins
Proteins that bind RNA molecules through RNA-recognition motifs and exert nuclear and cytoplasmic post-transcriptional functions including splicing, polyadenylation, mRNA stability and localization, editing, and translation
- Poison exon
Exon that when included in the mRNAs introduces a premature termination codon
- Nonsense-mediated mRNA decay
Gene expression regulatory mechanism that eliminates mRNA transcripts containing premature stop codons
- Filamin A
Actin-binding protein that is involved in the cross-linking of actin filaments, cytoskeleton remodelling, cell shape and cell migration
- Ninein
Protein involved in the anchoring of the microtubules minus-ends and in centrosomal functions
- Amyotrophic lateral sclerosis (ALS)
Progressive neurodegenerative disease where motor neuron functions are altered and die. In consequence, the brain is not capable to control muscle movement and patients lose ability to speak, eat, move, and breathe
- Fronto-temporal lobar degeneration (FTD)
Disease caused by a progressive damage and atrophy of the temporal and/or frontal lobes of the brain
- Sarcomere
Structural basic unit of a myofibril in striated muscles. Sarcomere consists of a dark band and the nearest half of the adjacent pale band. Sarcomeres are composed of myosin filaments (thick) and actin filaments (thin)
- Z-Line
Line that separates two adjacent sarcomeres
- M-Line
The M-line is the center of the sarcomere and is the anchor site for the thick myosin filaments
- Myoblast fusion
Myoblast cells fuse one with each other forming multinucleated myotubes that during development generate the muscle fibers (myofibers)
- JNK signalling
A signaling pathway activated by environmental stress, inflammatory cytokines, and growth factors. Upon its activation JNK translocates to the nucleus where it regulates transcription
Author biographies
Francisco E. Baralle studied Chemistry at the University of Buenos Aires and Medicine at the Universities of Buenos Aires and Naples. In 1974 he joined as a post doc the MRC Laboratory of Molecular Biology of Cambridge, UK. From 1977 to 1980 he was a staff scientist at the same institution. In 1980 he won a Tenured Lecturer position at the Sir William Dunn School of Pathology, Oxford. In 1990 he was named Director of the Trieste laboratory of the International Center for Genetic Engineering and Biotechnology (ICGEB, Trieste) and in 2004 he became the General Director of the ICGEB. His laboratory studies pre-mRNA splicing mechanisms, splicing defects in genetic diseases, and RNA-protein interactions, and in the last decade developed an interest in the role of hnRNPs in neurodegeneration.
Jimena Giudice received her Ph.D. in Biological Chemistry in the University of Buenos Aires, Argentina (2011). During that period, she trained in microscopy in the Max Planck Institute for Biophysical Chemistry (Germany) supported by a short-term EMBO fellowship. From 2011 to 2016 she trained in Baylor College of Medicine in Dr. Cooper’s lab and was awarded postdoctoral fellowships from the Pew Charitable Trusts and the American Heart Association. Since March 2016 she is a Tenure Track Assistant Professor in the Department of Cell Biology and Physiology in the School of Medicine at The University of North Carolina at Chapel Hill. Her lab studies alternative splicing and intracellular trafficking in development.
