

Abstract
WDR5 is a chromatin regulatory scaffold protein overexpressed in various cancers and a potential epigenetic drug target for the treatment of mixed lineage leukemia. Here we describe the discovery of potent and selective WDR5 WIN-site inhibitors using fragment-based methods and structure-based design. NMR-based screening of a large fragment library identified several chemically distinct hit series that bind to the WIN-site within WDR5. Members of a 6,7-dihydro-5H-pyrrolo[1,2-a]imidazole fragment class were expanded using a structure-based design approach to arrive at lead compounds with dissociation constants < 10 nM and micromolar cellular activity against an AML leukemia cell line. These compounds represent starting points for the discovery of clinically useful WDR5 inhibitors for the treatment of cancer.
Keywords: WDR5, fragment screening, structure-based design, mixed-lineage leukemia
Graphical Abstract

INTRODUCTION
WDR5 is part of a large family of WD40 repeat proteins1–3 that functions as a ubiquitous scaffold protein in multiple complexes found in epigenetic machinery and chromatin regulation.4 Based on its role in cell cycle regulation5 and reported prognostic expression level disease outcome relationships, WDR5 has become an increasingly important target of interest as a potential treatment strategy for a number of cancers, including mixed-lineage leukemia,6 neuroblastoma,7 breast,8 bladder,9 pancreatic,10 and colorectal cancer.11 WDR5 shares over 90% sequence identity among all vertebrates and consists of a characteristic circular barrel shaped 7-bladed β-propeller structure, with each blade interconnected through four stranded antiparallel β-sheets. Two major binding sites operate as the interfaces for complex formation. Mixed lineage leukemia 1 (MLL1) is one of the most well studied protein-protein interaction partners of WDR5 and belongs to a well characterized SET1 family of histone methyltransferases (HMTs), that mediates histone H3 Lysine 4 (H3K4) methylation and binds to WDR5 via a conserved arginine containing motif called the “WIN” or WDR5 interaction motif.12–14 Pediatric acute leukemia cases that harbor MLL1-rearrangements are caused by the chromosomal translocation of a single allele in the 11q23 MLL1 gene to one of more than 70 different partner genes and are associated with a poor prognosis.15–17 The most common fusion partners AF4, AF9, and ENL, account for more 70% of all diagnosed MLL leukemias which manifest as either acute lymphoid or acute myeloid leukemias.17,18 However, fusion partners lack the C-terminal catalytic SET domain with the proper required WIN-motif and thus are devoid of histone methyltransferase activity. Interestingly, early reports demonstrated that the wild-type MLL allele works in concert with the MLL fusion allele and is critical for leukemogenesis and maintenance of MLL-AF9 transformed cells.19,20 Consequently, strategies targeting the wild-type MLL complex and specifically WDR5 at the WIN-site have been pursued as a therapeutic strategy for the treatment of MLL harboring leukemia. Understanding the contributing factors of the MLL fusion protein and the wild-type MLL continues to be a highly active area of research and debate.21 In addition to MLL1, other SET1 family members include MLL2–4, SETd1A, and SETd1B. The full catalytic activity of MLL1 and SETd1A depends on a core complex that includes WDR5, RbBP5, ASH2L, and Dpy30 (WRAD complex). Within the WRAD complex, RbBP5 binds to the opposite side of WDR5 near the C-terminus at a shallow hydrophobic cleft between blades 5 and 6. This interface of WDR5 is electropositive in nature and is involved in additional important protein-protein interactions, including the MOF/HAT complex via KANSL2,22 and a direct interaction with MYC via its MbIIIb motif.23 Peptidomimetic compounds designed to mimic the MLL peptide residues within the WIN-site, such as MM-40124,25 and recently MM-589 (1, Figure 1),26 have been shown to bind to WDR5 with low nanomolar affinity at the WIN site, selectively inhibit MLL1 methyltransferase activity and induce leukemia cell growth arrest in MLL-r harboring cells. The findings described with 1 indicate that targeting the WDR5 WIN-site as a means to impact MLL-r leukemia as well as other cancer types dependent on WDR5 may be a viable path forward.
Figure 1.

Structures and properties of representative WDR5 WIN-site binders.
The first reported non-peptidomimetic class of small molecule WIN-site inhibitors of WDR5, represented by OICR-9429 (2, Figure 1),27,28 maintains 64 nM binding affinity to WDR5 and was shown to demonstrate reduced viability of primary human AML cells bearing C/EBPα mutations (GI50 ~5 μM). A series of analogs structurally related to 2 was recently described with improved binding affinity and promising HMT inhibition,29 but these compounds have similar limitations in their effects on cellular proliferation. Collectively, these results suggest that targeting the WIN-site of WDR5 may serve as a new therapeutic approach for the treatment of leukemias and other cancer types that depend on WDR5.
In an effort to discover more potent non-peptidic small molecules that inhibit WDR5 via the WIN-site, we pursued a fragment-based approach to identify novel hits and used structure-based design to optimize these hits for binding to WDR5. Our NMR-based fragment screen yielded multiple classes of hits. To determine how fragment hits bind to WDR5, we obtained crystal structures of the fragments bound to WDR5. On the basis of their potential drug-likeness and ligand efficiency, the most promising series of compounds were chosen for subsequent structure-based optimization. Optimized compounds were evaluated in cells harboring MLL-rearrangements for their anti-proliferative activity and for their functional inhibition of MLL1 methylation activity of nucleosomes in the presence of the purified WRAD MLL1 complex. This work resulted in the discovery of potent non-peptidic inhibitors of WDR5.
RESULTS AND DISCUSSION
Hit Identification.
Recombinant, 15N-labeled WDR5 (residues 22–334) was used to screen our fragment library (>13,800 compounds) by recording SOFAST 1H-15N HMQC spectra of WDR5. An initial HMQC spectrum of uniformly 15N-labeled WDR5 in complex with an unlabeled MLL1 10mer peptide Ac-ARTEVHLRKS-NH2 was obtained (Figure 2A), which showed peak shifts corresponding to amino acids in regions specific for the WIN-site of WDR5. To conduct the screen, the WDR5 protein was incubated with mixtures of 12 fragments. Fragment mixtures that caused similar peak shifts as the MLL1 peptide were identified as MLL WIN-site hits as illustrated in Figure 2B for a representative fragment mixture. Deconvolution of the mixtures by screening individual compounds generated 47 hits (0.34% hit rate), which can be divided into 12 classes based on their chemical scaffolds. Of the initial hits, 14 fragments that induced the largest chemical shift perturbations were selected for affinity determination by fluorescence polarization assay. Four fragments showed Ki values lower than 0.5 mM, and the other 10 were between 0.5 to 1 mM. Measurable fragment hits demonstrated ligand efficiency indices (LE) of between 0.20–0.39. A representative list of fragments is shown in Figure 3 and the remaining hits can be found in Supplemental Figure S1. Not surprisingly, many of the hits contained an arginine mimetic. For example, fragment hits F-2 through F-4 bear a basic amidine motif (calculated pKb ~11) with NMR based affinities between ~550–850 μM. The phenoxy ethers (F-2 and F-3) exhibited slightly improved affinities over biaryl F-4. Fragment hits F-1, F-5, and F-6 represent a distinct and interesting class and share a common bicyclic imidazole containing core. Fragment F-1 possesses a Ki value of 323 μM, and a relatively high LE of 0.34. Interestingly, the cyclic imidazole class is expected to have inherently weaker basicity relative to the amidine class (calculated pKb’s 5.4–5.9). The dihydro-pyrrolo-imidazole fragment F-1 was particularly interesting to us for two reasons: 1) F-1 represented the most potent member of the imidazole and amidine class and 2) F-1 maintained excellent ligand efficiency with no inherent H-bond donors. Therefore, based on structural novelty as a putative amidine mimetic and with potentially superior drug-like properties versus traditional amidine mimetics (e.g. reduced H-bond count and weak basicity), we prioritized fragment series F-1 for our initial structure-based design optimization campaign.
Figure 2.

1H-15N HMQC spectra of WDR5 with (red) and without (black) added ligand: A) WIN-peptide and B) representative fragment mixture. The NMR sample contained 2 mg/mL (~60 μM) 15N-labeled protein and 12-compound mixture of 670 μM of each ligand.
Figure 3.

Representative hit fragments that bind to WDR5 at the MLL-site with putative arginine (S2) mimics in blue. Ki values were obtained by Fluorescence Polarization Assay.
X-ray Structure of Fragment Co-Complex F-1 and WDR5.
Figure 4A depicts an X-ray co-crystal structure of the fragment F-1 bound to WDR5. The cyclic imidazole group of F-1 binds to WDR5 deeply in the central S2 pocket, where it normally accommodates the MLL1 peptide R3765 guanidine side chain.13 It is noted that the sp2-pyrrolidine moiety is not as puckered as observed in the energy-minimized unbound ligand structure due to limitations of the X-ray diffraction resolution and the Ligand Building and Optimization Workbench (eLBOW) program, which is used for generating non-standard ligands in Phenix. A WDR5 X-ray co-crystal structure with an MLL bound peptide with the major sub-pockets labeled is shown for reference in Figure 4B.12,30 As previously shown, this S2 interaction greatly contributes to the total binding affinity. A comparison of the F-1/WDR5 and MLL1-peptide arginine side-chain interactions with WDR5 is shown in Figures 4C and 4D. In the case of F-1, the cyclic imidazole group stacks with the aromatic ring of F133 and F263, and the N3 imidazole nitrogen of F-1 appears to interact with the backbone C=O of C261 through an H-bond with an inter-atomic distance of 2.8 angstroms (Figure 4C). These interactions drive complex formation. The phenyl group of F-1 exits the S2 channel, towards the solvent and upper rim region, which can be utilized as a handle to grow the fragment toward sub-pockets within the central cavity that are normally occupied by the MLL1 peptide.
Figure 4.

X-ray co-crystal structure of fragment F-1 bound to WDR5 (PDB: 6D9X) and the reference MLL1 peptide bound to WDR5 (PDB: 3EG6) showing respective unoccupied and occupied pockets: A) F-1 left panel top view surface depiction; B) right panel top view MLL1 peptide surface depiction (only Ac-ARA-NH2 residues shown, yellow capped sticks); C) F-1 lower left panel, key residues and hydrogen bonding within S2 pocket (orange capped sticks); D) lower right panel MLL1 peptide (only Ac-ARA-NH2 shown) key residues and hydrogen bonding within S2 pocket (blue capped sticks).
S5 and S4 Pocket Optimization.
Based on the bound structure of the F-1 fragment hit, our initial efforts focused on growing from the pendant phenyl to explore occupation of the nearby upper rim S5 or S4 pockets of the WIN-site (Figure 4A).30 Both pockets are hydrophobic in nature and based on earlier studies involving peptidomimetics, these pockets contribute significantly to the WIN-peptide WDR5 interaction.
A high throughput FPA binding assay using MLL-derived fluorescent probes, similar to that reported by Wang and co-workers,6 was adopted to assess the binding affinity of analogs relative to F-1. Previously reported MLL peptides, including tripeptide Ac-ART-NH2 were utilized as initial controls and the data were fit to standard four-parameter curve fit (see Experimental and SI for further details). Independent experiments (n of 2 or more) were performed in replicate to generate the final average Ki value. Each assay plate performed with an average Z’ value >0.5 and the average Ki values reported fall within a 30% variance. The SAR and ligand efficiency (LE) metrics of the resulting compounds can be found in Table 1. Our efforts focused on amide, ether, and biaryl analogs using either the meta or para position to expand into the neighboring S5 and S4 pockets. As shown in Table 1, a simple para substituted biaryl 3a led to a ~40-fold increase in affinity with a Ki of 8.4 μM. The difluoro congener 3g, gave a further 7.5-fold boost in affinity, representing the most potent analog from this initial effort. Ligand efficiencies for both 3a and 3g were well maintained. Aryl ethers designed to probe the S5 and S4 pockets, including the para congeners 3b, 3d, 3e, and 3f demonstrated a preference for small, branched hydrophobic substituents (e.g. 3d i-PrO Ki = 29.7 μM, LE = 0.33). Similar meta-substituted analogs, although tolerated, were not as favorable (MeO 3j, i-PrO 3k, Ki > 100 μM) leading to an erosion of ligand efficiency in the case of 3k. N-anilido acetamides 3c and 3l were unproductive as polar moieties; however, bicyclic heterocycles 3h and 3i were moderately effective, and N-methyl indoline 3h was sufficiently soluble and potent to obtain a subsequent X-ray co-crystal structure bound to WDR5. As shown in Figure 5, we achieved partial occupation of the S4 pocket, in which the N-1 methyl group of 3h was in close proximity to A47 and A65. Importantly, the indoline ring backbone was found to point away from the S5 and S4 pockets with potential sp3 carbon vectors toward the unoccupied S1 or S7 pockets.
Table 1.
Optimization/SAR of aryl substitution pattern.
| Cmpd | Ar Structure | FPA Ki (μM)a | LEb |
|---|---|---|---|
| F-1 | 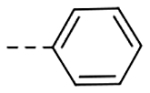 |
323 | 0.34 |
| 3a | 8.44 | 0.35 | |
| 3b | 61 | 0.36 | |
| 3c |  |
118 | 0.30 |
| 3d | 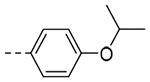 |
29.7 | 0.33 |
| 3e | 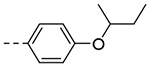 |
>125 | ND |
| 3f |  |
64.3 | 0.26 |
| 3g | 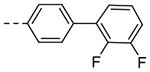 |
1.12 | 0.37 |
| 3h | 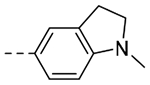 |
85.2 | 0.31 |
| 3i | 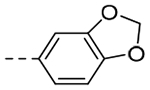 |
64.0 | 0.34 |
| 3j | 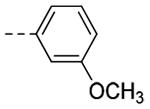 |
117 | 0.34 |
| 3k | 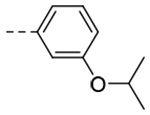 |
109 | 0.30 |
| 3l |  |
>125 | ND |
Ki values represent the average of two or more independent determinations conducted in replicate (CV < 0.3).
Ligand efficiency index, LE = 1.4*pKi / HAC.
Figure 5.

