

Abstract
We report here the synthesis and biological evaluation of phenylcarboxylic acid and phenylboronic acid containing HIV-1 protease inhibitors and their functional effect on enzyme inhibition and antiviral activity in MT-2 cell lines. Inhibitors bearing bis-THF ligand as P2 ligand and phenylcarboxylic acids and carboxamide as the P2’ ligands, showed very potent HIV-1 protease inhibitory activity. However, carboxylic acid containing inhibitors showed very poor antiviral activity compared to carboxamide-derived inhibitors which showed good antiviral IC50 value. Boronic acid-derived inhibitor with bis-THF as the P2 ligand showed very potent enzyme inhibitory activity, but it showed relatively reduced antiviral activity compared to darunavir in the same assay. Boronic acid-containing inhibitor with a P2-Crn-THF ligand also showed potent enzyme Ki but significantly reduced antiviral activity. We have evaluated antiviral activity against a panel of highly drug-resistant HIV-1 variants. One of the inhibitors maintained good antiviral activity against HIVDRVRP20 and HIVDRVRP30 viruses. We have determined high resolution X-ray structures of two synthetic inhibitors bound to HIV-1 protease and obtained molecular insight into the ligand-binding site interactions.
Keywords: HIV-1 protease inhibitors, brain penetration, Drug resistance, dimerization inhibitor, Structure-based design, synthesis, genetic barrier
Graphical Abstract
We report structure-based design, synthesis, biological evaluation and X-ray structural studies of a series of exceptionally potent HIV-1 protease inhibitors containing boronic acid-based P2’ –ligands to interact with active site residues in the S2’-subsite.
Introduction
Structure-based molecular design has become one of the most successful and widely used strategies for preclinical drug development. Over the years, it has led to numerous first-in-class approved drugs and preclinical candidates.1,2 Structure-based design strategies have played a key role in the design and development of HIV-1 protease inhibitor drugs.3,4 HIV-1 protease inhibitors are an important component of current antiretroviral therapy (ART) which is responsible for dramatic improvement of life expectancy and mortality rates of HIV/AIDS patients in the developing nations. HIV-1 protease is an aspartic acid protease which plays a critical role in viral replication.5,6 HIV-1 protease inhibitor (PI) drugs block HIV-1 protease and generate morphologically immature and noninfectious virions.7,8 Darunavir (1, Figure 1), the latest FDA-approved PI, has been highly efficacious in suppressing HIV-1 replication and showing significant clinical benefits to HIV/AIDS patients.9,10 Darunavir has emerged as a widely used first-line therapy for rescue treatment.11,12 Its exceptional resistance profile is due to a dual mechanism of action as it inhibits HIV-1 protease and inhibits dimerization of HIV-1 protease monomers.13
Figure 1.

Structures of HIV-1 protease inhibitors 1–6.
Darunavir was developed by structure-based design strategies with particular emphasis on maximizing active site interactions, especially promoting strong hydrogen bonding interactions with the HIV-1 backbone atoms in the S2 and S2’ subsites.14,15 In this context, we created a stereochemically defined 3(R),6a(R)-bis-tetrahydrofuranyl urethane as the P2 ligand to form hydrogen bonds with the Asp29 and Asp30 backbone NHs in the S2-subsite. Also, the P2’−4-aminobenzene sulfonamide was incorporated to interact with the backbone atoms in the S2’ subsite.16,17 These inhibitor design strategies targeting the protein backbone may be responsible for darunavir’s exceptional antiviral activity against highly multidrug-resistant HIV-1 variants.18,19 However, DRV-resistant HIV-1 variants have emerged, and development of novel antiviral drugs with a high genetic barrier against multidrug-resistant HIV-1 variants including DRV-resistant variants is urgently needed.20,21 In this context, we investigated further optimization of structural elements of darunavir that would enhance backbone interactions in the active site of HIV-1 protease.
The X-ray structure of darunavir-bound HIV-1 protease [PDB 1D : 4HLA] revealed that the P2’ 4-aminosulfonamide ligand is involved in water-mediated hydrogen bonding interactions with Gly48’ backbone amide NH through two crystallographic water molecules.17,22 Based upon this observation, we speculated that the modification of amine functionality with a carboxylic acid or with a carboxamide group would replace one of these two crystallographic water molecules closer to the amine functionality. Such modified ligands could not only maintain strong hydrogen bonds with backbone NH of Asp30, but also the carboxylic acid derivative can form a water-mediated hydrogen bond with the Gly48’ NH located in flap region of HIV-1 protease. Indeed, the resulting modified inhibitors 3 and 4 exhibited very potent enzyme inhibitory activity with Ki values of 12.9 pM and 8.9 pM, respectively.22 Interestingly, carboxylic acid-derived inhibitor 3 did not exhibit any appreciable antiviral activity in MT-2 cells. However, inhibitor 4 with a carboxamide derivative showed antiviral IC50 value of 93 nM in MT-2 cells.22 This result is not surprising as it has been reported that intracellular concentration of PIs are a dynamic balance between influx, efflux, and sequestration.23,24 Since DRV has been shown to serve as a transport substrate of P-glycoprotein, the carboxylic acid derivatives are expected to show similar efflux profiles.25 Thus, we have assayed intracellular concentrations of inhibitor 3 and 4 in MT-2 and MT-4 cells. Interestingly, both cell lines showed very low uptake of carboxylic acid-derived inhibitor 3.22 The sequestration and influx properties of PIs are important issues that need serious attention for PI design. We have determined X-ray crystal structures of inhibitor 3 and 4 with HIV-1 protease complexes. Our structural analysis revealed enhanced hydrogen bonding interactions in the S2’ subsite compared to DRV-HIV-1 protease complex.22 A stereoview of the active site interactions of inhibitor-bound HIV-1 protease is shown in Figure 2. As can be seen, the P2’ 4-carboxylic acid ligand is involved in water-mediated hydrogen bonding interactions with Gly48’ backbone amide NH located in the flap region of HIV-1 protease.17,22 More recently, Raines and co-workers reported the HIV-1 protease inhibitor 5 with a phenyl boronic acid as the P2’ ligand.26 This inhibitor also exhibited very potent enzyme inhibitory activity compared to darunavir. Furthermore, X-ray crystal structure of inhibitor 5-bound HIV-1 protease revealed that boronic acid is involved in enhanced hydrogen bonding interaction including a water-mediated hydrogen bonding interaction with Gly48’ similar to inhibitor 3 and 4.26 While boronic acid containing inhibitor 5 displayed potent enzyme inhibitory activity, assessment of its antiviral activity in cell-based assays was not carried out. Thus, the intriguing question is whether boronic acid-derived inhibitors show better antiviral potency compared to carboxylic acid-derived compounds. In the present studies, we have synthesized a selected group of HIV-1 protease inhibitors containing boronic acid as the P2’ ligand and evaluated their enzyme inhibitory activity as well as antiviral activity compared to darunavir. Furthermore, we have evaluated antiviral activity against a panel of highly darunavir-resistant HIV-1 variants for compounds 5 and 6 with darunavir control. To assess ligand-binding site interactions, we have determined a high resolution X-ray structure of inhibitor 6-bound HIV-1 protease.
Figure 2.

A stereoview of inhibitor 3-bound X-ray structure of HIV-1 protease. The major orientation of the inhibitor is shown. The inhibitor carbon atoms are shown in green, water molecules are red spheres, and the hydrogen bonds are indicated by dotted lines (PDB ID: 4I8W).