Footnotes
Competing interests
None
References
- 1.Pan Q, Shai O, Lee LJ, Frey BJ & Blencowe BJ Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet 40, 1413–1415 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Wang ET et al. Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim M-S et al. A draft map of the human proteome. Nature 509, 575–581 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scotti MM & Swanson MS RNA mis-splicing in disease. Nat. Rev. Genet 17, 19–32 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalsotra A et al. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc. Natl. Acad. Sci. U. S. A 105, 20333–20338 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bebee TW, Cieply BW & Carstens RP Genome-wide activities of RNA binding proteins that regulate cellular changes in the epithelial to mesenchymal transition (EMT). Adv. Exp. Med. Biol 825, 267–302 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Pradella D, Naro C, Sette C & Ghigna C EMT and stemness: flexible processes tuned by alternative splicing in development and cancer progression. Mol. Cancer 16, 8. doi: 10.1186/s12943-016-0579-2. (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chabot B & Shkreta L Defective control of pre-messenger RNA splicing in human disease. J. Cell Biol 212, 13–27 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh RK & Cooper TA Pre-mRNA splicing in disease and therapeutics. Trends in Molecular Medicine 18, 472–482 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinez NM et al. Alternative splicing networks regulated by signaling in human T cells. RNA 18, 1029–1040 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giudice J et al. Alternative splicing regulates vesicular trafficking genes in cardiomyocytes during postnatal heart development. Nat. Commun 5, 3603 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhate A et al. ESRP2 controls an adult splicing programme in hepatocytes to support postnatal liver maturation. Nat. Commun 6, 8768 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dillman AA et al. mRNA expression, splicing and editing in the embryonic and adult mouse cerebral cortex. Nat. Neurosci 16, 1–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh RK et al. Rbfox2-coordinated alternative splicing of Mef2d and Rock2 controls myoblast fusion during myogenesis. Mol. Cell 55, 592–603 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Llorian M et al. The alternative splicing program of differentiated smooth muscle cells involves concerted non-productive splicing of post-transcriptional regulators. Nucleic Acids Res. 44, 8933–8950 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fu XD Towards a splicing code. Cell 119, 736–738 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Barash Y et al. Deciphering the splicing code. Nature 465, 53–59 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Fu X-D & Ares M Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet 15, 689–701 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tilgner H et al. Deep sequencing of subcellular RNA fractions shows splicing to be predominantly co-transcriptional in the human genome but inefficient for lncRNAs. Genome Res. 22, 1616–1625 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vargas DY et al. Single-molecule imaging of transcriptionally coupled and uncoupled splicing. Cell 147, 1054–1065 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kornblihtt AR et al. Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat. Rev. Mol. Cell Biol 14, 153–65 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Perales R & Bentley D ‘Cotranscriptionality’: the transcription elongation complex as a nexus for nuclear transactions. Molecular Cell 36, 178–191 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neugebauer KM On the importance of being co-transcriptional. J. Cell Sci 115, 3865–3871 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Fiszbein A et al. Alternative splicing of G9a regulates neuronal differentiation. Cell Rep. 14, 2797–2808 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Luco RF, Allo M, Schor IE, Kornblihtt AR & Misteli T Epigenetics in alternative pre-mRNA splicing. Cell 144, 16–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ip JY et al. Global impact of RNA polymerase II elongation inhibition on alternative splicing regulation. Genome Res. 21, 390–401 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Q et al. The splicing regulator PTBP2 controls a program of embryonic splicing required for neuronal maturation. Elife 3, e01201 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Licatalosi DD et al. Ptbp2 represses adult-specific splicing to regulate the generation of neuronal precursors in the embryonic brain. Genes Dev. 26, 1626–1642 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quesnel-Vallières M, Irimia M, Cordes SP & Blencowe BJ Essential roles for the splicing regulator nSR100/SRRM4 during nervous system development. Genes Dev. 29, 746–759 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raj B et al. Cross-regulation between an alternative splicing activator and a transcription repressor controls neurogenesis. Mol. Cell 43, 843–850 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Zhang X et al. Cell-type-specific alternative splicing governs cell fate in the developing cerebral cortex. Cell 166, 1147–1162 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Makeyev EV, Zhang J, Carrasco MA & Maniatis T The microRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol. Cell 27, 435–448 (2007).This mechanistic study shows that regulation of a microRNA controls PTBP1 expression, that in turns regulates splicing of the exon 10 in PTBP2, inclusion of which allows PTBP2 protein expression, and consequently the non-neuronal to neuronal-specific splicing transition.