X-ray co-crystal structure of fragment analog 3h (magenta capped sticks) bound to WDR5 with surface display and unoccupied S1 and S7 pockets labeled along with S4 pocket A47 and A65 residues highlighted (atom color capped sticks) (PDB: 6DAI).
S7 Pocket Optimization.
In an effort to further improve binding affinity, we wished to incorporate an amide or similar polar bioisostere from the pendant phenyl to engage the WIN-site in additional backbone hydrogen bonding interactions while also providing a linker and vector to reach the neighboring S7 and potentially S1 pockets (Figure 4A).
In addition to the well conserved hydrogen bonding interactions found between key S2 residues and the arginine side chain of reported MLL and H3 peptide complexes, an additional conserved water-mediated hydrogen bond can be found with the backbone NH of C261. Multiple docking studies suggested this interaction could be targeted directly from F-1 while providing a suitable linker to reach the S7 pocket. Thus, based upon our docking studies, we pursued a series of tolyl amides from the meta-position of F-1 rather than the para-position, which initially appeared to have a more appropriate vector, due to the capacity to participate in a direct H-bond with C261 while also reaching the S7 pocket.
SAR results from a survey of meta-tolyl amides (both secondary and tertiary) are shown in Table 2. Docking suggested that benzamide 4a might engage C261 as predicted and occupy the S7 pocket; a region of WDR5 partially occupied by the MLL1 peptide H3769 imidazole side-chain which is defined by the aromatic residues F133, F149, and Y191. Inhibitor 4a afforded modest inhibition of WDR5 with a Ki = 7.21 μM and although this resulted in a slight loss in LE, the observed level of overall binding affinity was deemed encouraging considering the lack of success from simple meta-substituted analogs explored previously (Table 1, meta examples Ki > 100 μM, e.g. 3l). With this finding in mind, an X-ray co-crystal structure of 4a bound to WDR5 was obtained to confirm the binding orientation (Figure 6).
Table 2.
| Cmpd | R1 | R2 | FPA Ki (μM)a | LEb |
|---|---|---|---|---|
| 4a | H |  |
7.21 | 0.29 |
| 4b | H |  |
7.54 | 0.29 |
| 4c | H |  |
0.665 | 0.32 |
| 4d | H |  |
2.60 | 0.31 |
| 4e | H |  |
3.13 | 0.29 |
| 4f | H |  |
17.6 | 0.26 |
| 4g | H |  |
0.118 | 0.35 |
| 4h | H | 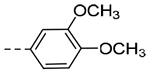 |
0.690 | 0.30 |
| 4i | H | 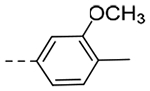 |
0.095 | 0.36 |
| 4j | H | 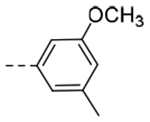 |
0.184 | 0.34 |
| 4k | H |  |
3.91 | 0.31 |
| 4l | H |  |
4.13 | 0.31 |
| 4m | H | 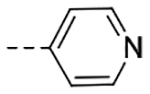 |
1.38 | 0.33 |
| 4n | H | 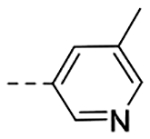 |
0.924 | 0.33 |
| 4o | CH3 | 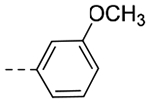 |
0.473 | 0.32 |
| 4p | -i-Pr | 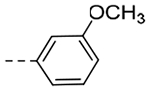 |
0.503 | 0.30 |
| 4qc | -CH2-c-Pr | 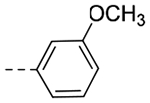 |
0.219 | 0.30 |
| 4rd | -CH2-c-Bu | 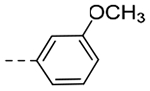 |
1.26 | 0.26 |
| 4sc | -CH2-c-Pr |  |
0.480 | 0.29 |

Ki values represent the average of two or more independent determinations conducted in replicate (CV < 0.3).
Ligand efficiency index, LE = 1.4*pKi / HAC.
c-Pr = cyclopropyl,
c-Bu = cyclobutyl.
Figure 6.

X-ray co-crystal structure of 4a bound to WDR5 (PDB: 6DAK): A) 4a (blue carbon capped sticks) with semi-transparent surface display with key S7 residues and unoccupied pockets highlighted; B) 4a engaged in bidentate-type H-bond interaction with C261 (white carbon capped sticks).
The structure revealed that 4a is indeed bound in an orientation with the amide engaging in a direct H-bond with C261. The C261 backbone C=O N-(3) imidazole nitrogen and the 4a amide C=O C261 NH interaction maintain interatomic distances of 2.6–2.9 Å (Figure 6B). The unsubstituted benzamide phenyl ring of 4a was found to be nearly co-planar with the amide and was readily accommodated in a groove defined by the S7 site defined by K259 and Y260 residues between the blade 5 and 6 loop structure. Further exploration of the secondary amide SAR indicates that the saturated cyclohexyl amide 4b was similarly tolerated. Secondary benzamide SAR of the meta and para positions within mono or disubstituted congeners (4c–e, 4h–j) exhibited a preference for a 3,4-disubstitution pattern affording the first fragment-based analog with an affinity below 100 nM (4i, Ki = 95 nM). Notably the LE index significantly increased within this series of analogs vs. 4a, suggesting a favorable interaction between the ligand and protein, particularly for inhibitors 4c and 4i (LE >0.31). Unsubstituted pyridyl (4k–4m) and 5-methyl substituted pyridyl analogs (4n) were 2–7 fold more potent vs. phenyl 4a. Acetamide 4f displayed weaker affinity vs. 4a (2.4-fold). Interestingly, within the acetamide backbone, the 3,4-dichloro derivative 4g provided a significant, more than two orders of magnitude boost in affinity (~150-fold). Tertiary amides were also explored in an attempt to develop a vector towards the neighboring S1 and S4 pockets. Methyl (4o) and isopropyl (4p) congeners afforded only slight improvements in potency (less than 2-fold). Larger cycloalkyl methyl derivatives (4q–r) were also examined, with cyclopropyl methyl 4q retaining a favorable LE and overall 3-fold improvement vs. 4c. In an attempt to reduce lipophilicity, the 2-methoxy pyridyl analog 4s was found to be tolerated and retained binding affinity (Ki < 500 nM).
In an effort to understand the preferred bound orientation of the cyclopropyl methyl of 4q, an X-ray co-crystal structure of this inhibitor was obtained when bound to WDR5. From this structure, we hoped to further define a strategy to access one of the three upper rim pockets; potentially S1, S5, or S4. Figure 7 depicts a solvent accessible surface displaying 4q bound to WDR5 at the WIN-site. Two key structural features are noted in this structure relative to that of secondary benzamide 4a. First, the 3-methoxybenzamide ring binds in an orthogonal conformation now optimally filling the S7 pocket through favorable contacts with F133, F149, and P173. Secondly, the cyclopropyl methyl does not appear to reach any of the targeted neighboring pockets, but rather reinforces the low-energy orthogonal benzamide conformation and engages in contacts with neighboring Y260. This result is consistent with the SAR and indicated that an alternative approach would be required to target the remaining upper rim pockets.
Figure 7.

X-ray co-crystal structure of 4q bound to WDR5 (grey surface display) revealing orthogonal benzamide conformation filling S7 pocket and unoccupied S1, S5, and S4 pockets (PDB: 6DAR).
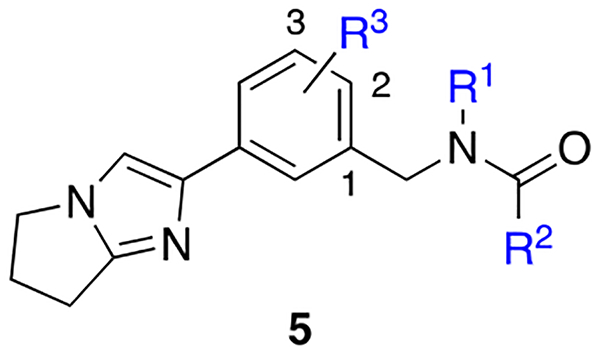
Combining S4 and S7 Binding Elements.
In an effort to target the nearest S4 pocket, we revisited series 3 (Table 1) and specifically the crystal structure involving 3h to apply a mix-and-match strategy in an attempt to co-occupy the S7 and S4 pockets through additional substitution of the central phenyl core. Results from this approach for a series of 2-substituted-1-benzyl-acetamides are found in Table 3.
Table 3.
Optimization/SAR of 3,4-disubstituted amides.c
| F | R1 | R2 | R3 | FPA Ki (μM)a | LEb |
|---|---|---|---|---|---|
| 5a | -CH2-c-Pr | 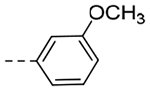 |
3-OMe | 1.46 | 0.25 |
| 5b | -CH2-c-Pr | 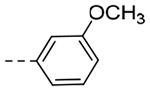 |
2-OMe | 0.148 | 0.29 |
| 5c | -CH2-c-Pr | 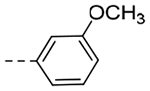 |
2-OEt | 0.0425 | 0.31 |
| 5d | -CH2-c-Pr |  |
2-O-i-Pr | 0.0276 | 0.30 |
| 5e | H |  |
2-O-i-Pr | 0.0761 | 0.32 |
| 5f | -CH2-c-Pr |  |
2-O-c-Bu | 0.0228 | 0.30 |
| 5g | H |  |
2-O-c-Bu | 0.105 | 0.31 |
| 5h | H |  |
2-N(CH3)2 | 0.185 | 0.32 |
| 5i | H |  |
2-N(CH3)2 | 0.0519 | 0.33 |
| 5j | H |  |
 (1,2) |
0.0207 | 0.32 |
| 5k | H |  |
2-O-i-Pr | 0.00989 | 0.35 |
| 5l | -CH2-c-Pr |  |
2-O-i-Pr | 0.0131 | 0.31 |
| 5m | H |  |
2-O-c-Bu | 0.00282 | 0.37 |
| 5n | -CH2-c-Pr |  |
2-O-c-Bu | 0.0289 | 0.29 |
| 5o | -CH2-c-Pr |  |
2-O-c-Bu | 0.00353 | 0.32 |
| 5p | H |  |
2-O-c-Bu | 0.0164 | 0.33 |
Ki values represent the average of two or more independent determinations conducted in replicate (CV < 0.3).
Ligand efficiency index, LE = 1.4*pKi / HAC.
c-Pr = cyclopropyl, i-Pr = isopropyl, c-Bu = cyclobutyl.
Employing the prior SAR from the first series (Table 1), methoxy substitution at either the 3-position (5a, 7-fold loss vs. 4q) or 2-position (5b) confirmed a preference for a 1,2 disubstitution pattern as predicted (5b Ki = 148 nM) based on the structure involving 3h. In the context of the tertiary amide 5b, ether homologs and small cycloalkyl derivatives were examined at the 2-position in order to more optimally fill the S4 pocket. Results from this study demonstrated an orderly improvement in affinity from 5c (O-Et, 3.5-fold improved vs. 5b) to 5d (O-i-Pr, 5.5-fold improved vs. 5b), demonstrating the first examples of inhibitors within the F-1 series with affinities less than 100 nM. O-Cyclobutyl derivative 5f was comparable in affinity to 5d. Within the secondary amide series, a similar trend was noted favoring O-cyclopropyl (5e Ki = 76 nM vs. 5g Ki =105 nM). Alternate 2-amino dialkyl derivatives designed to fill the proximal S4 pocket were also examined and were generally less favored. For example, N,N’-dimethyl 5h was 2.4-fold less potent relative to O-isopropyl congener 5e. Hybrid analogs of the homologated 3,4-dichlorophenyl acetamide identified within series 4 (e.g. 4g Ki = 118 nM) furnished secondary amides 3i–3k, and 3m. Addition of S4 binding within the acetamide series proved to be additive. Once again, small alkyl and cycloalkyl groups were preferred with affinity maintained below 100 nM. Interestingly, O-cyclobutyl exhibited the highest affinity within the dichloroacetamide series and the overall highest LE (secondary amide 5m Ki = 2.8 nM). Conformational analysis indicated that the additional strain within the tertiary acetamide was likely contributing to the divergent SAR observed upon cyclopropyl methyl introduction (e.g. 5m vs 5n, 10-fold loss). Returning to the benzamide 3,4-disubstituted series (5o-5p), inhibitor 5o demonstrated the expected additive SAR of the tertiary amide, affording an inhibitor with single digit nanomolar potency, comparable to 5m.
Further Optimization via the Incorporation of Conformational Constraints.
Based on the counterproductive results obtained from the tertiary acetamide class, lack of direct access to S4 pocket from the amide, and the general steric congestion of 1,2-disubstituted systems within series 5, we designed a series of cyclic constraints intended to simultaneously occupy the S4 pocket and restrict the benzylic methylene closer to the bioactive conformation in order to optimally position the amide toward the S7 pocket. Our basic strategy is outlined in Figure 8 with the bound conformation of 4a shown for reference.
Figure 8.

Strategy constraining the bioactive conformation 4a.
Select inhibitors and resulting profiles from this optimization campaign are shown below in Figure 9. Using this strategy, compound 6a was designed and synthesized resulting in a ~50-fold improvement in binding affinity based on the Ki determination relative to the direct acyclic comparator 4g. Moreover, benzamide indane 6b resulted in a ~20-fold improvement in affinity relative to comparator 4i. Based on the level of enhanced potency achieved at this juncture and the reported and confirmed Kd of the FITC-10mer peptide developed by the Wang laboratory,6 the FPA assay was expected to have an IC50 and resulting Ki limit near 0.5–1 nM (10mer-Thr probe Kd ~1 nM). To circumvent this issue we developed a stable Anti-His TR-FRET assay to allow for improved sensitivity below the nanomolar level in order to rank compounds based on potency (see Experimental and SI for details). The FPA and TR-FRET determined Ki values for series 6 analogs are shown in Figure 9 for comparison.
Figure 9.

Indane and tetrahydronaphthalene constrained analogs profile summary.
Expansion of the indane core to a tetrahydronaphthalene resulted in a modest 2–3-fold loss in affinity. Introduction of a 2-substituted pyridine ring in the S7 as before gave an inhibitor with modest affinity suggesting that alternate sites for pyridine ring incorporation will be necessary in order to modulate the physicochemical properties while maintaining potency. The pseudo-symmetrical benzamide 6e was the most potent inhibitor from this effort, bottoming out the prior FPA assay and giving a Ki of 0.90 nM in our TR-FRET assay. Inhibitor 6e represents an overall ~360,000-fold improvement in affinity relative to starting fragment F-1. In an effort to develop refined heterocyclic design elements within S7 and confirm the binding orientation and stereochemistry of the indane ring system, an X-ray co-crystal structure of 6b bound to WDR5 was determined. Shown in Figure 10 is a solvent accessible surface of 6b within the WIN binding site that highlights the amino acid residues of the S4 and S7 pockets.
Figure 10.

