

SUMMARY
More than 8,000 genes are turned on or off as progenitor cells produce the seven classes of retinal cell types during development. Thousands of enhancers are also active in the developing retinae, many having features of cell– and developmental stage–specific activity. We studied dynamic changes in the 3D chromatin landscape important for precisely orchestrated changes in gene expression during retinal development by ultra-deep in situ Hi-C analysis on murine retinae. We identified developmental stage–specific changes in chromatin compartments and enhancer–promoter interactions. We developed a machine learning–based algorithm to map euchromatin/heterochromatin domains genome-wide and overlaid it with chromatin compartments identified by Hi-C. Single-cell ATAC-seq and RNA-seq were integrated with our Hi-C and previous ChIP-seq data to identify cell– and developmental stage–specific super-enhancers (SEs). We identified a bipolar neuron–specific core regulatory circuit SE upstream of Vsx2, whose deletion in mice led to loss of bipolar neurons.
Graphical Abstract
eTOC
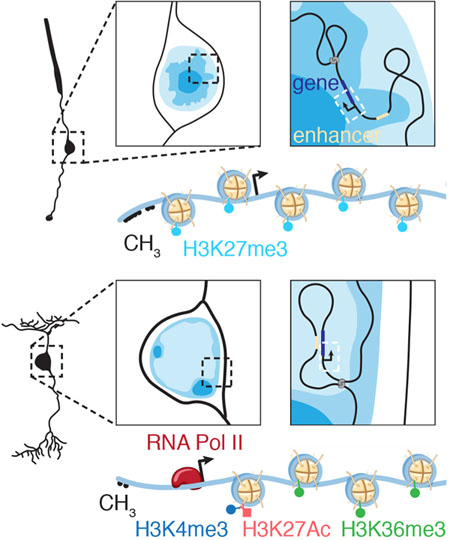
Norrie, Lupo et al. show in this article how the chromatin landscape in the mouse retina changes during development in coordination with transcriptional programs. They use those data to identify enhancers that are cell– and developmental–stage specific.
INTRODUCTION
We previously analyzed DNA methylation [whole-genome bisulfite sequencing (WGBS)], histone modifications [chromatin immunoprecipitation with DNA sequencing (ChIP-seq) for 8 marks], chromatin accessibility [assay for transposase-accessible chromatin using sequencing (ATAC-seq)], and transcriptional regulation through enhancer interactions (Brd4, CTCF, RNA-PolII, and ChIP-seq) in the developing murine retina (Aldiri et al., 2017). By integrating those data with RNA-seq data, we identified thousands of putative developmental stage–specific enhancers (Aldiri et al., 2017). The most striking developmental transitions involve large clusters of enhancers called super-enhancers (SEs) (Aldiri et al., 2017; Hnisz et al., 2013). Indeed, more than a third of 1,768 SEs in the murine retina were developmental stage-specific and 62% were conserved in developing human retinae (Aldiri et al., 2017). Putative target genes for SEs were identified by correlating developmental stage-specific super-enhancer activity with expression of nearby genes (Aldiri et al., 2017). Many of these SEs are located far (10–200 kb) from their putative developmentally regulated promoters and would require looping of the intervening chromatin to regulate gene expression.
Previous chromatin conformation capture (Hi-C) studies on developing human and mouse brain, human hematopoietic cell types, and differentiated human embryonic stem cells (ESCs) show that gene activation by distal enhancers is accompanied by changes in the 3D chromatin landscape (Benavente et al., 2013; Bonev et al., 2017; Freire-Pritchett et al., 2017; Javierre et al., 2016; Mumbach et al., 2017; Ong and Corces, 2011; Schoenfelder et al., 2010; Smallwood and Ren, 2013; Won et al.). Specifically, loops are formed that bring promoters and enhancers in close proximity (Rao et al., 2014). In addition to identifying chromatin loops, Hi-C can be used to delineate chromatin domain structure and compartmentalization (Lieberman-Aiden et al., 2009; Rao et al., 2014). Densely packed compartments (compartment B) have more interactions in Hi-C datasets than do more diffuse compartments (compartment A). Moreover, genomic regions in compartment A tend to be gene rich, have higher mRNA expression, and are enriched for histone modifications associated with active transcription (Lieberman-Aiden et al., 2009). The A/B compartments can be cell-type specific and undergo changes during development (Dixon et al., 2015; Fortin and Hansen, 2015; Lieberman-Aiden et al., 2009; Rao et al., 2014).
Chromatin compartmentalization and nuclear localization is particularly important in the murine retina. Most nuclei have diffuse, transcriptionally active, and gene-rich euchromatin regions (similar to compartment A) in their centers and more condensed heterochromatin (similar to compartment B) tethered to the nuclear lamina. However, rod photoreceptors of nocturnal species such as mice have an inverted nuclear organization, with centrally located heterochromatin and euchromatin adjacent to the nuclear lamina (Solovei et al., 2009). This pattern of nuclear chromatin organization optimizes vision in low light by reducing light scattering as it passes through the outer nuclear layer (Solovei et al., 2009). The difference in refractive indices of euchromatin and heterochromatin, combined with the inverted pattern, creates a series of aligned converging lenses in the outer nuclear layer of nocturnal species. Therefore, in addition to the importance of nuclear organization in gene regulation, this reflects the substantial influence of nuclear organization on cell and tissue adaptation and function.
Here we extend our previous analysis of chromatin accessibility and histone and DNA modifications during retinal development to include ultra-deep in situ Hi-C analysis of the developing mouse retina and purified rod photoreceptors. These data were used to analyze promoter–enhancer interactions, chromatin domain structure, and compartmentalization. To compare A/B compartments to nuclear euchromatin or heterochromatin domains in rod photoreceptors, we performed fluorescence in situ hybridization (FISH) combined with immunofluorescence (IF) of H3K4me3 for 264 genomic loci. Those data, combined with our previous epigenetic data, were used to train and validate a machine learning–based algorithm to predict euchromatin/heterochromatin nuclear localization in murine rod photoreceptors. Most genomic regions in compartment A and compartment B are predicted to be euchromatic and heterochromatic, respectively. All data have been integrated into a freely available cloud-based viewer. Genomic regions transition between compartments A/B and euchromatin/heterochromatin during retinal development. We combined single-cell ATAC-seq and single-cell RNA-seq with Hi-C data and our previous epigenetic data to identify cell-type– and developmental stage–specific SEs in the murine retina. We validated a bipolar neuron–specific super-enhancer for Vsx2 by deleting the SE in mice and showing that bipolar neurons are no longer formed. Taken together, these data demonstrate the importance of performing integrated analysis of the structure and organization of chromatin to identify cell-type– and developmental stage–specific regulatory elements during neurogenesis.
RESULTS
Mapping Chromatin Domains During Retinogenesis
To elucidate chromatin domains, compartmentalization, and promoter–enhancer interactions during retinal development, we performed ultra-deep in situ Hi-C on replicate embryonic day (E) 14.5, postnatal day (P) 0, and adult murine retinal samples (Rao et al., 2014). We also analyzed green fluorescent protein–positive (GFP+; rod photoreceptors) and GFP– cells (cone, bipolar, horizontal, and ganglion cells, and Müller glia) from Nrl-GFP mice (Akimoto et al., 2006) (Figures S1A and S1B). In total, more than 62 billion read pairs were sequenced and compared to 1.7 billion read pairs of Hi-C data published previously on the mouse cortex, fibroblasts, and murine ESCs (Table S1). Previous murine datasets contained 225–348 million pairwise contacts and had a map resolution of ~5.5 kb. Our retinal dataset contains 4.9–8.6 billion pairwise contacts and has a map resolution of 350–600 bp (Table S1). Data were analyzed using Juicer (Durand et al., 2016) and can be visualized on our cloud-based viewer (https://pecan.stjude.cloud/proteinpaint/study/retina_hic_2018).
To evaluate the quality and reproducibility of our retinal Hi-C data, we used HiC-Spector (Yardimci et al., 2019). Quality control measures and reproducibility of our data were similar to those of previously published datasets using these methods (Table S1; Figures S1C–S1G). As expected, most contacts were within 1 Mb (Figure S1D) and E14.5 and P0 samples were more similar to each other than to adult retina samples (Figures S1E–S1G) (Bonev et al., 2017; Crane et al., 2015; Rao et al., 2014).
Next, we identified topologically associating domains (TADs) and compare our data with previously published Hi-C data for the mouse cortex (Dixon et al., 2012) (Table S1). At E14.5, 2434 Mb of the genome was assigned to TADs, which was similar to that in P0 (2381 Mb), adult mouse retina (2216 Mb), and murine cortex (2285 Mb). The number of TADs was similar for E14.5 (3690), P0 (3912), and cortex (3756), but there was a slight increase in the number of TADs in the adult retina (5290) due to the highly condensed chromatin in rod nuclei (Table S1).
Although the TADs are largely conserved across cell types, the deeper coverage of our dataset allowed us to assign TAD boundaries and identify chromatin contacts in regions of the genome with lower coverage in the previous murine cortex dataset. For example, we identified a region on murine chromosome 4 containing several developmentally regulated SEs and genes implicated in retinal development and disease that were not assigned to a TAD in the previous cortex dataset (Figures 1A–1D) (Christiansen et al., 2016; Jordan et al., 2015; Perkowski and Murphy, 2011). Although most contacts were within 1 Mb (Figure S1D), we also identified some longer-range interactions. For example, our Hi-C data showed that a genomic region more than 8 Mb away and spanning multiple TADs was predicted to be in close proximity with the rhodopsin gene Rho in rod photoreceptors, which was confirmed by 2-color DNA FISH (Figures S2A and S2B).
Figure 1. Hi-C Analysis of Developing Murine Retinae.

A) Interaction map of chromosome 4 for mouse cortex from previously published data and E14.5, P0, and adult retinae in this study. Chromosome distance is given in megabases (0–156 Mb). B) Magnified view of the shaded region on chromosome 4 from (A) spanning 149–154 Mb. TAD boundaries assigned in the mouse cortex dataset and developmental SEs in the retina are shown as gray and blue bars, respectively. The highlighted region (yellow) was not assigned to a TAD in the cortex but contains several developmentally regulated genes in the murine retina. ChIP-seq for H3K36me3 and H3K27me3 as well as ATAC-seq and whole-genome bisulfite sequencing (WGBS) are shown below each stage of retinal development. C) ChromHMM and expression of Icmt throughout retinal development. D) ChIP-seq, ATAC-seq, and WGBS for Icmt in the adult retina (P21). Scales are indicated on the left of each track. Abbreviations: Chr, chromosome; TAD, topologically associating domain; SE, super-enhancer; FPKM, fragments per kilobase of million reads.
Dynamic Chromatin Compartments During Retinal Development
Previous studies show that the genome can be partitioned into A/B compartments based on Hi-C contact patterns that correlate with histone modifications and transcription (Dixon et al., 2015; Fortin and Hansen, 2015; Lieberman-Aiden et al., 2009; Rao et al., 2014). We identified A/B compartments across our Hi-C datasets and overlaid our ChIP-seq data. At E14.5, the promoter/enhancer mark H3K4me1 had the highest overlap with compartment A (79.6%) and the repressed chromatin mark H3K9me3 had the highest overlap with compartment B (Table S2). A similar trend was observed in the adult retina (Table S2). We also analyzed the overlap of chromHMM states with A/B compartments; the empty state (state 9) was excluded from this analysis. In E14.5 and adult retinae, chromHMM states associated with active transcription (states 2, 5, and 7) had the highest overlap with compartment A (Table S2) and states associated with polycomb repression and heterochromatin (states 4, 8, and 10) had the highest overlap with compartment B (Table S2). Interestingly, the proportion of all active states associated with promoters, gene bodies, and enhancers in compartment A from E14.5 to adult retinae increased (Table S2). In parallel, the proportion of polycomb repressed states in compartment B from E14.5 to adult retinae also increased. Consistent with previous studies, our data suggest that transitions in compartmental localization (A->B and B->A) during development may be associated with changes in gene expression (Lieberman-Aiden et al., 2009; Rao et al., 2014). Of 566 genes with changes in gene expression and compartment localization during development, 51% (290/566) were upregulated genes that transitioned from compartment B to A and were enriched in pathways involved in nervous system development (Table S2). Genes downregulated during development transitioned from compartment A to B and were enriched in pathways important for chromosome segregation during mitosis (Table S2).
Mapping Euchromatin and Heterochromatin Domains in Rod Photoreceptors
By gene density, gene expression, histone modifications, and chromHMM states, compartment A resembles euchromatin and compartment B resembles heterochromatin. Some loci that transition between A/B compartments may have differential localization to facultative heterochromatin during development (Allshire and Madhani, 2018). An individual genomic locus can be localized to a euchromatin or heterochromatin domain by DNA FISH, but no method can assign subnuclear domain localization in individual cell types using molecular data such as WGBS, ChIP-seq, chromHMM, or Hi-C.