Synthesis of carboxylic and boronic acid-based inhibitors
For our investigation regarding the effect on enzyme inhibitory and antiviral activity for inhibitors containing 4-phenylcarboxylic acid and 4-phenylboronic acid as the P2’ ligand, we have synthesized a set of selected inhibitors. The synthesis of 4-phenylcarboxylic acid derivatives 11 and 13 is shown in Scheme 1. Commercially available (S,S)-3(t-Boc-amino)-1,2-epoxy-4-phenylbutane 7 was converted to hydroxyethylamine sulfonamide derivative 8 with a diacetoxymethyl group as P2’ ligand as described by us previously.27 Exposure of the acetate derivative 8 to K2CO3 in methanol at 23 °C for 30 min provided the corresponding aldehyde. Pinnick oxidation28 of the resulting aldehyde with sodium chlorite in t-butanol in the presence of 2- methyl-2-butene and phosphate buffer afforded the carboxylic acid derivative 9 in 86% yield over 2-steps. For the conversion of carboxylic acid derivative 9 to inhibitor 11 Boc-group was removed by exposure to trifluoroacetic acid (TFA) in CH2Cl2 at 0 °C to 23 °C for 3 h. Reaction of the resulting amine with known29 activated carbonate of tetrahydropyranofuran 10 in the presence of diisopropylethylamine (DIPEA) in CH3CN at 23 °C for 10 d afforded inhibitor 11 in 37% yield over 2-steps. For the synthesis of carboxylic acid derivative 13, Boc derivative 9 was treated with TFA and the resulting amine was reacted with crown-THF carbonate 1230 as described above to provide 13 in 80% yield over 2 steps.
Scheme 1.

Synthesis of PIs 11, and 13. Reagents and chemicals. (a) K2CO3, MeOH, 23 °C, 30 min; (b) NaClO2, 2-methyl-2-butene, NaHPO4, tBuOH-H2O, 23 °C, 3 h (86% for 2-steps); (c) TFA, CH2Cl2, 0 °C to 23 °C, 3 h; (d) 10, DIPEA, CH3CN, 23 °C, 12 d (37–80% for 2-steps).
For convenient synthesis of inhibitors containing 4-phenylboronic acid, we first prepared 4-(chlorosulfonyl)-phenyl boronic acid. As shown in Scheme 2, commercially available 4-bromophenyl sulfonyl chloride 14 was treated with NaOMe in methanol at 0 °C to 23 °C for 12 h to provide the corresponding methyl sulfonate derivative in 62% yield.31 Reaction of the resulting 4-bromosulfonate derivative with nBuLi in THF at −78 °C for 10 min followed by addition of B(OMe)3 at −78 °C for 20 min provided boronic acid derivative 15 in 45% yield. Treatment of 15 with triethylamine in methanol 60 °C for 24 h followed by reaction of the resulting triethylammonium salt with thionyl chloride, heated at 80 °C for 24 h furnished, sulfonyl chloride 16 in 89% yield.32 This sulfonyl chloride was then reacted with the known hydroxyethylamine derivative 1727,33 in CH2Cl2 in the presence of triethylamine at 0 °C to 23 °C for 1 h to furnish hydroxyethylsulfonamide dipeptide isostere 18 with a 4-phenylboronic acid as the P2’ ligand in 82% yield.
Scheme 2.

Synthesis of boronic acid derivative 18. Reagents and chemicals. (a) NaOMe, MeOH, 0 °C to 23 °C, 12 h (62%); (b) nBuLi, THF, −78 °C, B(OMe)3, 30 min (45%); (c) Et3N, MeOH, 60 °C for 24 h; (d) SOCl2, 80 °C for 24 h (89% for 2-steps); (e) Et3N, CH2Cl2, 0 °C to 23 °C, 1 h (82%).
Synthesis of inhibitors with boronic acid as the P2’ ligand is shown in Scheme 3. Boc-derivative 18 was treated with TFA in CH2Cl2 at 0 °C for 30 min to provide the corresponding amine. Reaction of the resulting amine with known activated bis-THF carbonate derivative 1929 in the presence of DIPEA in CH3CN at 23 °C for 10 days furnished inhibitor 5 in 47% yield. Similarly, reaction of resulting amine with activated crown-THF carbonate 12 and activated Tp-THF carbonate 10 afforded inhibitors 6 and 20 in 55% and 51% yields, respectively.
Scheme 3.

Synthesis of PIs. Reagents and chemicals. (a) TFA, CH2Cl2, 0 °C, 30 min; (b) carbonate 19, DIPEA, CH3CN, 23 °C, 10 d, (47% yield for 2-steps).
Results and Discussion
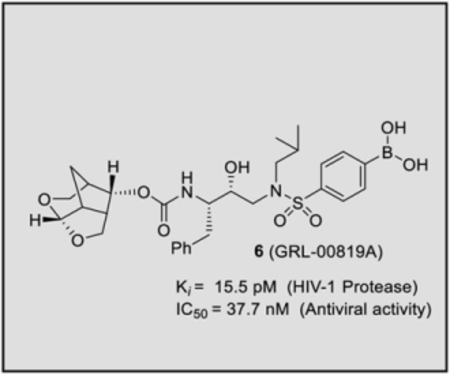
Our structure based design strategies resulted in a range of potent PIs with intriguing structural features, particularly PIs with cyclic ether-derived P2 ligands.3,14 Through X-ray structural analysis, we have shown that these PIs make extensive interactions particularly with backbone atoms in the active site. In general, these PIs exhibited exceptional enzyme affinity.14 Even though various factors such as cell penetration and flux, intracellular sequestration may affect antiviral activity, many of these PIs also showed very potent antiviral activity.3 Furthermore, they maintained robust antiviral activity against a wide range of highly multidrug-resistant HIV-1 variants. For the current studies, we designed a selected set of PIs containing carboxylic acid and boronic acid functionalities in the P2’-ligand. These functional groups can form more hydrogen bonds in the S2’-subsite than the aromatic amine functionality of darunavir.9 The structure and activity of these PIs are shown in Table 1. We first evaluated these PIs in an enzyme-inhibitory assay.34 As shown, inhibitor 3 with a P2-carboxylic acid showed a Ki value of 12.9 pM. We subsequently determined antiviral activity of these PIs in MT-2 and MT-4 human-T-lymphoid cells exposed to HIVLAI.35 Interestingly, in this cell-based antiviral assay, inhibitor 3 did not exhibit appreciable antiviral activity with IC50 values of 1 μM and 1 mM, in MT-2 and MT-4 cells, respectively. Carboxamide derivative 4 showed very potent enzyme Ki and antiviral IC50 values of 93 nM and 38 nM in MT-2 and MT-4, respectively. Both carboxylic acid containing inhibitors 11 and 13 showed excellent enzyme inhibitory activity (Ki 7.6 and 3.2 pM, respectively) however, they did not show antiviral activity. Boronic acid derivative 5 has been shown to have very potent enzyme inhibitory activity of 0.5 pM.26 However, there is no report of its antiviral activity in a cell-based assay. We have evaluated the boronic acid PI 5 in an antiviral assay with MT-2 cells. It turned out that boronic acid derivative 5 showed improved antiviral IC50 value of 48.9 nM compared to carboxylic derivatives 3 and 4 (entry 2, table 1). We prepared PI 6 containing a crown-like THF ligand as P2 ligand in combination with boronic acid P2’ ligand. This inhibitor exhibited an enzyme inhibitory Ki value of 15.5 pM and antiviral activity of 37.7 nM (entry 6). Inhibitor 20 with a (3aS,4S,7aR)-hexahydro-4H-furo[2,3-b]pyran as the P2 ligand also showed very potent enzyme Ki but much reduced antiviral IC50 value of (120 nM) compared to DRV (entry 7).