X-ray co-crystal structure surface display of 6b bound to WDR5 at the WIN-site. The residues that form the S7 and S4 pockets are labeled (PDB: 6DAS).
Structure 6b confirmed the pre-oriented R-configuration for the benzylic benzamide as well as favorable interactions within S7 between the 3-methoxy-4-methyl groups and the F149, P173, and Y191 residues. In the S4 pocket the indane displayed favorable interactions with L321 and I305 residues.
Biological Activity.
Since the WDR5-MLL1 interaction requires the full WRAD complex to achieve robust H3K4 histone methyl transferase (HMT) activity, compounds that target this interaction are expected to inhibit methylation activity. Assessment of WRAD-mediated MLL1 H3K4 methylation thus serves as a useful orthogonal and functional measure of PPI inhibition of a native WDR5 containing multi-protein complex. To this end, potent compounds from series 6 were profiled for their functional inhibition of the WRAD mediated MLL1 H3K4 histone methyl transferase (HMT) activity. HMT activity studies were performed at Reaction Biology Corp. using purified recombinant human MLL1 complex (MLL1 aa 3745–3969 plus WRAD), purified HeLa oligonucleosomes as substrate, and S-adenosyl-L-[methyl-3H]methionine (SAM) as the methylation cofactor.31 MLL1 HMT inhibition potency rank order tracked with affinity rank order (6e > 6a > 6b >> 6c-6d); even though weak potency was observed with IC50 values in the micromolar range. A large functional to biochemical shift of ~2800-fold (HMT IC50 / TR-FRET Ki) based on the TR-FRET competition assay was observed.
Compounds from the bicyclic series were also profiled for their cellular anti-proliferative activity using human acute leukemia MV4–11 cancer cells. Similarly, a significant shift was observed in a 7-day anti-proliferative growth inhibition assay, with measured GI50 values of 7 μM or greater (rank order 6e ~ 6a > 6b > 6c-6d). Importantly, compound 6e was found to have reasonable intrinsic permeability in a MDR1 expressing MDCK cell line (A-B Papp = 9.6 * 10−6 cm/sec) with no apparent active efflux (B-A/A-B ratio = 1.22), indicating that cellular penetration is unlikely a barrier that would account for the weak cellular activity observed.32 Based on the suboptimal functional inhibition and weak cellular activity demonstrated by analogs 6a–6e, we concluded that compounds with much better binding affinity will be required in order to achieve adequate functional and cellular inhibition of the fully intact multimeric WDR5-containing protein complex from this imidazole-based series of inhibitors. The origin of the functional and cellular shifts observed is not fully understood at this time; however, the permeability exhibited by 6e and the comparable cellular and functional activity across the examples tested suggest perhaps the disconnect is more likely related to the artificial nature of the equilibrium binding assays and or a property inherent to the scaffold. For example, the FPA and TR-FRET binding assays are conducted using purified non-complexed WDR5. In contrast, in the HMT and cellular context the inhibitor ligand must compete against the multi-valent WRAD complex in HMT, as well as WRAD and additional protein partners within the full cellular milieu. Secondly, it conceivable that physicochemical properties, such as basicity of the S2 cyclic imidazole for example,33 may not be fully optimal for performing in a more native context or reaching WDR5 efficiently within the nuclear target organelle. Recently, Wang and co-workers described a series of highly potent and basic guanidine containing S2 peptidomimetics which achieve sub-nanomolar affinity with functional HMT inhibition below 100 nM.26 Interestingly, they also exhibit a large functional to biochemical shift (MLL1 IC50 / Ki ~300–600); however, the shift is somewhat lower than that observed for the imidazole S2 system described here.
Chemical Synthesis.
Synthesis of final compounds in Tables 1 and 2 began from 2-(2-oxopyrrolidin-1-yl)acetamide 7 (Scheme 1). Cyclization using phosphoryl bromide or chloride gave key cyclic haloimidazole intermediates 8a–b. Treatment of 8a or 8b under Suzuki cross-coupling conditions using catalytic PdCl2(dppf).CH2Cl2 in a binary acetonitrile-water solvent mixture with aqueous K2CO3 affords 3a-3l or precursors 9–10. Deprotection of 9a afforded benzyl amine 9b in quantitative yield. Acylation readily afforded final compounds 4a–4n. In order to obtain tertiary amides 4o-4s reductive amination was performed using aldehyde 10. The resulting intermediate amides were then treated with sodium hydride in DMF with various alkylating agents allowing access to tertiary amides 4o-4s.
Scheme 1.

Synthesis of precursor 8 and series 3–4.
(a) POBr3, 70 °C, 1 h or POCl3, 85 °C, 15 h (65–70%); (b) PdCl2(dppf)CH2Cl2, ArB(OH)2 or ArBpin, CH3CN, aq. K2CO3, 95 °C, 1–16h; (c) 3e-3f phenol alkylation: RX, CsCO3, DMF, rt, 12 h or 3c, 3l aniline acetylation: acetyl chloride, CH2Cl2, rt, 2 h; (d) b using 3-(N-Boc-aminomethyl)phenylboronic acid (96%); (e) TFA, CH2Cl2 (quant.); (f) R2COCl, Et3N, CH2Cl2, rt, 2 h or R2CO2H, HATU, DMF, rt, 16–40 h; (g) b using 3-formylphenylboronic acid (47%); (h) R1NH2, NaBH(OAc)3, HOAc, DCE, rt, 2 h; (i) NaH, R1X, DMF, 0 °C to rt, 1 h.
Synthesis of compounds in Table 3 begins similarly from bromide 8b (Scheme 2). Suzuki cross-coupling with aldehydes 11 affords biaryl intermediates of type 12. Reductive amination and acylation affords specified compounds of series 5. Alternatively, nitriles 14 were utilized to afford, after coupling and reduction, primary amines of type 15. Final acylation proceeds smoothly to give the remaining specified analogs 5.
Scheme 2.