To develop a genome-wide map of euchromatin/heterochromatin localization, we took advantage of the unique inverted organization of murine rod photoreceptor nuclei. These nuclei have a dense central core of constitutive heterochromatin containing telomeres and centromeres, surrounded by a more diffuse zone of facultative heterochromatin and then a region of euchromatin adjacent to the nuclear envelope (Figures 2A–2C) (Solovei et al., 2009; Wang et al., 2018). To study the subnuclear localization of developmentally regulated genes and enhancers in rod photoreceptors, we performed DNA FISH and immunofluorescence (IF) analysis to detect H3K4me3 for marking the euchromatin domain (Figures 2D and 2E). We selected constitutively expressed genes, retinal cell type–specific genes, retinal progenitor genes, silent genes, and repressed genes for our initial FISH/IF analysis (Table S3). Localization of individual loci to nuclear domains was analyzed by 3D confocal imaging, and an automated image segmentation and scoring algorithm was developed for retinal FISH/IF (Wang et al., 2018) (Table S3; Supplemental Information). Genes expressed in rods (e.g., Pde6a, Rho, Crx, Nrl, and Nr2e3) colocalized with H3K4me3 in the euchromatin domain (Figure 2D; Table S3). Our initial FISH/IF analysis identified several retinal progenitor genes (e.g., Ascl1, Hells, Sfrp1, NeuroD4, and Sox11) in the facultative heterochromatin domain of rods and also a subset of other repressed genes (Figures 2E–2I; Table S3). To validate the localization, we measured distance from two genes (Gria3 and Grik1) to the nuclear periphery and each chromatin domain border (Table S3).
Figure 2. Mapping of Euchromatin and Heterochromatin in Rod P-hotoreceptors.

A) Electron micrograph of a murine rod photoceptor nucleus. B) Confocal micrograph of a DAPI-stained rod photoreceptor nucleus. C) Confocal micrograph of a rod photoreceptor nucleus immunostained for H3K4me3 (green) and counterstained with DAPI (blue). D,E) FISH/IF for Pde6a and Hells (red) with H3K4me3 (green) and counterstained with DAPI (blue). F) ChromHMM of the genomic region spanning Ascl1 (gray). Euchromatin (green) and heterochromatin (purple) predictions shown below the ChromHMM. G) FISH for Ascl1 (green) counterstained with DAPI (blue). H,I) ChIP-seq and WGBS tracks for Ascl1 in E14.5 and adult retinae. J) ChromHMM, Hi-C, and euchromatin/heterochromatin predictions for the genomic region spanning Car10 and Sp2 (gray), which are predicted to reside in the same TAD defined from previously published cortex Hi-C. Expression of Car10 is shown at the right of the ChromHMM. K) Two-color FISH for the Car10 (green) and Sp2 (red). Gray bars in (J) indicate coordinates of FISH probes. L) Stacked bar plot of the quantification of Car10 and Sp2 localized to the rod photoreceptor domains. Numbers of nuclei scored are indicated below each bar. Scale bars: 1 µm. Abbreviations: c-het, constitutive heterochromatin; Chr, chromosome; EM, electron micrograph; ESC, embryonic stem cells; euc, euchromatin; f-het, facultative heterochromatin; FPKM, fragments per kilobase per million reads; SE, superenhancer; TAD, topologically associated domain.
To extend these observations to the entire genome, we developed a machine learning–based algorithm to predict euchromatin/heterochromatin localization (Supplemental Information). The model was trained using FISH/IF data from 103 loci (Table S3), and genomic features were extracted from chromHMM data (Aldiri et al., 2017). Initially, the modeling was constrained to TAD boundaries from the mouse cortex (Dixon et al., 2012). This robustly predicted the TAD-based euchromatin/heterochromatin status with 89% accuracy (10-fold cross validation, Supplemental Information). Inclusion of additional genomic (LINE/SINE repeats), epigenomic (WGBS, ATAC-seq), or transcriptome (RNA-seq) data did not significantly improve performance (89% accuracy in 10-fold cross-validation; Supplemental Information). Most TADs were either euchromatin or heterochromatin, but some had a mixture of both. Therefore, we removed the TAD boundary constraint (Supplemental Information). To independently validate the euchromatin/heterochromatin predictive model, we performed FISH/ IF for an additional 161 loci and achieved 89% accuracy (Table S3). The predictive model has been extended to E14.5 retinae and added to the cloud-based viewer (Figure 2F). This model revealed that all rod and housekeeping genes were predicted to be localized to the euchromatin domain in adult retina. However, 14% (32/246) of retinal progenitor genes and 53% (307/692) of non-rod cell type–specific genes (e.g., Grik2, Gria3, Ctn4, Epha5, Cav1) were predicted to be localized to the heterochromatin domain of rods. Similarly, 21% (88/410) of cancer genes (e.g., Myc, Myb, Foxp1, Kit, Cdkn2a, Cdk6) and 13% (37/277) of stress response genes (e.g., Hspa13, Dnajc5b, Nell, Eno1b) were predicted to be localized to the heterochromatin domain of rods. Overall, there was a general agreement between our euchromatin/heterochromatin modeling and A/B compartment identification by Hi-C (Figure 3A–D). In E14.5 and adult retinae, 82% and 85% of euchromatin, respectively, was assigned to compartment A. In E14.5 and adult retinae, 95% and 96% of heterochromatin, respectively, was assigned to compartment B.
Figure 3. Comparison of compartment A/B to euchromatin and heterochromatin domains.

A,B) Stack bar plot of proportion of genes that are up or downregulated during retinal development that are in compartment A (red) or B (blue). C,D) Violin plot of genes in compartment A or B relative to euchromatin score. A score of 0 is neutral with positive numbers indicating euchromatin and negative numbers indicating heterochromatin. E,F) Hi-C contact frequency plot for the same region of chromosome 3 (57.2–63.5 Mb) in E14.5 and adult retina showing transitions across compartments (A/B) and chromatin states (euchromatin/heterochromatin). Plots of ChIP-seq, ATAC-seq and whole genome bisulfite sequencing are shown below the plot with the gene map indicated. The ChromHMM for one gene (Mbnl1) is shown at the bottom with developmental expression in FPKM.
Examples exist of developmental transitions between euchromatin and heterochromatin that overlap with A/B compartment transitions (Figure 3E,F; Table S3) as well as cell type–specific localization of genes to heterochromatin (Figure 2J; Table S4). For example, Car10 is expressed from the euchromatin domain in bipolar neurons (Kim et al., 2008b) but is sequestered in the facultative heterochromatin domain of rods (Tables S3 and S4; Figure 2J). Importantly, although Car10 and Sp2 are in the same TAD, they were predicted to be in different compartments (Figure 2J). Car10 was predicted to be in compartment B and in facultative heterochromatin, whereas Sp2 was predicted to be in compartment A and in euchromatin in rods (Figure 2J). FISH/IF confirmed the difference in subnuclear localization of Car10 and Sp2, showing that a single TAD can span nuclear domains in rod photoreceptors (Figures 2K and 2L). In total, 6.2% (245/3918) of genes downregulated during retinal development were sequestered to heterochromatin in the adult rods, and 9.3% (4/43) of G2/M-phase cell cycle genes and 4.6% (11/235) of retinal progenitor cell genes showed the same pattern (Tables S3 and S4). Of genes upregulated during retinal development, 7.6% (331/4313) were predicted to be in heterochromatin in E14.5 retinal progenitor cells; 7.3% (15/190) of rod genes showed a similar pattern. Strikingly, 15.4% (82/533) of genes upregulated during retinal differentiation and having corresponding changes in chromHMM initiated in the heterochromatin domain in E14.5 retinal progenitor cells and transitioned to euchromatin in adult retina (Tables S3 and S4; Figure 3).
Inversion of Nuclear Organization
Our integrated dataset showed that some genomic loci undergo developmental transitions in their histone and DNA modifications, DNA looping, and subnuclear localization. However, many regions are more stable throughout development. To determine if the physical association of heterochromatin with the nuclear lamina is essential for proper gene regulation in the retina, we performed RNA-seq analysis on Lamin B receptor (Lbr)–mutant mouse retinae. In the developing mouse retina, the Lbr protein helps tether heterochromatin to the nuclear periphery in newly postmitotic cells (Solovei et al., 2013). A previously described 2-bp insertion in Lbr (Lbric-J) in homozygous-mutant mice results in severe developmental defects and death around P8–P12 (Shultz et al., 2003). Heterozygous mutations in human LBR are associated with the Pelger–Huët anomaly, characterized by defects in nuclear morphology and chromatin distribution (Hoffmann et al., 2002). Occasionally, some Lbric-J/ic-J mice survive to adulthood, but their retinae have not been analyzed (Solovei et al., 2013).
We harvested retinae from wild-type (WT), Lbric-J-heterozygous, and Lbric-J-knockout mice at P12 and maintained retinal explants in culture for 2 weeks to complete retinal differentiation. We also analyzed retinae obtained from one litter of mice surviving to P21 in our analysis. There was significant increase in the amount of heterochromatin not tethered to the nuclear lamina in the inner nuclear layer (INL) of Lbric-J/ic-J mice (Figures 4A,B). Areas of the nucleus and individual domains of heterochromatin that remained tethered to the nuclear lamina in INL cells of Lbric-J/ic-J mice did not significantly differ from those in WT littermates (Figures 4C,D). However, the area of individual untethered heterochromatin domains was significantly increased in Lbric-J/ic-J mice (Figure 3E; p=0.042). As expected, the organization of euchromatin and heterochromatin in rods was unaffected (Figure 4A) but 3D electron microscopy confirmed that heterochromatin was untethered in INL cells of Lbric-J/ic-J mice (Figure 4F). RNA-seq analysis of P21 Lbric-J/ic-J, Lbr+/ic-J, and Lb+/+ littermates demonstrated that very few genes were deregulated in the absence of Lbr (Table S5).
Figure 4. Untethered Heterochromatin in Lbr-Deficient Retinae.
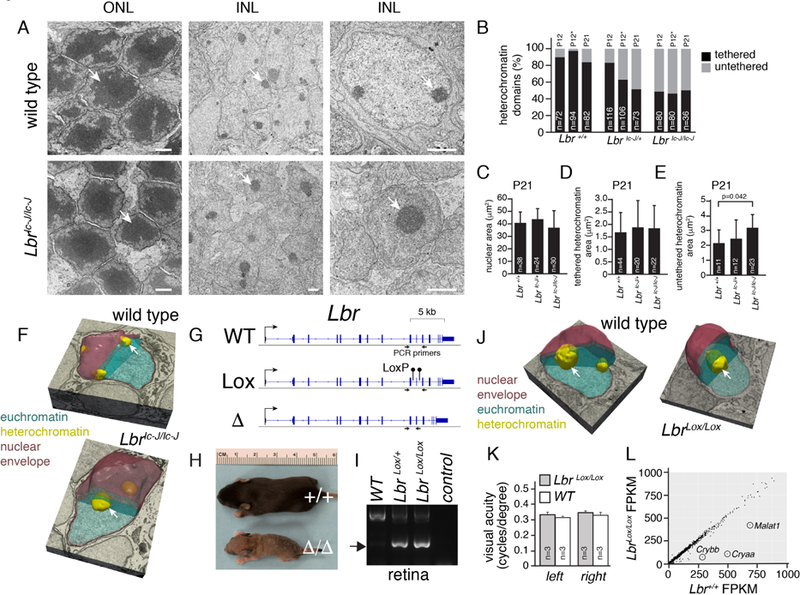
A) Electron micrographs of WT and Lbric-J/ic-J mouse retinae. Arrows indicate the heterochromatin domain in rods found in the outer nuclear layer (ONL) and neurons and glia found in the inner nuclear layer (INL). B) Stacked bar plot of the percentage of heterochromatin domains tethered or untethered to the nuclear lamina. The number of domains scored is indicated on each bar. The (*) indicates scoring on samples harvested at P12 and maintained in culture for 9 days. C-E) Bar plots of nuclear area (C), area of tethered heterochromatin domains (D), and untethered heterochromatin domains (E) from electron micrographs for Lbr+/+, Lbr+/ic-J, and Lbric-J/ic-J INL cells. F) 3D electron microscopy of bipolar nuclei in WT and Lbric-J/ic-J P21 retinae showing the difference in heterochromatin tethering (arrows). G) Genomic map of Lbr and location of PCR primers flanking the gRNA sequences used to produce LbrLox and Lbr∆ mouse strains with CRISPR-Cas9. H) Photo of P8 littermates with WT Lbr+/+ or deleted Lbr∆/∆ alleles. I) Photograph of an agarose gel with PCR products from retinal genomic DNA showing that the LbrLox allele can be recombined (arrow) in the Vsx2-Cre;LbrLox/+ or Vsx2-Cre;LbrLox/+ retina. J) 3D electron microscopy of bipolar nuclei in WT and LbrLox/Lox P21 retinae showing the difference in heterochromatin tethering (arrows). K) Barplot of visual acuity of WT and LbrLox/Lox adult mice measured on consequtive days over one week. Mean and standard deviation is plotted for 3 animals each. L) Scatterplot of gene expression for WT and LbrLox/Lox adult retina. Scale bars: 1 µm.