- 33.Boutz PL et al. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev. 21, 1636–1652 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spellman R, Llorian M & Smith CWJ Crossregulation and functional redundancy between the splicing regulator PTB and its paralogs nPTB and ROD1. Mol. Cell 27, 420–434 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raj B et al. A global regulatory mechanism for activating an exon network required for neurogenesis. Mol. Cell 56, 90–103 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fogel BL et al. RBFOX1 regulates both splicing and transcriptional networks in human neuronal development. Hum Mol Genet 21, 4171–4186 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gehman LT et al. The splicing regulator Rbfox1 (A2BP1) controls neuronal excitation in the mammalian brain. Nat. Genet 43, 706–711 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee JA et al. Cytoplasmic Rbfox1 regulates the expression of synaptic and autism-related genes. Neuron 89, 113–128 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jensen KB et al. Nova-1 regulates neuron-specific alternative splicing and is essential for neuronal viability. Neuron 25, 359–371 (2000). [DOI] [PubMed] [Google Scholar]
- 40.Yano M, Hayakawa-Yano Y, Mele A & Darnell RB Nova2 regulates neuronal migration through an RNA switch in Disabled-1 signaling. Neuron 66, 848–858 (2010).Shows that in the brain the balance between DAB1 splicing isoforms is developmentally controlled by NOVA2 and that this regulates neuronal migration.
- 41.Giampietro C et al. The alternative splicing factor Nova2 regulates vascular development and lumen formation. Nat. Commun 6, 8479 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Forster E et al. Emerging topics in Reelin function. Eur. J. Neurosci. 31, 1511–1518 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beffert U et al. Reelin-mediated signaling locally regulates protein kinase B/Akt and glycogen synthase kinase 3beta. J. Biol. Chem 277, 49958–49964 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Kim K, Nam J, Mukouyama Y & Kawamoto S Rbfox3-regulated alternative splicing of Numb promotes neuronal differentiation during development. J. Cell Biol 200, 443–458 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Traunmuller L, Gomez AM, Nguyen T-M & Scheiffele P Control of neuronal synapse specification by a highly dedicated alternative splicing program. Science 352, 982–986 (2016).This study demonstrates in vivo that the RNA-binding protein SLM2 controls specification of synases through alternative splicing of neurexin-1.
- 46.Iijima T et al. SAM68 regulates neuronal activity-dependent alternative splicing of neurexin-1. Cell 147, 1601–1614 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zibetti C et al. Alternative splicing of the histone demethylase LSD1/KDM1 contributes to the modulation of neurite morphogenesis in the mammalian nervous system. J. Neurosci 30, 2521–2532 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mauger O et al. Alternative splicing regulates the expression of G9A and SUV39H2 methyltransferases, and dramatically changes SUV39H2 functions. Nucleic Acids Res. 43, 1869–1882 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Laurent B et al. A specific LSD1/KDM1A isoform regulates neuronal differentiation through H3K9 demethylation. Mol. Cell 57, 957–970 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lister R et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–22 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hodges E et al. High definition profiling of mammalian DNA methylation by array capture and single molecule bisulfite sequencing. Genome Res. 19, 1593–1605 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choi JK Contrasting chromatin organization of CpG islands and exons in the human genome. Genome Biol. 11, R70 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oberdoerffer S A conserved role for intragenic DNA methylation in alternative pre-mRNA splicing. Transcription 3, 106–109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Amit M et al. Differential GC content between exons and introns establishes distinct strategies of splice-site recognition. Cell Rep. 1, 543–556 (2012). [DOI] [PubMed] [Google Scholar]
- 55.Gelfman S, Cohen N, Yearim A & Ast G DNA-methylation effect on cotranscriptional splicing is dependent on GC architecture of the exon-intron structure. Genome Res. 23, 789–799 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gelfman S et al. Changes in exon-intron structure during vertebrate evolution affect the splicing pattern of exons. Genome Res. 22, 35–50 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kucharski R, Maleszka J, Foret S & Maleszka R Nutritional control of reproductive status in honeybees via DNA methylation. Science 319, 1827–1830 (2008). [DOI] [PubMed] [Google Scholar]