Synthesis of series 5 analogs.
(a) PdCl2(dppf)CH2Cl2, ArB(OH)2 or ArBpin, CH3CN, aq. K2CO3, 95 °C, 1–16 h; (b) R1NH2, NaBH(OAc)3, HOAc, DCE, rt, 2 h; (c) R2COCl, Et3N, CH2Cl2, rt, 2 h or R2CO2H, HATU, DMF, rt, 16–40 h; (d) PdCl2(dppf)CH2Cl2, ArB(OH)2 or ArBpin, CH3CN, aq. K2CO3, 95 °C, 1–16 h; (e) LiAlH4, THF, 0–60 °C.
Synthesis of compounds in the indane and tetrahydronaphthalene series employed an Ellman sulfinamide reduction strategy to set the benzylic stereochemistry.34,35 Based upon the structural information provided from X-ray complexes 4a and 4q the R-configuration was targeted via the appropriate auxiliary.
The construction of the target compounds proceeds with the pinacol boryl indanone (16a) or tetralone (16b) and a Suzuki cross-coupling using 8b to install the S2 cyclic dihydro-pyrrolo-imidazole moiety resulting in 17a and 17b. Condensation of each ketone with (R)-(+)-2-methyl-2-propanesulfinamide using Ti(OEt)4 afforded the chiral sulfinamides, which upon reduction in the same pot, gave diastereomers 18a and 18b in good yield and high purity (>97% diastereomeric excess by NMR) after column chromatography. Subsequent deprotection and acylation furnished the desired constrained enantiopure analogs 6a–6e.
CONCLUSION
In summary, we have discovered potent WIN-site WDR5 inhibitors using a fragment-based approach. Optimization of the fragment hits was rapidly achieved utilizing multiple co-crystal structures to guide design resulting in several potent inhibitors. The imidazole inhibitors displayed single digit nanomolar inhibition displacing WIN peptides from binding to WDR5, demonstrate dose-dependent inhibition of H3K4 methylation activity, and display moderate growth inhibition against an MLL-r harboring cell line. The most potent inhibitor described (6e), represents an overall ~360,000-fold improvement in affinity relative to starting fragment F-1. The key dihydro-pyrrolo-imidazole that binds in the S2 pocket lacks an intrinsic hydrogen bond donor, maintains less than five rotatable bonds, and displays a favorable ligand efficiency and molecular weight profile (LE > 0.35, MW < 450). This work highlights the power of unbiased fragment-based screening and the resulting identification of a novel dihydro-pyrrolo-imidazole substructure as an arginine side-chain mimetic for the S2 channel. Prior WDR5 WIN-site inhibitors such as 1 and 2 are currently limited to S2 binders bearing either highly basic guanidines or piperidine substructures; both of which potentially face challenges associated with cell permeability and/or pharmacological promiscuity. In addition to fundamental differences within the S2 arginine mimetic portion, the inhibitors described herein occupy the S7 pocket rather than the S1 pocket, as observed in the currently reported WDR5 inhibitor complexes. The WDR5 inhibitors described here provide an excellent starting point for the discovery and design of future WIN-site inhibitors targeting WDR5-dependent cancers.
EXPERIMENTAL SECTION
General Chemistry.
All chemical reagents and reaction solvents were purchased from commercial suppliers and used as received. Proton nuclear magnetic resonance (1H NMR) spectra were recorded at either 400 MHz or 600 MHz on a Bruker spectrometer, as stated. For 1H NMR spectra, chemical shifts are reported in parts per million (ppm) and are reported relative to residual non-deuterated solvent signals. Coupling constants are reported in hertz (Hz). The following abbreviations (or a combination, thereof) are used to describe splitting patterns: s, singlet; d, doublet; t, triplet; q, quartet; pent, pentet; m, multiplet; br, broad. All compounds were of 95% purity or higher, unless otherwise noted, as measured by analytical reversed-phase HPLC. Analytical HPLC was performed on an Agilent 1200 series system with UV detection at 214 and 254 nm, along with evaporative light scattering detection (ELSD). Low-resolution mass spectra were obtained on an Agilent 6140 mass spectrometer with electrospray ionization (ESI). LCMS experiments were performed with the following parameters: Phenomenex Kinetex 2.6 μm XB-C18 100 Å, LC column 50 × 2.1 mm; 2 min gradient, 5%–95% MeCN in H2O, and 0.1% TFA or 0.1% formic acid. Analytical thin layer chromatography (TLC) was performed on Kieselgel 60 F254 glass plates precoated with a 0.25 mm thickness of silica gel. TLC plates were visualized with UV light and iodine. Silica gel chromatography was performed using a Teledyne Isco Combiflash® Rf system. Preparative reversed-phase HPLC was performed on a Gilson instrument equipped with a Phenomenex Kinetex C18 column, using varying concentrations of MeCN in H2O, and 0.1% TFA.
Fragment hits F-2–F6 were purchased from commercial vendors and assayed without further purification.
General Procedure A: Suzuki Coupling
The aryl boronic acid or pinacol ester (2 eq), Pd[P(tBu)3]2 (5 mol%) and Cs2CO3 (1.5 eq) were added to a solution of aryl halide (1 eq) in 1,4-dioxane (0.25 M) under an Ar atmosphere, then stirred at 90 °C for 12–16 h. The reaction mixture was filtered through celite, washing with CH2Cl2, concentrated in vacuo and purified by flash column chromatography.
General Procedure B: Suzuki Coupling
The representative aryl halide (1 eq), aryl boronic acid or pinacol ester (2 eq), PdCl2(dppf).CH2Cl2 (5 mol %) were taken in CH3CN (0.25 M) and K2CO3 (2.5 M aq. solution, 3 eq) under an Ar atmosphere, then stirred at 95 °C for 1–16 h. The reaction mixture was filtered through Celite, washing with EtOAc, concentrated in vacuo, and purified by flash column chromatography.
General Procedure C: Amide Formation
Acyl chloride (1.3 eq) and Et3N (2.0 eq) were added to a solution of amine (1.0 eq) in CH2Cl2 (0.5 M) at 0 °C under Ar atmosphere, then stirred at room temperature for 2 h. The reaction was quenched with water and extracted with CH2Cl2. The combined organic layers were dried (Na2SO4), filtered and concentrated. The crude residue was purified by flash column chromatography.
General Procedure D: HATU Coupling
The corresponding amine (1 eq), carboxylic acid (1.1 eq), HATU (1.2 eq) and DIPEA (5 eq) were combined in DMF (0.25 M) and stirred at r.t. for 16–40 h. Upon complete reaction mixture was diluted with EtOAc and washed with water, sat. aq. NaHCO3, and sat. aq. NaCl. The concentrated crude material was purified by flash column chromatography.
General Procedure E: Reductive Amination
To a solution of aldehyde (1 eq) in DCE (0.25 M) was added amine (2 eq) and AcOH (1.5 eq), followed by NaBH(OAc)3 (1 eq) and the reaction mixture was stirred at room temperature for 2 h. The reaction was quenched qith sat. aq. NaHCO3 and extracted with CH2Cl2. The combined organic layers were dried (Na2SO4), filtered, concentrated in vacuo and purified by flash column chromatography.
General Procedure F: O-Alkylation
Alkyl bromide (2.0 eq) and Cs2CO3 (2.0 eq) were added to a solution of alcohol (1.0 eq) in DMF (0.1 M). The reaction mixture was stirred at room temperature for 12 h. The reaction was quenched with water and extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with water (x 5), dried (MgSO4), filtered, concentrated in vacuo and purified by flash column chromatography.
General Procedure G: Nitrile Reduction
Lithium aluminium hydride (5.0 eq) was added to a solution of aryl nitrile (1.0 eq) in THF (0.1 M) at 0 °C. The reaction mixture was stirred at 60 °C. After 2 h, the reaction mixture was quenched with 2 N NaOH solution. The organic layer was dried (MgSO4), filtered and concentrated. The residue was used for next step without further purification.
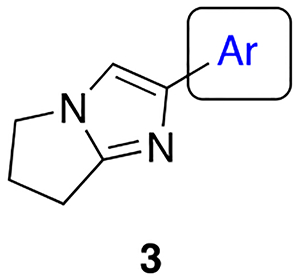
2-Phenyl-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (F-1).
The title compound (25 mg, 20% yield) was prepared following General Procedure A using phenylboronic acid (171 mg, 1.40 mmol) and 8a (100 mg, 0.70 mmol): 1H NMR (400 MHz, CDCl3): δH 7.74 (d, J = 7.7 Hz, 2H), 7.35 (t, J = 7.5 Hz, 2H), 7.20 (t, J = 7.2 Hz, 1H), 7.16 (s, 1H), 3.99 (t, J = 6.9 Hz, 2H), 2.92 (t, J = 7.7 Hz, 2H), 2.60 (pent, J = 7.2 Hz, 2H); LCMS (ESI): Rt = 0.16 min, m/z = 185.2 [M+H]+; > 98% (215, 254 nm).
2-([1,1’-Biphenyl]-4-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (3a).
The title compound (31.5 mg, 28% yield) was prepared following General Procedure B using 4-biphenylboronic acid (127 mg, 0.64 mmol) and 8b (80.0 mg, 0.73 mmol): 1H NMR (400 MHz, CDCl3): δH 7.83 (dd, J = 8.3, 1.5 Hz, 2H), 7.64–7.60 (m, 4H), 7.43 (t, J = 7.4 Hz, 2H), 7.32 (t, J = 7.6 Hz, 1H), 7.21 (s, 1H), 4.02 (t, J = 7.1 Hz, 2H), 2.70 (t, J = 7.1 Hz, 2H), 2.63 (pent, J = 7.2 Hz, 2H); LCMS (ESI): Rt = 0.80 min, m/z = 261.1 [M+H]+; > 98% (215, 254 nm).
2-(4-Methoxyphenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (3b).
The title compound (53.4 mg, 47% yield) was prepared following General Procedure B using 4-methoxyphenylboronic acid (162 mg, 1.07 mmol) and 8b (100 mg, 0.70 mmol): 1H NMR (400 MHz, CD3OD): δH 7.70 (d, J = 7.0 Hz, 2H), 7.11 (s, 1H), 6.95 (d, J = 8.7 Hz, 2H), 4.15 (t, J = 7.1 Hz, 2H), 3.24 (t, J = 7.1 Hz, 2H), 2.74 (pent, J = 7.2 Hz, 2H); LCMS (ESI): Rt = 0.611 min, m/z = 215.2 [M+H]+; > 98% (215, 254 nm).
N-(4-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)phenyl)acetamide (3c).
Step 1: 4-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)aniline The title compound (190 mg, 89% yield) was prepared following General Procedure B using 4-aminophenylboronic acid (352 mg, 1.60 mmol) and 8b (200 mg, 1.07 mmol): LCMS (ESI): Rt = 0.083 min, m/z = 200.2 [M+H]+; > 98% (215, 254 nm). Step 2: N-(4-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)phenyl)acetamide The title compound (12.8 mg, 14% yield) was prepared following General Procedure C using acetyl chloride (35 μL, 0.49 mmol) and 4-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)aniline (45 mg, 0.38 mmol): 1H NMR (400 MHz, CD3OD): δH 7.62 (d, J = 8.3 Hz, 2H), 7.54 (d, J = 8.5 Hz, 2H), 7.38 (s, 1H), 4.08 (t, J = 7.1 Hz, 2H), 2.92 (t, J = 7.4 Hz, 2H), 2.65 (pent, J = 7.3 Hz, 2H), 2.12 (s, 3H); LCMS (ESI): Rt = 0.190, m/z = 242.2 [M+H]+; > 98% (215, 254 nm).
2-(4-Isopropoxyphenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (3d).
The title compound (15.8 mg, 12% yield) was prepared following General Procedure B using 4-isoproxyphenylboronic acid (144 mg, 0.80 mmol) and 8b (100 mg, 0.70 mmol): 1H NMR (400 MHz, CDCl3): δH 7.70 (d, J = 8.6 Hz, 2H), 7.06 (s, 1H), 6.89 (d, J = 8.7 Hz, 2H), 4.55 (hept, J = 6.1 Hz, 1H), 4.01 (t, J = 7.2 Hz, 2H), 2.98 (t, J = 7.5 Hz, 2H), 2.65 (pent, J = 7.0 Hz, 2H), 1.34 (d, J = 6.0 Hz, 6H); LCMS (ESI): Rt = 0.738 min, m/z = 243.2 [M+H]+; > 98% (215, 254 nm).
2-(4-(sec-Butoxy)phenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (3e).
Step 1: 4-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)phenol The title compound (202 mg, 94% yield) was prepared following General Procedure A using 4-hydroxyphenylboronic acid (221 mg, 1.60 mmol) and 8a (200 mg, 1.07 mmol): LCMS (ESI): Rt = 0.125 min, m/z = 201.1 [M+H]+; > 98% (215, 254 nm). Step 2: 2-(4-(sec-Butoxy)phenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole The title compound (6.5 mg, 13% yield) was prepared following General Procedure F using 2-bromobutane (43.5 μL, 0.40 mmol) and 4-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)phenol (40 mg, 0.20 mmol): 1H NMR (400 MHz, CD3OD): δH 7.71 (d, J = 7.7 Hz, 2H), 7.09 (s, 1H), 6.91 (d, J = 7.7 Hz, 2H), 4.34–4.29 (m, 1H), 4.11 (t, J = 7.5 Hz, 2H), 3.16 (t, J = 7.2 Hz, 2H), 2.71 (pent, J = 7.5 Hz, 2H), 1.67–1.57 (m, 2H), 1.30 (d, J = 6.0 Hz, 3H), 0.98 (d, J = 7.5 Hz, 3H); LCMS (ESI): Rt = 0.829 min, m/z = 256.8 [M+H]+; > 98% (215, 254 nm).
2-(4-(Benzyloxy)phenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (3f).
Step 1: 4-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)phenol The title compound (202 mg, 94% yield) was prepared following General Procedure B using 4-hydroxyphenylboronic acid (221 mg, 1.60 mmol) and 8b (200 mg, 1.07 mmol): LCMS (ESI): Rt = 0.125 min, m/z = 201.1 [M+H]+; > 98% (215, 254 nm). Step 2: 2-(4-(Benzyloxy)phenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole The title compound (9.3 mg, 16% yield) was prepared following General Procedure F using benzyl bromide (47.5 μL, 0.40 mmol) and 4-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)phenol (40 mg, 0.20 mmol): 1H NMR (400 MHz, CDCl3): δH 7.71 (d, J = 8.3 Hz, 2H), 7.44 (d, J = 7.1 Hz, 2H), 7.39 (t, J = 7.1 Hz, 2H), 7.32(t, J = 7.1 Hz, 1H), 7.08 (s, 1H), 7.00 (d, J = 8.3 Hz, 2H), 5.08 (s, 2H), 4.06 (t, J = 7.2 Hz, 2H), 3.06 (t, J = 7.5 Hz, 2H), 2.66 (pent, J = 7.0 Hz, 2H); LCMS (ESI): Rt = 0.85 min, m/z = 291.1 [M+H]+; > 98% (215, 254 nm).
