

Abstract
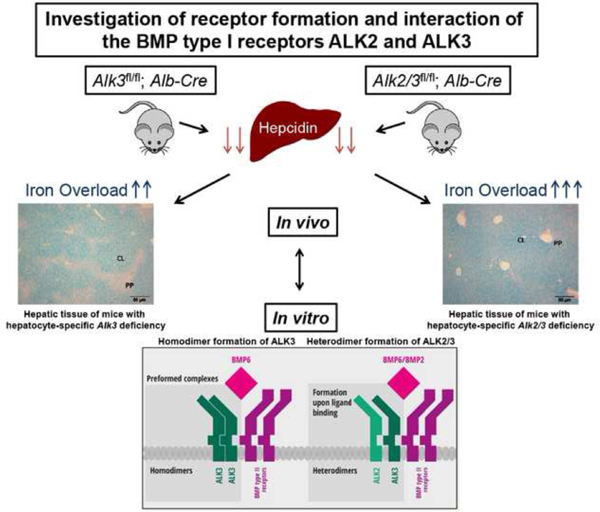
The bone morphogenetic protein (BMP) type I receptors ALK2 and ALK3 are essential for expression of hepcidin, a key iron regulatory hormone. In mice, hepatocyte-specific Alk2 deficiency leads to moderate iron overload with periportal liver iron accumulation, while hepatocyte-specific Alk3 deficiency leads to severe iron overload with centrilobular liver iron accumulation and a more marked reduction of basal hepcidin levels. The objective of this study was to investigate whether the two receptors have additive roles in hepcidin regulation. Iron overload in mice with hepatocyte-specific Alk2 and Alk3 (Alk2/3) deficiency was characterized and compared to hepatocyte-specific Alk3 deficient mice. Co-immunoprecipitation studies were performed to detect the formation of ALK2 and ALK3 homodimer and heterodimer complexes in vitro in the presence and absence of ligands. The iron overload phenotype of hepatocyte-specific Alk2/3-deficient mice was more severe than that of hepatocyte-specific Alk3-deficient mice. In vitro co-immunoprecipitation studies in Huh7 cells showed that ALK3 can homodimerize in absence of BMP2 or BMP6. In contrast, ALK2 did not homodimerize in either the presence or absence of BMP ligands. However, ALK2 did form heterodimers with ALK3 in the presence of BMP2 or BMP6. ALK3-ALK3 and ALK2-ALK3 receptor complexes induced hepcidin expression in Huh7 cells. Our data indicate that: (I) ALK2 and ALK3 have additive functions in vivo, as Alk2/3 deficiency leads to a greater degree of iron overload than Alk3 deficiency; (II) ALK3, but not ALK2, undergoes ligand-independent homodimerization; (III) the formation of ALK2-ALK3 heterodimers is ligand-dependent and (IV) both receptor complexes functionally induce hepcidin expression in vitro.
Keywords: Iron overload, Liver, Ferroportin, Hepcidin, BMP type I receptor
Graphical Abstract
Introduction
The bone morphogenetic protein (BMP) signaling pathway is essential for the induction of hepcidin, the main regulator of systemic iron homeostasis. Hepcidin is induced by iron, BMPs, and inflammation.[1, 2] The BMP receptor complex is a tetramer consisting of two type I and two type II receptors [3], although the identity of each receptor in the complex has not yet been determined. Binding of ligands to the receptor complex activates SMAD signaling. The SMAD complex, composed of Smad1/5/8 and Smad4, translocate to the nucleus and, upon binding to the two BMP response elements (BMP-RE1 and 2) in the hepcidin promoter, induces hepcidin expression.[4] The dominant ligand is BMP6, which is produced in liver endothelial cells.[5] In addition to the BMP-SMAD signaling pathway, hepcidin expression is also induced by cytokines, especially IL-6, via the JAK/STAT3 signaling pathway. Phosphorylated STAT3 binds to the STAT3 response element (STAT3-RE) located downstream of BMP-RE1 in the hepcidin promoter.[6]
The BMP type I receptors ALK2 and ALK3 are expressed in hepatocytes and are required for optimal hepcidin induction. Mice with hepatocyte-specific deficiency of Alk2 develop moderate iron overload.[7] Iron- and BMP-mediated hepcidin induction is impaired in Alk2-deficient mice, while IL-6-mediated hepcidin induction is intact.[8] In contrast, hepatocyte-specific Alk3-deficient mice develop severe iron overload, hepcidin deficiency, and impaired hepcidin induction by iron, BMPs, and IL-6.[8, 9] The distribution of iron accumulation in the liver differs between hepatocyte-specific Alk2- and Alk3-deficient mice: iron accumulates in a periportal distribution in hepatocyte-specific Alk2-deficient mice and in a centrilobular distribution in hepatocyte-specific Alk3-deficient mice.[7]
In zebrafish, orthologues of ALK2 and ALK3 have been shown to assemble into a heteromeric receptor complex.[10] Whether mammalian ALK2 and ALK3 form a heterodimeric complex in hepatocytes and thereby induce hepcidin expression has yet to be determined. In this study, the iron overload phenotype of hepatocyte-specific Alk2/3-deficient mice was characterized to determine if ALK2 and ALK3 have distinct or redundant functions in systemic iron homeostasis. Compared to mice with hepatocyte-specific Alk3 deficiency, mice with hepatocyte-specific Alk2/3 deficiency developed a more severe iron overload phenotype with increased liver iron content and a distinctive liver iron accumulation pattern that correlated with ferroportin expression pattern in male and female mice. Extrahepatic iron content in the kidney, pancreas and heart of female mice with hepatocyte-specific Alk2/3 deficiency was higher than in mice with hepatocyte-specific Alk3 deficiency. Co-immunoprecipitation assays were used to elucidate receptor dimerization and the role of dimerization in hepcidin regulation. The in vitro studies indicate that ALK3 is the dominant BMP type I receptor in terms of hepcidin regulation. We present the first evidence of homo- and hetero-dimerization of the BMP type I receptors in a human hepatocyte cell line. ALK3 forms homodimers in the absence of ligand. Heterodimers are formed with ALK2 upon binding of BMP2 or BMP6. Both ALK3-ALK3 and ALK2-ALK3 complexes are able to induce hepcidin expression in vitro.