To overcome the lethality of the Lbric-J/ic-J mice, we introduced LoxP sites into Lbr by CRISPR-Cas9 (Figure 4G and Supplemental Information). We identified founders with a deletion between the two gRNA target sites (Lbr∆ or with two loxP sites integrated on the same allele [LbrLox]). The Lbr∆ mice were intracrossed to produce Lbr∆/∆, Lbr∆/+, and Lbr+/+ mice. The Lbr ∆/∆ mice were indistinguishable from Lbric-J/ic-J mice (Fig. 4A). The LbrLox founders were intracrossed and mated to a mouse strain with a retinal-specific Cre transgene (Vsx2-Cre). The Cre transgene is expressed in retinal progenitor cells throughout retinal development in a mosaic pattern and could recombine the LbrLox allele in the retina (Figure 3I) (Rowan and Cepko, 2004; Rowan and Cepko, 2005). Adult LbrLox/Lox;Vsx2-Cre mice were analyzed by 3D electron microscopy and the proportion of INL nuclei with tethered and untethered heterochromatin was scored in a blinded manner in triplicate (Figure 4J; Table S6; and Supplemental Information). In LbrLox/Lox;Vsx2-Cre retinae, 57% (28/49) of INL nuclei had untethered, 20% (10/49) had a mixture of tethered and untethered, and 23% (11/49) had tethered heterochromatin (Figure 4J; Table S6). The proportion of untethered heterochromatin is similar to the fraction of cells that express Cre (Rowan and Cepko, 2004; Rowan and Cepko, 2005). In Lbr+/+;Vsx2-Cre retinal INL cells, 97% (41/42) had tethered or a mixture of tethered and untethered heterochromatin (Table S6). Scoring of single sections yielded data similar to those for Lbric-J/ic-J retinae (Table S6). Measurement of visual acuity by the OptoMotry system revealed that Vsx2-Cre;LbrLox/Lox mice had a normal optomotor response under bright-light (cone) conditions (Figure 4K). RNA-seq analysis also showed very few difference in gene expression for LbrLox/Lox;Vsx2-Cre mice (Figure 4L; Table S5). Taken together, these data suggest that euchromatin and heterochromatin domains are determined as part of cell type–specific differentiation programs in the retina, and changes in attachment to the nuclear lamina have little impact on gene expression or neuronal function.
Identification of Promoter–Enhancer Interactions
Next, we used Hi-C and CTCF ChIP-seq datasets to identify local chromatin contacts and long-distance chromatin loops. Identifying chromatin loops from Hi-C data depends on the threshold and map resolution for the Hi-C dataset and cell-type complexity of the sample. For example, we identified more chromatin loops in our ultra-deep Hi-C dataset than in previous mouse cortex data due to deeper coverage (Table S1). aggregate peak analysis (APA) analysis showed that peaks to lower-left intensity ratios were higher for our datasets than for previously published data (Figure S3). Using strict criteria for stable DNA-looping predictions from Hi-C data (Supplemental Information) (Durand et al., 2016), we identified promoter–enhancer loops dynamically regulated during development, in a manner that correlated with gene expression (Table S7). For example, there were changes in the 3D chromatin landscape of the Sox2 promoter and a previously identified downstream SE (Bonev et al., 2017) (Figure 5A,B). Our Hi-C data predicted a looping interaction in retinal progenitor cells expressing the gene, but this interaction was absent at later developmental stages (Figure 5A,B). Two-color FISH for Sox2 and its enhancer revealed that the enhancer is sequestered to the facultative heterochromatin domain in rod photoreceptors, whereas the promoter and gene body remain in the euchromatin domain (Figure 5C). Both the enhancer and promoter were inactive in the adult retina when the gene was not expressed (Figure 5D,E).
Figure 5. Identification of Enhancer–Promoter Looping Interactions.

A) ChromHMM and expression of Sox2 during retinal development. B) Hi-C contact map of the region on chromosome 3 spanning Sox2 and downstream SE. Circles and yellow highlight the looping interaction between Sox2 and the downstream SE. ChIP-seq for H3K36me3, H3K27med, Brd4, and H3K27Ac is shown below the Hi-C contact map, and ATAC-seq and WGBS at each developmental stage are also shown. An E14.5/P0-specific predicted looping interaction from the Hi-C is indicated by the purple line and purple boxes. C) Two-color FISH for Sox2 (red) and SE (green) is shown for a representative E14.5 retinal progenitor cell and an adult rod photoreceptor. D,E) ChIP-seq, ATAC-seq, and WGBS for Sox2 and SE at E14.5 and P21 (adult). Scale bars: 1 µm. Abbreviations: Chr, chromosome; ESC, embryonic stem cells; FPKM, fragments per kilobase per million reads; SE, superenhancer.
To determine if additional changes occurred in promoter–enhancer interactions that did not meet our strict criteria for stable loop formation (Durand et al., 2016), we analyzed promoter–enhancer contacts from Hi-C data to identify those correlating with changes in expression (Table S7). We found that 25% (39/156) of rod genes, including Rho, Crx, Aipl1, Rp1, Prom1, and Pde6a, had increased promoter–enhancer interactions that correlated with gene expression (Table S7). Similarly, 16% (38/235) of progenitor genes, including Pcna, Dkk3, Foxn4, Cdk1, Tuba1b, and Sfrp2, had decreased promoter–enhancer contacts during retinogenesis, as genes were silenced. Our Hi-C data identified dynamic looping interactions and more subtle differences in promoter–enhancer contacts associated with transcriptional changes during retinal development.
Cell type– and Developmental Stage–Specific Core Regulatory Circuit SEs
Developmentally regulated SEs had the highest proportion of looping interactions from our Hi-C data (Table S7). However, cellular complexity remains a challenge in identifying SE chromatin looping interactions with promoters in developing and mature retinae. It was not technically feasible to purify sufficient numbers of individual retinal cell types for Hi-C analysis other than rod photoreceptors analyzed herein. While the rod and non-rod Hi-C data were useful for identifying chromatin loops in these cell populations (Figure 6) we were unable to identify robust promoter–enhancer or SE interactions in low-abundance retinal cell types (e.g., bipolars, cones, horizontal and ganglion cells, and Müller glia). To more accurately identify cell– and developmental stage–specific SE–promoter interactions in the adult retina, we performed single-cell ATAC-seq (scATAC-seq) and single-cell RNA-seq (scRNA-seq). We overlapped the scATAC-seq data with our previously identified developmentally regulated SEs and integrated the data with the scRNA-seq (Aldiri et al., 2017). We identified 297 cell type–specific SEs that had corresponding gene expression; 40 were in rods, 106 in cones, 67 in bipolar cells, 41 in Müller glia and 43 in amacrine cells (Figure S4 and Table S8). Nearly one-third (12/37) of the previously identified core regulatory circuit super-enhancers (CRC-SEs) in the postnatal retina had cell type specific ATAC-seq peaks and RNA expression (Table S8).
Figure 6. Identification of cell-type specific looping from Hi-C data.

A) ChromHMM for developing retina for the Nrxn3 gene along with FPKM data on the right. B) Hi-C contact frequency plot for rods and non-rods for the region shown in (A). The yellow highlight indicates a region with a clear difference in contact frequency consistent with a difference in expression and chromatin state for Nrxn3 in rods and non-rods. C) tSNE plot of single cell RNA-seq with each cell type highlighted in different colors and the number of each cell type in parentheses. Nrxn3 is expressed in all non-rods as shown in the single cell heatmap for Nrxn3. Representative examples of rod (Pde6a) and cone (Pde6h) genes are shown for comparison.
One of the genes with a putative cell type–specific core regulatory circuit super enhancer (CRC-SE) was Vsx2 (Figure 7A; Table S8). The paired-type homeobox gene is expressed in retinal progenitor cells throughout development, and its mutations lead to microphthalmia in humans and mice (Bar-Yosef et al., 2004; Faiyaz-Ul-Haque et al., 2007; Ferda Percin et al., 2000; Green et al., 2003). Vsx2 is also expressed in mature bipolar neurons and at low levels in Müller glia (Rowan and Cepko, 2004; Rowan and Cepko, 2005). How this precise temporal and spatial pattern of expression is achieved during retinal development remains poorly understood. We discovered a CRC-SE that was developmental stage-specific and consistent with bipolar cell expression of Vsx2 (Figure 7A). Indeed, the region within the CRC-SE that is most conserved across species contains a Vsx2 consensus-binding site (Figure 7B). We confirmed binding of Vsx2 to this site by ChIP-seq using two different antibodies (Figure 7C). Single-cell RNA-seq confirmed that Vsx2 is expressed in bipolar neurons and Müller glia in the adult retina (Figure 7D), and single-cell ATAC-seq revealed that chromatin at the Vsx2 CRC-SE, where Vsx2 binds, is open in bipolar cells but not other cell types (Figures 7E–G). The resolution of Hi-C was insufficient to identify a looping interaction between CRC-SE and the promoter, because the enhancer is active in only a small subset (<10%) of cells. However, single-cell ATAC-seq provided additional evidence that a cell- and developmental stage–specific CRC-SE exists for Vsx2.
Figure 7. Identification of a Bipolar Neuron–Specific CRC-SE.

A) Genomic map of the murine Vsx2 locus showing a 2.5-kb region previously identified in heterologous reporter assays (Rowan and Cepko, 2004, 2005) as containing candidate bipolar neuron–specific regulatory element (black bar). The 32-kb putative Vsx2 CRC-SE is indicated with genomic coordinates from mm9 with evolutionary conservation across species. Arrow indicates region of highest evolutionary conservation centered an ATAC-seq and Brd4 peak. B) Evolutionary conservation of a Vsx2 binding site at the arrow indicated in (A). C) ChIP-seq using two different antibodies for Vsx2 showing binding at the Vsx2 consensus binding site in (A). There were no other peaks in the promoter or CRC-SE. D) tSNE plot of single-cell RNA-seq from WT adult retina showing major cell types and number of cells. Expression of Vsx2 in Müller glia and bipolar cells is shown in red. E) T-distributed stochastic neighbor embedding (tSNE) plot of single-cell ATAC-seq for adult retina with representative number of nuclei. F) Trace of single-cell ATAC-seq for the rhodopsin gene (Rho). G) Trace of single-cell ATAC-seq for the Vsx2 promoter and CRC-SE. The promoter is active in bipolar cells and Müller glia, whereas the CRC-SE is active only in bipolar neurons. The yellow highlight and dashed line indicate the position of the Vsx2 consensus binding site. H) Bar plot of gene expression from RNA-seq analysis of Lin52 and Vsx2 for the Vsx2-SE+/+, Vsx2-SE+/∆, and Vsx2-SE ∆/ ∆ retinae. Each bar is the mean and SD for the number of animals indicated. I) Bar plot of gene expression from RNA-seq analysis of 11 representative bipolar neuron–specific genes spanning subtypes of bipolar neurons. Each bar is the mean and SD of biological replicates. J) Photograph of a Vsx2orJ/orJ mouse with microphthalmia and a Vsx2-SE ∆/ ∆ mouse with normal eye size (arrows). K) ChIP-seq for the Vsx2-SE ∆/ ∆ and Vsx2-SE+/+ adult retina. Abbreviations: CRC-SE, core regulatory circuit super-enhancer; SE, super-enhancer; tSNE, T-distributed stochastic neighbor embedding (tSNE) plot.
To determine if the conserved region is required for bipolar neuron–specific expression, we deleted the CRC-SE in mice by CRISPR–Cas9. In total, three independent sublines were generated that could be distinguished by their deletion junctions (Supplemental Information). Individual sublines were intracrossed to produce litters with WT (Vsx2-SE+/+), heterozygous (Vsx2-SE+/∆), or homozygous (Vsx2-SE ∆/∆) deletion of the Vsx2 CRC-SE. RNA-seq confirmed significant downregulation of Vsx2 and other bipolar neuron–specific genes in the adult retina (Shekhar et al., 2016) (Figures 7H and 7I; Table S9). Mice did not have microphthalmia, suggesting that deletion of the Vsx2 CRC-SE did not disrupt expression in retinal progenitor cells (Figure 7J). We performed ChIP-seq and chromHMM for the Vsx2-SE ∆/ ∆ retinae as we did previously for developing murine retinae (Aldiri et al., 2017) and added those data to our cloud-based viewer. There was a decrease in active chromatin marks and an increase in H3K27me3 at the Vsx2 promoter in Vsx2-SE ∆/ ∆ retinae (Figures 7K).
To extend our bulk RNA-seq analysis, we performed single-cell transcriptome analysis of adult Vsx2-SE+/+ and Vsx2-SE ∆/ ∆ retinae in biological replicates (Figures 8A and 8B). Bipolar cells were absent in Vsx2-SE ∆/ ∆ retinae, which is consistent with data from bulk RNA-seq analysis showing virtually every known bipolar-specific gene was downregulated (Table S9).
Figure 8. Mice Lacking the Vsx2 CRC-SE Are Blind Due to Loss of Bipolar Neurons.

A) tSNE plot of single-cell RNA sequencing for 2 adult Vsx2-SE+/+ and 2 adult Vsx2-SE ∆/ ∆ littermates. Cell types were assigned based on known gene expression profiles from previous studies. B) Distribution of cells from each library in the tSNE plot from (A). The bipolar cluster (box) lacks cells from the Vsx2-SE ∆/ ∆ strain. C) Bar plot of visual acuity measured by the optomotor response for 8-week-old Vsx2-SE+/+, Vsx2-SE+/ ∆, and Vsx2-SE ∆/ ∆ mice. Data represent means and SD for 2 mice for each genotype measured on 4 successive days. D) Confocal micrographs of DAPI-stained adult retinae. Arrows indicate disruption in the outer plexiform layer. E) Confocal micrographs of adult retinae immunostained using antibodies for specific retinal cell types. Side-by-side images with the immunostaining (black and white) and DAPI overlay (color) are shown. F,G) Representative electron micrographs (F) and 3D tracing (G) of rod spherules from Vsx2-SE+/+and Vsx2-SE ∆/ ∆ mice (H,I). Horizontal cell processes can be identified by the presence of synaptic vesicles (blue); bipolar dendrites (orange) lack synaptic vesicles. Scale bars: 25 µm. Abbreviations: GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer.