Table 1.
Structures and activity of carboxylic and boronic acid containing inhibitors.
| Entry | Inhibitor | Ki (pM)a | IC50 (nM)c |
|---|---|---|---|
| 1 | 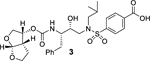 |
12.9 | >1000 |
| 2 | 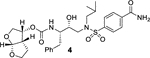 |
8.9 | 93 |
| 3 | 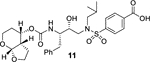 |
7.6 | >1000 |
| 4 | 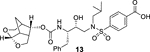 |
3.2 | >1000 |
| 5 | 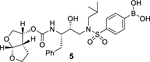 |
0.5b | 48.9 |
| 6 | 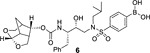 |
15.5 | 37.7 |
| 7 | 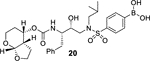 |
2.1 | 120 |
| 8 | 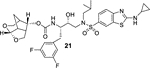 |
14 | 0.017 |
Ki values represents at least 5 data points. Standard error in all cases was less than 7%. Darunavir (DRV) exhibited Ki = 16 pM.
reported value.
Values are means of at least three experiments in MT-2 cells. Standard error in all cases was less than 5%. DRV exhibited IC50 = 1.6 nM.
Clinically, DRV is used widely for the treatment of naïve and experienced HIV/AIDS patients. However, heavily-ART regimen experienced patients have been showing treatment-failure with approved PIs, including DRV.35,36 Therefore, we have examined inhibitors 5 and 6 against DRV-resistant HIV-1 variants and compared their antiviral activity against DRV and one of our preclinical inhibitors, 21. For these studies, MT-4 cells (1×104) are exposed to wild type HIV-1 and three highly DRV-resistant HIV-1 variants, HIVDRVR20, HIVDRVRP30, and HIVDRVRP51.35,37 These variants were selected by propagating cells with increasing concentrations of DRV and they are highly resistant to all currently approved PIs, including DRV and nucleoside-RT inhibitors, such as tenofovir. Antiviral activity of selected PIs was determined using p24 assays. The results are shown in Table 2. As can be seen, FDA approved PI, LPV was unable to block the replication of these highly DRV-resistant variants. Inhibitor 5, resulting from incorporation of boronic acid in place of amine in DRV, showed relatively more potent antiviral activity (IC50 value of 219 nM) against DRV-resistant HIV-1 variants selected in the presence of DRV over 20 viral passages (HIVDRVRP20). Compound 5 lost only 4-fold activity compared to its IC50 values against HIV-1WT. Interestingly, DRV exhibited 164-fold loss of antiviral activity (IC50 525.8 nM) compared to its IC50 against HIV-1WT. Boronic acid derivative 5 displayed antiviral IC50 of 946 nM (19-fold loss over HIV-1WT) compared to DRV (IC50 601 nM, 187-fold over HIV-1WT). Both compound 5 and DRV did not exhibit any appreciable antiviral activity against the most highly DRV-resistant HIV-1 variants, HIVDRVRP51. Inhibitor 6 with a crown-like THF (Crn-THF) as the P2-ligand displayed improved antiviral activity compared to inhibitor 5 with a bis-THF P2 ligand. It showed antiviral IC50 value of 71 nM against HIVDRVRP20 variants and IC50 value of 532 nM against HIVDRVRP30 variants. Its fold-changes of antiviral activity compared to HIV-1WT are significantly lower than compound 5 or DRV. Compound 6, however, exhibited antiviral IC50 > 1 μM (>1000-fold change) against highly resistant HIV variants HIVDRVRP51. One of our recent inhibitors, 21, showed exceptional antiviral activity against all highly DRV-resistant HIV-1 variants.38,39 Both inhibitors 5 and 6 showed no appreciable cytotoxicity in MT-2 cells, with CC50 values of >100 μM, similar to DRV. However, selectivity index (CC50/IC50) were >2,040 and >2,650 compared to 34,480 for DRV and 2,473,684 for compound 21.
Table 2.
Antiviral activity of novel compounds against highly DRV-resistant HIV-1 variants.
| Mean IC50 in nM (fold-change) | |||||
|---|---|---|---|---|---|
| LPV | 1 (DRV) | 5 | 6 | 21 (GRL-14213) | |
| cHIVLAIWT | 13 | 3.2 | 48.9 | 37.7 | 0.017 |
| HIVDRVRP20 | >1000 (>77) | 525.8 (164) | 219.3 (4) | 71.2 (2) | 0.0024 |
| HIVDRVRP30 | >1000 (>77) | 601 (187) | 946.4 (19) | 532.3 (14) | 0.14 |
| HIVDRVRP51 | >1000 (>77) | 5429.7 (1696) | >1000 | >1000 | 1.3 |
Numbers in parentheses represent fold changes in IC50s for each isolate compared to the IC50s for wild-type cHIVLAIWT. All assays were conducted in triplicate, and the data shown represent mean values (±1 standard deviation) derived from the results of three independent experiments. DRV, Darunavir; LPV, Lopinavir.
X-ray description
To gain molecular insight into the ligand-binding site interactions of inhibitors 6 (GRL-008–19A) and 20 (GRL-031–19A), we co-crystallized these inhibitors with wild-type HIV-1 protease and the X-ray structures were refined at 1.33 and 1.13 Å resolution with R-factors of 13.7% and 12.5%, respectively. These structures contain a single PR dimer and the inhibitor binds in the active site cavity in two mutually exclusive orientations related by 180° rotation with major/minor occupancies of 60/40% and 65/35% for inhibitors 6 and 20, respectively. The overall dimer structures are very similar to the HIV-1 protease with DRV (2IEN)16 with root mean square difference (RMSD) for Cα of 0.20 and 0.12 Å for inhibitors 6 and 20, respectively. Similarly, the two new structures of inhibitors 6 and 20 are alike (Cα RMSD of 0.14 Å). The urethane NH of both inhibitors forms hydrogen bonding interactions with the carbonyl oxygen of Gly27. In addition, these inhibitors show water-mediated interactions that connect the inhibitor carbonyl oxygen and sulfonamide oxygen with the amides of Ile50 and 50’ in the flaps. Similar interactions are shared by the protease-darunavir complex and other protease inhibitors.4,16 The key interactions of inhibitor 6 and inhibitor 20 with HIV-1 protease are highlighted in Figure 3.
Figure 3.

A. The X-ray structure of inhibitor 6-bound HIV-1 protease (carbon atoms, turquoise; PDB ID: 6U7O). B. The X-ray structure of inhibitor 20-bound HIV-1 protease (carbon atoms, orange; PDB ID: 6U7P). The major orientations of the inhibitors are shown. All water molecules are red spheres, the hydrogen bonds are shown by dotted lines and bond lengths are shown in the S2’-subsite.