2-(2’,3’-Difluoro-[1,1’-biphenyl]-4-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (3g).
The title compound (7 mg, 13% yield) was prepared following General Procedure A using 2,3-difluorophenylboronic acid (58 mg, 0.37 mmol) and 8a (40 mg, 0.18 mmol): 1H NMR (400 MHz, CD3OD): δH 7.88 (s, 1H), 7.79 (d, J = 8.5 Hz, 2H), 7.71 (d, J = 8.2 Hz, 2H), 7.35–7.26 (m, 3H), 4.34 (t, J = 7.3 Hz, 2H), 3.27 (t, J = 7.8 Hz, 2H), 2.84 (pent, J = 7.8 Hz, 2H); LCMS (ESI): Rt = 0.88 min, m/z = 297.1 [M+H]+; > 98% (215, 254 nm).
5-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-1-methylindoline (3h).
The title compound (43 mg, 34% yield) was prepared following General Procedure B using (1-methylindolin-5-yl)boronic acid (208 mg, 0.80 mmol) and 8b (100 mg, 0.70 mmol): 1H NMR (400 MHz, CDCl3): δH 7.50 – 7.47 (m, 2H), 7.00 (s, 1H), 6.47 (d, J = 8.0 Hz, 1H), 4.00 (t, J = 6.8 Hz, 2H), 3.31 (t, J = 8.4 Hz, 2H), 2.98–2.94 (m, 4H), 2.77 (s, 3H), 2.61 (pent, J = 7.4 Hz, 2H); LCMS (ESI): Rt = 0.13 min, m/z = 240.1 [M+H]+; > 98% (215, 254 nm).
2-(Benzo[d][1,3]dioxol-5-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (3i).
The title compound (26 mg, 18% yield) was prepared following General Procedure A using 3,4-(methylenedioxy)phenylboronic acid (233 mg, 1.40 mmol) and 8a (100 mg, 0.70 mmol): 1H NMR (400 MHz, CDCl3): δH 7.26–7.22 (m, 2H), 7.04 (s, 1H), 6.80 (d, J = 8.1 Hz, 1H), 5.94 (s, 2H), 3.99 (t, J = 7.1 Hz, 2H), 2.92 (t, J = 8.4 Hz, 2H), 2.60 (pent, J = 7.4 Hz, 2H); LCMS (ESI): Rt = 0.132 min, m/z = 229.1 [M+H]+; > 98% (215, 254 nm).
2-(3-Methoxyphenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (3j).
The title compound (14.8 mg, 11% yield) was prepared following General Procedure A using 3-methoxyphenylboronic acid (171 mg, 1.40 mmol) and 8a (100 mg, 0.70 mmol): 1H NMR (400 MHz, CDCl3): δH 7.37–7.36 (m, 1H), 7.31–7.23 (m, 2H), 7.17 (s, 1H), 6.80–6.77 (m, 1H), 4.02 (t, J = 7.2 Hz, 2H), 3.86 (s, 3H), 2.96 (t, J = 8.4 Hz, 2H), 2.62 (pent, J = 7.5 Hz, 2H); LCMS (ESI): Rt = 0.128 min, m/z = 215.1 [M+H]+; > 98% (215, 254 nm).
2-(3-Isopropoxyphenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (3k).
The title compound (51 mg, 39% yield) was prepared following General Procedure B using 3-isoproxyphenylboronic acid (144 mg, 0.80 mmol) and 8b (100 mg, 0.70 mmol); 1H NMR (400 MHz, CDCl3): δH 7.36 (d, J = 1.2 Hz, 1H), 7.30 (d, J = 7.5 Hz, 1H), 7.27–7.23 (m, 1H), 7.16 (s, 1H), 6.78 (d, J = 8.0 Hz, 1H), 4.69–4.63 (m, 1H), 4.03 (t, J = 7.5 Hz, 2H), 3.00 (t, J = 7.5 Hz, 2H), 2.64 (pent, J = 6.7 Hz, 2H), 1.34 (d, J = 6.1 Hz, 6H); LCMS (ESI): Rt = 0.757 min, m/z = 243.2 [M+H]+; > 98% (215, 254 nm).
N-(3-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)phenyl)acetamide (3l).
Step 1: 3-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)aniline The title compound (253 mg, 79% yield) was prepared following General Procedure B using 3-aminobenzeneboronic acid (329 mg, 2.41 mmol) and 8b (300 mg, 1.60 mmol): LCMS (ESI): Rt = 0.083 min, m/z = 200.2 [M+H]+; > 98% (215, 254 nm). Step 2: 3-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)aniline The title compound (12.8 mg, 14% yield) was prepared following General Procedure C using acetyl chloride (35.1 μL, 0.49 mmol) and 3-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)aniline (50 mg, 0.38 mmol): 1H NMR (400 MHz, CD3OD): δH 7.75 (s, 1H), 7.46 (d, J = 8.1 Hz, 1H), 7.42 (d, J = 7.7 Hz, 1H), 7.35 (s, 1H), 7.27 (t, J = 8.0 Hz, 1H), 4.04 (t, J = 7.3 Hz, 2H), 2.88 (t, J = 7.4 Hz, 2H), 2.63 (pent, J = 7.1 Hz, 2H), 2.13 (s, 3H); LCMS (ESI): Rt = 0.517 min, m/z = 242.2 [M+H]+; > 98% (215, 254 nm).
N-(3-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)benzamide (4a).
The title compound (53 mg, 45% yield) was prepared following General Procedure C using benzoyl chloride (52.4 μL, 0.45 mmol) and intermediate 9b (80 mg, 0.38 mmol): 1H NMR (400 MHz, CDCl3): δH 7.84–7.80 (m, 3H), 7.60 (d, J = 7.8 Hz, 1H), 7.48–7.44 (m, 1H), 7.42 – 7.37 (m, 1H), 7.31 (t, J = 7.6 Hz, 1H), 7.23 (d, J = 7.6 Hz, 1H), 7.12 (s, 1H), 6.82 (s, 1H), 4.64 (d, J = 5.6 Hz, 2H), 3.97 (t, J = 7.2 Hz, 2H), 2.88 (t, J = 7.6 Hz, 2H), 2.59 (pent, J = 7.4 Hz, 2H); LCMS (ESI): Rt = 0.711 min, m/z = 318.1 [M+H]+; > 98% (215, 254 nm).
N-(3-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)cyclohexanecarboxamide (4b).
The title compound (49 mg, 40% yield) was prepared following General Procedure C using cyclohexanecarbonyl chloride (75.4 μL, 0.56 mmol) and intermediate 9b (80 mg, 0.38 mmol): 1H NMR (400 MHz, CD3OD): δH 7.58–7.56 (m, 1H), 7.36 (s, 1H), 7.29–7.27 (m, 2H), 7.12 (d, J = 7.6 Hz, 1H), 4.36 (s, 2H), 4.06 (t, J = 7.2 Hz, 2H), 2.88 (t, J = 7.6 Hz, 2H), 2.62 (pent, J = 7.6 Hz, 2H), 2.27–2.18 (m, 1H), 1.81–1.78 (m, 4H), 1.52–1.22 (m, 6H); LCMS (ESI): Rt = 0.991 min, m/z = 324.1 [M+H]+; > 98% (215, 254 nm).
N-(3-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-3-methoxybenzamide (4c).
The title compound (49 mg, 38% yield) was prepared following General Procedure C using 3-methoxybenzoyl chloride (79.2 μL, 0.56 mmol) and intermediate 9b (80 mg, 0.38 mmol): 1H NMR (400 MHz, CD3OD): δH 7.70 (s, 1H), 7.57 (d, J = 7.7 Hz, 1H), 7.45–7.40 (m, 2H), 7.38–7.34 (m, 2H), 7.31 (t, J = 7.7 Hz, 1H), 7.22 (d, J = 7.7 Hz, 1H), 7.09 (dd, J = 8.7, 2.9 Hz, 1H), 4.59 (s, 2H), 4.04 (t, J = 6.9 Hz, 2H), 3.84 (s, 3H), 2.88 (t, J = 7.7 Hz, 2H), 2.62 (pent, J = 7.7 Hz, 2H); LCMS (ESI): Rt = 0.714 min, m/z = 348.1 [M+H]+; > 98% (215, 254 nm).
N-(3-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-3-methylbenzamide (4d).
The title compound (19 mg, 15% yield) was prepared following General Procedure C using 3-methylbenzoyl chloride (74.2 μL, 0.56 mmol) and intermediate 9b (80 mg, 0.38 mmol): 1H NMR (400 MHz, CD3OD): δH 7.69–7.67 (m, 2H), 7.65–7.62 (m, 1H), 7.57 (d, J = 7.7 Hz, 1H), 7.37 – 7.29 (m, 4H), 7.21 (d, J = 7.9 Hz, 1H), 4.59 (s, 2H), 4.57 (s, 1H), 4.03 (t, J = 6.7 Hz, 2H), 2.87 (t, J = 7.6 Hz, 2H), 2.62 (pent, J = 7.4 Hz, 2H), 2.39 (s, 3H); LCMS (ESI): Rt = 0.752 min, m/z = 332.2 [M+H]+; > 98% (215, 254 nm).
N-(3-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-4-methoxybenzamide (4e).
The title compound (61.5 mg, 47% yield) was prepared following General Procedure C using 4-methoxybenzoyl chloride (76.3 μL, 0.56 mmol) and intermediate 9b (80 mg, 0.38 mmol): 1H NMR (400 MHz, CD3OD): δH 7.83 (d, J = 8.5 Hz, 2H), 7.66–7.49 (m, 2H), 7.39–7.36 (m, 1H), 7.34–7.20 (m, 2H), 6.98 (d, J = 9.2 Hz, 2H), 4.58 (s, 2H), 4.03 (t, J = 7.1 Hz, 2H), 3.84 (s, 3H), 2.87 (t, J = 7.6 Hz, 2H), 2.62 (pent, J = 7.1 Hz, 2H); LCMS (ESI): Rt = 0.737 min, m/z = 348.1 [M+H]+; > 98% (215, 254 nm).
N-(3-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-2-phenylacetamide (4f).
The title compound (22.4 mg, 17% yield) was prepared following General Procedure C using phenylacetyl chloride (80 μL, 0.60 mmol) and intermediate 9b (80 mg, 0.38 mmol): 1H NMR (400 MHz, CD3OD): δH 7.56 (d, J = 8.0 Hz, 1H), 7.52 (s, 1H), 7.34–7.27 (m, 4H), 7.26–7.21 (m, 3H), 7.09 (d, J = 7.6 Hz, 1H), 4.38 (s, 2H), 4.37 (s, 1H), 4.04 (t, J = 7.1 Hz, 2H), 3.56 (s, 2H), 2.87 (t, J = 7.7 Hz, 2H), 2.62 (pent, J = 7.4 Hz, 2H); LCMS (ESI): Rt = 0.731 min, m/z = 332.1 [M+H]+; > 98% (215, 254 nm).
2-(3,4-Dichlorophenyl)-N-(3-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)acetamide (4g).
The title compound (57 mg, 30% yield) was prepared following General Procedure D using 3,4-dichlorophenylacetic acid (144 mg, 0.70 mml) and intermediate 9b (100 mg, 0.47 mmol): 1H NMR (400 MHz, CD3OD): δH 7.62–7.56 (m, 1H), 7.55–7.44 (m, 4H), 7.36–7.23 (m, 3H), 7.18–7.09 (m, 1H), 4.41 (s, 2H), 4.12–4.02 (m, 2H), 3.57 (s, 2H), 2.90 (dd, J = 8.3, 6.8 Hz, 2H), 2.72–2.55 (m, 2H); LCMS (ESI): Rt = 0.766 min, m/z = 400.0 [M+H]+; > 98% (215, 254 nm).
N-(3-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-3,4-dimethoxybenzamide (4h).
The title compound (12 mg, 14% yield) was prepared following General Procedure D using 3,4-dimethoxybenzoic acid (70.4 mg, 0.47 mmol) and intermediate 9b (50 mg, 0.23 mmol): 1H NMR (400 MHz, CD3OD): δH 7.67 (s, 1H), 7.57 (d, J = 7.7 Hz, 1H), 7.53–7.47 (m, 2H), 7.39 (s, 1H), 7.32 (t, J = 7.7 Hz, 1H), 7.23 (d, J = 7.5 Hz, 1H), 7.01 (d, J = 8.5 Hz, 1H), 4.59 (s, 2H), 4.58 (s, 1H), 4.06 (t, J = 7.1 Hz, 2H), 3.87 (s, 6H), 2.89 (t, J = 7.7 Hz, 2H), 2.64 (pent, J = 7.4 Hz, 2H); LCMS (ESI): Rt = 0.661 min, m/z = 378.1 [M+H]+; > 98% (215, 254 nm).
N-(3-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-3-methoxy-4-methylbenzamide (4i).
The title compound (40 mg, 28% yield) was prepared following General Procedure D using 3,4-dimethoxybenzoic acid (79 mg, 0.48 mmol) and intermediate 9b (99 mg, 0.40 mmol). 1H NMR (400 MHz, CD3OD): δH 7.67 (s, 1H), 7.57 (d, 1H, J = 7.9 Hz), 7.41–7.39 (m, 2H), 7.33–7.30 (m, 2H), 7.24 (d, 1H, J = 7.7 Hz), 7.20 (d, 1H, J = 7.9 Hz), 4.60 (s, 2H), 4.08 (t, 2H, J = 7.2 Hz), 3.89 (s, 3H), 2.91 (t, 2H, J = 7.2 Hz), 2.67–2.63 (m, 2H), 2.23 (s, 3H); LCMS (ESI): Rt = 1.15 min, m/z = 362.1 [M+H]+; > 98% (215, 254 nm).
N-(3-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-3-methoxy-5-methylbenzamide (4j).
The title compound (38 mg, 26% yield) was prepared following General Procedure D using 3,4-dimethoxybenzoic acid (79 mg, 0.48 mmol) and intermediate 9b (99 mg, 0.40 mmol). 1H NMR (400 MHz, CD3OD): δH 7.67 (s, 1H), 7.57 (d, 1H, J = 7.6 Hz), 7.39 (s, 1H), 7.32 (t, 1H, J = 7.6 Hz), 7.27 (s, 1H), 7.22 (d, 1H, J = 7.6 Hz), 6.29 (s, 1H), 4.58 (s, 2H), 4.08 (t, 2H, J = 7.0 Hz), 3.82 (s, 3H), 2.89 (t, 2H, J = 7.0 Hz), 2.84 (q, 2H, J = 7.0 Hz), 2.37 (s, 3H); LCMS (ESI): Rt = 1.08 min, m/z = 362.1 [M+H]+; > 98% (215, 254 nm).
N-(3-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)picolinamide (4k).
The title compound (15.4 mg, 10% yield) was prepared following General Procedure D using 2-picolinic acid (115 mg, 0.94 mmol) and intermediate 9b (100 mg, 0.47 mmol): 1H NMR (400 MHz, CD3OD): δH 8.63 (d, J = 3.8 Hz, 1H), 8.13–8.10 (m, 1H), 7.96 (t, J = 7.8 Hz, 1H), 7.68 (s, 1H), 7.59–7.53 (m, 2H), 7.44 (d, J = 1.3 Hz, 1H), 7.34 (t, J = 7.6 Hz, 1H), 7.27 (d, J = 7.6 Hz, 1H), 4.64 (s, 2H), 4.08 (t, J = 7.1 Hz, 2H), 2.93 (t, J = 7.7 Hz, 2H), 2.65 (pent, J = 7.4 Hz, 2H); LCMS (ESI): Rt = 0.665 min, m/z = 319.1 [M+H]+; > 98% (215, 254 nm).
N-(3-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)nicotinamide (4l).
The title compound (23.5 mg, 16% yield) was prepared following General Procedure C using isonicotinoyl chloride hydrochloride (125 mg, 0.70 mmol) and intermediate 9b (100 mg, 0.47 mmol): 1H NMR (400 MHz, CD3OD): δH 8.68 (d, J = 4.9 Hz, 2H), 7.82 (d, J = 5.4 Hz, 2H), 7.69 (s, 1H), 7.58 (d, J = 7.8 Hz, 1H), 7.37 (s, 1H), 7.32 (t, J = 7.6 Hz, 1H), 7.22 (d, J = 7.6 Hz, 1H), 4.61 (s, 2H), 4.04 (t, J = 6.9 Hz, 2H), 2.87 (t, J = 7.4 Hz, 2H), 2.62 (pent, J = 7.4 Hz, 2H); LCMS (ESI): Rt = 0.517 min, m/z = 319.1 [M+H]+; >98% (215, 254 nm).
N-(3-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)isonicotinamide (4m).
The title compound (20.5 mg, 14% yield) was prepared following General Procedure C using nicotinoyl chloride hydrochloride (125 mg, 0.70 mmol) and intermediate 9b (100 mg, 0.47 mmol): 1H NMR (400 MHz, CD3OD): δH 9.09 (s, 1H), 9.02 (d, J = 1.7 Hz, 1H), 8.68–8.65 (m, 2H), 8.38–8.35 (m, 2H), 7.68 (s, 1H), 7.58–7.56 (m, 1H), 7.54–7.48 (m, 2H), 7.47 (s, 1H), 7.35 (t, J = 7.6 Hz, 1H), 7.28 (t, J = 7.8 Hz, 1H), 4.62 (s, 2H), 4.09 (t, J = 7.1 Hz, 2H), 2.95 (t, J = 8.0 Hz, 2H), 2.65 (pent, J = 7.8 Hz, 2H); LCMS (ESI): Rt = 0.095 min, m/z = 319.1 [M+H]+; > 98% (215, 254 nm).
N-(3-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-5-methylnicotinamide (4n).
The title compound (45 mg, 48% yield) was prepared according to General Procedure D, using 5-methylpyridine-3-carboxylic acid (45 mg, 0.32 mmol) and intermediate 9b (60 mg, 0.28 mmol). 1H NMR (400 MHz, CD3OD): δH 9.19 (t, J = 5.7 Hz, 1H), 8.88 (d, J = 1.8 Hz, 1H), 8.55 (d, J = 1.3 Hz, 1H), 8.09 (s, 1H), 7.71 (s, 1H), 7.57 (d, J = 7.7 Hz, 1H), 7.51 (s, 1H), 7.27 (t, J = 7.7 Hz, 1H), 7.12 (d, J = 7.7 Hz, 1H), 4.50 (d, J = 5.7 Hz, 2H), 3.98 (t, J = 7.2 Hz, 2H), 3.16 (d, J = 6.0 Hz, 2H), 2.74 (d, J = 7.2 Hz, 2H), 2.36 (s, 3H); LCMS Rt = 0.60 min, m/z = 333.1 [M+H]+; > 98% (215, 254 nm).
N-(3-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-3-methoxy-N-methylbenzamide (4o).