We measured visual acuity by the OptoMotry system and found that Vsx2-SE ∆/ ∆ mice lacked optomotor response under bright-light (cone) conditions (Figure 8C). We then immunostained retinal vibratome sections from each of the three genotypes, using antibodies specific for proteins found in rods (rhodopsin), cones (cone arrestin), bipolar neurons (PKCα, Go α, Vsx2), Müller glia (glutamine synthetase), reactive Müller glia (GFAP), horizontal neurons (calbindin), and amacrine cells (Pax6, calretinin, TH). Bipolar cells were absent and the outer plexiform layer (OPL) was disrupted (Figures 8D, 8E, and S5). This phenotype resembled retinae from a recent study in which Wnt5a/b signaling was disrupted between photoreceptors and bipolar neurons at the OPL and is consistent with the loss of bipolar neurons (Sarin et al., 2018). Transmission electron microscopy confirmed the absence of bipolar cell bodies, and 3D electron microscopy was used to reconstruct rod and cone terminals in Vsx2-SE+/+ and Vsx2-SE ∆/ ∆ mice. In total, 509 and 490 10-nm sections were collected and analyzed from the Vsx2-SE+/+ OPL and Vsx2-SE ∆/ ∆ OPL, respectively. Across 30 rod spherules and six cone pedicles, there was no evidence of bipolar dendrites in Vsx2-SE ∆/ ∆ retinae (Figures 8F–I). Together, these data indicate that deletion of a developmental stage– and cell type–specific CRC-SE leads to complete absence of bipolar cells in the murine retina.
DISCUSSION
We build upon our previous epigenetic profiling studies of the murine retina to include analysis of chromatin structure, compartmentalization, and looping using ultradeep in situ Hi-C analysis. Although TAD boundaries are largely conserved across development and brain regions (cortex vs. retina), dynamic changes occur in the localization of genes to chromatin compartments (A/B). We developed a machine learning–based algorithm to predict euchromatin/heterochromatin localization in murine rod photoreceptors, and showed an overlap between euchromatin and compartment A and between heterochromatin and compartment B. A subset of developmentally regulated genes could transition between compartments and subnuclear domains (euchromatin/heterochromatin) during development, and individual TADs could span euchromatin and heterochromatin in rods. We also used Hi-C data to identify promoter–enhancer interactions, which were effective for genes such as Sox2 or Nrxn3 that occur in most cells, but not as effective for less abundant cell populations in the retina such as bipolar neurons. To overcome this barrier, we integrated single-cell ATAC-seq and single-cell RNA-seq with epigenetic and Hi-C data to further refine putative cell– and developmental stage–specific promoter–enhancer interactions. This integrated approach identified a CRC-SE for Vsx2 that was specific to bipolar neurons. Deletion of this CRC-SE in mice led to complete absence of bipolar cells, with no effect on retinal progenitor cells or Müller glia. These data highlight the value of integrating bulk and single-cell data to identify promoter–enhancer interactions important for neurogenesis.
Ultradeep Hi-C Analysis
Hi-C studies have been extensively performed on human cell lines and tissues, but are more limited in the developing murine central nervous system (CNS)(Dixon et al., 2012). Our dataset is more than 20-fold deeper than the previous embryonic mouse cortex dataset, which allowed us to identify previously undetected features of the chromatin landscape of the murine CNS. Our data also provide insights into developmental changes in chromatin domains, compartments, and looping. Pearson correlation, insulation score, and eigenvector hierarchical clustering showed that E14.5 and P0 retinae were more similar to each other than to adult retinae, which is consistent with our understanding of retinal development. Retinal progenitor cells are present at E14.5 and P0, and although postmitotic cells are present at P0, they are still immature relative to those in the adult retina. In addition, the chromatin in murine rod photoreceptors undergoes dramatic condensation, which further distinguishes Hi-C data on adult retina from those in E14.5 and P0 retinae. The amount of genome assigned to TADs was similar across samples, but some algorithms identified more TADs in the adult retina, likely due to high levels of chromatin compaction in rod nuclei, which make up ~80% of adult retinae.
One important metric of Hi-C data quality is the cis/trans ratio (Lieberman-Aiden et al., 2009). A low cis/trans ratio may indicate that local chromatin conformation is poorly preserved and the majority of junctions detected in the Hi-C library sequencing are due to random ligation of genomic fragments. The cis/trans ratio in our dataset is higher than those of previous studies (Bonev et al., 2017; Rao et al., 2014) and this may reflect the unique structure of the retina. Specifically, the tissue can usually be isolated in less than a minute and fixation is very rapid because it is a thin sheet of tissue. In contrast, other brain regions are more difficult to isolate and penetration of the fixative can be slow and uneven.
Chromatin Compartments and Subnuclear Localization
Chromatin compartments (A/B) are defined by local interactions by Hi-C analyses, whereas euchromatin and heterochromatin domains are defined by chromatin compaction visualized as the intensity of staining with dyes that bind DNA. More intense staining represents more compacted chromatin, whereas lighter staining indicates more open, accessible chromatin, where active transcription is thought to occur. Although some genomic regions are preferentially localized to euchromatin or heterochromatin across cell types, facultative heterochromatin is likely developmental stage– and cell–type specific. However, no methods are available to map euchromatin and facultative chromatin domains genome-wide, and previous studies postulate that all genes are localized to the euchromatin domain irrespective of their expression in rod photoreceptors (Solovei et al., 2009).
We used the unique inverted nuclear organization of murine rod nuclei and developed a protocol for FISH/IF to localize genes and enhancers to euchromatin or facultative heterochromatin. These data were then used to train a machine learning–based algorithm for analyzing our extensive epigenetic and transcriptomic data to identify euchromatin and heterochromatin domains in rod nuclei. These data were validated with another group of FISH/IF studies and were 89% accurate. Discordant loci were at domain boundaries and often involved very large genes with their promoter in the euchromatin domain and gene body in the facultative heterochromatin domain. Contrary to previous studies (Solovei et al., 2009), we found that some genes and regulatory elements are localized to facultative heterochromatin in a developmental stage– and cell type–specific manner.
Our map of euchromatin and heterochromatin enabled the comparison of A/B compartments from Hi-C with subnuclear localization in rod photoreceptors. Overall, there was concordance between compartment A and euchromatin and compartment B and heterochromatin. Notably, a subset of developmentally regulated genes transitioned from euchromatin (compartment A) to heterochromatin (compartment B) and vice versa. Resolution of the euchromatin/heterochromatin map was higher than for A/B compartment analysis, because data (ChIP-seq and chromHMM) used to generate the model are of higher resolution than for Hi-C. However, our euchromatin/heterochromatin map and A/B compartment analysis have the same shortcomings. In the adult retina, the map is a mixture of all cell types. Therefore, future studies need to focus on epigenetic profiling of individual cell populations to extend our modeling beyond rod photoreceptors.
Euchromatin and Heterochromatin Localization in the Retina
Conventional nuclei have centrally located euchromatin with heterochromatin tethered to the nuclear lamina. However, this structure is inverted in rod nuclei of nocturnal species such as mice (Solovei and Joffe, 2010; Solovei et al., 2009). Refractive indices of centrally located heterochromatin in nocturnal rod nuclei and surrounding euchromatin are different, such that nuclei act as collecting lenses to capture and focus low-level light, which is important for night-time vision (Solovei and Joffe, 2010; Solovei et al., 2009). However, in diurnal species, rod nuclei have a more conventional nuclear organization. A mechanism to explain this difference across species is Lbr expression: nocturnal rods lack Lbr expression, which frees the heterochromatin to coalesce in nucleus center. To convert conventional nuclei of the INL to inverted nuclei, we generated an LbrLox mouse strain and conditionally inactivated Lbr in the developing retina. Overall, vision was unaffected and there were very few changes in gene expression, suggesting that localization of genes to euchromatin or facultative heterochromatin involves a process distinct from tethering heterochromatin to the nuclear lamina. Thus, changes in Lbr expression can readily convert nocturnal rods to diurnal rods without altering their developmental programs. More subtle effects of converting INL nuclei to an inverted structure may be revealed under stress and disease conditions. In addition, the heterochromatin domains that are normally tethered to the nuclear lamina of INL nuclei coalesced into a single large domain in the center of the nucleus in the absence of Lbr mimicking the structure of rod nuclei.
Developmental Stage– and Cell Type–Specific Enhancers
Ultra-deep in situ Hi-C was very useful for mapping chromatin domains, compartments, and looping interactions in most retinal cells but not as useful to identify cell type– and developmental stage–specific interactions between promoters and enhancers. Indeed, even for the most abundant cell type (rod photoreceptors) it was not feasible to perform Hi-C on replicate samples for the purified cells so those data should be used for qualitative comparisons only. However, by combining single-cell ATAC-seq and RNA-seq with SE profiling, we successfully identified cell-type– and developmental stage–specific SEs in the developing retina. This is essential to better understand the role of transcription factors (TFs) during retinal development, because they often have complex expression patterns, and gene deletions have dramatic phenotypes that are difficult to interpret. In contrast to mutating the gene, deletion of an adjacent cell-type– and developmental stage–specific SE may allow researchers to study the role of that TF at a particular developmental stage in an individual cell type.
Our analysis of the Vsx2 CRC-SE exemplifies the advantage of our approach. Vsx2 is expressed in retinal progenitor cells, mature bipolar neurons, and Müller glia. Mutations in VSX2/Vsx2 cause microphthalmia in humans and mice, because it is required in retinal progenitor cells (Bar-Yosef et al., 2004; Faiyaz-Ul-Haque et al., 2007; Ferda Percin et al., 2000; Green et al., 2003). However, analyzing the role of Vsx2 in Müller glia and bipolar neurons has been more challenging, because it is difficult to separate the primary effect on retinal progenitor cells from subsequent effects on bipolar neurons and Müller glia. However, by deleting a developmental stage– and cell–type specific CRC-SE for bipolar expression of Vsx2, we showed that Vsx2 is required for bipolar cell development and this did not affect retinal progenitor cell proliferation or Müller glial cell development.
We also further define the regulatory elements for Vsx2 gene expression. Previous studies show that a bacterial artificial chromosome spanning Vsx2 and 55 kb of the upstream sequence is sufficient to confer retinal progenitor, bipolar, and Müller glial cell expression of a reporter gene in transgenic mice (Rowan and Cepko, 2004; Rowan and Cepko, 2005). Other studies report that a proximal promoter element that contributes to retinal progenitor cell expression and an upstream element sufficient for bipolar neuron expression (Kim et al., 2008a; Rowan and Cepko, 2005). However, none of these elements have been shown to be required in vivo. Moreover, the bipolar-specific upstream element does not overlap with the Vsx2 binding site or the bipolar cell–specific ATAC-seq peak. This may reflect the large size of the CRC-SE and binding of multiple bipolar cell–specific TFs. That is, elements within the Vsx2 CRC-SE may be sufficient to confer bipolar cell–specific expression in transient electroporation studies because they contain TF binding sites, but the complex regulation of genes such as Vsx2 in the context of chromatin must be studied in vivo. Further dissection of the Vsx2 CRC-SE is required to refine smaller domains required for bipolar cell– specific expression, and additional studies are required to dissect the elements specific to retinal progenitor cells and Müller glial cells.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael A. Dyer (michael.dyer@stjude.org). Mouse lines generated from this study are being deposited at the Jackson Laboratory.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Wild type mice
All animal procedures and protocols were approved by the St. Jude Laboratory Animal Care and Use Committee. All studies conform to federal and local regulatory standards. Mice were housed on ventilated racks on a standard 12 hour light-dark cycle. Wild type C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME). For timed pregnancy, individual male mice were housed with 4 females in a single cage. Plugged/pregnant females (identified by visual examination and/or palpation) were isolated and embryos or pups were harvested at the appropriate time. Both males and females were combined for this study.