Materials and Methods
Animals
The current study was carried out in strict accordance with the recommendations and approval of the institutional ethics committee of the North Rhine-Westphalian Agency for Nature, Environment, and Consumer Protection (permit no. Az.84–02.05.2012.181). Mice with homozygous loxP-flanked (“floxed”) Alk2 (Alk2fl/fl)[7, 11] and Alk3 alleles (Alk3fl/fl)[7, 12, 13] on a C57BL/6 background were bred with B6.Cg-Tg(Alb-Cre)21Mgn/J mice to obtain homozygous animals (Alk2/3fl/fl) with and without a Cre recombinase driven by the hepatocyte-specific albumin promoter. Mice were held in individually ventilated cages and fed a standard diet with an iron content of 198 ppm. To generate mice for this study, Alk3fl/fl and Alk2/3fl/fl female mice were bred with Alk3fl/fl; Alb-Cre and Alk2/3fl/fl; Alb-Cre male mice, respectively. Twelve-week-old littermates with hepatocyte-specific deficiency of Alk3 (Alk3fl/fl; Alb-Cre) were compared to mice with hepatocyte-specific Alk2 and Alk3 (Alk2/3fl/fl; Alb-cre) deficiency of the same gender.
Cell culture
The human hepatocellular carcinoma cell line Huh7 was a kind gift from Martina U. Muckenthaler (Heidelberg, Germany). Cells were cultured in high glucose DMEM (Sigma-Aldrich, Hamburg, Germany) supplemented with 10% (v/v) fetal calf serum (heat inactivated, Capricorn, Ebsdorfergrund, Germany), 2 mM glutamine, 100 units/mL penicillin, and 100 µg/mL streptomycin (Sigma-Aldrich, Hamburg, Germany). Cells were grown at 37°C and 5% CO2.
Hematologic and iron parameters
Retro-orbital blood draw was performed under deep ketamine/xylazine anesthesia, and all efforts were made to minimize suffering. Serum iron concentrations, unsaturated iron binding capacity (UIBC), and transferrin saturation were determined using the Iron/UIBC Kit (Sekisui Diagnostics, Charlottetown, Canada) following the manufacturer’s protocol. Complete blood counts were obtained using a scil Vet abc Plus+™ analyzer (Viernheim, Germany). Non-heme tissue iron levels were determined as previously described.[14]
Total reflection x-ray fluorescence (TXRF).
Total liver iron content was determined by total reflection x-ray fluorescence (TXRF). The liver samples were weighed and digested with nitric acid over night at 95°C. Dried samples were re-dissolved in purified water and aliquots of 100 µL were mixed with the same volume of the 10 mg/L arsenic standard solution. Measurements were carried out on a S2-PICOFOX instrument (Bruker Nano, Berlin, Germany) with an air-cooled molybdenum anode for X-ray generation. The excitation settings were 50 kV and 750 µA and quartz glass disks were used as sample carriers. As internal standard, arsenic with a concentration of 10 mg/L was applied. Aliquots of 5 µL of the samples were placed on the sample carriers and evaporated to dryness. The analysis was performed by signal integration over 500 seconds. For the determination, the signal of iron (Kα1 = 6.405 keV) and arsenic (Kα1 = 10.543 keV), as internal standard, were used. Quantification was performed by the Bruker Spectra software (version 6.1.5.0) and based on the known concentration of the internal arsenic standard.
Hepatic mRNA levels
RNA was extracted from tissue using Trizol® (Sigma, Hamburg, Germany) and homogenized with an ultrasonic homogenizer. MMLV-reverse transcriptase (Sigma, Hamburg, Germany) was used to synthesize cDNA. Quantitative RT-PCR was performed on a Bio-Rad CFX Connect™ Real-Time-PCR system using iTaq™ Universal SYBR® Green Supermix (Bio-Rad, Munich, Germany). The relative CT method was used to normalize the levels of target transcripts to 18S rRNA levels. Primer pairs used for semi-quantitative RT-PCR are listed in Table S1.
Hepcidin expression analysis
Huh7 cells were seeded in six-well plates at a density of 1.5×105 cells/well and were transfected 15 hours later with a total of 2.5 µg DNA using the TransIt®-LT1 transfection reagent (MoBiTec, Gottingen, Germany). Twenty-four hours after transfection, cells were harvested and RNA was isolated using Trizol® (Sigma, Hamburg, Germany). Hepcidin levels were measured as described above.
For BMP6 treatment, 24 hours after transfection, cells were serum starved (cell growth medium with 0.1% (v/v) FCS) for three hours and stimulated with 10 ng BMP6 for 90 minutes at 37°C. RNA was isolated afterwards and used for cDNA synthesis.
Tissue iron staining
Paraffin-embedded tissue sections were stained with Perls’ Prussian blue stain to detect non-heme iron. Sections were stained for 15 minutes in 5% (w/v) potassium ferrocyanide with 5% (v/v) hydrochloric acid. Subsequently, slides were stained with 0.1% nuclear-fast red (Chroma Technology, Olching, Germany) in a 5% (w/v) aluminum sulfate (Merck, Darmstadt, Germany) solution. The results of histological staining were documented using an Olympus BX51P microscope and ColorView Soft Imaging System with the analySIS software (Olympus, Hamburg, Germany).
Immunohistochemistry
Paraffin-embedded tissue sections were deparaffinized and rehydrated. Heat-mediated antigen retrieval was performed using Tris/EDTA buffer at a pH of 8. Sections were blocked in PBS with 1% BSA and 10% FCS for one hour and then incubated with rabbit-anti-mouse MTP1/FPN IgG (dilution 1:50; Alpha Diagnostics, Cologne, Germany) at 4°C in a humid chamber overnight. After three washing steps, sections were incubated with FITC-conjugated goat-anti-rabbit IgG (dilution 1:1000; Sigma-Aldrich, Hamburg, Germany). Autofluorescence was reduced with 10% (w/v) Soudan Black B and sections were mounted with Immu-Mount (Thermo Fisher Scientific, Darmstadt, Germany). Slides were examined using an Olympus BX50 microscope and results were documented with Cell^F software (Olympus, Hamburg, Germany).
Plasmids
Eukaryotic expression plasmids (pcDNA3) encoding human ALK2 or ALK3 with an N-terminal 3xFLAG tag (3xFLAG-ALK3, 3xFLAG-ALK2) and a plasmid encoding human ALK3 with a C-terminal HA tag (ALK3-HA) under the control of a CMV promoter were provided by Patricio Leyton and Donald Bloch (Boston, USA). The plasmid encoding human ALK2 with a C-terminal HA tag (ALK2-HA) was provided by Kohei Miyazono (Tokyo, Japan).