The hydrogen bond interactions between HIV-1 protease and the two inhibitors were compared with those for DRV. Inhibitors 6 and 20 differ only at the P2-group where the former boasts a Crn-THF while the latter contains a tetrahydropyrano-tetrahydrofuran (Tp-THF) ligand.29,30 Both oxygen atoms of the Crn-THF in inhibitor 6 formed a network of hydrgen bonds with the backbone amide NHs of Asp30 and Asp29 in the S2 subsite. These interactions are very similar to what was observed in the X-ray strucrure of the Crn-THF containing inhibitor 21, described recently.38,39 For inhibitor 20, both oxygens of the Tp-THF ligand also formed hydrogen bonds with the backbone amide NHs of Asp30 and Asp29. These interactions are comparable to the equivalent interactions with the bis-THF of DRV.[16,17]
The P2′ position of both inhibitors 6 and 20 harbors a phenylboronic acid moiety in place of the 4-aminosulfonamide of DRV. In both crystal structures, the boronic acid is aligned in plane with the phenyl group with less than 2° difference in torsion angle. The boronic acid introduces an additional water-mediated hydrogen bond with the Gly48′ amide located on the flexible flap of HIV-1 protease. This interaction is similar to the carboxylic acid derivative 3 as shown in Figure 2. The other hydroxyl group of the boronic acid is within hydrogen bonding distance to both main chain amide of Asp30′ (2.7–2.9 Å), although the bond angle departs from the ideal in the crystal structures. It appears that inhibitor 6 makes additional water-mediated interactions in the S2’-site of the active site. Overall, the inhibitor-HIV-1 protease hydrogen bonding interactions resemble those in DRV-HIV-1 protease with the exception of the new water-mediated interaction of boronic acid at P2’ and Gly48′, which is likely to stabilize the mobile flaps. These new interactions are likely to account for the improved enzyme affinity for inhibitors 6 and 20. These inhibitors showed better antiviral activity compared to the corresponding carboxylic acid derivatives likely due to improved efflux profiles.
Conclusions
In conclusion, we have designed and synthesized a selected set of HIV-1 protease inhibitors containing benzene carboxylic acid and benzene boronic acid as the P2’-ligands. These ligands are designed to form enhanced hydrogen bonding interactions with the backbone atoms in the S2’-subsite of HIV-1 protease. Inhibitor 3 with P2 bis-THF and P2’-benzene carboxylic acid showed potent enzyme inhibitory Ki, but showed no antiviral activity due to poor cell penetration. However, carboxamide derivative 4 displayed excellent enzyme inhibitory activity and good antiviral activity (IC50 of 93 nM). The corresponding boronic acid derivative 5 showed excellent enzyme Ki and improvement of antiviral IC50 value (49 nM) over the carboxamide derivative. Carboxylic acid and boronic acid containing inhibitors 6 and 13 in combination with a crown-like THF as the P2 ligand also showed very potent enzyme inhibitory activity. Carboxylic acid derivative 13, however, showed poor antiviral activity. Boronic acid-derived inhibitors displayed potent enzyme activity. They exerted much better antiviral activity in cell-based assay compared to the corresponding carboxylic acid derivatives possibly due to higher efflux profiles. However, DRV with a P2’-aniline ligand displayed significantly more potent antiviral activity compared to boronic acids. We have obtained high resolution X-ray structures of inhibitor 6-HIV-1 protease and inhibitor 20-HIV-1 protease complexes. Our structural studies established that both carboxylic acid and boronic acid containing inhibitors are involved in significantly enhanced hydrogen bonding interactions and water-mediated hydrogen bonding interactions in the S2’-site. Inhibitor 5, while less potent than DRV against HIVLAIWT, exerted comparable antiviral activity against highly DRV-resistant HIV-1 variants, HIVDRVRP20 and HIVDRVP30. Inhibitor 6, with a Crn-THF ligand as the P2-ligand, displayed improved antiviral activity compared to bis-THF derived inhibitor 5. Interestingly, fold-change of antiviral activity for boronic acid-derived inhibitors are significantly lower compared to DRV against these highly DRV-resistant HIV-1 variants. Only inhibitor 21 showed potent antiviral activity against HIVDRVRP51. Further design of new PIs using the current molecular insight is in progress.
Experimental Section
General
All reactions were carried out under an argon atmosphere in either flame or oven-dried (120 °C) glassware. All reagents and chemicals were purchased from commercial suppliers and used without further purification unless otherwise noted. Anhydrous solvents were obtained as follows: Dichloromethane from calcium hydride, diethyl ether and tetrahydrofuran from Na/Benzophenone, methanol and ethanol from activated magnesium under argon. All purification procedures were carried out with reagent grade solvents (purchased form VWR) in air. TLC analysis was conducted using glass-backed Thin-Layer Silica Gel Chromatography Plates (60 Å, 250 μm thickness, F-254 indicator). Column chromatography was performed using 230–400 mesh, 60 Å pore diameter silica gel. 1H, 13C NMR spectra were recorded at room temperature on a Bruker AV800, DRX-500, ARX-400. Chemical shifts (δ values) are reported in parts per million, and are referenced to the deuterated residual solvent peak. NMR data is reported as: δ value (chemical shift, J-value (Hz), integration, where s = singlet, d = doublet, t = triplet, q = quartet, brs = broad singlet). LRMS and HRMS spectra were recorded at the Purdue University Department of Chemistry Mass Spectrometry Center.
4-(N-((2R,3S)-3-((tert-Butoxycarbonyl)amino)-2-hydroxy-4-phenylbutyl)-N-isobutylsulfamoyl)benzoic acid (9):
To a stirred solution of acetal derivative 8 (100 mg, 0.165 mmol) in methanol (6 mL) at 23 °C, potassium carbonate (34 mg, 0.247 mmol) was added. The resulting mixture was stirred at 23 °C for 30 min. The solvent was evaporated under reduced pressure, and the product was extracted with ethyl acetate, dried over anhydrous sodium sulfate and concentrated. The crude product was purified by flash chromatography to provide the corresponding aldehyde (80 mg, 96%).
To a stirred solution of above aldehyde (80 mg, 0.158 mmol) at 23 °C in t-BuOH (3.0 mL), were added NaH2PO4 (65 mg, 0.476 mmol), 2-methyl-2-butene (170 μL, 1.58 mmol), NaClO2 (43 mg, 0.476 mmol) and H2O (1.0 mL). The resulting reaction mixture was allowed stir for 3 h at 23 °C. Then the reaction mixture was extracted with EtOAc and washed with H2O, brine solution, dried over Na2SO4, filtered, and concentrated under reduced pressure to yield crude residue which was purified by column chromatography over silica gel (5% MeOH/CH2Cl2) to afford acid inhibitor 9 (75 mg, 90%) as an amorphous solid. 1H-NMR (400 MHz, Methanol-d4) δ 8.17 (d, J = 8.5 Hz, 2H), 7.92 (d, J = 8.5 Hz, 2H), 7.27 – 7.18 (m, 4H), 7.15 (m, 1H), 3.71 (ddd, J = 9.3, 7.3, 2.5 Hz, 1H), 3.58 (ddd, J = 11.0, 7.3, 3.8 Hz, 1H), 3.43 (dd, J = 15.1, 2.6 Hz, 1H), 3.18 – 3.02 (m, 3H), 2.95 (dd, J = 13.7, 6.8 Hz, 1H), 2.54 (dd, J = 13.8, 10.6 Hz, 1H), 2.08 – 1.97 (m, 1H), 1.28 (s, 9H), 0.91 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H); 13C-NMR (100 MHz, Methanol-d4) δ 166.7, 156.5, 143.3, 138.7, 134.2, 129.9, 128.9, 127.7, 127.1, 125.6, 78.5, 72.4, 56.7, 55.4, 52.0, 35.8, 27.2, 26.3, 18.9. LRMS-ESI (m/z): 521.2 [M+H]+.