Generation of Vsx2-CRC-SE deletion mice
A total of 65 pups were generated from 5 separate days of injections. On each day, ten 3–4 week old C57BL/6J female mice from Jackson Labs were superovulated with 5 units of gonadotrophin from pregnant mare’s serum (PMSG from ProSpec) and 48 hours later, with 5 units of human chorionic gonadotrophin (hCG from Sigma). After overnight mating with C57BL/5J males, the females were euthanized and oocytes were harvested from the ampullae. The protective cumulus cells were removed using hyaluronidase, and the oocytes were washed and graded for fertilization by observing the presence of two pronuclei. A mixture provided by the Center for Advanced Genome Engineering of 100 ng/ul Cas9 mRNA and 25–50 ng/ul each of two guide RNAs, ZZ30.mVsx2.g7 (5’ GCAGGCCATGTGCTCGTCGA 3’) and ZZ31.Vxs2.g16 (5’ CAGGGTGCAGGCTGACAACG 3’), was injected into the cytoplasm of the oocytes. gRNAs were designed to have at least 3bp of mismatch between the target site and any other site in the mouse genome. gRNAs were tested prior to embryo injection for activity in mouse N2A cells using targeted next generation sequencing as previously described(Sentmanat et al., 2018). They were then returned to culture media (M16 from Millipore or A-KSOM from Millipore) and later the same day transferred to day 0.5 pseudopregnant fosters (7–10 week old CD-1 females from Charles River Laboratories mated to vasectomized CD-1 males). Pups were born after 19 days gestation and genotyped using PCR and Illumina Mi-Seq. Positive animals were weaned at day 21 and at 6 weeks of age, they were back-crossed to C57BL/6J mice. The primers used for genotyping are shown below with Mi-Seq adaptors (underlined):
Forward:
5’-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGGCTCTGACCTTCCTGGAAGCCCCGC-3’
Reverse:
5’-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGCTCAGGAGGTTACAAGGAGGTGTAG-3’
The primers span 32 kb and will not give a PCR product for the wild type allele but will give a 300 bp product in the deleted allele. We also generated a set of primers internal to the deletion to serve as a wild type primer set to distinguish heterozygous and homozygous deleted mice. Those internal wild type primers will produce a PCR product of 149 bp and are:
Forward:
5’-CATAACTGGCTGTATTCTGTGTGACTC-3’
Reverse:
5’-CTTACATCCTTTGACCCTGGCTATG-3’
The deletion junctions for the 3 sublines are:
Vsx2–3-3: 5’-TCTAC/CCTGCACCCTGAAATCA-3’
Vsx2–59-12: 5’-TCTACCCT/CACCCTGAAATCA-3’
Vsx2–23-3: 5’-TCTACCCT/GCACCCTGAAATCA-3’
Generation of Lbr-flox mice
Lbr floxed mice were generated using CRISPR-Cas9 technology. Briefly, C57BL/6J (Jackson Laboratories, Bar Harbor, ME) fertilized zygotes were injected with 20ng/ul of each sgRNA (Synthego), 60ng/ul SpCas9 protein (Berkeley Microlab), 25ng/ul of each ssODN donor (IDT) into the pronucleus. Founder mice were genotyped by targeted next generation sequencing. Editing construct sequences and relevant primers are listed in below.
| Name | Sequence (5’ to 3’) |
|---|---|
| mLbr.5’.sgRNA | GGAACUUUAUUUGGATCAGG |
| mLbr.5’LoxP.ssODN.anti | ttatttttgagatgaggtctcactatgtagtcctggctggcctggaactttatttggatcGGATCC ATAACTTCGTATAATGTATGCTATACGAAGTTATaggagggcctcaaactcacagaaatcctcctgcatctgcctgacaagtgctcagattaaa |
| mLbr.5’.NGS.F partial Illumina adaptors (upper case) |
CACTCTTTCCCTACACGACGCTCTTCCGATCTcaggctgcaagttatttggttctg |
| mLbr.5’.NGS.R partial Illumina adaptors (upper case) |
GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTgatcttgagagctcacttcacc |
| mLbr.3’.sgRNA | GGUUCCUUAGGCUCUUGUGA |
| mLbr.3’LoxP.ssODN.sense | actgttgctccattctttcatcttggtctcctctcactatgctggttccttaggctcttgGGATCCATAACTTCGTATAGCATACATTATACGAAGTTATtgacggtgtcgcccttttgaagagcactgttcgtaagaccatcattccgttgttctcctg |
| mLbr.5’.NGS.F partial Illumina adaptors (upper case) |
CACTCTTTCCCTACACGACGCTCTTCCGATCTcccttattggggttcattggtg |
| mLbr.3’.NGS.R partial Illumina adaptors (upper case) |
GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTccaccagagaacaagacacctc |
METHOD DETAILS
Vision Testing
The OptoMotry system from CerebralMechanics was used to measure the optomotor response of Vsx2-SE∆/∆ mice. Briefly, a rotating cylinder covered with a vertical sine wave grating was calculated and drawn in virtual three-dimensional (3-D) space on four computer monitors facing to form a square. Vsx2 mice standing unrestrained on a platform in the center of the square tracked the grating with reflexive head and neck movements. The spatial frequency of the grating was clamped at the viewing position by repeatedly recentering the cylinder on the head. Acuity was quantified by increasing the spatial frequency of the grating until an optomotor response could not be elicited. Contrast sensitivity was measured at spatial frequencies between 0.1 and 0.45 cyc/deg.
In total, 2 Vsx2-SE∆/∆, 3 Vsx2-SE+/∆ and 2 Vsx2-SE+/+ were analyzed along with 2 C57Bl/6J mice. The tester was blinded to genotype until after testing was complete. Mice were acclimated to the device for 1 week and then tested on 4 successive days the following week.
Immunostaining
For immunostaining, retinae from 9.4 week old littermates were isolated and fixed in 4% PFA overnight at 4°C. Retinae were washed 3x with PBS and then embedded in 4% LMP agarose in PBS for vibratome sectioning at 50 µm. They were incubated in block solution for 1 hr at room temperature and then in primary antibody in block solution overnight at 4°C.
The antibodies were:
| Antibody | Company | Catalog number | Dilution |
|---|---|---|---|
| Calbindin | Sigma | C9848 | 1:100 |
| Calretinin | Chemicon International | MAB1568 | 1:100 |
| Pax6 (mo) | DSHB | Pax6 | 1:500 |
| PSD95 | Affinity Bioreagents | MA1–046 | 1:100 |
| GFAP | Sigma | G3893 | 1:100 |
| Bassoon | Stressgen | VAM-PS003 | 1:500 |
| HPC-1 | Sigma | S0664 | 1:500 |
| Rhodopsin | Custom-made | N/A | 1:500 |
| Brn3a | Santa Cruz | sc-8429 | 1:1000 |
| PKC-a | Upstate | 05–154 | 1:5000 |
| GS | BD Pharmingen | 610518 | 1:100 |
| TH | Pel-Freez | P40101 | 1:500 |
| Ribeye | Custom-made | N/A | 1:100 |
| Cone A | Millipore | AB15282 | 1:5000 |
| Recoverin | Millipore | AB5585 | 1:5000 |
| Piccolo | Synaptic Systems | 142002 | 1:100 |
| GaO | Santa Cruz | sc-387 | 1:5000 |
| Vsx2 | Exalpha | X1180P | 1:500 |
Vibratome sections were washed twice in PBS and then incubated in secondary antibody for 1 hour at room temperature. All secondary antibodies were incubated a dilution of 1:500 in the appropriate block solution. We used donkey anti-mouse (Vector Labs BA-2000), goat anti-rabbit (Vector Labs BA-1000) and rabbit anti-sheep (Vector Labs BA-6000). After secondary antibody, they were washed twice with PBS and and incubated with ABC reagent (Vector Laboratories, Cat. # PK6100) for 30 minutes. We then used tyramide Cy3 (PerkinElmer, Cat. # FP1046) for 10 minutes at room temperature and washed 2x in PBS followed by DAPI at 1:1000 in PBS. Slices were mounted in Prolong gold reagent and imaged on a Zeiss LSM700 confocal microscope.
Flow Cytometry
Cells were evaluated on a FACS Aria Fusion (Becton Dickinson) flow cytometer. Cell death was assessed using a forward scatter versus DAPI plot. Data was analyzed using Diva software (Becton Dickinson).
Sequencing
RNA-Seq
RNA was extracted from each iPS cell lines by using RNeasy Plus Mini kit (Qiagen, 74134) or Direct-zol kit (Zymo Research, R2050). Libraries were prepared from ~500 ng total RNA with the TruSeq Stranded Total RNA Library Prep Kit according to the manufacturer’s directions (Illumina). Paired-end 100-cycle sequencing was performed on HiSeq 2000 or HiSeq 2500 sequencers according to the manufacturer’s directions (Illumina.)
Hi-C
The in situ Hi-C protocol was performed using retinal cells, with minor modification to the original protocol (Rao et al., 2014). Two to five million cells were crosslinked with 1% formaldehyde for 10 min at room temperature, digested with 125 units of MboI overnight, and labeled with biotinylated nucleotides and proximity ligation. After reverse crosslinking, ligated DNA was purified and sheared to 300–500 bp by using Covaris LE220. DNA fragments containing ligation junctions were pulled down with streptavidin beads, followed by Illumina-compatible library construction and paired-end sequencing.
ChIP antibody validation
All antibodies were independently validated and biological replicates were performed for each mark at each stage of development. For most antibodies, we also validated the data using an independent antibody. All antibody validation data and SOPs for ChIP are freely available through the CSTN (www.stjude.org/cstn). Specifically, for the H3K27me3 mark, we have two antibodies with two replicates each at every stage. To estimate the consistency between antibodies, we first called domains using SICER as above, then at each stage for each antibody domains overlapping common regions between two replicates have been merged, at last we merged the domains from two antibodies and counted extend reads for each sample overlapping these merged domains. At each stage, we draw the correlation plot between each pair of samples as (Fig. S1), as most of the correlation coefficient is > 0.9 (except one outlier at P7 as replicate 1 for AB3), we think both antibodies are working consistently and we used AB1 in the final analysis since it had better signal to noise profile.
ChIP-Seq
All antibodies were independently validated, and those validation data and protocols are available through the Childhood Solid Tumor Network website (https://www.stjude.org/CSTN/) (Stewart et al., 2016).
Freshly isolated retinae or cells were cross-linked for 10 min in 1% ChIP-seq–grade formaldehyde (Thermo scientific # 28906) in 1X PBS at room temperature. Glycine was added to a final concentration of 0.125 M to stop the cross linking reaction. The retinal tissue was washed in 1X PBS and then dispersed into a single cell suspension using a dounce homogenizer with 7 ml pestle (Fisher scientific # 06–435A). The nuclei were isolated and prepared for shearing using TruChIP chromatin shearing kit (Covaris #520127) according to manufacturer’s protocol. After shearing, ChIP was performed using the iDeal ChIP-seq kit (Diagenode # C01010051). Ten percent of chromatin from each ChIP reaction was used as an input. After de-crosslinking, DNA was extracted using MinElute PCR-purification kit (QIAGEN #28006), quantified using the Quant-iT PicoGreen ds DNA assay (Life Technologies #Q33120) and subjected to qPCR analysis and library construction for sequencing. The RNA-Pol II, Brd4 and H3K9me3 ChIP was performed by Active Motif.
Libraries were prepared from 5–10 ng DNA by using the NEBNext ChIP-Seq Library Prep Reagent Set for Illumina with NEBNext High-Fidelity 2× PCR Master Mix according to the manufacturer’s instructions (New England Biolabs) with the following modifications: a 1:1 Ampure cleanup was added after adaptor ligation a total of 2 times. The Ampure size-selection step prior to PCR was eliminated. The 72 °C extension step of the PCR was lengthened to 45 s. Completed libraries were analyzed for insert-size distribution on a 2100 BioAnalyzer High Sensitivity kit (Agilent Technologies) or Caliper LabChip GX DNA High Sensitivity Reagent Kit (PerkinElmer). Libraries were quantified using the Quant-iT PicoGreen ds DNA assay (Life Technologies) and Kapa Library Quantificaiton kit (Kapa Biosystems) or low-pass sequencing on a MiSeq nano kit (Illumina). Fifty-cycle single-end sequencing was performed on an Illumlina HiSeq 2500.
Retina Dissociation
Retina dissociation buffer was prepared by adding 40 U papain (Worthington CAT#LS003119) to 400 µL of papain buffer and incubating at 37°C for 15 min. Retinas were individually dissected in retinal explant media (REM) and placed on ice. Four hundred microliters of buffer was added to each retina and incubated at 37°C. To dissociate the retina, tubes were agitated twice at 5 min intervals and 40 µL of DNAse solution (DS) was added and incubated at 37°C for an additional 5 min. The cell suspension was filtered through a 40-µm cell strainer (Falcon CAT#352340) and the filter was washed with PBS to bring the total volume to 1.4 mL.
Single cell RNA-seq and ATAC-seq
Retina from two wild type C57B6 mice and 2 Vsx2-SE KO mice (all between 8–9 weeks old) were dissected and each pair of retina was dissociated with 40U papain (Worthington CAT#LS003119) in 400uL of papain buffer (1mM L-cysteine with 0.5mM EDTA in PBS −/−) at 37°C for 10 minutes with aggitation every 5 minutes. 40 µl of Dnase was added and the cells were incubated for an additional 5 min in at 37 °C. The cells were filtered through a 40-µm mesh cell strainer and added to a 5 ml of BSA cushion medium (4% BSA in Retinal Explant media) and spun at 500g for 10 min at 4 °C to clear any debris. The supernatant was aspirated and the cells were resuspended in 400uL of retinal explant media and the concentration was determined by hemocytometer.
Approximately 10,000 cells from each sample were taken and loaded onto the 10x chromium controller for single cell RNA sequencing analysis which was completed according to the 10x genomics protocol.
The remaining cells were further diluted to a concentration of 1 million cells / mL in PBS + 0.04% BSA according to the 10X genomics nuclei Isolation for single cell ATAC sequencing protocol. The cells were spun down at 300g for 5 min at 4°C. 100 ul of Cold Lysis Buffer (10mM Tris 7.4, 10mM NaCl, 3mM MgCl2, 0.1% Tween-20, 0.1%NP40, 0.01% Digitonin, 1% BSA) was added for 2 min on ice and immediately washed with 1mL chilled Wash Buffer (10mM Tris 7.4, 10mM NaCl, 3mM MgCl2, 0.1% Tween-20, 1% BSA) and spun at 500g for min at 4°C. The cells were resuspended in Diluted Nuclei Buffer from the Single cell ATAC kit and the concentration was determined by hemocytometer.
Approximately 10,000 nuclei from each sample were taken and for the transposition reaction and further processed according to the 10X genomics single cell ATAC protocol.