Sodium hydride (60% dispersion in mineral oil, 14 mg, 0.36 mmol) was added to a solution of 4c (62 mg, 0.18 mmol) in anhydrous DMF (1.8 mL) at 0 °C under an inert atmosphere. After stirring for 5 min, iodomethane (22 μL, 0.36 mmol) was added and the mixture stirred for 1 h. The reaction was quenched with water and extracted with EtOAc (3 × 10 mL). Combined organic layers were washed with water (5 × 10 mL), dried (MgSO4), filtered and purified by flash column chromatography to afford the title compound (24 mg, 37% yield). 1H NMR (400 MHz, CD3OD): δH 8.05–7.70 (m, 3H), 7.63–7.30 (m, 2H), 7.04 (s, 4H), 4.83 (s, 2H), 4.68–4.53 (m, 2H), 3.85 (s, 3H), 3.81–3.71 (m, 2H), 3.15 – 3.01 (m, 2H), 2.96 (s, 3H); LCMS (ESI): Rt = 0.792 min, m/z = 362.3 [M+H]+; > 98% (215, 254 nm).
N-(3-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-N-isopropyl-3-methoxybenzamide (4p).
Step 1: N-(3-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)propan-2-amine. The title compound (55 mg, 35% yield) was prepared following General Procedure E using isopropylamine (100 μL, 1.22 mmol) and 10 (130 mg, 0.61 mmol): LCMS Rt = 0.277 min, m/z = 256.2 [M+H]+; > 98% (215, 254 nm). Step 2: N-(3-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-N-isopropyl-3-methoxybenzamide. The title compound (53.5 mg, 64% yield) was prepared following General Procedure C using 3-methoxybenzoyl chloride (45.4 μL, 0.32 mmol) and (N-(3-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)propan-2-amine (55 mg, 0.22 mmol): 1H NMR (400 MHz, CD3OD): δH 7.69 (s, 1H), 7.59–7.54 (m, 2H), 7.39 (s, 1H), 7.36–7.32 (m, 2H), 7.27–7.21 (m, 1H), 7.05–7.00 (m, 2H), 4.72 (s, 2H), 4.05 (t, J = 6.9 Hz, 2H), 3.83 (s, 3H), 3.66–3.61 (m, 1H), 2.89 (t, J = 7.4 Hz, 2H), 2.64 (pent, J = 7.4 Hz, 2H), 1.15 (d, J = 5.4 Hz, 6H); LCMS (ESI): Rt = 0.886 min, m/z = 390.2 [M+H]+; > 98% (215, 254 nm).
N-(Cyclopropylmethyl)-N-(3-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-3-methoxybenzamide (4q).
Step 1: 1-Cyclopropyl-N-(3-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)methanamine. The title compound (70 mg, 35% yield) was prepared following General Procedure E using cyclopropylmethylamine (131 μL, 1.51 mmol) and intermediate 10 (160 mg, 0.75 mmol). LCMS (ESI): Rt = 0.099 min, m/z = 268.2 [M+H]+; > 98% (215, 254 nm). Step 2: N-(Cyclopropylmethyl)-N-(3-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-3-methoxybenzamide. The title compound (101 mg, 96% yield) was prepared following General Procedure C using 3-methoxybenzoyl chloride (55 μL, 0.39 mmol) and 1-cyclopropyl-N-(3-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)methanamine (70 mg, 0.26 mmol): 1H NMR (400 MHz, CD3OD): δH 7.60–7.53 (m, 2H), 7.41–7.33 (m, 3H), 7.03 (d, J = 7.6 Hz, 1H), 7.00–6.97 (m, 3H), 4.93 (s, 2H), 4.07 (t, J = 6.6 Hz, 2H), 3.83 (s, 3H), 3.39 (t, J = 7.2 Hz, 2H), 2.93–2.87 (m, 2H), 2.64 (pent, J = 7.2 Hz, 2H), 0.95–0.89 (m, 1H), 0.27 – 0.20 (m, 2H), 0.02– −0.01 (m, 2H); LCMS (ESI): Rt = 0.898 min, m/z = 402.2 [M+H]+; > 98% (215, 254 nm).
N-(Cyclobutylmethyl)-N-(3-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-3-methoxybenzamide (4r).
Step 1: 1-Cyclobutyl-N-(3-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)methanamine. The title compound (135 mg, 51% yield) was prepared following General Procedure E using cyclobutylamine hydrochloride (286 mg, 2.36 mmol) and 3-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzaldehyde (200 mg, 0.94 mmol). LCMS (ESI): Rt = 0.101 min, m/z = 282.2 [M+H]+; > 98% (215, 254 nm). Step 2: N-(Cyclobutylmethyl)-N-(3-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-3-methoxybenzamide. The title compound (40.5 mg, 21% yield) was prepared following General Procedure C using 3-methoxybenzoyl chloride (97.4 μL, 0.69 mmol) and 1-cyclobutyl-N-(3-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)methanamine (130 mg, 0.46 mmol). 1H NMR (400 MHz, CD3OD): δH 7.84 (s, 1H), 7.66–7.53 (m, 3H), 7.41–7.36 (m, 2H), 7.04–6.89 (m, 3H), 4.70 (s, 1H), 4.59 (s, 1H), 4.33 (t, J = 6.9 Hz, 2H), 3.83 (s, 3H), 3.72–3.55 (m, 2H), 3.27 (t, J = 7.4 Hz, 2H), 2.84 (pent, J = 7.4 Hz, 2H), 2.64–2.55 (m, 1H), 2.13–1.79 (m, 4H), 1.68–1.59 (m, 2H); LCMS (ESI): Rt = 0.940 min, m/z = 416.2 [M+H]+; > 98% (215, 254 nm).
N-(Cyclopropylmethyl)-N-(3-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-2-methoxyisonicotinamide (4s).
The title compound (34 mg, 28% yield) was prepared following General Procedure D using 2-methoxyisonicotinic acid (92 mg, 0.60 mmol) and 1-cyclopropyl-N-(3-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)methanamine (80 mg, 0.30 mmol): 1H NMR (400 MHz, CD3OD): δH 8.24–8.17 (m, 2H), 7.67–7.22 (m, 5H), 7.06–6.98 (m, 1H), 6.87–6.81 (m, 1H), 4.92 (s, 1H), 4.61 (s, 1H), 4.06 (t, J = 6.9 Hz, 2H), 3.95 (s, 2H), 3.88 (s, 1H), 3.41–3.39 (m, 1H), 3.09 (d, J = 7.3 Hz, 1H), 2.92–2.87 (m, 2H), 2.64 (pent, J = 7.3 Hz, 2H), 1.14–0.89 (m, 1H), 0.54–0.47 (m, 2H), 0.25–0.24 (m, 1H), 0.03–0.02 (m, 1H); LCMS (ESI): Rt = 0.832 min, m/z = 403.1 [M+H]+; > 98% (215, 254 nm).
N-(Cyclopropylmethyl)-N-(3-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-5-methoxybenzyl)-3-methoxybenzamide (5a).
Step 1: 3-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-5-methoxybenzaldehyde. The title compound (200 mg, 77% yield) was prepared following General Procedure B using 3-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde (364 mg, 1.39 mmol) and intermediate 8b (200 mg, 1.07 mmol). LCMS (ESI): Rt = 0.511 min, m/z = 243.1 [M+H]+; > 98% (215, 254 nm). Step 2: 1-Cyclopropyl-N-(3-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-5-methoxybenzyl)methanamine The title compound (80 mg, 33% yield) was prepared following General Procedure E using cyclopropylmethylamine (143 μL, 1.65 mmol) and 3-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-5-methoxybenzaldehyde (200 mg, 0.83 mmol). LCMS (ESI): Rt = 0.318 min, m/z = 298.2 [M+H]+; > 98% (215, 254 nm). Step 3: N-(Cyclopropylmethyl)-N-(3-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-5-methoxybenzyl)-3-methoxybenzamide The title compound (30 mg, 26% yield) was prepared following General Procedure C using 3-methoxybenzoyl chloride (57 μL, 0.40 mmol) and 1-cyclopropyl-N-(3-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-5-methoxybenzyl)methanamine (80 mg, 0.27 mmol): 1H NMR (400 MHz, CD3OD): δH 7.39 (s, 1H), 7.36–7.08 (m, 3H), 7.05–6.92 (m, 4H), 4.61 (s, 2H), 4.04 (t, J = 7.1 Hz, 2H), 3.89–3.74 (m, 6H), 3.78–3.63 (m, 2H), 3.20–3.06 (m, 1H), 2.87 (t, J = 7.5 Hz, 2H), 2.62 (pent., J = 7.3 Hz, 2H), 0.49 (d, J = 26.1 Hz, 2H), 0.13 (d, J = 102.8 Hz, 2H); LCMS (ESI): Rt = 0.808 min, m/z = 432.1 [M+H]+; > 98% (215, 254 nm).
N-(Cyclopropylmethyl)-N-(5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-methoxybenzyl)-3-methoxybenzamide (5b).
Step 1: 5-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-methoxybenzaldehyde The title compound (190 mg, 82% yield) was prepared following General Procedure B using 2-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde (364 mg, 1.39 mmol) and intermediate 8b (180 mg, 0.96 mmol): LCMS (ESI): Rt = 0.476 min, m/z = 243.0 [M+H]+; > 98% (215, 254 nm). Step 2: 1-Cyclopropyl-N-(5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-methoxybenzyl)methanamine The title compound (90 mg, 39% yield) was prepared following General Procedure E using cyclopropylmethylamine (136 μL, 1.57 mmol) and 5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-methoxybenzaldehyde (190 mg, 0.78 mmol). LCMS (ESI): Rt = 0.420 min, m/z = 298.2 [M+H]+; > 98% (215, 254 nm). Step 3: N-(Cyclopropylmethyl)-N-(5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-methoxybenzyl)-3-methoxybenzamide The title compound (60 mg, 46% yield) was prepared following General Procedure C using 3-methoxybenzoyl chloride (64 μL, 0.45 mmol) and 1-cyclopropyl-N-(5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-methoxybenzyl)methanamine (90 mg, 0.30 mmol). 1H NMR (400 MHz, CD3OD): δH 7.60 (s, 1H), 7.41–7.25 (m, 2H), 7.08–6.88 (m, 5H), 4.61 (s, 2H), 4.10–4.01 (m, 2H), 3.87–3.82 (m, 2H), 3.78 (s, 3H), 3.68 (s, 3H), 3.19–3.09 (m, 1H), 2.92–2.84 (m, 2H), 2.64 (pent., J = 7.4 Hz, 2H), 0.60–0.34 (m, 2H), 0.39– −0.13 (m, 2H); LCMS (ESI): Rt = 0.693 min, m/z = 432.1 [M+H]+; > 98% (215, 254 nm).
N-(Cyclopropylmethyl)-N-(5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-ethoxybenzyl)-3-methoxybenzamide (5c).
Step 1: 5-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-ethoxybenzaldehyde The title compound (300 mg, 88% yield) was prepared following General Procedure B using 2-ethoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde (443 mg, 1.60 mmol) and intermediate 8b (250 mg, 1.34 mmol). LCMS (ESI): Rt = 0.582 min, m/z = 257.1 [M+H]+; > 98% (215, 254 nm). Step 2: 1-Cyclopropyl-N-(5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-ethoxybenzyl)methanamine The title compound (38 mg, 10% yield) was prepared following General Procedure E using cyclopropylmethylamine (203 μL, 2.34 mmol) and 5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-ethoxybenzaldehyde (300 mg, 1.17 mmol). LCMS (ESI): Rt = 0.564 min, m/z = 312.1 [M+H]+; > 98% (215, 254 nm). Step 3: N-(Cyclopropylmethyl)-N-(5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-ethoxybenzyl)-3-methoxybenzamide The title compound (17.5 mg, 32% yield) was prepared following General Procedure C using 3-methoxybenzoyl chloride (25.7μL, 0.18 mmol) and 1-cyclopropyl-N-(5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-ethoxybenzyl)methanamine (38 mg, 0.12 mmol): 1H NMR (400 MHz, CD3OD): δH 7.70–7.66 (m, 1H), 7.58 (dd, J = 8.5, 2.0 Hz, 1H), 7.41–7.28 (m, 2H), 7.14 (d, J = 7.8 Hz, 1H), 7.09–7.01 (m, 1H), 6.98 (d, J = 7.5 Hz, 1H), 6.95–6.90 (m, 1H), 4.29 (s, 2H), 4.03 (t, J = 7.4 Hz, 2H), 4.20–4.08 (m, 2H), 3.83 – 3.81 (m, 3H), 3.46–3.42 (m, 1H), 3.23 (t, J = 8.0 Hz, 2H), 2.81 (pent, J = 7.4 Hz, 2H), 1.47 (t, J = 6.0 Hz, 3H), 0.53–0.48 (m, 2H), 0.28–0.23 (m, 2H), 0.06–0.01 (m, 2H); LCMS (ESI): Rt = 0.814 min, m/z = 446.1 [M+H]+; > 98% (215, 254 nm).
N-(Cyclopropylmethyl)-N-(5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-isopropoxybenzyl)-3-methoxybenzamide (5d).
Step 1: 5-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-isopropoxybenzaldehyde The title compound (300 mg, 83% yield) was prepared following General Procedure B using 2-isopropoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde (484 mg, 1.67 mmol) and intermediate 8b (250 mg, 1.34 mmol). LCMS (ESI): Rt = 0.642 min, m/z = 271.1 [M+H]+; > 98% (215, 254 nm). Step 2: 1-Cyclopropyl-N-(5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-isopropoxybenzyl)methanamine The title compound (150 mg, 31% yield) was prepared following General Procedure E using cyclopropylmethylamine (257 μL, 2.96 mmol) and 5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-isopropoxybenzaldehyde (400 mg, 1.48 mmol). LCMS (ESI): Rt = 0.587 min, m/z = 326.1 [M+H]+; > 98% (215, 254 nm). Step 3: N-(Cyclopropylmethyl)-N-(5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-isopropoxybenzyl)-3-methoxybenzamide The title compound (38 mg, 36% yield) was prepared following General Procedure C using 3-methoxybenzoyl chloride (48.6 μL, 0.35 mmol) and 1-cyclopropyl-N-(5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-isopropoxybenzyl)methanamine (75 mg, 0.23 mmol): 1H NMR (400 MHz, CD3OD): δH 7.64–7.54 (m, 2H), 7.48 (s, 1H), 7.23 (s, 1H), 7.07–6.91 (m, 4H), 4.61 (s, 2H), 4.05 (t, J = 7.1 Hz, 2H), 3.66 (s, 3H), 3.50–3.40 (m, 2H), 3.19–3.08 (m, 1H), 2.91–2.82 (m, 2H), 2.64 (pent, J = 7.3 Hz, 2H), 1.44–1.20 (m, 6H), 0.59–0.40 (m, 2H), 0.30– −0.06 (m, 3H); LCMS (ESI): Rt = 0.857 min, m/z = 460.1 [M+H]+; > 98% (215, 254 nm).
N-(5-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-isopropoxybenzyl)-3-methoxybenzamide (5e).