Co-Immunoprecipitations
Huh7 cells were seeded in 10 cm dishes at a density of 16×103 cells/cm2 and were transfected 16 hours later with a total of 15 µg DNA using the TransIT®-LT1 transfection reagent (MoBiTec, Goettingen, Germany). Twenty-four hours after transfection, cells were harvested and lysed in NET-buffer (150mM NaCl; 5mM EDTA; 10mM Tris-HCL, pH 7.4; 0.8% (v/v) Triton X-100). Immunoprecipitation assays were performed using 750 µg of protein lysates and either ANTI-FLAG® M2 Affinity Gel or EZview™ Red Anti-HA Affinity Gel (Sigma-Aldrich, Hamburg, Germany) according to the manufacturers’ instructions. Adherent proteins were eluted with non-reducing SDS sample buffer and separated by electrophoresis and used for immunoblot analysis.
Ligand stimulation and crosslinking
Twenty-four hours after transfection, cells were washed with PBS and serum-starved by incubation in tissue culture medium supplemented with 0.1% (v/v) FCS. Cells were incubated with either 25 ng BMP6 and BMP2 or 100 ng IL-6 (both R&D Systems, Wiesbaden, Germany) for 90 minutes at 4°C. Cross-linking in BMP-treated cells was performed as described previously.[15]
After washing with ice cold PBS, cells were harvested in 200 µl PBS and incubated with 10 µl of 100 mM DSS ((Disuccinimidyl suberate) Thermo Fisher Scientific, Darmstadt, Germany) in DMSO for 30 minutes at RT. The reaction was stopped with Tris at a final concentration of 20 mM and incubated at RT for 15 min. Cells were lysed in a mild lysis buffer (150mM NaCl; 5mM EDTA; 10mM Tris-HCL, pH 7.4; 0.8% (v/v) Triton X-100) and protein extracts were subsequently used in co-immunoprecipitation assays.
Protein analysis
Liver tissue samples were prepared using RIPA (50mM Tris, pH 8; 150mM NaCl; 1% (v/v) Triton X-100; 0.5% (v/v) Sodium deoxycholate; 0.1% (v/v) SDS) buffer containing protease and phosphatase inhibitors (Sigma-Aldrich, Hamburg, Germany). Proteins were separated by electrophoresis using 4%–10% bis-tris gels and blotted on nitrocellulose membranes (GE Healthcare, Freiburg, Germany). Membranes were incubated with antibodies directed against HA (Catalog No: H6908), FLAG (Catalog No: F7425), TfR1 (Catalog No: SAB2502166) (all from Sigma-Aldrich, Hamburg, Germany) and ferritin heavy chain (Cell Signaling Technology, Leiden, Netherlands, Catalog No: 4393). Antibodies directed against α-tubulin (Sigma-Aldrich, Hamburg, Germany, Catalog No: T5168) were used as a control to demonstrate equal loading of total protein. Washed membranes were incubated with horseradish peroxidase (HRP)–conjugated anti-rabbit or anti-mouse IgG (New England Biolabs, Frankfurt, Germany). Membranes were incubated with ECL-Plus (Bio-Rad, Munich, Germany), and chemiluminescence was detected using a ChemiDoc™ XRS+ (Bio-Rad, Munich, Germany).
Statistical analysis
All values are expressed as mean±SD. Data were analyzed using One-way Anova and non-parametric Mann-Whitney U tests with two tailed p- values. Differences were considered statistically significant when levels were p ≤.05 (*) or p≤.01 (**).
Results
Hepatocyte-specific deficiency of Alk2/3 leads to centrilobular hepatocellular iron accumulation with expansion into the periportal area.
To determine the iron overload phenotype of mice with hepatocyte-specific Alk2/3 deficiency compared to hepatocyte-specific Alk3 deficiency, twelve-week old mice with Alk2/3fl/fl (control) and Alk2/3fl/fl; Alb-Cre (hepatocyte-specific Alk2/3 deficiency) were generated and compared to Alk3fl/fl (control) and Alk3fl/fl; Alb-Cre (hepatocyte-specific Alk3 deficient) mice. In Alk3fl/fl; Alb-Cre mice, hepatic Alk3 mRNA levels were reduced by 93% compared to control mice. In Alk2/3fl/fl; Alb-Cre mice, hepatic Alk2 mRNA levels were reduced by 84%, and hepatic Alk3 mRNA levels were reduced by 90% compared to control mice (Supplementary Figure 1 A–B). Histological liver sections were prepared from male mice and stained with Perls’ Prussian blue stain to determine hepatic, non-heme iron accumulation. Control mice did not accumulate iron in the liver (Figure 1 A–B, left panels). Male mice with a hepatocyte-specific Alk2 deficiency are characterized by a moderate iron accumulation phenotype [7] and presented with iron accumulation in the periportal area of the liver (Figure 1C, left panel [7]). Male mice with a hepatocyte-specific Alk3 deficiency had centrilobular iron accumulation with iron-free periportal areas as previously described (Figure 1D, left panel).[7] Interestingly, male mice with hepatocyte-specific Alk2/3 deficiency showed iron accumulation not only in the centrilobular, but also in the periportal area (Figure 1E, left panel). These results indicate that the iron distribution in the liver of hepatocyte-specific Alk2/3-deficient mice is more severe than that in hepatocyte-specific Alk3-deficient mice.
Figure 1. Hepatic iron accumulation pattern and ferroportin expression in periportal and centrilobular areas of male mice with hepatocyte-specific Alk3 or Alk2/3 deficiency.

Left panels show representative formalin-fixed paraffin sections of livers stained with Prussian blue (n=4). Middle and right panels show representative sections of livers stained with a fluorescently tagged antibody against ferroportin. Blue staining (Dapi) corresponds to the nucleus of cells (n=4). Marked areas are shown as magnifications at the right side of each immunofluorescence image. Liver sections of (A) Alk3fl/fl, (B) Alk2/3fl/fl, (C) Alk2fl/fl; Alb-Cre (D) Alk3fl/fl; Alb-Cre, and (E) of Alk2/3fl/fl; Alb-Cre mice are shown. Periportal=PP; centrilobular=CL. Arrowhead indicates Kupffer cells, arrow indicates hepatocytes.