Sequences were analyzed first using the cell ranger pipeline (v3.0.2 and v1.0.1 for RNA and ATAC respectively) aligned to mm10 reference data (v3.0.0 and v1.0.1 for RNA and ATAC respectively) Single cell RNA sequencing ananlysis was run as individual datasets in cellranger. Cluster average gene expression and differential expression analysis were done using the R program Seurat (Butler et al., Nature Biotechnology 2018) by combining both wildtype datasets. Wildtype and SEKO replicates of single cell ATAC sequencing was run through cell ranger as a single dataset and the peaks were analyzed using the 10x genomics Loupe Browser.
| Sample | Number of Cells | Mean Reads/Cell | Median Genes/Cell | Number of Reads (mil) | Total Genes Detected |
|---|---|---|---|---|---|
| WT1 | 4584 | 19207 | 768 | 88.05 | 17795 |
| WT2 | 6322 | 15736 | 788 | 99.49 | 17995 |
| SEKO1 | 4977 | 18956 | 720 | 94.35 | 16449 |
| SEKO2 | 6676 | 14398 | 743 | 96.12 | 17444 |
| Sample | Number of Cells | Median Frag/Cell | Fraction of fragments overlapping any target region | Fraction of fragments overlapping enhancer regions | Fraction of fragments overlapping promoter regions | Number of Reads (mil) |
|---|---|---|---|---|---|---|
| WT | 20098 | 2194 | 77.2 | 18.7 | 45.7 | 165.20 |
| SEKO | 20108 | 2015 | 77.6 | 18.7 | 46.2 | 140.43 |
Computational Analysis
Single-cell ATAC-seq data analysis
Single-cell ATAC-seq(scATAC) data were first pre-processed by 10X genomics’ pipeline Cell Ranger ATAC. After reviewing the tSNE plot and cluster assignment from Cell Ranger ATAC pipeline, we manually assigned each cluster to Rods, Cones, Bipolar, Muller, Amacrine or Glycinergic amacrine cells. Using the UMI information provided by 10X we then split the scATAC reads into these cell types. For data of each cell type, we extract nucleosome free reads and call peaks by MACS2(version 2.1.1.20160309, parameters “--extsize 200 --nomodel --keep-dup all -q 0.05”). To get cell type special scATAC peaks, for each cell type, we first excluded peaks if the closest gene assigned to it doesn’t have a scATAC peak at promoter (TSS +/− 2kb). We then excluded scATAC peaks could appeared in two or more cell types. At last, we assigned retina development Super-Enhancers to these cell type specific scATAC peaks and provide table of their assigned genes, the gene’s expression level and analysis results from scRNA.
Hi-C data analysis
All HiC data have been processed by the Juicer pipeline (version 1.5)(Durand et al., 2016) against mouse genome mm9. One of our adult mouse retinal samples was sequenced on the Illumina GAIIx and all other samples were sequenced on the Illumina Novaseq. We confirmed the Hi-C profiles were reproducible before merging the files using HiC-spector (version 201706028 from github)(Yan et al., 2017) (Fig. S1C). Mouse cortex raw data were downloaded from GEO: GSE35156. All 5 samples had more than 4.9 billion contacts and 3 samples had >7.5 billion contacts providing a resolution less than 1 kb. We plot distance of anchors from deduplicated contacts output by Juicer and observed similar trend as contact per million total contacts reduced along the increasing distance (Fig. S1D). We then extract contact counts at 100KB or 250KB resolution from the hic file by straw (from Juicer pipeline), we excluded anchors distance more far than 5 bins and calculated their pearson correlation coefficient. Heatmap of their hierarchical clustering shown the Adult sample is closer to Nrl-GFP adult Rod cells or non-Nrl-GFP adult cells than E14.5 or P0 cells (Fig. S1E).
Next we called TAD boundaries and insulation scores by matrix2insulation.pl (Crane et al., 2015) and merged TAD boundaries from 5 samples, then we extract insulation scores for each sample at the merged boundaries and calculated the pearson correlation coefficient. We observed similar trend (Fig. S1F). Consistently, eigenvector values calculated by Juicer at 100KB resolution also shown similar clustering (Fig. S1G).
For compartment analysis, we first excluded windows with eigenvector value between - 0.01 and 0.01. Then we merged windows with the same sign of eigenvector values so we have two set of regions either were positive eigenvector region or negative. At last, for each chromosome, we calculated the total size of H3K4me3 retina development peaks overlap positive/negative eigenvector regions and assign the one overlap with larger H3K4me3 regions as Compartment A (the active compartment, mostly euchromatin). The other eigenvector regions were assigned as compartment B (the repress compartment, mostly heterochromatin).
Stable loops were identified using HiCCUPS(default parameters) from Juicer at 5kb and 10kb resolution for the highest quality (MAPQ > 30) contacts, 370~640 loops were called from each sample and those that were less than 3 Mb were included in the visualization online. To confirm the loops called were good, we first used APA from Juicer to plot the genome-wide aggregate plot at 5KB resolution for the loops called, the profile and Peak to Lower Left(P2LL) ratio indicated the contact were indeed enriched for the loops called except mCortex sample that were low resolution (Fig. S3). We then plot APA using loops from one sample but Hi-C contacts from another sample, we always observed lower P2LL value compared to using Hi-C contacts of itself, that indicated the loops called were sample specific (data not shown).
To ensure we didn’t missed any loops, we also called loops with less stringent cutoffs for HiCCUPS(lower FDR to 2%, lower sum threshold for merge to 0.05, lower horizontal and vertical threshold for merge to 1.25, lower donut threshold for merge to 1.5, or lower all these parameters). By assuming a loop have CTCF peaks would be more likely to be a real loop, we checked the true positive rate and find only lower all these parameters could call many more loops, although along with lower true positive rate.
For contact based analysis, we called and merged H3K4me3, H3K27ac peaks from all retina development stages and generated all combinations between H3K4me3 peaks and H3K27ac peaks within 1Mb by windowBed from bedtools(version 2.24.0) (Quinlan and Hall, 2010). Later, Hi-C raw contact after remove duplicates from Juicer were converted to pgl format and counted for each H3K4me3-H3K27ac peak pair by Pgltools(Greenwald et al., 2017). We also provide normalized counts by library size as total counts summaried for all peak pairs. Observed the anticorrelation of counts and distance between H3K4me3 and H3K27ac peaks, we further provide normalized counts by peak sizes of both H3H4me3 and H3K27ac peaks and log10 of distance between midpoint of peaks.
Computational analysis of TAD boundaries is dependent on the computational pipeline(Forcato et al., 2017). We chose Armatus because it had the best overlap of TAD boundaries and CTCF binding sites in our dataset. The TADs called by Armatus using both normalized method(KR:Knight-Ruiz, VC_SQRT:square root of vanilla coverage) had a similar trend of increased TAD number over development. 100K resolution achieved the best balance between TAD number and the enrichment of CTCF sites at TAD boundaries. We also noticed some large regions enriched for Hi-C contacts that were not called as TADs either by KR or VC_SQRT, so we developed a “vote” mechanism to merge the TADs called by KR to VC_SQRT only if the TADs called by KR did not overlap with TADs called by VC_SQRT.
An epigenetic-based classifier for euchromatin/heterochromatin domain prediction
We integrated global epigenetic signatures of multiple histone marks and transcription factors of retina cells at various developmental stages and explored the feasibility of predicting the euchromatin/heterochromatin domain status from epigenetic and transcriptomic features. Although the experimental euchromatin mark (H3K4me3) was included in the epigenetic modeling, our chromHMM model estimated that only 1.80% (range: 1.47–2.15%) of the genome have strong H3K4me3 signal (States 1 and 2), which is substantially lower from the experimentally estimated fraction of euchromatin domains. Therefore, H3K4me3 alone is not sufficient to classify euchromatin and heterochromatin on a genome-wide scale.
Previous studies have found that TADs overlap with linear chromatin domains defined by histone modifications and TAD boundaries are somewhat conserved across cell types. As a first step, we evaluated the performance of a predictive model using features extracted from chromHMM modeling to determine the euchromatin/heterochromatin status of predefined TADs (based on mouse cortex). The analysis of 103 genomic regions that were analyzed by FISH suggested that ChromHMM based features provided accurate and robust prediction of TAD-based euchromatin/heterochromatin status (accuracy: 0.89 +/− 0.09 in 10-fold cross validation). Inclusion of additional genomic/epigenomic/transcriptomic features did not improve performance (0.89 +/− 0.09). The euchromatin/heterochromatin prediction improved to 0.95 when we removed the TAD boundary constraint and this is consistent with sub-TAD regions of euchromatin and heterochromatin. On an independent validation set of 161 genes/regions we achieved an accurace of 0.89.
Based on the accuracy of the original model, we next generated a model based on all 264 (103+161) genes/regions collected in P21 retinal cells, which was used to derive the genomewide probability of being euchromatin using ChromHMM features in a sliding-window of 200kb with a step size of 20kb. We further applied the p21 model to epigenetic data collected on E14.5 retina cells to evaluate whether the classifier is robust in data collected independently. Although the selected regions were biased towards euchromatin domains in both training (68%) and validation (70%) sets, the genomwide prediction revealed that majority of genome found were in heterochromatin regions for both E14.5 (62.9%) and P21 (65.4%) cells. P21 cells packaged more genomic regions to heterochromatin domains. The predicted euchromatin/heterochromatin topology is consistent with the high-resolution HiC data, providing independent validation of the modeling data. Further result suggested that while most TADs were dominated by one chromatin type (heterochromatin or euchromatin), occasionally these two domains co-existed in a single TAD. Moreover, although most TADs are relatively stable among different developmental stages, a subset of them displayed dynamic boundaries.
ChromHMM features:
For a defined region (TAD or sub-TAD region) in the genome, 11 features were derived, representing the fraction of the region covered by each of the 11 chromatin states.
Other TAD level features analyzed:
Distance from nearest TAD boundary
TAD size
Epigenetic state of TAD
Distance from nearest expressed gene (FPKM > 10)
Distance from nearest enhancer
LINE repeat density at gene + promoter
SINE repeat density at gene + promoter
LINE repeat density in region
SINE repeat density in region
ATAC-Seq peak density at gene + promoter
ATAC-Seq peak density in region
Expression level of targeted gene
DNA methylation of targeted gene (promoter and whole gene)
We generated an ensemble classifier combining three component models: Linear SVM (Scholkopf et al., 2000), PCA Logistic Regression L2 (Fan et al., 2008; Tipping and Bishop, 1999), and Random Forest (Breiman, 2001). For each component, a model was trained to predict the probability of a given region in euchromatin. The prediction from the ensemble classifier is the average of the individual probabilities of each component. The algorithms were implemented in Sci-Kit Learn (Pedregosa, 2011).
Using the 103 training genes/regions, we evaluated multiple feature sets using 10-fold cross validation (CV): 1) All Features, 2) Features based only on ChromHMM[1–11] for TAD regions, 3) Features based only on ChromHMM[1–11] for probe regions, and 4) Features without Chrom_HMM[1–11]. We performed 10-Fold Cross Validation and report the results of the different feature sets.
| Feature Set | CV Accuracy (mean +/− stdev) |
|---|---|
| ALL | 0.885 +/− 0.092 |
| TAD Level ChromHMM features | 0.885 +/− 0.092 |
| Regional Level ChromHMM features | 0.866 +/− 0.104 |
| ALL without chromHMM features | 0.827 +/− 0.098 |
We trained an ensemble classifier using the regional-level ChromHMM features on the entire training set (103 training genes/regions) to predict chromatin state. The performance of the classifier was evaluated on a separate independent validation set of 161 genes/regions, not seen in the classifier building step.
We generated a final classifier using all 264 genes/regions (combined from training and validation sets) in P21 retina cells and predicted the probability of being in euchromatin for genome-wide regions in both P21 and E14.5 retinas using a sliding window of 200kb with a step size of 20kb.
RNA-Seq Analysis
FASTQ sequences were mapped to the mouse mm9 (MGSCv37 from Sanger) or human genome hg19(GRCh37) by StrongARM, developed for the PCGP (Downing et al., 2012). To estimate the FPKM values, we first downloaded GTF files(mouse vM7, human v23) from GENCODE (Downing et al., 2012) website and liftOver them to mm9 or hg19 by CrossMap version 0.1.5 (Zhao et al., 2014). Then only “transcript” and “exon” records from the GTF files were retained for compatible of feed to cuffdiff (from Cufflinks package version 2.1.1) along with default parameters and “--frag-bias-correct --multi-read-correct -time-series “.
Four gene sets (Rod, Progenitor, G2/M, Housekeeping) that were previously identified and validated by single-cell gene expression analysis (Cherry et al., 2009, Trimarchi et al., 2007 and Trimarchi et al., 2008) were clustered by their correlation of log2 (FPKM) across mouse development and the largest cluster has been used for Fig.2.