Step 1: 5-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-isopropoxybenzonitrile The title compound (730 mg, 73% yield) was prepared following General Procedure B using 2-isopropoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzonitrile (1.72 g, 5.99 mmol) and intermediate 8b (700 mg, 3.74 mmol): LCMS (ESI): Rt = 0.745 min, m/z = 268.2 [M+H]+; > 98% (215, 254 nm). Step 2: (5-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-isopropoxyphenyl)methanamine The title compound (610 mg, 82% yield) was prepared following General Procedure G using 5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-isopropoxybenzonitrile (730 mg, 2.73 mmol): LCMS (ESI): Rt = 0.629 min, m/z = 272.3 [M+H]+; > 98% (215, 254 nm). Step 3: N-(5-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-isopropoxybenzyl)-3-methoxybenzamide The title compound (29 mg, 10% yield) was prepared following General Procedure C using 3-methoxybenzoyl chloride (155 μL, 1.11 mmol) and (5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-isopropoxyphenyl)methanamine (200 mg, 0.74 mmol): 1H NMR (400 MHz, CD3OD): δH 7.57–7.53 (m, 2H), 7.45–7.43 (m, 2H), 7.40–7.34 (m, 2H), 7.11–7.08 (m, 1H), 6.99 (d, J = 8.2 Hz, 1H), 4.71–4.65 (m, 1H), 4.59 (s, 2H), 4.03 (t, J = 7.6 Hz, 2H), 3.83 (s, 3H), 2.88 (t, J = 7.6 Hz, 2H), 2.62 (pent, J = 7.6 Hz, 2H), 1.35 (d, J = 6.0 Hz, 6H); LCMS (ESI): Rt = 0.891 min, m/z = 406.3 [M+H]+; > 98% (215, 254 nm).
N-(2-Cyclobutoxy-5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-N-(cyclopropylmethyl)-3-methoxybenzamide (5f).
Step 1: 2-Cyclobutoxy-5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzaldehyde The title compound (300 mg, 83% yield) was prepared following General Procedure B using 2-cyclobutoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzaldehyde (629 mg, 2.08 mmol) and intermediate 8b (190 mg, 1.02 mmol). LCMS (ESI): Rt = 0.808 min, m/z = 283.3 [M+H]+; > 98% (215, 254 nm). Step 2: N-(2-Cyclobutoxy-5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-1-cyclopropylmethanamine The title compound (120 mg, 53% yield) was prepared following General Procedure E using cyclopropylmethylamine (257 μL, 2.96 mmol) and 2-cyclobutoxy-5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzaldehyde (190 mg, 0.67 mmol). LCMS (ESI): Rt = 0.733 min, m/z = 338.3 [M+H]+; > 98% (215, 254 nm). Step 3: N-(2-Cyclobutoxy-5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-N-(cyclopropylmethyl)-3-methoxybenzamide The title compound (35 mg, 36% yield) was prepared following General Procedure C using 3-methoxybenzoyl chloride (52.5 μL, 0.31 mmol) and N-(2-cyclobutoxy-5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-1-cyclopropylmethanamine (70 mg, 0.21 mmol). 1H NMR (400 MHz, CD3OD): δH 7.66–7.44 (m, 2H), 7.41–7.27 (m, 2H), 7.26 (s, 1H), 7.12–6.90 (m, 3H), 4.62 (s, 2H), 4.04 (t, J = 7.1 Hz, 2H), 3.84 (d, J = 4.1 Hz, 2H), 3.66 (s, 3H), 3.50–3.42 (m, 2H), 3.20–3.08 (m, 1H), 2.95–2.81 (m, 1H), 2.64 (q, J = 7.4 Hz, 2H), 2.48 (d, J = 27.8 Hz, 2H), 2.10 (d, J = 66.4 Hz, 2H), 1.83 (dd, J = 46.1, 19.8 Hz, 2H), 0.50 (d, J = 31.6 Hz, 2H), 0.32– −0.07 (m, 2H); LCMS (ESI): Rt = 1.018 min, m/z = 472.4 [M+H]+; > 98% (215, 254 nm).
N-(2-Cyclobutoxy-5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-3-methoxybenzamide (5g).
Step 1: 2-Cyclobutoxy-5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzonitrile The title compound (1.00 g, quant.) was prepared following General Procedure B using 2-cyclobutoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzonitrile (2.02 g, 6.74 mmol) and intermediate 8b (700 mg, 3.74 mmol). LCMS (ESI): Rt = 0.933 min, m/z = 280.3 [M+H]+; > 98% (215, 254 nm). Step 2: (2-Cyclobutoxy-5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)phenyl)methanamine The title compound (500 mg, 49% yield) was prepared following General Procedure G using 2-cyclobutoxy-5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzonitrile (1.00 g, 3.58 mmol). LCMS (ESI): Rt = 0.677 min, m/z = 284.3 [M+H]+; > 98% (215, 254 nm). Step 3: N-(2-Cyclobutoxy-5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-3-methoxybenzamide The title compound (55 mg, 31% yield) was prepared following General Procedure C using 3-methoxybenzoyl chloride (89.3 μL, 0.64 mmol) and (2-cyclobutoxy-5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)phenyl)methanamine (120 mg, 0.42 mmol). 1H NMR (400 MHz, CDCl3): δH 7.64–7.62 (m, 2H), 7.37–7.36 (m, 2H), 7.06 (s, 1H), 7.02– 6.99 (m, 1H), 6.74 (d, J = 7.7 Hz, 2H), 4.73 – 4.66 (m, 3H), 3.97 (t, J = 7.2 Hz, 2H), 3.83 (s, 3H), 2.99 (t, J = 7.7 Hz, 2H), 2.59 (pent, J = 7.3 Hz, 2H), 2.51 – 2.44 (m, 2H), 2.22 – 2.12 (m, 2H), 1.90 – 1.82 (m, 1H), 1.76 – 1.66 (m, 1H); LCMS (ESI): Rt = 1.024 min, m/z = 418.3 [M+H]+; > 98% (215, 254 nm).
N-(5-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-(dimethylamino)benzyl)-3-methoxybenzamide (5h).
Step 1: 5-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-(dimethylamino)benzonitrile The title compound (200 mg, 27% yield) was prepared following General Procedure B using 2-(dimethylamino)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzonitrile (1.50 g, 5.53 mmol) and intermediate 8b (550 mg, 2.94 mmol). LCMS (ESI): Rt = 0.712 min, m/z = 253.2 [M+H]+; > 98% (215, 254 nm). Step 2: 2-(Aminomethyl)-4-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-N,N-dimethylaniline The title compound (200 mg, quant.) was prepared following General Procedure G using 5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-(dimethylamino)benzonitrile (200 mg, 0.79 mmol): LCMS (ESI): Rt = 0.493 min, m/z = 257.2 [M+H]+; > 98% (215, 254 nm). Step 3: N-(5-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-(dimethylamino)benzyl)-3-methoxybenzamide The title compound (92 mg, 61% yield) was prepared following General Procedure C using 3-methoxybenzoyl chloride (98.8 μL, 0.59 mmol) and 2-(aminomethyl)-4-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-N,N-dimethylaniline (100 mg, 0.39 mmol): 1H NMR (400 MHz, CD3OD): δH 7.60 (d, J = 1.9 Hz, 1H), 7.55 (dd, J = 8.4, 1.6 Hz, 1H), 7.46 – 7.44 (m, 2H), 7.37 (t, J = 7.5 Hz, 1H), 7.22 (s, 1H), 7.18 (d, J = 8.2 Hz, 1H), 7.10 – 7.08 (m, 1H), 4.72 (s, 2H), 4.01–3.96 (m, 2H), 3.83 (s, 3H), 2.83 (t, J = 7.0 Hz, 2H), 2.73 (s, 6H), 2.58 (pent, J = 7.0 Hz, 2H); LCMS (ESI): Rt = 0.673 min, m/z = 391.1 [M+H]+; > 98% (215, 254 nm).
2-(3,4-Dichlorophenyl)-N-(5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-(dimethylamino)benzyl)acetamide (5i).
The title compound (65 mg, 38% yield) was prepared following General Procedure D using 3,4-dichlorophenylacetic acid (120 mg, 0.59 mmol) and 2-(aminomethyl)-4-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-N,N-dimethylaniline (100 mg, 0.39 mmol): 1H NMR (400 MHz, CD3OD): δH 7.53 (dd, J = 7.5, 2.2 Hz, 2H), 7.44 (d, J = 8.4 Hz, 1H), 7.39 (d, J = 2.2 Hz, 1H), 7.27 (dd, J = 8.3, 2.2 Hz, 1H), 7.13 (d, J = 8.3 Hz, 1H), 7.05 (s, 1H), 4.50 (s, 2H), 4.04 (t, J = 6.9 Hz, 2H), 3.58 (s, 2H), 2.87 (t, J = 7.5 Hz, 2H), 2.66–2.61 (m, 8H); LCMS (ESI): Rt = 0.744 min, m/z = 443.2 [M+H]+; > 98% (215, 254 nm).
2-(3,4-Dichlorophenyl)-N-(5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-morpholinobenzyl)acetamide (5j).
Step 1: 5-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-morpholinobenzonitrile The title compound (390 mg, 50% yield) was prepared following General Procedure B using 2-morpholino-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzonitrile (1.68 g, 5.35 mmol) and intermediate 8b (550 mg, 2.94 mmol). LCMS (ESI): Rt = 0.600 min, m/z = 295.2 [M+H]+; > 98% (215, 254 nm). Step 2: (5-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-morpholinophenyl)methanamine The title compound (400 mg, quant.) was prepared following General Procedure G using 5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-morpholinobenzonitrile (390 mg, 1.32 mmol). LCMS (ESI): Rt = 0.542 min, m/z = 299.3 [M+H]+; > 98% (215, 254 nm). Step 3: 2-(3,4-Dichlorophenyl)-N-(5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-morpholinobenzyl)acetamide The title compound (60.5 mg, 47% yield) was prepared following General Procedure D using 3,4-dichlorophenylacetic acid (82.5 mg, 0.40 mmol) and (5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-morpholinophenyl)methanamine (80 mg, 0.27 mmol): 1H NMR (400 MHz, CD3OD): δH 7.56 (dd, J = 8.3, 2.1 Hz, 1H), 7.51 (d, J = 1.9 Hz, 1H), 7.45 (d, J = 8.2 Hz, 1H), 7.43 (d, J = 2.1 Hz, 1H), 7.27 – 7.25 (m, 2H), 7.19 (d, J = 8.3 Hz, 1H), 4.52 (s, 2H), 4.16 (t, J = 7.0 Hz, 2H), 3.79 – 3.77 (m, 4H), 3.57 (s, 2H), 3.02 (t, J = 7.5 Hz, 2H), 2.89 – 2.86 (m, 4H), 2.71 (pent, J = 7.0 Hz, 2H); LCMS (ESI): Rt = 0.878 min, m/z = 485.2 [M+H]+; > 98% (215, 254 nm).
2-(3,4-Dichlorophenyl)-N-(5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-isopropoxybenzyl)acetamide (5k).
The title compound (120 mg, 36% yield) was prepared following General Procedure D using 3,4-dichlorophenylacetic acid (227 mg, 1.11 mmol) and (5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-isopropoxyphenyl)methanamine (200 mg, 0.74 mmol): 1H NMR (400 MHz, CDCl3): δH 7.50–7.44 (m, 4H), 7.39 (d, J = 2.3 Hz, 1H), 7.23 (dd, J = 8.3, 2.1 Hz, 1H), 7.08 (d, J = 8.7 Hz, 1H), 4.75–4.68 (m, 1H), 4.38 (s, 2H), 4.29 (t, J = 7.5 Hz, 2H), 3.21 (t, J = 7.8 Hz, 2H), 2.99 (s, 2H), 2.83–2.80 (m, 2H), 1.33 (d, J = 5.9 Hz, 6H); LCMS (ESI): Rt = 0.982 min, m/z = 458.2 [M+H]+; > 98% (215, 254 nm).
N-(Cyclopropylmethyl)-2-(3,4-dichlorophenyl)-N-(5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-isopropoxybenzyl)acetamide (5l).
The title compound (16 mg, 20% yield) was prepared following General Procedure D using 3,4-dichlorophenylacetic acid (47.3 mg, 0.23 mmol) and 1-cyclopropyl-N-(5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2-isopropoxybenzyl)methanamine (50 mg, 0.15 mmol): 1H NMR (400 MHz, CDCl3): δH 7.44–7.42 (m, 1H), 7.33 (d, J = 8.1 Hz, 1H), 7.24–7.16 (m, 2H), 7.09–7.07 (m, 1H), 7.04–7.00 (m, 1H), 6.80 (d, J = 8.8 Hz, 1H), 4.77 (s, 2H), 4.06 (s, 2H), 4.25–4.20 (m, 2H), 3.34 (t, J = 7.8 Hz, 2H), 3.21–3.18 (m, 3H), 2.78–2.71 (m, 2H), 1.34 (d, J = 5.6 Hz, 6H), 0.56–0.51 (m, 2H), 0.49–0.44 (m, 1H), 0.22–0.18 (m, 2H); LCMS (ESI): Rt = 1.112 min, m/z = 512.3 [M+H]+; > 98% (215, 254 nm).
N-(2-Cyclobutoxy-5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-2-(3,4-dichlorophenyl)acetamide (5m).
The title compound (130 mg, 92% yield) was prepared following General Procedure D using 3,4-dichlorophenylacetic acid (92.3 mg, 0.45 mmol) and (2-cyclobutoxy-5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)phenyl)methanamine (85 mg, 0.45 mmol): 1H NMR (400 MHz, CD3OD): δH 7.50 (dd, J = 7.3, 2.1 Hz, 2H), 7.46 (d, J = 8.1 Hz, 1H), 7.40 (d, J = 2.1 Hz, 1H), 7.30 (dd, J = 8.3, 2.1 Hz, 1H), 7.16 (s, 1H), 6.79 (d, J = 8.5 Hz, 1H), 4.70 (pent, J = 7.0 Hz, 1H), 4.38 (s, 2H), 4.08 (t, J = 7.0 Hz, 2H), 3.55 (s, 2H), 2.93 (t, J = 7.5 Hz, 2H), 2.66 (pent, J = 7.5 Hz, 2H), 2.47–2.40 (m, 2H), 2.15–2.01 (m, 2H), 1.86–1.63 (m, 3H); LCMS (ESI): Rt = 1.255 min, m/z = 470.2 [M+H]+; > 98% (215, 254 nm).
N-(2-cyclobutoxy-5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-N-(cyclopropylmethyl)-2-(3,4-dichlorophenyl)acetamide (5n).
The title compound (59 mg, 76% yield) was prepared following General Procedure D using 3,4-dichlorophenylacetic acid (45.6 mg, 0.22 mmol) and N-(2-cyclobutoxy-5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-1-cyclopropylmethanamine (50 mg, 0.15 mmol): 1H NMR (400 MHz, CD3OD): δH 7.35–7.22 (m, 2H), 7.21–7.10 (m, 2H), 7.12–7.00 (m, 2H), 6.95–6.90 (m, 1H), 6.60 (dd, J = 18.0, 8.5 Hz, 1H), 4.58–4.53 (m, 2H), 4.51 (s, 2H), 3.92–3.75 (m, 2H), 3.73–3.61 (m, 2H), 3.60–3.52 (m, 2H), 3.14–3.12 (m, 1H), 2.69–2.61 (m, 2H), 2.43 (pent., J = 7.3 Hz, 2H), 2.37–2.23 (m, 2H), 2.04–1.86 (m, 2H), 1.75–1.44 (m, 2H), 0.46–0.19 (m, 2H); LCMS (ESI): Rt = 1.133 min, m/z = 524.3 [M+H]+; > 98% (215, 254 nm).
N-(2-cyclobutoxy-5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-N-(cyclopropylmethyl)-3-methoxy-4-methylbenzamide (5o).
The title compound (110 mg, 76% yield) was prepared following General Procedure C using 3-methoxy-4-methylbenzoic acid (74 mg, 0.44 mmol) and N-(2-cyclobutoxy-5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-1-cyclopropylmethanamine (100 mg, 0.30 mmol): 1H NMR (400 MHz, CD3OD): δH 7.64–7.47 (m, 2H), 7.26 (s, 1H), 7.22–7.13 (m, 1H), 6.94 (dd, J = 7.5, 1.2 Hz, 1H), 6.89–6.79 (m, 2H), 4.64 (s, 2H), 4.04 (t, J = 6.9 Hz, 2H), 3.86–3.85 (m, 1H), 3.57–3.46 (m, 3H), 3.17–3.13 (m, 1H), 2.87 (t, J = 8.0 Hz, 2H), 2.63 (pent, J = 7.5 Hz, 2H), 2.54–2.38 (m, 3H), 2.25–2.17 (m, 5H), 2.06–1.99 (m, 1H), 1.85–1.70 (m, 2H), 0.57–0.42 (m, 2H), 0.30–0.24 (m, 1H), 0.04– −0.01 (m, 1H); LCMS (ESI): Rt = 0.743 min, m/z = 486.3 [M+H]+; > 98% (215, 254 nm).
N-(2-Cyclobutoxy-5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)-3-methoxy-4-methylbenzamide (5p).