- 58.Lyko F et al. The honey bee epigenomes: Differential methylation of brain DNA in queens and workers. PLoS Biol. 8, e1000506 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Foret S, Kucharski R & Pellegrini M DNA methylation dynamics, metabolic fluxes, gene splicing, and alternative phenotypes in honey bees. Proc. Natl. Acad. Sci. U. S. A 109, 4968–4973 (2012).Refs 57–59 demonstrate how the connection between alternative DNA-methylation and splicing impacts development of honeybees into queens or workers
- 60.Shukla S et al. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 479, 74–79 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Phillips JE & Corces VG CTCF: Master Weaver of the Genome. Cell 137, 1194–1211 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sun S et al. ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nat. Commun 6, 6171 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bentmann E, Haass C & Dormann D Stress granules in neurodegeneration - Lessons learnt from TAR DNA binding protein of 43 kDa and fused in sarcoma. FEBS Journal 280, 4348–4370 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Tollervey JR et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci 14, 452–458 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Janssens J & Van Broeckhoven C Pathological mechanisms underlying TDP-43 driven neurodegeneration in FTLD-ALS spectrum disorders. Hum. Mol. Genet 22, R77–R87 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Scotter EL, Chen HJ & Shaw CE TDP-43 proteinopathy and ALS: insights into disease mechanisms and therapeutic targets. Neurotherapeutics 12, 352–363 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ling JP et al. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 349, 650–655 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Coady TH & Manley JL ALS mutations in TLS / FUS disrupt target gene expression. Genes Dev. 29, 1696–1706 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Eréndira Avendaño-Vázquez S et al. Autoregulation of TDP-43 mRNA levels involves interplay between transcription, splicing, and alternative polyA site selection. Genes Dev. 26, 1679–1684 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.De Conti L et al. TDP-43 affects splicing profiles and isoform production of genes involved in the apoptotic and mitotic cellular pathways. Nucleic Acids Res. 43, 8990–9005 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weyn-Vanhentenryck SM et al. HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell Rep. 6, 1139–1152 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bill BR, Lowe JK, DyBuncio CT & Fogel BL Orchestration of neurodevelopmental programs by RBFOX1: Implications for autism spectrum disorder. Int. Rev. Neurobiol 113, 251–267 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Voineagu I et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Irimia M et al. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell 159, 1511–1523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lacovich V et al. Tau isoforms imbalance impairs the axonal transport of the Amyloid Precursor Protein in human neurons. J. Neurosci 37, 58–69 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vuong CK, Black DL & Zheng S The neurogenetics of alternative splicing. Nat. Rev. Neurosci 17, 265–281 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Buljan M et al. Tissue-specific splicing of disordered segments that embed binding motifs rewires protein interaction networks. Mol. Cell 46, 871–883 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ellis JD et al. Tissue-specific alternative splicing remodels protein-protein interaction networks. Mol. Cell 46, 884–892 (2012). [DOI] [PubMed] [Google Scholar]
- 79.Giudice J & Cooper TA RNA-binding proteins in heart development. Adv. Exp. Med. Biol 825, 389–429 (2014). [DOI] [PubMed] [Google Scholar]
- 80.Wang ET et al. Antagonistic regulation of mRNA expression and splicing by CELF and MBNL proteins. Genome Res. 25, 858–871 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gao C et al. RBFox1-mediated RNA splicing regulates cardiac hypertrophy and heart failure. J. Clin. Invest 126, 195–206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gallagher TL et al. Rbfox-regulated alternative splicing is critical for zebrafish cardiac and skeletal muscle functions. Dev. Biol 359, 251–261 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Salomonis N et al. Alternative splicing in the differentiation of human embryonic stem cells into cardiac precursors. PLoS Comput. Biol 5, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Giudice J, Loehr J, Rodney GG & Cooper TA Alternative splicing of Snap23, Tmed2, Trip10, and Cltc regulates myofiber structure and skeletal muscle physiology. Cell Rep. 17, 1923–1933 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Labeit S & Kolmerer B Titins: giant proteins in charge of muscle ultrastructure and elasticity. Science 270, 293–296 (1995). [DOI] [PubMed] [Google Scholar]