To consider the possibility that the observed differences in the iron accumulation pattern could be due to differences in the distribution of ferroportin (Fpn) in the liver, immunohistochemical analysis of hepatic Fpn expression was performed. In control male mice, very low levels of ferroportin were detected in the centrilobular and periportal areas of the liver (Figure 1 A–B, four panels on the right with magnification of the marked areas). In hepatocyte-specific Alk2 deficiency immunoreactive Fpn was detected mainly in Kupffer cells located in the periportal area of the liver (Figure 1C, first and second panel from the right). The hepatocyte-specific Alk3 in male mice resulted in an increase in immunoreactive Fpn in hepatocytes and Kupffer cells located in the periportal region (Figure 1D, first and second panel from the right). Hepatocyte-specific Alk2/3-deficient male mice had an increase in immunoreactive Fpn in hepatocytes and Kupffer cells in both, the periportal and centrilobular distribution (Figure 1D, right four panels). These results suggest that the pattern of iron accumulation in the liver may be dependent on the Fpn expression pattern.
Mice with hepatocyte-specific deficiency of Alk2 and Alk3 developed a more severe iron overload phenotype compared to Alk3-deficient mice.
As the histological iron accumulation and Fpn expression patterns were distinct in Alk3fl/fl; Alb-Cre and Alk2/3fl/fl; Alb-Cre male mice, the iron status of all genotypes were determined next. The iron overload phenotype was severe in both genotypes as indicated by increased serum iron levels, transferrin saturation, and hemoglobin levels, as well as well a decrease in the UIBC in Alk3fl/fl; Alb-Cre and 14 Alk2/3fl/fl; Alb-Cre male mice when compared to controls (Figure 2 A–C, Supplementary Figure 2A). In order to determine the difference between mice with hepatocyte-specific Alk3 compared with Alk2/3 deficiency, non heme iron content and total iron content (Figure 2 D–E) were measured. The hepatic iron content was higher in mice with hepatocyte specific Alk2/3 deficiency than in Alk3 deficiency. Splenic iron content was decreased in both Alk3fl/fl; Alb-Cre and Alk2/3fl/fl; Alb-Cre male mice when compared to control mice (Supplementary Figure 2B).
Figure 2. Hepatocyte-specific Alk2/3-deficient male mice develop more severe iron overload than hepatocyte-specific Alk3-deficient mice.

(A) Serum iron levels from Alk3fl/fl and Alk3fl/fl; Alb-Cre (white bars) and Alk2/3fl/fl and Alk2/3fl/fl; Alb-Cre mice (black bars) (one-way Anova: **p=0.0008; **p≤0.008; n=5–7), (B) transferrin saturations (one-way Anova: ***p=0.008; **p≤0.008; ***p=0.0008; n=5–7), (C) hemoglobin values (one-way Anova **p=0.0078; *p=0.04; **p=0.009; n=5–7) (D) non-heme liver iron levels (*p=0.0177; **p≤0.008; n=5–7), and (E) total liver iron content (**p≤ 0.001; n=4–6) are shown.
The levels of transferrin receptor (TfR1), which is a protein required for iron uptake that is negatively regulated by iron, were similarly decreased in both Alk3fl/fl; Alb-Cre and Alk2/3fl/fl male mice (Supplementary Figure 3 A–B). Levels of ferritin, an iron storage protein abundantly expressed in iron overload, were increased in Alk3fl/fl; Alb-Cre and Alk2/3fl/fl; Alb-Cre male mice (Supplementary Figure 3 C–D). Despite the differences in non-heme liver iron content in Alk3fl/fl; Alb-Cre and Alk2/3fl/fl; Alb-Cre mice, ferritin levels were comparable between the two genotypes (Supplementary Figure 3 E–F). The data indicate that the amount of iron in the circulation and within the iron storage cells is very high in mice with hepatocyte-specific Alk3 deficiency. Hepatocyte-specific Alk2 deficiency enhances the iron load further, which can – in an already saturated system- only be observed in the liver iron content.
In iron overload caused by genetic mutations, hepatic hepcidin is absolutely or relatively decreased. Hepatic hepcidin mRNA expression in the liver analyzed, which was shown to correlate well with serum hepcidin levels. [18] Hepcidin mRNA expression was markedly reduced in Alk3fl/fl; Alb-Cre and Alk2/3fl/fl; Alb-Cre male mice compared to their corresponding controls (Figure 3 A). There was a trend towards lower hepatic hepcidin mRNA levels in mice with hepatocyte-specific Alk2/3 deficiency compared to hepatocyte-specific Alk3-deficient male mice, which reflects the small difference in the iron overload phenotype. Ferroportin expression in the small intestine and the liver was increased in Alk3fl/fl; Alb-Cre and Alk2/3fl/fl; Alb-Cre mice compared to their corresponding controls (Figure 3 B), which correlates with decreased hepatic hepcidin mRNA expression. As expected, ferroportin was not different between both iron overloaded genotypes.
Figure 3. Ferroportin expression is increased in hepatocyte-specific Alk3 and Alk2/3 deficient mice.

(A) Hepatic hepcidin mRNA levels were normalized to the gene 18S (**p≤0.0079; n=5–7). The actual CT value denoted in Alk3fl/fl mice as ‘1’ is 15.83. (B) Ferroportin expression of control as well as hepatocyte-specific Alk3 and Alk2/3 deficient mice was analyzed in the small intestine and the liver.
We next compared hepatic and extrahepatic iron loading in female mice with hepatocyte-specific Alk2/3 deficiency and hepatocyte-specific Alk3 deficiency. Female mice in general have less iron than male mice of the same age and differences in iron overload in different genotypes are therefore more pronounced and easier to detect.
In twelve week-old female mice, non-heme liver iron content and total liver iron content was higher in hepatocyte-specific Alk2/3 deficient mice compared to Alk3fl/fl; Alb-Cre (Figure 4 A–B). Female mice with hepatocyte-specific Alk2/3 deficiency presented extrahepatic iron accumulation in the kidney, the heart, and the pancreas, while hepatocyte-specific Alk3 deficient female mice did not have extrahepatic iron accumulation (Figure 4 C–E). These data indicate a more severe iron overload phenotype in Alk2/3fl/fl; Alb-Cre compared to Alk3fl/fl; Alb-Cre mice.
Figure 4. Female mice with hepatocyte-specific Alk2/3 deficiency exhibit extrahepatic iron accumulation in the kidney, heart and pancreas.

(A) non heme liver iron content (*p=0.01, **p=0.001, ***p≤0.0001; n=5–10), (B) total liver iron content (*p≤0.03, **p=0.008; n=4–5), (C) non heme renal iron content (*p=0.003, ***p=0.0007, n=5–8), and (D) non heme cardiac iron content (**p=0.001, ***p=0.0007, n=6–8) is shown. (E) The panels show representative formalin-fixed paraffin sections of pancreatic tissue stained with Prussian blue (n=4).