4-(N-((2R,3S)-3-(((((3aS,4S,7aR)-hexahydro-4H-furo[2,3-b]pyran-4-yl)oxy)carbonyl)amino)-2-hydroxy-4-phenylbutyl)-N-isobutylsulfamoyl)benzoic acid (11):
To a stirred solution of 9 (21 mg, 0.04 mmol) in CH2Cl2 (1.5 mL) was added TFA (0.5 mL) at 0 °C under argon atmosphere and the mixture was stirred at 23 °C for 3 h. Solvents were evaporated under reduced pressure, then CH2Cl2 (3 mL) was added and evaporated twice to give corresponding amine salt which was used for next step without further purification. The above amine salt (21 mg, 0.04 mmol) was dissolved in acetonitrile (1 mL) and N,N-diisopropylethylamine (28 μL, 0.16 mmol) was added. The resulting mixture was cooled to 0 °C and mixed carbonate 10 (10 mg, 0.03 mmol) was added to it. The resulting reaction mixture was allowed to warm to 23 °C and stir under argon for 12 d. After this period, the reaction mixture was diluted with a mixture of 10% MeOH in CH2Cl2 (10 mL), transferred to a separatory funnel, and washed with 1M HCl (2 mL). The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure to afford a crude solid which was purified by silica gel chromatography (5% MeOH/CH2Cl2) to yield the desired inhibitor 11 as an amorphous white solid (7.1 mg, 37%). Rf = 0.70 (silica plate, 20% MeOH/CH2Cl2). 1H-NMR (800 MHz, CD3OD) δ 8.17 (d, J = 8.1 Hz, 2H), 7.92 (d, J = 8.2 Hz, 2H), 7.30 – 7.14 (m, 5H), 6.97 (d, J = 8.6 Hz, 1H), 4.91 (s, 1H), 4.06 (t, J = 9.1 Hz, 1H), 3.96 – 3.68 (m, 4H), 3.44 (d, J = 15.1 Hz, 1H), 3.38 (t, J = 12.3 Hz, 1H), 3.16 (d, J = 14.0 Hz, 2H), 3.04 (dd, J = 15.4, 8.1 Hz, 1H), 2.97 – 2.91 (m, 1H), 2.53 (t, J = 12.2 Hz, 1H), 2.31 (s, 1H), 2.03 (s, 1H), 1.85 (p, J = 11.3, 10.5 Hz, 1H), 1.72 (q, J = 12.5 Hz, 1H), 1.65 (d, J = 13.4 Hz, 1H), 1.42 (d, J = 10.1 Hz, 1H), 1.37 (s, 1H), 0.93 (d, J = 5.6 Hz, 3H), 0.89 (d, J = 6.0 Hz, 3H). 13C-NMR (200 MHz, CD3OD) δ 157.8, 144.4, 140.3, 131.4, 130.5, 129.2, 128.6, 127.1, 102.4, 74.1, 71.0, 69.3, 61.8, 58.4, 57.3, 53.6, 44.8, 37.3, 27.9, 27.4, 23.2, 20.3. HRMS (ESI) m/z: [M + Na]+ Calcd for C29H38N2O9S 613.2190; Found 613.2174.
4-(N-((2R,3S)-3-(((((3S,7aS,8S)-Hexahydro-4H-3,5-methanofuro[2,3-b]pyran-8-yl)oxy)carbonyl)amino)-2-hydroxy-4-phenylbutyl)-N-isobutylsulfamoyl)benzoic acid (13):
To a stirred solution of 9 (25 mg, 0.048 mmol) in CH2Cl2 (1.5 mL) was added TFA (0.5 mL) at 0 °C under argon atmosphere and the mixture was stirred at 23 °C for 3 h. Solvents were evaporated under reduced pressure, then CH2Cl2 (3 mL) was added and evaporated twice to give corresponding amine which was used for next step without further purification. The above amine was dissolved in acetonitrile (1 mL) and then added DIPEA (33 μL, 0.187 mmol) at 0 °C under argon atmosphere. To this reaction mixture, activated crown-THF carbonate 12 (12 mg, 0.037 mmol) was added and stirred at 23 °C for 7 days. Volatiles were evaporated under reduced pressure and the crude residue was purified by column chromatography over silica gel to provide inhibitor 13 (23 mg) as an amorphous solid (80 % yield over two steps). 1H NMR (400 MHz, Methanol-d4) δ 8.16 (d, J = 8.5 Hz, 2H), 7.91 (d, J = 8.5 Hz, 2H), 7.28 – 7.20 (m, 5H), 7.16 (m, 1H), 5.33 (d, J = 6.2 Hz, 1H), 4.69 (dd, J = 9.1, 5.6 Hz, 1H), 3.95 (d, J = 11.3 Hz, 1H), 3.78 – 3.67 (m, 2H), 3.62 – 3.53 (m, 2H), 3.43 (dd, J = 15.1, 2.5 Hz, 1H), 3.38 (dd, J = 9.3, 5.9 Hz, 1H), 3.16 (ddd, J = 13.7, 5.6, 2.4 Hz, 2H), 3.03 (dd, J = 15.0, 8.4 Hz, 1H), 2.94 (dd, J = 13.6, 6.8 Hz, 1H), 2.68 – 2.60 (m, 2H), 2.54 (dd, J = 13.9, 10.8 Hz, 1H), 2.32 (q, J = 6.3 Hz, 1H), 2.02 (ddd, J = 12.7, 7.9, 6.3 Hz, 1H), 1.76 (d, J = 12.0 Hz, 1H), 1.49 (dt, J = 12.2, 3.9 Hz, 1H), 1.36 (m, 1H), 0.92 (d, J = 6.5 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H); 13C NMR (125 MHz, Methanol-d4) δ 166.7, 156.3, 143.0, 138.7, 134.3, 129.9, 128.9, 127.8, 127.1, 125.7, 104.2, 74.6, 72.6, 67.9, 59.5, 56.8, 56.0, 52.0, 44.6, 41.9, 37.3, 35.7, 26.4, 22.7, 18.9. LRMS-ESI (m/z): 603.2 [M+H]+. HRMS-ESI (m/z): [M+H]+ calcd for C30H39N2O9S, 603.2371; found 603.2360.
(4-(methoxysulfonyl)phenyl)boronic acid (15):
To a stirred solution of 4-bromophenylsulfonyl chloride (2.55 g, 10 mmol) in methanol (20 mL) was added NaOMe (810 mg, 15 mmol) at 0 °C. The reaction was stirred at 23 °C for 12 h and TLC showed the complete consumption of the starting material. The solvent was removed under vaccum and water was added to the residue. The mixture was extracted with a mixture (15:1) of CH2Cl2 and methanol. The combined organic layer was washed with brine, dried over Na2SO4 and filtered. The solvent was removed under reduced pressure and the residue was purified by silica gel column chromatography (ethyl acetate/hexane 1:20 to 1:15) to afford the corresponding methyl ester (1.56 g, 62% yield). 1H-NMR (400 MHz, CD3OD) δ 7.81 (s, 4H), 3.76 (s, 3H). 13C-NMR (100 MHz, CD3OD) δ 134.3, 132.4, 129.3, 128.5, 56.1. MS (ESI) calcd for C7H8BrO3S [M+H]+: 250.9, Found: 250.9, 252.9.