ChIP-Seq Analysis
We first employ BWA (version 0.5.9-r26-dev, default parameter) to align the ChIP-Seq reads to mouse genome mm9(MGSCv37 from Sanger), Picard(version 1.65(1160)) then have been used for marking duplicated reads. Then only non-duplicated reads with have been kept by samtools (parameter “-q 1 -F 1024” version 0.1.18 (r982:295)). We followed the ENCODE criterion to quality control (QC) the data that non-duplicated version of SPP (version 1.11) have been used to draw cross-correlation and calculated relative strand correlation value (RSC) under support of R (version 2.14.0) with packages caTools(version 1.17) and bitops(version 1.0–6) and estimated the fragment size. We required > 10M unique mapped reads for point-source factor (H3K4me2/3, H3K9/14Ac, H3K27Ac, CTCF, RNAPolII, BRD4) and RSC > 1. We required 20M unique mapped reads for broad markers (H3K9me3, H3K27me3, H3K36me3). We required 10M unique mapped reads for INPUTs and RSC < 1. We noticed H3K4me1 is point-source factor in some stages while broad in other stages so we QC H3K4me1 as broad markers. Then upon manually inspection, the cross-correlation plot generated by SPP, the best fragment size estimated (the smallest fragment size estimated by SPP in all our cases) were used to extend each reads and generate bigwig file to view on IGV (version 2.3.40). All profiles were manually inspected for clear peaks and good signal to noise separation. For mouse data, all data broad markers with RSC < 0.8 have biological replicates except several samples with one of antibody for H3K9me3(due to availability of outsourcing), we think the quality of those H3K9me3 are good since they show clear peaks and RSC are bigger than most of the H3K9me3 data available in ENCODE or Epigenomics Roadmap(details of QC matrix could be found in supplementary table S1).
To estimate the consistence between biological replicates, for point-source factors, MACS2 (version 2.0.9 20111102, option “nomodel” with “extsize” defined as fragment size estimated above) have been used to call peaks with FDR corrected p-value cutoff 0.05(strong peak set) and 0.5(weak peak set) separately, peaks within 100bp have been merged by bedtools (version 2.17.0). We required > 80% of strong peak set from either one replicate overlapping the weak peak set from the other replicate (median percentage 96.8%). For broad peaks, SICER (version 1.1 redundancy threshold 1, window size 200bp, effective genome fraction 0.86, gap size 600bp, FDR 0.00001 with fragment size defined above) has been used for domain calling and we required > 70% of domains called overlapping the domains called from the other replicate(median percentage 89.9%). After confirmed the consistency between replicates, we pool extended reads and generate the final bigwig track for visualization.
Chromatin State Modeling
Non-duplicated aligned reads were extend by fragment size define above and ChromHMM (version 1.10, with “-colfields 0,1,2,5 -center“ for BinarizeBed) was used for chromatin state modeling. To choose the state number we first modeling all mouse development stage together from 7 states to 33 states. We want choose the state number most correlated to the biological event as greatest up/down regulation of genes here. So we take the FPKM values from RNA-seq then remove all genes that are FPKM <1 across all stages of mouse development. Of those that remain, genes FPKM is <1 at a particular stage have been normalized to 1. Than for 311 genes down 10 fold from P0 to P21 and 366 genes up 10 fold from P0 to P21, we tested the significance that each state in each model occupied different percentage of gene and flank region (from −2kb of TSS to +2kb of TES) by t-test. Order the state and model by smallest p-value we found larger percentage of state 9 in model 11 at P0 is greatest associated with genes down-regulated 10 fold from P0 to P21 while larger percentage of state 1 in model 11 at P0 is greatest associated with genes up-regulated 10 fold. We repeat similar analysis for genes up/down 10 fold from E14.5 to P14, model 11 is still top in all the models so we choose HMM model with 11 states.
For better visualization of the dynamics of HMM state across stages, we normalized color intensity by the max total percentage of a state covered a gene and flanking region across mouse development and tumor. To determine the best region represent a gene, we first filtering annotated isoforms by TSS within 2kb of any H3K4me3/H3K4me2/H3K27Ac/H3K9–14Ac peaks at any development stages and then we selected the highest expressed isoform at any development stages estimated by cuffdiff or the longest isoform if no expression level estimated by cuffdiff. At last, we reduced the interval for a HMM state to half bar and the intensity to half of the normalized intensity if it didn’t ranked top 2 HMM state for a gene. For HMM states could be assigned by multiple genes, the max total percentage across genes have been used for these normalization.
For the HMM state in Human, after processing of ChIP-seq data similar to mouse, we applied the Mouse 11 states HMM model to human data. The selection of isoform is similar but using human data while the max total percentage used for normalization of intensity is based on mouse if there is mouse ortholog for that gene.
Imaging
Confocal Image Analysis of FISH Probes
Images were taken with the Zeiss LSM 700 confocal microscope using 63X lens. To map the FISH probe nuclear position, machine-learning methods were employed (Schindelin et al., 2012) to segment different nuclear region types (core: blue channel, DAPI-ring: blue channel, green ring: green channel, FISH spots: red channel) in each 2D plane of the z-stacks. The machine-learning classifier uses 85 feature images for each plane that are created based on texture, gray-scale intensity, and neighborhood information. Then, the 3DROIManager ImageJ plug-in (Ollion et al., 2013) was used to calculate the common surface area between segmented regions in the 3D segmented image. Values of common surface area to identify FISH spots inside each region were used.
3DEM fixation and processing
The samples for 3DEM were fixed in 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1M cacodylate buffer overnight and rinsed in same buffer. The tissue was stained using a modified heavy metal staining method (https://ncmir.ucsd.edu/sbem-protocol) and processed through a graded series of alcohol, propylene oxide and infiltrated in propylene oxide /epon gradients. The tissue was then infiltrated overnight in 100% Epon 812 resin and polymerized for 48 hours in a 70°C oven.
Preparation steps for focused ion beam scanning electron microscopy imaging
The sample block is mounted on an aluminum pin stub with a conductive silver epoxy, and sputter-coated with a thin layer (~ 60nm) of iridium to electrically ground the sample reducing charging. The block face is scanned to locate the ROI. The block is trimmed to the ROI using an ultramicrotome with a diamond knife. Next a relief is milled into the block face using a suitable ion beam current, and a protective cap is deposited using the carbon gas injection system. Fiducials are created to aid the computer vision pattern placement algorithm.
Focused ion beam scanning electron microscopy
The samples are imaged and processed for 3D data collection on a Helios G3 system. Imaging is normally at 2kV, 100pA, 5×5×5nm, ICD. Regions of interest are imaged utilizing Auto Slice and View 4 software package to automate serial sectioning and data collection processes.
Preparation steps for serial blockface scanning electron microscopy imaging
The sample block is mounted on an aluminum pin stub with a conductive silver epoxy, and sputter-coated with a thin layer (~ 30nm) of iridium to electrically ground the sample reducing charging. The block face is scanned to locate the ROI. The block is trimmed to the ROI using an ultramicrotome with a diamond knife.
Serial blockface scanning electron microscopy
The samples are imaged and processed for 3D data collection on a Teneo Volumescope. Imaging is performed at 1.65kV, 800pA, 15×15×50nm voxel.
Image segmentation and 3D reconstruction
We used the ilastik machine-learning platform(Sommer et al., 2011) to segment the representative nuclei and their heterochromatin domains in the 3D EM volumes. We then used Paraview to reconstruct the segmented nuclei in 3D(Ahrens et al., 2005).
Scoring Lbr-deficient heterochromatin in 2D electron microscopy
Images of retinae were taken on the Tecnai T20 and heterochromatin aggregate attachment was scored for each nucleus as tethered or untethered based on contact with the nuclear lamina. Nuclei and chromatin aggregates in the inner nuclear layer were traced by hand, and their areas quantified by ROI measurement in Fiji. Nuclei bearing classic features of horizontal cells or Müller glia were excluded. The same method was used to score the Lbr-lox heterochromatin.
Scoring Lbr-deficient heterochromatin in 3D electron microscopy
3D data from both KO and control retinas included stacks of 400 – 500 individual images or tiles to form a Tile Set, with each individual tile containing cross-sections of approximately 90 nuclei within the inner nuclear layer. Three Tile Sets from adjacent areas of both KO and control retinas were obtained and each tile analyzed individually in order to identify appropriate representative nuclei from horizontal, amacrine and bipolar cells (Müller cells were excluded in the analysis). Only nuclei whose complete circumference could be observed within the 400 – 500 images of a given Tile Set were scored. Each nucleus was scored as a) having all heterochromatin attached to the interior margin of the nuclear envelope (tethered), b) having all heterochromatin unattached/free-floating within the nucleus (un-tethered), or c) having a mixture of tethered and untethered heterochromatin. The total number of individual heterochromatin clumps within each nucleus was also determined.
Scoring of Vsx2-CRC-SE knockout
Individual images from a sequential, block-face of the outer retina from wild type (a total 509 images) and knockout samples (a total of 490 images) were analyzed. For analysis of spherules, three areas, each containing cross sections of a minimum of 10 spherules, provided a total of 30 spherules each for scoring wild type and knockout samples. For each of the three areas, the thirty adjacent images containing the full set of cross sections of the ten selected spherules were then examined in order to determine the presence or absence of synaptic vesicles within all invaginating processes. Because of the relatively large size of pedicles, the entire series of sequential, block-face images was used to select six representative examples of pedicles from each wild type and knockout sample. All adjacent images containing the full set of cross sections of pedicles from the six wild type and six knockout samples were analyzed for presence or absence of synaptic vesicles within all invaginating processes.
Combining immunofluorescence protein staining with DNA FISH
Freshly cut unfixed cryosections were fixed in 1% PFA in PBS for 5 minutes, followed by an additional fixation in 1% PFA and 0.05% NP40. Fixed samples were stored in 70% ethanol at - 20⁰C until needed. Following fixation slides were subjected to immunostaining by first blocking in 1% BSA and 2X SSC for 10 minutes followed by protein detection using various antibodies at appropriate dilutions in the same blocking solution. Detections were carried out at RT for 45 minutes followed by washing in PBS for 5 minutes. Appropriate secondary antibodies were applied using the same procedure as for primary antibodies. Slides were then fixed in 4% PFA and 0.5% Tween 20 and 0.5% NP40 for 10 minutes, followed by treatment in 0.2N HCl containing 0.5% Triton X-100 for 10 minutes. Slides were then denatured in 70% formamide and 2X SSC at 80⁰C for 10 minutes. Following denaturation, the slides were dehydrated in a graded ethanol series consisting of 70%, 80%, and 100% ethanol for 2 minutes each. Denatured probes were then applied to the slides under 22 X 22 mm coverslips at 37⁰C overnight in a solution containing 50% formamide, 2X SSC, and 10% dextran. Slides were then washed in 50% formamide and 2X SSC at 37⁰C for 5 minutes followed by mounting in Vectashield containing DAPI.
Confocal image modification
Brightness and contrast were modified for images presented in the figures for the FISH/IF studies. Raw original data are available for all datasets and probes.
Localization of FISH Spots to Nuclear Domains
We used Weka Trainable Segmentation(Arganda-Carreras et al., 2017) in FIJI(Schindelin et al., 2012) to segment FISH spots (one classifier per channel) and to segment chromatin domains (one classifier for three regions: f-heterochromatin, c-heterochromatin and euchromatin).
For single color FISH localization in FISH/IF experiments, we used 3D ROI manager plugin(Ollion et al., 2013) in FIJI to calculate voxel overlaps between FISH spots and chromatin domains. We considered a FISH spot included in a domain when voxels of a FISH spot overlap more than 90% with a domain and partially included when voxels of a fish spot overlap between 40% to 60% with a domain.
For measurements of distance for two-color FISH, we used 3D ROI manager plugin in FIJI to calculate the distances between the border of FISH spots within a cell in the two-color FISH images. We used gray-scale watershed(Sage and Unser, 2003) to segment individual cells using DAPI channel. In these images, we only included cells that contained exactly four FISH spots, two in red channel and two in green channel that overlap 90% or more, for distance measurements. Two FISH spots that have zero distance between their borders are touching pairs. We used the distance between spot borders and the following definition to classify cells (or interaction between spots in a cell):
positive: the cells with exactly two pairs of touching red-green FISH spots
negative: no touching spots
unknown: one touching pair
QUANTIFICATION AND STATISTICAL ANALYSIS
The statistical details of experiments can be found above in the METHODS DETAILS section for each type of analysis.
DATA AND CODE AVAILABILITY
All of the software for the analysis is freely available and is described above in the METHODS DETAILS section for each type of analysis. All sequence data has been deposited in the GEO database with the following accession number: GEO GSE87064.
ADDITIONAL RESOURCES
We have developed a new website so users can browse our integrated data for any genomic region. The site is: https://pecan.stjude.cloud/proteinpaint/study/retina_hic_2018
Supplementary Material
Table S8 related to Figure 7. Single cell RNA and ATAC-seq.
Table S9 related to Figure 8. Analysis of bipolar cell gene expression.
Table S1 related to Figure 1. Hi-C metrics and QC.
Table S2 related to Figure 1. Chromatin domain analysis.
Table S3 related to Figure 2. Subnuclear localization of genes and enhancers in rods.
Table S4 related to Figure 3. Dynamic chromatin compartment analysis.
Table S5 related to Figure 4. Gene expression analysis in Lbr-deficient retinae.
Table S6 related to Figure 4. Nuclear morphology in Lbr-deficient retinae.
Table S7 related to Figure 5. Looping interactions in the developing retina.
HIGHLIGHTS.
There are dynamic changes in chromatin compartments and looping during retinogenesis.
Euchromatin/heterochromatin localization can be predicted by a machine learning.
Vsx2 has a cell type– and stage–specific core regulatory circuit super-enhancer.
Vsx2 core regulatory circuit super-enhancer deletion eliminates bipolar neurons.