The results show that the iron overload phenotype in hepatocyte-specific Alk2/3-deficient mice compared to hepatocyte-specific Alk3-deficient mice was more severe as indicated by increased liver iron content as well as increased extrahepatic iron accumulation in female mice. In male mice, the more severe iron overload 19 phenotype is also supported by the histological panlobular iron accumulation, the corresponding Fpn expression pattern, and a trend towards decreased hepcidin expression in hepatocyte-specific Alk2/3-deficient mice compared to hepatocyte-specific Alk3-deficient mice. Therefore, ALK2 and ALK3 have additive roles in vivo.
ALK3 forms homodimeric complexes and heterodimeric complexes with ALK2 upon ligand binding.
The characterization of hepatocyte-specific Alk3- and Alk2/3-deficient mice revealed a cumulative effect of ALK2 and ALK3 on iron overload in vivo. As the goal was to determine BMP type I receptor ligand- independent and -dependent complex formation, in vitro co-immunoprecipitation assays were performed. The formation of hetero- and homo-dimeric complexes in the absence of added ligand was studied using co-immunoprecipitation assays in Huh7 cells co-transfected with ALK2-HA and ALK2-Flag, ALK3-HA and ALK3-Flag, or ALK2-Flag and ALK3-HA.
In the absence of any exogenous ligand, ALK3-HA co-immunoprecipitated with ALK3-Flag (Figure 5A). In contrast, ALK3 did not co-immunoprecipitate with ALK2 (Figure 5B), and ALK2-HA did not co-immunoprecipitate with ALK2-Flag (Figure 5C). To consider the possibility that the presence of exogenous ligands may alter receptor dimer formation, interactions between the BMP type I receptors after addition of BMPs or IL-6 were tested. ALK2-HA did not co-immunoprecipitate with ALK2-Flag, despite incubation of Huh7 cells with BMP6 or BMP2 (Figure 5 D–E).
Figure 5. ALK3 forms homodimers in the absence of exogenous ligand and ALK2/3 heterodimers in presence of BMP6.

Huh7 cells were transfected with plasmids as indicated. Co-immunoprecipitation was carried out with Flag beads or HA-Beads followed by western blotting with α-HA antibodies, as indicated. To test transfection and precipitation efficiency, blots were probed with α-Flag or α-HA antibody (n=3). Co-immunoprecipitation, without addition of ligand, of (A) ALK3-Flag and ALK3-HA, (B) ALK3-HA and ALK2-Flag, and (C) ALK2-Flag and ALK2-HA is shown. Co-immunoprecipitation, after addition of BMP6 or BMP2, of (D) ALK2-HA and ALK2-Flag, (E) ALK2-HA and ALK2-Flag, and (F) ALK2-Flag and ALK3-HA is shown. Input (In), Immunoprecipitation (Ip).
In contrast, ALK2-Flag co-immunoprecipitated with ALK3-HA in the presence of BMP6 (Figure 5F) and BMP2 (Supplementary Figure 4). As ALK3 homodimer formation was already observed in the absence of an exogenous ligand, ALK3 21 homodimers were also detected in the presence of BMP6 (data not shown). Stimulation of Huh7 cells with IL-6 did not result in detectable ALK2 homodimerization or heterodimer formation between ALK2 and ALK3 (Figure 6 A–B). ALK3 homodimerization, in contrast, was detected in presence of IL-6 without additional BMP ligand (Figure 6C).
Figure 6. Addition of IL-6 leads to formation of ALK3 homodimers but not ALK2 homodimers or ALK2/3 heterodimers.

Transfected cells were stimulated with IL-6 (100ng/mL) when indicated. Proteins were precipitated with Flag beads and analyzed by western blotting with α-HA antibody. Blots were probed with α-Flag antibody to test transfection efficiency. Co-immunoprecipitation of (A) ALK2-HA and ALK2-Flag, (B) ALK2-Flag and ALK3-HA, and (C) ALK3-HA and ALK3-Flag is shown. Input (In), Immunoprecipitation (Ip). (D) Hepcidin expression in Huh7 cells 24 hours after transfection is shown (*P=.05; **P≤.01) (N=3, n=4–6). (E) Stimulation of serum-starved cells with BMP6 (10 ng/ml) further increased hepcidin expression (*P=.05; **P≤.01) (N=3, n=4–6).
To test whether ALK2-ALK3 and ALK3-ALK3 receptor complexes are functional, hepcidin expression was analyzed in Huh7 cells after co-transfection with different tagged receptor complexes. Without the addition of BMP6, transfection of ALK2-ALK3 and ALK3-ALK3 increased hepcidin expression in Huh7 cells (Figure 6D). Addition of BMP6 to serum-starved cells increased hepcidin expression in cells transfected with an empty vector (pcDNA3). This expression was further increased when cells were transfected with ALK2-ALK3 or ALK3-ALK3. In contrast, transfection with ALK2-ALK2 did not further increase hepcidin expression (Figure 6E). In the absence of BMP6 or IL6, serum-starved cells transfected with either ALK2-ALK3 or ALK3-ALK3 did not increase hepcidin expression, suggesting that both receptor complexes require ligands for signaling (data not shown).
These in vitro data show that ALK3, but not ALK2, can form homodimers in the absence of BMP6 and IL-6. In the presence of BMP6, ALK3 can form both homodimers and heterodimers with ALK2. In the presence of ligands, the presence of both receptor complexes resulted in increased hepcidin expression.
Taken together with the in vivo data, the results suggest that ALK2 interacts with another iron regulatory protein (than ALK3 or HFE) to induce hepcidin expression, as mice with a hepatocyte-specific Alk2/3 deficiency present with a more severe iron overload phenotype compared to hepatocyte-specific Alk3 deficiency.
Discussion
The BMP-SMAD signaling pathway plays an important role in the regulation of hepcidin expression and systemic iron homeostasis. Deficiency of the BMP type I receptors ALK2 and ALK3 leads to iron overload in mice.[7] Because mice with hepatocyte-specific Alk2 deficiency and Alk3 deficiency have different degrees of iron overload and different patterns of hepatic iron accumulation, we investigated whether ALK2 and ALK3 might have additive functions in hepcidin regulation. The results revealed the development of a more severe iron overload phenotype in mice with hepatocyte-specific Alk2/3 deficiency than with either Alk2- or Alk3- deficiency alone. In vitro experiments revealed that ALK3 forms homodimeric complexes as well as ligand-dependent heterodimeric complexes with ALK2. In the presence of ligands, both receptor complexes were associated with increased hepcidin levels in a human hepatocyte cell line.