- 86.Bang ML et al. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ. Res 89, 1065–1072 (2001). [DOI] [PubMed] [Google Scholar]
- 87.Li S, Guo W, Dewey CN & Greaser ML Rbm20 regulates titin alternative splicing as a splicing repressor. Nucleic Acids Res. 41, 2659–2672 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Krüger M & Linke WA The giant protein titin: A regulatory node that integrates myocyte signaling pathways. Journal of Biological Chemistry 286, 9905–9912 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Guo W et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 18, 766–773 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Refaat MM et al. Genetic variation in the alternative splicing regulator RBM20 is associated with dilated cardiomyopathy. Hear. Rhythm 9, 390–396 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brauch KM et al. Mutations in Ribonucleic Acid Binding Protein Gene Cause Familial Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 54, 930–941 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Beqqali A et al. A mutation in the glutamate-rich region of RNA-binding motif protein 20 causes dilated cardiomyopathy through missplicing of titin and impaired Frank-Starling mechanism. Cardiovasc. Res 112, 452–463 (2016). [DOI] [PubMed] [Google Scholar]
- 93.Haas J et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J 36, 1123–1135a (2015). [DOI] [PubMed] [Google Scholar]
- 94.Charton K et al. Exploiting the CRISPR/CAS9 system to study alternative splicing in vivo: application to Titin. Hum. Mol. Genet. 25, 4518–4532 (2016).First study applying CRISPR-Cas9 editing technologies to investigate alternative splicing functions in vivo.
- 95.Beraldi R et al. Rbm20-deficient cardiogenesis reveals early disruption of RNA processing and sarcomere remodeling establishing a developmental etiology for dilated cardiomyopathy. Hum. Mol. Genet. 23, 3779–3791 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Maatz H et al. RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. J. Clin. Invest 124, 3419–3430 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lara-Pezzi E, Gómez-Salinero J, Gatto A & García-Pavía P The alternative heart: Impact of alternative splicing in heart disease. J. Cardiovasc. Transl. Res 6, 945–955 (2013). [DOI] [PubMed] [Google Scholar]
- 98.Mirtschink P et al. HIF-driven SF3B1 induces KHK-C to enforce fructolysis and heart disease. Nature 522, 444–449 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Castle JC et al. Expression of 24,426 human alternative splicing events and predicted cis regulation in 48 tissues and cell lines. Nat. Genet 40, 1416–1425 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Runfola V, Sebastian S, Dilworth FJ & Gabellini D Rbfox proteins regulate tissue-specific alternative splicing of Mef2D required for muscle differentiation. J. Cell Sci 128, 631–637 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hall MP et al. Quaking and PTB control overlapping splicing regulatory networks during muscle cell differentiation. RNA 19, 627–638 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sebastian S et al. Tissue-specific splicing of a ubiquitously expressed transcription factor is essential for muscle differentiation. Genes Dev. 27, 1247–1259 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Harper P Myotonic dystrophy. (2001). [Google Scholar]
- 104.Savkur RS, Philips AV & Cooper TA Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet 29, 40–47 (2001). [DOI] [PubMed] [Google Scholar]
- 105.Savkur RS et al. Insulin receptor splicing alteration in myotonic dystrophy type 2. Am. J. Hum. Genet 74, 1309–1313 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fugier C et al. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat. Med 17, 720–725 (2011). [DOI] [PubMed] [Google Scholar]
- 107.Kimura T et al. Altered mRNA splicing of the skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase in myotonic dystrophy type 1. Hum. Mol. Genet 14, 2189–2200 (2005). [DOI] [PubMed] [Google Scholar]