To a stirred solution of above ester (2.50 g, 10 mmol) in THF (20 mL) under argon at −78 °C, n-BuLi (1.6 M in hexane, 6.9 mL, 11 mmol) was added. The reaction was stirred at −78 °C for 10 min and B(OMe)3 (1.68 mL, 15 mmol) was added. The reaction was continued to stir at −78 °C for additional 30 min. The reaction was quenched with aqueous NH4Cl solution. The mixture was extracted with ethyl acetate/hexance (ethyl acetate/hexane 1:3) and the organic layer was discarded. The aqueous layer was adjusted to pH 6 and the mixture was extracted with a mixture (15:1) of CH2Cl2 and methanol. The combined organic layer was washed with brine, dried over Na2SO4 and filtrated. The solvent was removed under reduced pressure to obtain the crude boronic acid derivative 15 (970 mg, 45% yield). This was used for the next step without purification. 1H-NMR (400 MHz, CD3OD) δ 7.91 (br s, 2H), 7.84 (d, J = 8.0 Hz, 2H), 3.72 (s, 3H). 13C-NMR (100 MHz, CD3OD) δ 136.1, 134.1, 126.4, 55.8. MS (ESI) calcd for C7H11BO6S [M+H2O]+: 234.0, Found: 234.0; calcd for C7H9BNaO5S [M+Na]+: 239.0, Found: 239.0; calcd for C7H10BO5S [M+H]+: 217.0, Found: 217.0.
(4-(chlorosulfonyl)phenyl)boronic acid (16):
To a stirred solution of boronic acid derivative 15 (1.08 g, 5 mmol) in methanol (10 mL) at 23 °C, was added TEA (2.8 mL, 20 mmol). The resulting reaction mixture was heated to 60 °C for 24 h. After this period, the solvent was removed completely under vaccum and the residue was used for the next step without purification.
To a stirred solution of the above residue at 0 °C, SOCl2 (10 mL) was added. The reaction mixture was warmed to 23 °C and then heated to 80 °C for 24 h. After this period, the reaction mixture was concentrated under reduced pressure and the resulting residue was carefully quenched with crushed ice. The mixture was extracted with CH2Cl2. The combined organic layer was washed with brine, dried over Na2SO4 and filtrated. The solvent was removed under reduced pressure to give the title sulfonyl chloride 16 (980 mg slightly yellow soild, 89% yield over two steps). This sulfonyl chloride is sufficiently pure and was used for the next step without purification. An analytical sample was prepared by recrystalliztion in hexane. MP: decomposed at 110 °C. 1H-NMR (400 MHz, CD3OD) δ 8.06–8.01 (m, 2H), 7.91–7.97 (m, 2H). 13C-NMR (100 MHz, CD3OD) δ 144.9, 134.5, 125.3. MS (EI) calcd for C6H6ClO2S [M-B(OH)2+2H]+: 177.0, Found: 177.0, 179.0.
(4-(N-((2R,3S)-3-((tert-butoxycarbonyl)amino)-2-hydroxy-4-phenylbutyl)-N-isobutylsulfamoyl)phenyl)boronic acid (18):
To a stirred solution of amine 17 (336 mg, 1 mmol) in CH2Cl2 (10 mL) at 0 °C, sulfonyl chloride 16 (264 mg, 1.2 mmol) followed by Et3N (0.7 mL, 5 mmol) were added. The resulting reaction mixture was stirred at 23 °C for 1 h and TLC showed the complete consumption of the secondary amine. The reaction was quenched with ice water and adjust to pH 6 with 1 N HCl. The mixture was extracted with a mixture (15:1) of CH2Cl2 and methanol. The combined organic layer was washed with brine, dried over Na2SO4, and filtrated. After the solvent was removed under reduced pressure, the residue was purified by silica gel column chromatography (CH2Cl2 and methanol 50:1 to 15:1) to afford sulfonamide 18 (425 mg, 82% yield). 1H-NMR (400 MHz, CD3OD, two rotamers) δ 7.95–7.70 (m, 4H), 7.25–7.09 (m, 5H), 3.80–3.57 (m, 2H), 3.46–3.56 (m, 1H), 3.15–2.83 (m, 4H), 2.60–2.44 (m, 1H), 2.05–1.93 (m, 1H), 1.27, 1.15 (s, 9H), 0.88 (d, 6.4 Hz, 3H), 0.84 (d, 6.4 Hz, 3H). 13C-NMR (100 MHz, CD3OD, major rotamer) δ 156.4, 140.2, 138.7, 133.9, 129.3, 129.0, 127.7, 126.0, 125.9, 125.6, 78.6, 72.7, 57.2, 55.3, 52.4, 35.8, 27.3, 26.5, 19.1. MS (ESI) calcd for C21H30BN2O7S [M-C4H7]+: 465.2, Found: 465.1; calcd for C25H38BN2O7S [M+H]+: 521.3, Found: 521.3.
(4-(N-((2R,3S)-3-(((((3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl)oxy)carbonyl)amino)-2-hydroxy-4-phenylbutyl)-N-isobutylsulfamoyl)phenyl)boronic acid (5):
To a stirred solution of Boc-derivative 18 (21 mg, 0.04 mmol) in CH2Cl2 (1.5 mL) at 0 °C, TFA (0.5 mL) was added. The resulting reaction mixture was stirred at 0 °C for 30 min. The reaction mixture was concentrated under reduced pressure and ice water (2 mL) was added to the residue. The pH was adjusted to neutral with 2.5 N NaOH. The mixture was extracted with a mixture (15:1) of CH2Cl2 and methanol. The combined organic layer was washed with brine, dried over Na2SO4 and filtrated. The solvent was removed under reduced pressure, the resulting crude amine was used for the next step without purification.
To a stirred solution of the above amine in CH3CN (1.0 mL), DIPEA (70 μL, 10 equiv) followed by mixed carbonate 19 (13 mg, 0.04 mmol) were added. The resulting reaction mixture was stirred at 23 °C for 10 days. After this period, the solvent was removed under reduced pressure and 1 N HCl was added to the residue and the pH was adjusted to about 6. The mixture was extracted with a mixture (15:1) of CH2Cl2 and methanol. The combined organic layer was washed with brine, dried over Na2SO4 and filtrated. The solvent was removed under reduced pressure and the residue was purified by silica gel column chromatography (CH2Cl2: methanol:HCO2H, 100:2:1 to 100:5:1) to afford inhibitor 5 (11 mg, 47% yield) as an amorphous solid. The proton spectrum is identical with the reported one.3 Analytical data: 1H-NMR (400 MHz, CD3OD) δ 7.87–7.77 (m, 4H), 7.26–7.16 (m, 5H), 5.59 (d, J = 5.2 Hz, 1H), 4.97–4.91 (m, 1H), 3.94 (dd, J = 9.6, 6.0 Hz, 1H), 3.83–3.64 (m, 5H), 3.45 (dd, J = 14.8, 2.4 Hz, 1H), 3.23–3.10 (m, 2H), 3.01–2.84 (m, 3H), 2.53 (dd, J = 14.0, 10.8 Hz, 1H), 2.09–1.99 (m, 1H), 1.56–1.47 (m, 1H), 1.38–1.32 (m, 1H), 0.94 (d, J = 6.4 Hz, 3H), 0.88 (d, J = 6.4 Hz, 3H). 13C-NMR (200 MHz, CD3OD) δ 157.8, 157.7, 140.3, 135.1, 130.5, 129.3, 127.4, 127.2, 110.8, 74.6, 74.5, 72.1, 70.6, 58.9, 57.4, 53.9, 46.9, 37.1, 28.0, 27.0, 20.5, 20.4. MS (ESI) calcd for C27H38BN2O9S [M+H]+: 577.2, Found: 577.3.