The iron accumulation pattern in the liver of iron-overloaded mice is different between experimental models. Seventy percent of blood flow enters the liver through the portal vein, and 30% via the hepatic artery. This mixed venous-arterial blood carries iron absorbed from the diet and iron released from macrophages after phagocytosis of senescent red blood cells. As blood passes through the hepatic sinusoids, the accumulation of iron occurs predominantly within periportal fields.[16, 17] The periportal distribution of iron accumulation was observed in Tfr2-, Hfe-, and Alk2-deficient mice.[7, 18, 19] In contrast, Hjv deficiency in male mice leads to centrilobular liver iron accumulation with iron levels that are similar to those observed in mice with hepatocyte-specific Alk3 deficiency.[7, 20] Panlobular iron overload, in contrast, was observed in mice with hepatocyte-specific Alk2/3 deficiency and in male mice with hepatocyte-specific hepcidin (Hamp) deficiency.[21]
Of note, mice in these various studies received different diets (Supplemental Table 3), so that the data have to be interpreted with caution.[7, 20–23] Our work suggests that ALK2 and ALK3, in combination with other factors such as HJV, TfR1 or TfR2, are required for appropriate regulation of hepcidin expression. ALK3 forms homodimers and heterodimers with ALK2. ALK2 itself plays also a functional role and might interact with another iron regulatory protein such as HJV, TfR1 or TfR2. If one or more of these factors are deficient, hepcidin deficiency and iron overload result. Hepatocyte-specific Alk2/Alk3 deficiency reaches higher iron overload than Alk3 deficiency alone. Of note, hepatocyte-specific Alk2 deficiency causes a moderate iron overload only.
Single cell analysis of hepatic gene expression demonstrated that absolute ALK2 mRNA levels are lower than ALK3 mRNA levels. However, both, ALK2 and ALK3, exhibit panlobular expression patterns (Supplementary Figure 5, data from the analysis of Halpern et al. [24]). At the protein level, ALK3 might be predominantly expressed in the periportal area of the liver. Hepatocyte-specific ALK3 deficiency leads to severe decrease in hepcidin expression and a subsequent increase in ferroportin expression in the periportal area of the liver. As a consequence, iron would be transported into the centrilobular area and accumulates there. In mice with hepatocyte-specific Alk2/3 deficiency the FPN expression pattern is panlobular. The loss of both, Alk2 and Alk3, causes the expression of Fpn at similar levels in both periportal and centrilobular areas and iron accumulates in both areas. Future research in the next years will hopefully provide the definite answer to whether the expression pattern of Alk2 and ALK3 are involved in hepatic iron accumulation, once commercially available, reliable antibodies can detect ALK2 and ALK3.
Our data are in line with the hypothesis of Ramey et al.[25], who suggested that differences in iron accumulation patterns may be due to the expression pattern of Fpn. In female Hamp-deficient mice, Fpn is mainly expressed in Kupffer cells as well as in hepatocytes located in the periportal area of the liver. Iron is exported from the periportal hepatocytes into the hepatic sinusoids so that iron is available for centrilobular hepatocytes. Because hepcidin causes Fpn internalization and degradation, severe hepcidin deficiency leads to an increase in periportal Fpn protein levels.[25, 26] A periportal liver iron accumulation might be caused by either sufficient hepcidin levels that lead to the degradation of ferroportin, as observed in Hfe KO mice, or the expression of ferroportin in Kupffer cells, but not in hepatocytes, as it is observed in female Hjv KO mice.[25, 27] Hepatocyte-specific Alk2 deficient mice still express sufficient levels of hepcidin, so that ferroportin expression is only observed in Kupffer cells surrounding the periportal area of the liver, which causes periportal iron accumulation. In Alk2/3fl/fl; Alb-Cre, hepcidin levels are severely reduced, so that Fpn exhibits a panlobular expression in hepatocytes and Kupffer cells.
Prior to this study, the dimerization of BMP type I receptors in the context of mammalian iron homeostasis was poorly understood. In COS7 cells, the BMP receptors ALK3, ALK6, and BMPRII assemble into homomeric as well as heteromeric receptor complexes. These complexes form either before ligand binding or upon ligand binding.[15, 28] The results of the current study suggest that ALK3 exist as homodimeric complexes in the absence of ligand and assemble into heterodimeric complexes with ALK2 upon exposure to BMP. ALK2, in contrast, does not form homodimers in the presence of BMP2, BMP6, or IL-6. One caveat to in vitro overexpression systems is that they may not detect transient or weak interactions and may not be generalizable to other cell or animal systems.[29]
Ligands other than BMP6 or IL-6 may trigger BMP type I receptor formation. Latour et al. hypothesized that another ligand could replace BMP6 to induce hepcidin expression.[18] BMP2 is essential for hepcidin regulation and its deficiency leads to an iron overload phenotype [30] similar to that observed in hepatocyte-specific Alk2-deficient mice.[7] Our in vitro assays did not detect formation of a homodimeric ALK2 complex formation in the presence of BMP2. Our in vivo data, on the other hand, reveal a more severe iron overload phenotype in Alk2/3-deficient mice than in Alk3-deficient mice, which suggest that ALK2 is able to induce hepcidin expression in the absence of ALK3. We speculate that another ligand or receptor can compensate for Alk3 deficiency. A previous publication showed that ALK2 interacts with HJV.[31] This interaction might be important for hepcidin induction via ALK2 in the absence of ALK3. However, Healey et al. used surface plasmon resonance studies to show that HJV does not bind directly to BMP type I receptors.[32]
Previous studies revealed distinct roles for ALK2 and ALK3 in iron homeostasis. ALK2 is required for BMP- and iron-mediated hepcidin induction, while ALK3 is important not only for BMP- and iron-mediated, but also for basal hepcidin expression and IL-6-mediated hepcidin induction.[7, 8]
Recently, a working model in which two different pathways induce hepcidin by iron status were proposed by Latour and colleagues. [18] The first pathway senses extracellular differic transferrin concentration; the second pathway senses intracellular iron concentrations. The first pathway uses HJV and a BMP ligand, presumably BMP2, and eventually includes TfR2 and HFE to induce hepcidin. The second pathway requires BMP6 that binds to preformed BMP receptor complexes to induce hepcidin expression. [18] Based on prior findings [18, 31, 32] and the novel results presented here, we propose the following model in which the formation of either ALK3 homodimers or ALK2-ALK3 heterodimers plays a key role in BMP-mediated signaling. In particular, formation of an ALK2-ALK3 complex might be an essential step in HJV/BMP signaling and be required for BMP- and iron-mediated hepcidin induction. The assembly of ALK3 homodimers may be essential for basal as well as IL-6-mediated hepcidin induction (Figure 7, proposed model).