- 108.Freyermuth F et al. Splicing misregulation of SCN5A contributes to cardiac conduction delay and heart arrhythmia in myotonic dystrophy. Nat. Commun 7, 11067 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Eizirik DL et al. The human pancreatic islet transcriptome: Expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet. 8, e1002552 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Villate O et al. Nova1 is a master regulator of alternative splicing in pancreatic beta cells. Nucleic Acids Res. 42, 11818–11830 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cnop M et al. RNA sequencing identifies dysregulation of the human pancreatic islet transcriptome by the saturated fatty acid palmitate. Diabetes 63, 1978–1993 (2014). [DOI] [PubMed] [Google Scholar]
- 112.Lin JC et al. RBM4 promotes pancreas cell differentiation and insulin expression. Mol Cell Biol 33, 319–327 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sen S, Jumaa H & Webster NJG Splicing factor SRSF3 is crucial for hepatocyte differentiation and metabolic function. Nat. Commun 4, 1336 doi: 10.1038/ncomms2342 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pihlajamäki J et al. Expression of the splicing factor gene SFRS10 is reduced in human obesity and contributes to enhanced lipogenesis. Cell Metab. 14, 208–218 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Elizalde M et al. Splicing regulator SLU7 is essential for maintaining liver homeostasis. J. Clin. Invest. 124, 2909–2920 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Carreira-Rosario A et al. Repression of Pumilio protein expression by Rbfox1 promotes germ cell differentiation. Dev. Cell 36, 562–571 (2016).Report showing how nuclear and cytoplasmic isoforms of RBFOX1 contribute to germ cell differentiation
- 117.Slaidina M & Lehmann R Translational control in germline stem cell development. J. Cell Biol 207, 13–21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Soumillon M et al. Cellular source and mechanisms of high transcriptome complexity in the mammalian testis. Cell Rep 3, 2179–2190 (2013). [DOI] [PubMed] [Google Scholar]
- 119.Ramsköld D, Wang ET, Burge CB & Sandberg R An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput. Biol 5, e1000598 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Schmid R et al. The splicing landscape is globally reprogrammed during male meiosis. Nucleic Acids Res. 41, 10170–10184 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zagore LL et al. RNA binding protein Ptbp2 is essential for male germ cell development. Mol. Cell. Biol 35, 4030–4042 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Paronetto MP et al. Sam68 marks the transcriptionally active stages of spermatogenesis and modulates alternative splicing in male germ cells. Nucleic Acids Res. 39, 4961–4974 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Luco RF et al. Regulation of alternative splicing by histone modifications. Science 327, 996–1000 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Iwamori N et al. MRG15 is required for pre-mRNA splicing and spermatogenesis. Proc. Natl. Acad. Sci. U. S. A 113, E5408–E5415 (2016).This study nicely demonstrates how the interplay between epigenetic information and splicing impacts on mouse spermatogenesis
- 125.Gaudreau M-C, Heyd F, Bastien R, Wilhelm B & Möröy T Alternative splicing controlled by heterogeneous nuclear ribonucleoprotein L regulates development, proliferation, and migration of thymic pre-T cells. J. Immunol 188, 5377–5388 (2012). [DOI] [PubMed] [Google Scholar]
- 126.Shankarling G, Cole BS, Mallory MJ & Lynch KW Transcriptome-wide RNA interaction profiling reveals physical and functional targets of hnRNP L in human T cells. Mol. Cell. Biol. 34, 71–83 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yarosh CA et al. TRAP150 interacts with the RNA-binding domain of PSF and antagonizes splicing of numerous PSF-target genes in T cells. Nucleic Acids Res. 43, 9006–9016 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ajith S et al. Position-dependent activity of CELF2 in the regulation of splicing and implications for signal-responsive regulation in T cells. RNA Biol. 13, 569–581 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mallory MJ et al. Induced transcription and stability of CELF2 mRNA drives widespread alternative splicing during T-cell signaling. Proc. Natl. Acad. Sci. U. S. A 112, E2139–E2148 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ni T et al. Global intron retention mediated gene regulation during CD4+ T cell activation. Nucleic Acids Res. 44, 6817–6829 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Martinez NM et al. Widespread JNK-dependent alternative splicing induces a positive feedback loop through CELF2-mediated regulation of MKK7 during T-cell activation. Genes Dev. 29, 2054–2066 (2015).Refs 128, 129, and 131 provide evidence of a positive feedback loop in T-cell activation comprising CELF2 splicing factor, JNK signalling pathway, and MKK7 alternative splicing.