(4-(N-((2R,3S)-3-(((((3S,7aS,8S)-Hexahydro-4H-3,5-methanofuro[2,3-b]pyran-8-yl)oxy)carbonyl)amino)-2-hydroxy-4-phenylbutyl)-N-isobutylsulfamoyl)phenyl)boronic acid (6):
Following the procedure outlined for inhibitor 5, reaction of amine derived from Boc-derivative 18 (20 mg, 0.04 mmol) and activated crown-THF carbonate 12 (10 mg, 0.031 mmol) afforded inhibitor 6 (12.5 mg, 55% for 2-steps) as an amorphous solid. 1H-NMR (400 MHz, Methanol-d4) δ 7.79 (s, 5H), 7.30 – 7.21 (m, 4H), 7.19 – 7.13 (m, 1H), 5.34 (d, J = 6.2 Hz, 1H), 4.71 – 4.64 (m, 1H), 3.95 (d, J = 11.2 Hz, 1H), 3.81 – 3.67 (m, 2H), 3.61 – 3.54 (m, 2H), 3.47 – 3.35 (m, 2H), 3.26 – 3.20 (m, 1H), 3.17 (dd, J = 14.0, 3.4 Hz, 1H), 3.10 (dd, J = 13.6, 8.1 Hz, 1H), 3.02 – 2.80 (m, 2H), 2.73 – 2.60 (m, 2H), 2.55 (dd, J = 13.8, 11.0 Hz, 1H), 2.32 (m, 1H), 2.06 – 1.96 (m, 1H), 1.76 (d, J = 12.0 Hz, 1H), 1.48 (dt, J = 12.1, 3.9 Hz, 1H), 0.91 (d, J = 6.6 Hz, 3H), 0.86 (d, J = 6.9 Hz, 3H); 13C-NMR (200 MHz, Methanol-d4) δ 156.4, 138.8, 133.7, 129.1, 128.8, 127.9, 126.0, 125.8, 104.3, 74.7, 73.0, 72.1, 68.0, 59.7, 57.4, 56.1, 56.0, 55.0, 53.4, 52.5, 48.1, 44.7, 42.0, 37.4, 35.8, 26.6, 22.8, 19.1, 19.0. LRMS-ESI (m/z): 603.2 [M+H]+. HRMS-ESI (m/z): [M+Na]+ calcd for C29H39BN2O9SNa, 624.2398; found 624.2393.
(4-(N-((2R,3S)-3-(((((3aS,4S,7aR)-hexahydro-4H-furo[2,3-b]pyran-4-yl)oxy)carbonyl)amino)-2-hydroxy-4-phenylbutyl)-N-isobutylsulfamoyl)phenyl)boronic acid (20):
Following the procedure outlined for inhibitor 5, reaction of amine salt derived from Boc-derivative 18 (21 mg, 0.04 mmol) and activated carbonate 10 (13.6 mg, 0.04 mmol) afforded inhibitor 20 (12 mg, 51%) as an amorphous solid. 1H-NMR (400 MHz, CD3OD, two isomers) δ 7.79 (br s, 4H), 7.31–7.09 (m, 5H), 4.92–4.86 (m, 2H), 4.05 (t, J = 7.6 Hz, 1H), 3.84–3.70 (m, 4H), 3.46–3.32 (m, 2H), 3.21–3.06 (m, 2H), 3.02–2.84 (m, 2H), 2.57–2.49 (m, 1H), 2.35–2.25 (m, 1H), 2.07–1.95 (m, 1H), 1.92–1.79 (m, 1H), 1.78–1.60 (m, 2H), 1.46–1.36 (m, 1H), 0.91 (d, 6.4 Hz, 3H), 0.86 (d, 6.4 Hz, 3H). 13C-NMR (100 MHz, CD3OD, major isomer) δ 156.2, 139.7, 138.8, 133.6, 129.2, 129.0, 127.7, 125.9, 125.6, 100.9, 72.8, 69.4, 67.8, 60.2, 57.3, 55.7, 55.6, 52.4, 43.3, 35.7, 26.5, 25.8, 21.7, 18.9. HRMS (ESI) calcd for C28H39BN2O9SNa [M+Na]+: 612.2398, Found: 612.2391.
Determination of X-ray structures of HIV-1 protease-inhibitor complexes for inhibitors 6 and 20:
HIV-1 PR optimized for bacterial expression was purified as described.40 PR at 4.2 mg/mL was complexed with inhibitor at 5:1 molar ratio. Crystals of protease-inhibitor complexes were grown by hanging drop vapor diffusion with well solutions of 1.4 M sodium chloride, 0.1 M sodium acetate buffer pH 6.0 and 1.3 M sodium chloride, 0.1 M sodium acetate pH 6.2 reservoir solution for inhibitors 6 and 20, respectively. Streak-seeding was employed to induce crystallization for inhibitor 6. Crystals were cryo-cooled to 90 K and 1.0 Å wavelength X-ray diffraction data was collected on beamline 22-ID at SER-CAT, Advanced Photon Source, Argonne National Lab (Chicago, IL, USA). Data were processed and scaled using HKL2000.41 The structures were solved by molecular replacement in PHASER42 in CCP4i suite,43,44 using PR-APV (PDB: 3NU3)45 as an isomorphous model and refined with COOT,46,47 and Refmac5 in CCP4i, using anisotropic displacement parameters (B-factors).48 JLignad49 in CCP4i, was used to generate refinement restraints for inhibitors. Crystallographic statistics are supplied in Table 1. Coordinates and structure factors for inhibitors 6 (GRL-008–19A) and 20 (GRL-031–19A) were deposited in the Protein Data Bank,50 with accession codes codes 6U7O and 6U7P.
Supplementary Material
Acknowledgements
The present research was supported by grants from the National Institutes of Health AI150466 (AKG) and AI150461 (ITW). DWK is supported by Georgia State University Molecular Basis of Disease Fellowship. This work was also supported in part by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health (HM: R01AI121315); a Grant-in-Aid for Scientific Research (Priority Areas) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Monbu-Kagakusho), a Grant for Promotion of AIDS Research from the Ministry of Health, Welfare, and Labor of Japan; grants from Japan Agency for Medical Research and Development (AMED)(HM: JP15fk0410001 and JP18fk0410001); and grants from National Center for Global Health and Medicine Research Institute (HM). We thank the staff at the Southeast Regional-Collaborative Access Team (SER-CAT) at the Advanced Photon Source, Argonne National Laboratory, for assistance during X-ray data collection. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31–109-Eng-38. We also thank the Purdue University Center for Cancer Research, which supports the shared NMR and mass spectrometry facilities.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References:
- [1].Ghosh AK, Gemma S. Structure-based Design of Drugs and Other Bioactive Molecules: Tools and Strategies; Wiley-VCH; 2014: 337–354. [Google Scholar]
- [2].Hubbard RE, Structure-based Drug Discovery: An Overview RSC Publishing, 2006. [Google Scholar]
- [3].Ghosh AK, Osswald HL, Prato G, J. Med. Chem 2016, 59, 5172–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wlodawer A, Vondrasek J, Annu. Rev. Biophys. Biomol. Struct 1998, 27, 249–284. [DOI] [PubMed] [Google Scholar]
- [5].Broder S, Antiviral Res. 2010, 85, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Edmonds A, Yotebieng M, Lusiama J, Matumona Y, Kitetele F, Napravnik S, Cole SR, Van Rie A, Behets F, PLoS Med. 2011, 8, e1001044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Peng C, Ho BK, Chang TW, Chang NT. J. Virol 1989, 63, 2550–2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kohl NE, Emini EA, Schleif WA, Davis LJ, Heimbach JC, Dixon RA, Scolnick EM, Sigal IS. Proc. Natl. Acad. Sci. USA 1988, 85, 4686–4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ghosh AK, Dawson ZL, Mitsuya H, Bioorg. Med. Chem 2007, 15, 7576–7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].de Bethune MP, Sekar V, Spinosa-Guzman S, Vanstockem M, De Meyer S, Wigerinck P, Lefebvre E, Darunavir (Prezista, TMC114): from Bench to Clinic, Improving Treatment Options for HIV-infected Patients in Antiviral Drugs: from Basic Discovery through Clinical Trials, John Wiley & Sons, Inc., New Jersey, 2011, pp. 31e45. [Google Scholar]
- [11].Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents https://aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdf, accessed on March 31, 2018.