Figure 7. Proposed model: Three signaling pathways that induce hepcidin expression require ligand-dependent ALK2/ALK3 heterodimerization or ligand independent ALK3 homodimerization.

The left upper panel depicts a scheme of ligand mediated assembly of ALK2-ALK3 heterodimers. The upper middle panel indicates spontaneous preformation of ALK3 homodimers. In both signaling cascades, SMAD activation and phosphorylation leads to activation of the BMP-RE1 (BMP responsive element) and BMP-RE2 in the hepcidin promotor. The upper right panel depicts activation in inflammatory settings via IL-6. Intact BMP signaling and presence of ALK3 homodimers are required for IL-6 mediated hepcidin induction via STAT3. For IL-6 mediated hepcidin induction, only the BMP-RE1 and STAT3-RE are needed (modified according to Latour et al., Healey et al.[18, 32]; new findings highlighted in dark grey).
Conclusion
To conclude, characterization of Alk2/3fl/fl; Alb-Cre mice and in vitro analyses of ALK2/3 complex formation have revealed new insights into the regulation of iron homeostasis. Hepatocyte-specific Alk2/3 deficiency leads to a more marked iron overload phenotype and a different Fpn expression pattern than hepatocyte-specific Alk3 deficiency. ALK3 plays a dominant role in hepcidin induction in hepatocytes with the formation of homomeric ALK3 complexes as well as ligand-dependent ALK2-ALK3 heterodimeric complexes.
Supplementary Material
Acknowledgement:
The authors would like to thank Mark Fleming (Boston Children’s Hospital, Boston) for helpful discussions at the initiation of the project and discussions of the histological findings.
The authors thank Patricio Leyton (Massachusetts General Hospital and Harvard Medical School, Boston) for helpful comments on performance of Co-IP experiments and Kohei Miyazono (University of Tokyo, Japan) for kindly providing all mentioned plasmids.
Funding:
This study was supported by research funding from the German Research Foundation (Deutsche Forschungsgemeinschaft) to AUS (STE 1895/4–1 and STE 1895/4–2).
Public funding sources are reported as follows: FCH receives general funding from INSERM. MM is supported by the Deutsche Forschungsgemeinschaft (SFB1036). DBB is supported by NIH grant R01DK082971 and TBB by NIH grant DK110049. Information on support received for work outside the submitted work from commercial entities is reported by the following authors: TK receives a financial honorarium from Novartis. UK reports a conflict of interest in relation to other projects, which cannot be announced due to legal reasons. MM declares financial support from Novartis Pharma GmbH, Silence Therapeutics PLC and Vifor Pharma Ltd.
List of Abbreviations:
- BMP
Bone morphogenetic protein
- ALK2
Activin receptor type-1
- ALK3
Activin Receptor-like Kinase
- IL
Interleukin
- Smad
Small Mothers Against Decapentaplegic Homolog
- JAK
Janus kinase
- Stat3
Signal transducer and activator of transcription
- Cre
causes recombination
- FCS
fetal calf serum
- DSS
Disuccinimidyl suberate
- ECL
enhanced chemiluminescence
- mRNA
messenger RNA
- FPN
ferroportin
- TfR2
Transferrin receptor 2
- HFE
Hereditary hemochromatosis protein
- HAMP
Hepcidin
- HJV
Hemojuvelin
Footnotes
Disclosure of Conflict of interest:
The authors declare no competing financial interest related to the current manuscript.
Data availability statement:
The authors confirm that all relevant data are included in the paper and its supplementary files.
References
- [1].Michels K; Nemeth E; Ganz T; Mehrad B. Hepcidin and host defense against infectious diseases. PLoS Pathog 11:e1004998; 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Steinbicker AU; Muckenthaler MU. Out of balance--systemic iron homeostasis in iron-related disorders. Nutrients 5:3034–3061; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Knaus P; Sebald W. Cooperativity of binding epitopes and receptor chains in the BMP/TGFbeta superfamily. Biol. Chem 382:1189–1195; 2001. [DOI] [PubMed] [Google Scholar]
- [4].Verga Falzacappa MV; Casanovas G; Hentze MW; Muckenthaler MU. A bone morphogenetic protein (BMP)-responsive element in the hepcidin promoter controls HFE2-mediated hepatic hepcidin expression and its response to IL-6 in cultured cells. J. Mol. Med. (Berl) 86:531–540; 2008. [DOI] [PubMed] [Google Scholar]
- [5].Canali S; Zumbrennen-Bullough KB; Core AB; Wang CY; Nairz M; Bouley R; Swirski FK; Babitt JL. Endothelial cells produce bone morphogenetic protein 6 required for iron homeostasis in mice. Blood 129:405–414; 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Verga Falzacappa MV; Vujic Spasic M; Kessler R; Stolte J; Hentze MW; Muckenthaler MU. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood 109:353–358; 2006. [DOI] [PubMed] [Google Scholar]
- [7].Steinbicker AU; Bartnikas TB; Lohmeyer LK; Leyton P; Mayeur C; Kao SM; Pappas AE; Peterson RT; Bloch DB; Yu PB; Fleming MD; Bloch KD. Perturbation of hepcidin expression by BMP type I receptor deletion induces iron overload in mice. Blood 118:4224–4230; 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mayeur C; Lohmeyer LK; Leyton P; Kao SM; Pappas AE; Kolodziej SA; Spagnolli E; Yu B; Galdos RL; Yu PB; Peterson RT; Bloch DB; Bloch KD; Steinbicker AU. The type I BMP receptor Alk3 is required for the induction of hepatic hepcidin gene expression by interleukin-6. Blood 123:2261–2268; 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Steinbicker AU; Sachidanandan C; Vonner AJ; Yusuf RZ; Deng DY; Lai CS; Rauwerdink KM; Winn JC; Saez B; Cook CM; Szekely BA; Roy CN; Seehra JS; Cuny GD; Scadden DT; Peterson RT; Bloch KD; Yu PB. Inhibition of bone morphogenetic protein signaling attenuates anemia associated with inflammation. Blood 117:4915–4923; 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Little SC; Mullins MC. Bone morphogenetic protein heterodimers assemble heteromeric type I receptor complexes to pattern the dorsoventral axis. Nat. Cell Biol 11:637–643; 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Postic C; Shiota M; Niswender KD; Jetton TL; Chen Y; Moates JM; Shelton KD; Lindner J; Cherrington AD; Magnuson MA. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using cre recombinase. J. Biol. Chem 274:305–315; 1999. [DOI] [PubMed] [Google Scholar]