- 132.Cole BS et al. Global analysis of physical and functional RNA targets of hnRNP L reveals distinct sequence and epigenetic features of repressed and enhanced exons. RNA 21, 2053–2066 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rothrock C, House A & Lynch KW HnRNP L represses exon splicing via a regulated exonic splicing silencer. EMBO J. 24, 2792–2802 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yamamoto ML et al. Alternative pre-mRNA splicing switches modulate gene expression in late erythropoiesis. Blood 113, 3363–3370 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hou VC et al. Decrease in hnRNP A/B expression during erythropoiesis mediates a pre-mRNA splicing switch. EMBO J. 21, 6195–6204 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cheng AW et al. Muscleblind-like 1 (Mbnl1) regulates pre-mRNA alternative splicing during terminal erythropoiesis. Blood 124, 598–610 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Pimentel H et al. A dynamic alternative splicing program regulates gene expression during terminal erythropoiesis. Nucleic Acids Res. 42, 4031–4042 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pimentel H et al. A dynamic intron retention program enriched in RNA processing genes regulates gene expression during terminal erythropoiesis. Nucleic Acids Res. 44, 838–851 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yan Q et al. Systematic discovery of regulated and conserved alternative exons in the mammalian brain reveals NMD modulating chromatin regulators. Proc. Natl. Acad. Sci. U. S. A 112, 3445–3450 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Charizanis K et al. Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in Myotonic Dystrophy. Neuron 75, 437–450 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Pilaz L-J & Silver DL Post-transcriptional regulation in corticogenesis: how RNA-binding proteins help build the brain. Wiley Interdiscip. Rev. RNA 6, 501–515 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Raj B & Blencowe BJ Alternative splicing in the mammalian nervous system: recent insights into mechanisms and functional roles. Neuron 87, 14–27 (2015). [DOI] [PubMed] [Google Scholar]
- 143.Blech-Hermoni Y & Ladd AN RNA binding proteins in the regulation of heart development. Int. J. Biochem. Cell Biol 45, 2467–2478 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Van Den Hoogenhof MMG, Pinto YM & Creemers EE RNA Splicing regulation and dysregulation in the heart. Circulation Research 118, 454–468 (2016). [DOI] [PubMed] [Google Scholar]
- 145.Licatalosi DD Roles of RNA-binding proteins and post-transcriptional regulation in driving male germ cell development in the mouse. Adv Exp Med Biol 907, 123–151 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Martinez NM & Lynch KW Control of alternative splicing in immune responses: Many regulators, many predictions, much still to learn. Immunological Reviews 253, 216–236 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yabas M, Elliott H & Hoyne G The role of alternative splicing in the control of immune homeostasis and cellular differentiation. Int. J. Mol. Sci 17, E3. doi: 10.3390/ijms17010003 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Conboy JG RNA splicing during terminal erythropoiesis. Curr. Opin. Hematol In press, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bland CS et al. Global regulation of alternative splicing during myogenic differentiation. Nucleic Acids Res. 38, 7651–7664 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wells QS et al. Whole exome sequencing identifies a causal RBM20 mutation in a large pedigree with familial dilated cardiomyopathy. Circ. Cardiovasc. Genet 6, 317–326 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Echeverria GV & Cooper TA RNA-binding proteins in microsatellite expansion disorders: Mediators of RNA toxicity. Brain Research 1462, 100–111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Goodwin M et al. MBNL sequestration by toxic RNAs and RNA misprocessing in the Myotonic Dystrophy brain. Cell Rep. 12, 1159–1168 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Chau A & Kalsotra A Developmental insights into the pathology of and therapeutic strategies for DM1: Back to the basics. Developmental Dynamics 244, 377–390 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.