ACKNOWLEDGEMENTS
We thank Angie McArthur for editing the manuscript. This work was supported, in part, by Cancer Center Support (CA21765) from the NCI, grants to M.A.D from the NIH (EY014867 and EY018599 and CA168875), and ALSAC. M.A.D. was also supported by a grant from Alex’s Lemonade Stand Foundation for Childhood Cancer, the Tully Family Foundation, and the Peterson Foundation. The majority of this research was supported by the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ahrens James, Geveci Berk, Law, and Charles (2005). ParaView: An End-User Tool for Large Data Visualization. Visualization Handbook
- Akimoto M, Cheng H, Zhu D, Brzezinski JA, Khanna R, Filippova E, Oh EC, Jing Y, Linares JL, Brooks M, et al. (2006). Targeting of GFP to newborn rods by Nrl promoter and temporal expression profiling of flow-sorted photoreceptors. Proc Natl Acad Sci U S A 103, 3890–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldiri I, Xu B, Wang L, Chen X, Hiler D, Griffiths L, Valentine M, Shirinifard A, Thiagarajan S, Sablauer A, et al. (2017). The Dynamic Epigenetic Landscape of the Retina During Development, Reprogramming, and Tumorigenesis. Neuron 94, 550–568 e510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allshire RC, and Madhani HD (2018). Ten principles of heterochromatin formation and function. Nature reviews Molecular cell biology 19, 229–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arganda-Carreras I, Kaynig V, Rueden C, Eliceiri KW, Schindelin J, Cardona A, and Sebastian Seung H (2017). Trainable Weka Segmentation: a machine learning tool for microscopy pixel classification. Bioinformatics 33, 2424–2426. [DOI] [PubMed] [Google Scholar]
- Bar-Yosef U, Abuelaish I, Harel T, Hendler N, Ofir R, and Birk OS (2004). CHX10 mutations cause non-syndromic microphthalmia/ anophthalmia in Arab and Jewish kindreds. Hum Genet 115, 302–309. [DOI] [PubMed] [Google Scholar]
- Benavente CA, McEvoy JD, Finkelstein D, Wei L, Kang G, Wang YD, Neale G, Ragsdale S, Valentine V, Bahrami A, et al. (2013). Cross-species genomic and epigenomic landscape of retinoblastoma. Oncotarget 4, 844–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonev B, Mendelson Cohen N, Szabo Q, Fritsch L, Papadopoulos GL, Lubling Y, Xu X, Lv X, Hugnot JP, Tanay A, and Cavalli G (2017). Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell 171, 557–572.e524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L (2001). Random Forests. Machine Learning 45, 5–32. [Google Scholar]
- Christiansen JR, Pendse ND, Kolandaivelu S, Bergo MO, Young SG, and Ramamurthy V (2016). Deficiency of Isoprenylcysteine Carboxyl Methyltransferase (ICMT) Leads to Progressive Loss of Photoreceptor Function. The Journal of neuroscience : the official journal of the Society for Neuroscience 36, 5107–5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane E, Bian Q, McCord RP, Lajoie BR, Wheeler BS, Ralston EJ, Uzawa S, Dekker J, and Meyer BJ (2015). Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature 523, 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, et al. (2015). Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downing JR, Wilson RK, Zhang J, Mardis ER, Pui CH, Ding L, Ley TJ, and Evans WE (2012). The Pediatric Cancer Genome Project. Nature genetics 44, 619–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Shamim MS, Machol I, Rao SS, Huntley MH, Lander ES, and Aiden EL (2016). Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell systems 3, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faiyaz-Ul-Haque M, Zaidi SH, Al-Mureikhi MS, Peltekova I, Tsui LC, and Teebi AS (2007). Mutations in the CHX10 gene in non-syndromic microphthalmia/anophthalmia patients from Qatar. Clin Genet 72, 164–166. [DOI] [PubMed] [Google Scholar]
- Fan RE, Chang KW, Hsieh CJ, Wang XR, and Lin CJ (2008). A Library for Large Linear Classification. Journal of Machine Learning Research 9, 1871–1874. [Google Scholar]
- Ferda Percin E, Ploder LA, Yu JJ, Arici K, Horsford DJ, Rutherford A, Bapat B, Cox DW, Duncan AM, Kalnins VI, et al. (2000). Human microphthalmia associated with mutations in the retinal homeobox gene CHX10. Nature genetics 25, 397–401. [DOI] [PubMed] [Google Scholar]
- Filippova D, Patro R, Duggal G, and Kingsford C (2014). Identification of alternative topological domains in chromatin. Algorithms for molecular biology : AMB 9, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcato M, Nicoletti C, Pal K, Livi CM, Ferrari F, and Bicciato S (2017). Comparison of computational methods for Hi-C data analysis. Nature methods 14, 679–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin JP, and Hansen KD (2015). Reconstructing A/B compartments as revealed by Hi-C using long-range correlations in epigenetic data. Genome biology 16, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire-Pritchett P, Schoenfelder S, Varnai C, Wingett SW, Cairns J, Collier AJ, Garcia-Vilchez R, Furlan-Magaril M, Osborne CS, Fraser P, et al. (2017). Global reorganisation of cis-regulatory units upon lineage commitment of human embryonic stem cells. eLife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green ES, Stubbs JL, and Levine EM (2003). Genetic rescue of cell number in a mouse model of microphthalmia: interactions between Chx10 and G1-phase cell cycle regulators. Development 130, 539–552. [DOI] [PubMed] [Google Scholar]
- Greenwald WW, Li H, Smith EN, Benaglio P, Nariai N, and Frazer KA (2017). Pgltools: a genomic arithmetic tool suite for manipulation of Hi-C peak and other chromatin interaction data. BMC Bioinformatics 18, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, and Young RA (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann K, Dreger CK, Olins AL, Olins DE, Shultz LD, Lucke B, Karl H, Kaps R, Muller D, Vaya A, et al. (2002). Mutations in the gene encoding the lamin B receptor produce an altered nuclear morphology in granulocytes (Pelger-Huet anomaly). Nature genetics 31, 410–414. [DOI] [PubMed] [Google Scholar]
- Javierre BM, Burren OS, Wilder SP, Kreuzhuber R, Hill SM, Sewitz S, Cairns J, Wingett SW, Varnai C, Thiecke MJ, et al. (2016). Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters. Cell 167, 1369–1384.e1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan VK, Zaveri HP, and Scott DA (2015). 1p36 deletion syndrome: an update. The application of clinical genetics 8, 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DS, Matsuda T, and Cepko CL (2008a). A core paired-type and POU homeodomain-containing transcription factor program drives retinal bipolar cell gene expression. The Journal of neuroscience : the official journal of the Society for Neuroscience 28, 7748–7764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DS, Ross SE, Trimarchi JM, Aach J, Greenberg ME, and Cepko CL (2008b). Identification of molecular markers of bipolar cells in the murine retina. The Journal of comparative neurology 507, 1795–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumbach MR, Satpathy AT, Boyle EA, Dai C, Gowen BG, Cho SW, Nguyen ML, Rubin AJ, Granja JM, Kazane KR, et al. (2017). Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nature genetics 49, 1602–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollion J, Cochennec J, Loll F, Escude C, and Boudier T (2013). TANGO: a generic tool for high-throughput 3D image analysis for studying nuclear organization. Bioinformatics 29, 1840–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong CT, and Corces VG (2011). Enhancer function: new insights into the regulation of tissue-specific gene expression. Nature reviews Genetics 12, 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F (2011). Machine Learning in Python. Journal of Machine Learning Research 12, 2825–2830. [Google Scholar]
- Perkowski JJ, and Murphy GG (2011). Deletion of the mouse homolog of KCNAB2, a gene linked to monosomy 1p36, results in associative memory impairments and amygdala hyperexcitability. The Journal of neuroscience : the official journal of the Society for Neuroscience 31, 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, and Aiden EL (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan S, and Cepko CL (2004). Genetic analysis of the homeodomain transcription factor Chx10 in the retina using a novel multifunctional BAC transgenic mouse reporter. Dev Biol 271, 388–402. [DOI] [PubMed] [Google Scholar]
- Rowan S, and Cepko CL (2005). A POU factor binding site upstream of the Chx10 homeobox gene is required for Chx10 expression in subsets of retinal progenitor cells and bipolar cells. Dev Biol 281, 240–255. [DOI] [PubMed] [Google Scholar]
- Sage D, and Unser M (2003). Teaching Image-Processing Programming in Java. IEEE Signal Processing Journal online
- Sarin S, Zuniga-Sanchez E, Kurmangaliyev YZ, Cousins H, Patel M, Hernandez J, Zhang KX, Samuel MA, Morey M, Sanes JR, and Zipursky SL (2018). Role for Wnt Signaling in Retinal Neuropil Development: Analysis via RNA-Seq and In Vivo Somatic CRISPR Mutagenesis. Neuron 98, 109–126.e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nature methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfelder S, Clay I, and Fraser P (2010). The transcriptional interactome: gene expression in 3D. Current opinion in genetics & development 20, 127–133. [DOI] [PubMed] [Google Scholar]
- Scholkopf B, Smola AJ, Williamson RC, and Bartlett PL (2000). New support vector algorithms. Neural computation 12, 1207–1245. [DOI] [PubMed] [Google Scholar]
- Sentmanat MF, Peters ST, Florian CP, Connelly JP, and Pruett-Miller SM (2018). A Survey of Validation Strategies for CRISPR-Cas9 Editing. Sci Rep 8, 888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar K, Lapan SW, Whitney IE, Tran NM, Macosko EZ, Kowalczyk M, Adiconis X, Levin JZ, Nemesh J, Goldman M, et al. (2016). Comprehensive Classification of Retinal Bipolar Neurons by Single-Cell Transcriptomics. Cell 166, 1308–1323.e1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultz LD, Lyons BL, Burzenski LM, Gott B, Samuels R, Schweitzer PA, Dreger C, Herrmann H, Kalscheuer V, Olins AL, et al. (2003). Mutations at the mouse ichthyosis locus are within the lamin B receptor gene: a single gene model for human Pelger-Huet anomaly. Human molecular genetics 12, 61–69. [DOI] [PubMed] [Google Scholar]
- Smallwood A, and Ren B (2013). Genome organization and long-range regulation of gene expression by enhancers. Current opinion in cell biology 25, 387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovei I, and Joffe B (2010). Inverted nuclear architecture and its development during differentiation of mouse rod photoreceptor cells: a new model to study nuclear architecture. Genetika 46, 1159–1163. [PubMed] [Google Scholar]
- Solovei I, Kreysing M, Lanctot C, Kosem S, Peichl L, Cremer T, Guck J, and Joffe B (2009). Nuclear architecture of rod photoreceptor cells adapts to vision in mammalian evolution. Cell 137, 356–368. [DOI] [PubMed] [Google Scholar]
- Solovei I, Wang AS, Thanisch K, Schmidt CS, Krebs S, Zwerger M, Cohen TV, Devys D, Foisner R, Peichl L, et al. (2013). LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 152, 584–598. [DOI] [PubMed] [Google Scholar]
- Sommer C, Strahle U, Kothe FA, and Hamprecht FA (2011). Ilastik: Interactive Learning and Segmentation Toolkit. Eighth IEEE International Symposium on biomedical Imaging Proceedings, 230–233. [Google Scholar]
- Stewart E, Federico S, Karlstrom A, Shelat A, Sablauer A, Pappo A, and Dyer MA (2016). The Childhood Solid Tumor Network: A new resource for the developmental biology and oncology research communities. Dev Biol 411, 287–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipping ME, and Bishop CM (1999). Mixtures of probabilistic principal component analyzers. Neural computation 11, 443–482. [DOI] [PubMed] [Google Scholar]
- Wang L, Hiler D, Xu B, AlDiri I, Chen X, Zhou X, Griffiths L, Valentine M, Shirinifard A, Sablauer A, et al. (2018). Retinal Cell Type DNA Methylation and Histone Modifications Predict Reprogramming Efficiency and Retinogenesis in 3D Organoid Cultures. Cell Rep 22, 2601–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won H, de la Torre-Ubieta L, Stein JL, Parikshak NN, Huang J, Opland CK, Gandal MJ, Sutton GJ, Hormozdiari F, Lu D, et al. (2016). Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 538, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan KK, Yardimci GG, Yan C, Noble WS, and Gerstein M (2017). HiC-spector: a matrix library for spectral and reproducibility analysis of Hi-C contact maps. Bioinformatics 33, 2199–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yardimci GG, Ozadam H, Sauria MEG, Ursu O, Yan KK, Yang T, Chakraborty A, Kaul A, Lajoie BR, Song F, et al. (2019). Measuring the reproducibility and quality of Hi-C data. Genome biology 20, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Sun Z, Wang J, Huang H, Kocher JP, and Wang L (2014). CrossMap: a versatile tool for coordinate conversion between genome assemblies. Bioinformatics 30, 1006–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S8 related to Figure 7. Single cell RNA and ATAC-seq.
Table S9 related to Figure 8. Analysis of bipolar cell gene expression.
Table S1 related to Figure 1. Hi-C metrics and QC.
Table S2 related to Figure 1. Chromatin domain analysis.
Table S3 related to Figure 2. Subnuclear localization of genes and enhancers in rods.
Table S4 related to Figure 3. Dynamic chromatin compartment analysis.
Table S5 related to Figure 4. Gene expression analysis in Lbr-deficient retinae.
Table S6 related to Figure 4. Nuclear morphology in Lbr-deficient retinae.
Table S7 related to Figure 5. Looping interactions in the developing retina.
Data Availability Statement
All of the software for the analysis is freely available and is described above in the METHODS DETAILS section for each type of analysis. All sequence data has been deposited in the GEO database with the following accession number: GEO GSE87064.