- [12].Hué S, Gifford RJ, Dunn D, Fernhill E, Pillay D, J. Virol 2009, 83, 2645–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hayashi H, Takamune N, Nirasawa T, Aoki M, Morishita Y, Das D, Koh Y, Ghosh AK, Misumi S, Mitsuya H, Proc. Natl. Acad. Sci 2014, 111, 12234–12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ghosh AK, Anderson DD, Weber IT, Mitsuya H, Angew. Chem. Int. Ed 2012, 51, 1778–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ghosh AK, Chapsal B, Weber IT, Mitsuya H, Acc. Chem. Res 2008, 41, 78–86. [DOI] [PubMed] [Google Scholar]
- [16].Tie Y, Boross PI, Wang Y-F, Gaddis L, Hussian AK, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT, J. Mol. Biol 2004, 338, 341–352. [DOI] [PubMed] [Google Scholar]
- [17].Kovalevsky AY, Liu F, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT, J. Mol. Biol 2006, 363, 161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang Y-F, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H, Antimicrob. Agents Chemother 2003, 47, 3123–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].De Meyer S, Azijn H, Surleraux D, Jochmans D, Tahri A, Pauwels R, Wigerinck P, de Bethune MP, Antimicrob. Agents Chemother 2005, 49, 2314–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sterrantino G, Zaccarelli M, Colao G, Baldanti F, Di Giambenedetto S, Carli T, Maggiolo F, Zazzi M, Infection 2012, 40, 311–318. [DOI] [PubMed] [Google Scholar]
- [21].De Meyer S, Lathouwers E, Dierynck I, De Paepe E, Van Baelen B, Vangeneugden T, Spinosa-Guzman S, Lefebvre E, Picchio G, de Béthune M-P, AIDS, 2009, 23, 1829–1840. [DOI] [PubMed] [Google Scholar]
- [22].Yedidi RS, Maeda K, Fyvie WS, Steffey M, Davis DA, Palmer I, Aoki M, Kaufman JD, Stahl SJ, Garimella H, Das D, Wingfield PT, Ghosh AK, Mitsuya H, Antimicrob. Agents and Chemother 2013, 57, 4920–4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhou S-F, Xenobiotica 2008, 38, 802–832. [DOI] [PubMed] [Google Scholar]
- [24].Hoggard PG, Owen A, Antimicrob. Chemother 2003, 51, 493–496. [DOI] [PubMed] [Google Scholar]
- [25].Fujimoto H, Higuchi M, Watanabe H, Koh Y, Ghosh AK, Mitsuya H, Tanoue N, Hamada A, Saito H. Biol. Pharm. Bull 2009, 32, 1588–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Windsor IW, Palte MJ, Lukesh III JC, Gold B, Forest KT, Raines RT, J. Am. Chem. Soc 2018, 140, 14015–14018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ghosh AK, Sridhar PR, Leshchenko S, Hussain AK, Li J, Kovalevsky AY, Walters DE, Wedekind J, Grum-Tokars V, Das D, Koh Y, Maeda K, Gatanaga H, Weber IT, Mitsuya H, J. Med. Chem 2006, 49, 5252–5261. [DOI] [PubMed] [Google Scholar]
- [28].Bal BS, Childers WE, Pinnick HW, Tetrahedron 1981, 37, 2091–2096. [Google Scholar]
- [29].Ghosh AK, Chapsal BD, Parham GL, Steffey M, Agniswamy J, Wang Y-F, Amano M, Weber IT, Mitsuya H J. Med. Chem 2011, 54, 5890–5901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ghosh AK, Rao KV, Nyalapatla PR, Osswald HL, Martyr CD, Aoki M, Hayashi H, Agniswamy J, Wang YF, Bulut H, Das D, Weber IT, Mitsuya H, J. Med. Chem 2017, 60, 4267–4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kunst E, Gallier F, Dujardin G, Yusubov MS, Kirschning A Org. Lett 2007, 9, 5199–5202. [DOI] [PubMed] [Google Scholar]
- [32].Sulfonyl chloride 16 is quite stable. The preliminary X-ray structure shows mixed conformational isomers. Further refinement is in progress.
- [33].Ghosh AK, Fyvie WS, Brindisi M, Steffey M, Agniswamy J, Wang Y-F, Aoki M, Amano M, Weber IT, Mitsuya H. ChemMedChem 2017, 12, 1942–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Toth MV, Marshall GR, Int. J. Pept. Protein Res 1990, 36, 544–550. [DOI] [PubMed] [Google Scholar]
- [35].Koh Y, Amano M, Towata T, Danish M, Leshchenko-Yashchuk S, Das D, Nakayama M, Tojo Y, Ghosh AK, Mitsuya H, J. Virol 2010, 84, 11961–11969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].De Meyer S, Lathouwers E, Dierynck I, De Paepe E, Van Baelen B, Vangeneugden T, Spinosa-Guzman S, Lefebvre E, Picchio G, de Béthune MP, AIDS 2009, 23, 1829–1840. [DOI] [PubMed] [Google Scholar]
- [37].Smart BE, J. Fluor. Chem 2001, 109, 3–11. [Google Scholar]
- [38].Aoki M, Hayashi H, Rao KV, Das D, Higashi-Kuwata N, Bulut H, Aoki-Ogata H, Takamatsu Y, Yedidi RS, Davis DA, Hattori S.-i., Nishida N, Hasegawa K, Takamune N, Nyalapatla PR, Osswald HL, Jono H, Saito H, Yarchoan R, Misumi S, Ghosh AK, Mitsuya H, eLife 2017, 6, e28020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ghosh AK, Rao KV, Nyalapatla PR, Kovela S, Brindisi M, Osswald HL, Sekhara Reddy B, Agniswamy J, Wang YF, Aoki M, Hattori SI, Weber IT and Mitsuya H, ChemMedChem 2018, 13, 803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sayer JM, Agniswamy J, Weber IT, Louis JM Protein Sci. 2010, 19, 2055–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Otwinowski Z, Minor W, Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods in Enzymology, 276: Macromolecular Crystallography, Part A; Carter CW Jr., Sweet RM, Eds.; Academic Press: New York, 1997; pp 307–326. [DOI] [PubMed] [Google Scholar]
- [42].McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ, J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS, Acta Crystallogr., Sect. D: Biol. Crystallogr 2011, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Potterton E, Briggs P, Turkenburg M, Dodson E, Acta Crystallogr., Sect. D: Biol. Crystallogr 2003, 59, 1131–1137. [DOI] [PubMed] [Google Scholar]
- [45].Shen C-H, Wang Y-F, Kovalevsky AY, Harrison RW, Weber IT, FEBS J. 2010, 277, 3699–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Emsley P, Lohkamp B, Scott WG, Cowtan K, Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Emsley P, Cowtan K , Acta Crystallogr., Sect. D: Biol. Crystallogr 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- [48].Murshudov GN, Vagin AA, Dodson Acta EJ. Crystallogr. D Biol. Crystallogr 1997, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- [49].Lebedev AA, Young P, Isupov MN, Moroz OV, Vagin AA, Murshudov Acta GN. Crystallogr. D. Biol. Crystallogr 2012, 68, 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE, Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.