- [12].Sauer B. Functional expression of the cre-lox site-specific recombination system in the yeast saccharomyces cerevisiae. Mol. Cell. Biol 7:2087–2096; 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mishina Y; Hanks MC; Miura S; Tallquist MD; Behringer RR. Generation of Bmpr/Alk3 conditional knockout mice. Genesis 32:69–72; 2002. [DOI] [PubMed] [Google Scholar]
- [14].Torrance JD; Bothwell TH. Torrance JD & bothwell TH. (1980) tissue iron stores cook JD. eds. methods in hematology. iron :90–115 churchill livingstone new york, NY: In: Churchill Livingstone New York, NY, ed. Methods in Hematology. New York: Churchill Livingstone New York, NY; 1980: 90–115. [Google Scholar]
- [15].Gilboa L; Nohe A; Geissendorfer T; Sebald W; Henis YI; Knaus P. Bone morphogenetic protein receptor complexes on the surface of live cells: A new oligomerization mode for serine/threonine kinase receptors. Mol. Biol. Cell 11:1023–1035; 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wallace DF; Summerville L; Crampton EM; Frazer DM; Anderson GJ; Subramaniam VN. Combined deletion of hfe and transferrin receptor 2 in mice leads to marked dysregulation of hepcidin and iron overload. Hepatology 50:1992–2000; 2009. [DOI] [PubMed] [Google Scholar]
- [17].Wright TL; Brissot P; Ma WL; Weisiger RA. Characterization of non-transferrin-bound iron clearance by rat liver. J. Biol. Chem 261:10909–10914; 1986. [PubMed] [Google Scholar]
- [18].Latour C; Besson-Fournier C; Meynard D; Silvestri L; Gourbeyre O; Aguilar-Martinez P; Schmidt PJ; Fleming MD; Roth MP; Coppin H. Differing impact of the deletion of hemochromatosis-associated molecules HFE and transferrin receptor-2 on the iron phenotype of mice lacking bone morphogenetic protein 6 or hemojuvelin. Hepatology 63:126–137; 2016. [DOI] [PubMed] [Google Scholar]
- [19].Wallace DF; Summerville L; Subramaniam VN. Targeted disruption of the hepatic transferrin receptor 2 gene in mice leads to iron overload. Gastroenterology 132:301–310; 2007. [DOI] [PubMed] [Google Scholar]
- [20].Niederkofler V; Salie R; Arber S. Hemojuvelin is essential for dietary iron sensing, and its mutation leads to severe iron overload. J. Clin. Invest 115:2180–2186; 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zumerle S; Mathieu JR; Delga S; Heinis M; Viatte L; Vaulont S; Peyssonnaux C. Targeted disruption of hepcidin in the liver recapitulates the hemochromatotic phenotype. Blood 123:3646–3650; 2014. [DOI] [PubMed] [Google Scholar]
- [22].Kent P; Wilkinson N; Constante M; Fillebeen C; Gkouvatsos K; Wagner J; Buffler M; Becker C; Schumann K; Santos MM; Pantopoulos K. Hfe and hjv exhibit overlapping functions for iron signaling to hepcidin. J. Mol. Med. (Berl) 93:489–498; 2015. [DOI] [PubMed] [Google Scholar]
- [23].Meynard D; Kautz L; Darnaud V; Canonne-Hergaux F; Coppin H; Roth MP. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat. Genet 41:478–481; 2009. [DOI] [PubMed] [Google Scholar]
- [24].Halpern KB; Shenhav R; Matcovitch-Natan O; Toth B; Lemze D; Golan M; Massasa EE; Baydatch S; Landen S; Moor AE; Brandis A; Giladi A; Stokar-Avihail A; David E; Amit I; Itzkovitz S. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 542:352–356; 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ramey G; Deschemin J; Durel B; Canonne-Hergaux F; Nicolas G; Vaulont S. Hepcidin targets ferroportin for degradation in hepatocytes. Haematologica 95:501–504; 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhang Z; Zhang F; Guo X; An P; Tao Y; Wang F. Ferroportin1 in hepatocytes and macrophages is required for the efficient mobilization of body iron stores in mice. Hepatology 56:961–971; 2012. [DOI] [PubMed] [Google Scholar]
- [27].Huang FW; Pinkus JL; Pinkus GS; Fleming MD; Andrews NC. A mouse model of juvenile hemochromatosis. J. Clin. Invest 115:2187–2191; 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Nohe A; Hassel S; Ehrlich M; Neubauer F; Sebald W; Henis YI; Knaus P. The mode of bone morphogenetic protein (BMP) receptor oligomerization determines different BMP-2 signaling pathways. J. Biol. Chem 277:5330–5338; 2002. [DOI] [PubMed] [Google Scholar]
- [29].Mueller TD. RGM co-receptors add complexity to BMP signaling. Nat. Struct. Mol. Biol 22:439–440; 2015. [DOI] [PubMed] [Google Scholar]
- [30].Koch PS; Olsavszky V; Ulbrich F; Sticht C; Demory A; Leibing T; Henzler T; Meyer M; Zierow J; Schneider S; Breitkopf-Heinlein K; Gaitantzi H; Spencer-Dene B; Arnold B; Klapproth K; Schledzewski K; Goerdt S; Geraud C. Angiocrine Bmp2 signaling in murine liver controls normal iron homeostasis. Blood 129:415–419; 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Pagani A; Colucci S; Bocciardi R; Bertamino M; Dufour C; Ravazzolo R; Silvestri L; Camaschella C. A new form of IRIDA due to combined heterozygous mutations of TMPRSS6 and ACVR1A encoding the BMP receptor ALK2. Blood 129:3392–3395; 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Healey EG, Bishop B, Elegheert J, Bell CH, Padilla-Parra S, Siebold C, Repulsive guidance molecule is a structural bridge between neogenin and bone morphogenetic protein, Nat. Struct. Mol. Biol, 22 (2015), pp. 458–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.