The title compound (32 mg, 26% yield) was prepared following General Procedure D using 3-methoxy-4-methylbenzoic acid (47 mg, 0.28 mmol) and (2-cyclobutoxy-5-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)phenyl)methanamine (80 mg, 0.28 mmol): 1H NMR (400 MHz, CD3OD): δH 7.56 (d, J = 1.8 Hz, 2H), 7.51 (d, J = 8.4, 2.1 Hz, 1H), 7.42 (d, J = 1.3 Hz, 1H), 7.38 (dd, J = 7.7, 1.3 Hz, 1H), 7.21–7.19 (m, 2H), 6.80 (d, J = 8.4 Hz, 1H), 4.75 (pent, J = 6.9 Hz, 1H), 4.60 (s, 2H), 4.00 (t, J = 6.9 Hz, 2H), 3.88 (s, 3H), 2.84 (t, J = 7.9 Hz, 2H), 2.60 (pent, J = 7.9 Hz, 2H), 2.51–2.44 (m, 2H), 2.23 (s, 3H), 2.19–2.12 (m, 2H), 1.87–1.69 (m, 2H); LCMS (ESI): Rt = 0.972 min, m/z = 432.1 [M+H]+; > 98% (215, 254 nm).
(R)-2-(3,4-Dichlorophenyl)-N-(6-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2,3-dihydro-1H-inden-1-yl)acetamide (6a).
Prepared according to General Procedure D, combining intermediate 19a (45 mg, 0.19 mmol) and 3,4-dichlorophenylacetic acid (42 mg, 0.21 mmol). Purification by flash column chromatography (12 g, 0 – 3.5% MeOH/CH2Cl2) afforded tan solid (47 mg, 0.11 mmol, 58%). 1H NMR (400 MHz, CD3OD) δH 7.56 – 7.52 (m, 2H), 4.48 (d, 1H, J = 7.4 Hz), 7.41 (s, 1H), 7.28 (d, 1H, J = 7.4 Hz), 7.23–7.19 (m, 2H), 5.37 (t, 1H, J = 7.5 Hz), 4.05 (t, 2H, J = 7.0 Hz), 3.55 (d, 2H, J = 4.2 Hz), 3.02–2.94 (m, 1H), 2.87 (t, 2H, J = 7.0 Hz), 2.84–2.80 (m, 1H), 2.63 (dt, 2H, J = 7.0, 7.0 Hz), 2.56–2.47 (m, 1H), 1.93–1.81 (m, 1H); LCMS (ESI): Rt = 1.04 min, m/z = 418.1 [M+H]+; > 95% (215, 254 nm).
(R)-N-(6-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2,3-dihydro-1H-inden-1-yl)-3-methoxy-4-methylbenzamide (6b).
Prepared by according to General Procedure D, combining intermediate 19a (45 mg, 0.19 mmol) and 3-methoxy-4-methylbenzoic acid (34 mg, 0.21 mmol). Purification by flash column chromatography affords title compound (44 mg, 60%). 1H NMR (400 MHz, CD3OD) δH 7.53 (m, 2H), 7.45 (d, J = 8.2 Hz, 1H), 7.40 (s, 1H), 7.27 (d, J = 8.2 Hz, 1H), 7.18 (m, 2H), 5.36 (m, 1H), 4.04 (t, J = 6.7 Hz, 2H), 3.54 (m, 2H), 2.97 (m, 1H), 2.86 (m, 3H), 2.62 (m, 2H), 2.49 (m, 1H), 1.86 (m, 1H); LCMS (ESI): Rt = 0.89 min, m/z = 388 [M+H]+; > 95% (214, 254 nm).
(R)-N-(7-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-1,2,3,4-tetrahydronaphthalen-1-yl)-3-methoxy-4-methylbenzamide (6c).
Prepared according to General Procedure D, combining 19b (39 mg, 0.13 mmol) and 3-methoxy-4-methylbenzoic acid (25 mg, 0.15 mmol). Purification by flash column chromatography affords title compound (36 mg, 0.09 mmol, 67%). 1H NMR (400 MHz, CD3OD) δH 7.59 (s, 1H), 7.50 (dd, 1H, J = 8.0, 1.5 Hz), 7.43 (d, 1H, J = 1.5 Hz), 7.39 (dd, 1H, J = 8.0, 1.5 Hz), 7.25 (s, 1H), 7.18 (d, J = 8.0 Hz, 1H), 7.10 (d, J = 8.0 Hz, 1H), 5.34 (t, 1H, J = 6.2 Hz), 4.00 (t, 2H, J = 7.1 Hz), 3.88 (s, 3H), 2.89–2.77 (m, 4H), 2.59 (p, 2H, J = 7.1 Hz), 2.22 (s, 3H), 2.15–2.09 (m, 1H), 2.05 – 1.84 (m, 3H); LCMS (ESI): Rt = 0.90 min, m/z = 402 [M+H]+; > 95% (215, 254 nm).
(R)-N-(6-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2,3-dihydro-1H-inden-1-yl)-2-methylisonicotinamide (6d).
Prepared according to General Procedure D, combining 19a (35 mg, 0.13 mmol) and 2-methylisonicotinic acid (19 mg mg, 0.14 mmol). Purification by flash column chromatography afforded title compound (29 mg, 0.08 mmol, 62%). 1H NMR (400 MHz, DMSO-d6) δH 9.00 (d, J = 8.4 Hz, 1H), 8.56 (d, J = 5.1 Hz, 1H), 7.71 (s, 1H), 7.62 (m, 1H), 7.55 (m, 2H), 7.48 (s, 1H), 7.20 (d, J = 7.7 Hz, 1H), 5.55 (m, 1H), 3.92 (t, J = 7.0 Hz, 2H), 2.93 (m, 1H), 2.84 (m, 1H), 2.71 (m, 2H), 2.52 (s, 3H), 2.46 (m, 3H), 1.98 (m, 1H); LCMS (ESI): Rt = 0.64 min, m/z =359 [M+H]+; > 95% (215, 254 nm).
(R)-N-(6-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2,3-dihydro-1H-inden-1-yl)-3,5-dimethoxy-4-methylbenzamide (6e).
Prepared according to General Procedure D, combining 19a (40 mg, 0.13 mmol) and 3,5-dimethoxy-4-methylbenzoic acid (27 mg, 0.14 mmol). Purification by flash column chromatography afforded title compound (43 mg, 0.10 mmol, 80%). 1H NMR (400 MHz, CD3OD) δH 7.62 (s, 1H), 7.57 (d, 1H, J = 7.8 Hz), 7.32 (s, 1H), 7.24 (d, 1H, 7.8 Hz), 7.17 (s, 1H), 5.89 (t, 1H, J = 7.6 Hz), 4.02 (t, 2H, J = 7.2 Hz), 3.87 (s, 6H), 3.19–3.02 (m, 2H), 2.96–2.89 (m, 1H), 2.84 (t, 2H, J = 7.2 Hz), 2.66–2.59 (m, 2H), 2.07–2.02 (m, 1H), 2.07 (s, 3H); LCMS (ESI): Rt = 0.94 min, m/z = 418 [M+H]+; > 95% (215, 254 nm).
2-Chloro-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (8a).
2-(2-Oxopyrrolidin-1-yl)acetamide (2.0 g, 14.1 mmol) was taken in POCl3 (5.25 mL, 56.3 mmol) in a sealed tube and heated to 85 °C for 15 h. The slurry was cooled to 0 °C and quenched with cold water (10 mL), followed by sat. aq. NaHCO3 (10 mL). Solid potassium carbonate was added until the solution was basic. The mixture was extracted with CH2Cl2 (3 × 20 mL), the combined organics were washed with sat. aq. NaCl, dried (MgSO4) and concentrated in vacuo. The combined material from the three preparations was purified by flash column chromatography (gradient 0–20% MeOH in DCM), to afford the title compound as an off-white solid (1.30 g, 9.1 mmol, 65%): 1H NMR (400 MHz, CDCl3) δH 6.78 (s, 1H), 3.97 (t, 2H, J = 7.2 Hz), 2.87 (t, 2H, J = 7.6 Hz), 2.55 (pent, 2H, J = 7.5 Hz); LCMS (ESI): Rt = 0.10 min, m/z = 143.1 [M+H]+; >98% (215, 254 nm).
2-Bromo-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (8b).
2-(2-Oxopyrrolidin-1-yl)acetamide (2.0 g, 14.1 mmol) was taken in POBr3 (4.44 g, 15.5 mmol) in a sealed tube and heated to 70 °C for 1 h. The reaction was run in triplicate (6.0 g, 42.2 mmol). The slurry was cooled to 0 °C and quenched with cold water (10 mL), followed by sat. aq. NaHCO3 (10 mL). Solid potassium carbonate was added until the solution was basic. The mixture was extracted with CH2Cl2 (3 × 50 mL), the combined organics were washed with sat. aq. NaCl, dried (MgSO4) and concentrated in vacuo. The combined material from the three preparations was purified by flash column chromatography (80 g, gradient 0–20% MeOH in DCM), to afford the title compound as an off-white solid (5.54 g, 29.6 mmol, 70%): 1H NMR (400 MHz, CD3OD) δH 7.05 (s, 1H), 4.02 (t, 2H, J = 7.1 Hz), 2.83 (t, 2H, J = 7.8 Hz), 2.58 (dt, 2H, J = 7.8, 7.1 Hz); LCMS (ESI): Rt = 0.09 min, m/z = 187.1 [M+H]+; >98% (215, 254 nm).
tert-Butyl (3-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzyl)carbamate (9a).
The title compound (160 mg, 0.51 mmol, 96%) was prepared according to General Procedure B, reacting intermediate 8b (100 mg, 0.53 mmol) with 3-(N-Boc-aminomethyl)phenylboronic acid (268 mg, 1.07 mmol). 1H NMR (400 MHz, CDCl3) δH 7.70–7.51 (m, 2H), 7.30–7.25 (m, 2H), 7.07 (s, 1H), 4.29–4.23 (m, 2H), 3.99 (t, 2H, J = 6.3 Hz), 3.46 (s, 1H), 2.96 (t, 2H, J = 6.0 Hz), 2.59 (m, 2H), 1.45 (s, 9H); LCMS (ESI): Rt = 0.78 min, m/z = 314.2 [M+H]+; > 98% (215, 254 nm). (3-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)phenyl)methanamine (9b). TFA (1 mL) was added to a solution of 9a (160 mg, 0.510 mmol) in CH2Cl2 (2 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 1 h. The reaction mixture was concentrated to obtain the desired product as the TFA salt. The crude mixture was used for next step without further purification. LCMS (ESI): Rt = 0.257 min, m/z = 214.2 [M+H]+; > 98% (215, 254 nm).
3-(6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)benzaldehyde (10).
The title compound (480 mg, 47% yield) was prepared following General Procedure B using 3-formylphenylboronic acid (1.08 g, 7.22 mmol) and 8b (900 mg, 4.81 mmol): LCMS, >98% (215, 254 nm), Rt = 0.10 min, m/z = 213.2 [M+H].
6-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-2,3-dihydro-1H-inden-1-one (16a).
6-Bromo-2,3-dihydro-1H-inden-1-one (2.0 g, 9.5 mmol), bis(pinacolato)diborane (3.1 g, 12.3 mmol), PdCl2(dppf).CH2Cl2 (390 mg, 0.47 mmol) and potassium acetate (2.8 g, 28 mmol) were taken in toluene (40 mL) under an inert atmosphere and heated to 90 °C for 12 h. The cooled mixture was diluted with EtOAc and filtered through celite, washing with further EtOAc. The filtrate was washed with water, sat. aq. NaCl and concentrated in vacuo. Purification by flash column chromatography (80 g, 0–20% EtOAc in hexanes) afforded a pale-yellow solid (2.35 g, 9.10 mmol, 96%). 1H NMR (400 MHz, DMSO-d6) δH 7.91 (d, 1H, J = 7.5 Hz), 7.89 (s, 1H), 7.60 (d, 1H, J = 7.5 Hz), 3.14–3.11 (m, 2H), 2.66–2.62 (m, 2H), 1.21 (s, 12H); LCMS (ESI): Rt = 1.03 min, m/z = 259.1 [M+H]+; > 98% (215, 254 nm).
7-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-3,4-dihydronaphthalen-1(2H)-one (16b).
7-Bromo-1-tetralone (1.0 g, 4.44 mmol), bis(pinacolato)diborane (1.47 g, 5.8 mmol), PdCl2(dppf).CH2Cl2 (181 mg, 0.22 mmol) and potassium acetate (1.31 g, 13.3 mmol) were taken in toluene (18 mL) under an inert atmosphere and heated to 90 °C for 12 h. The cooled mixture was diluted with EtOAc and filtered through celite, washing with further EtOAc. The filtrate was washed with water, sat. aq. NaCl and concentrated in vacuo. Purification by flash column chromatography (40 g, 0–25% EtOAc in hexanes) afforded a pale-yellow solid (1.01 g, 3.71 mmol, 84%). 1H NMR (400 MHz, CD3OD) δH 8.34 (s, 1H), 7.85 (d, 1H, J = 7.6 Hz), 7.32 (d, 1H, J = 7.6 Hz), 3.01 (t, 2H, J = 6.2 Hz), 2.66 (t, 2H, J = 6.2 Hz), 2.13 (pent., 2H, J = 6.2 Hz), 1.35 (s, 12H); LCMS (ESI): Rt = 1.11 min, m/z = 273 [M+H]+; > 95% (215, 254 nm).
6-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2,3-dihydro-1H-inden-1-one (17a).
Intermediate 8b (100 mg, 0.53 mmol), 16a (221 mg, 0.86 mmol), Pd(OAc)2 (12 mg, 0.05 mmol), PCy3.HBF4 (30 mg, 0.08 mmol) and K3PO4 (230 mg, 1.1 mmol) were combined and taken under an inert atmosphere. DME:Water (4 mL, 5:1) was added and the mixture heated to 105 °C for 16 h. The cooled mixture was diluted with EtOAc, filtered through celite, washing with further EtOAc; the filtrate was washed with water and concentrated in vacuo. Purification by flash column chromatography (12 g, 0–5% MeOH in CH2Cl2 solution) affords title compound (60 mg, 0.25 mmol, 47%). 1H NMR (400 MHz, DMSO-d6) δH 8.03 (dd, 1H, J = 8.0, 1.7 Hz), 7.92 (d, 1H, J = 1.7 Hz), 7.67 (s, 1H), 7.53 (d, 1H, J = 8.0 Hz), 3.97 (t, 2H, J = 7.0 Hz), 3.09 – 3.06 (m, 2H), 2.76 (t, 2H, J = 7.0 Hz), 2.66 – 2.62 (m, 2H), 2.55 – 2.49 (m, 2H); LCMS (ESI): Rt = 0.60 min, m/z = 239 [M+H]+; > 98% (215, 254 nm).
7-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-3,4-dihydronaphthalen-1(2H)-one (17b).
Intermediate 8b (250 mg, 1.43 mmol), 16b (640 mg, 2.3 mmol), PdCl2(dppf).CH2Cl2 (110 mg, 0.13 mmol) and Cs2CO3 (880 mg, 2.7 mmol) were combined and taken under an inert atmosphere. THF:Water (4 mL, 5:1) was added and the mixture heated under microwave irradiation at 95 °C for 1 h. The cooled mixture was diluted with EtOAc, filtered through celite, washing with further EtOAc; the filtrate was washed with water and concentrated in vacuo. Purification by flash column chromatography (12 g, 0–5% MeOH in CH2Cl2 solution) affords title compound (221 mg, 0.87 mmol, 65%). 1H NMR (400 MHz, CD3OD) δH 8.26 (d, 1H, J = 1.7 Hz), 7.85 (dd, 1H, J = 8.0, 1.7 Hz), 7.42 (s, 1H), 7.31 (d, 1H, J = 8.0 Hz), 4.08 (t, 2H, J = 7.1 Hz), 2.99 (t, 2H, J = 6.1 Hz), 2.67–2.59 (m, 4H), 2.13 (pent, 2H, J = 6.1 Hz); LCMS (ESI): Rt = 0.64 min, m/z = 253 [M+H]+; > 95% (215, 254 nm).
N-((R)-6-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2,3-dihydro-1H-inden-1-yl)-2,2-dimethyl-2-propane-2-sulfinamide (18a).
17a (410 mg, 1.72 mmol) and (R)-(+)-2-methyl-2-propanesulfinamide (417 mg, 3.44 mmol) were dissolved in THF (3.4 mL) under an inert atmosphere. Ti(OEt)4 (1.44 mL, 6.9 mmol) was added and the mixture heated to reflux for 18 h. To the cooled slurry was added further THF (2 mL), aiding dissolution. The mixture was cooled to −60 °C (dry ice/IPA) and NaBH4 (262 mg, 6.9 mmol) was added portion-wise and stirred for 0.5 h, then warmed to 0 °C and quenched by the drop-wise addition of MeOH until gas evolution ceased. The resulting slurry was filtered, washing with EtOAc, brine was added to the eluent and stirred vigorously for 1 h. The aqueous layer was separated and extracted with EtOAc (x2). Combined organics were dried (MgSO4) and concentrated in vacuo; purification by flash column chromatography (40 g, 0–5% MeOH in CH2Cl2) affording the product as a pale yellow solid (368 mg, 1.07 mmol, 62 %). 1H NMR (400 MHz, CD3OD) δH 7.77 (s, 1H), 7.68 (d, 1H, J = 8.0 Hz), 7.26–7.23 (m, 2H), 4.15 (t, 2H, J = 7.2 Hz), 3.05 (t, 2H, J = 7.2 Hz), 3.03–2.99 (m, 1H), 2.90–2.80 (m, 1H), 2.65 (p, 7.2 Hz), 2.56–2.46 (m, 1H), 2.12–2.03 (m, 1H), 1.25 (s, 9H); LCMS (ESI): Rt = 0.82 min, m/z = 344 [M+H]+; > 98% (215, 254 nm).
N-((R)-7-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-1,2,3,4-tetrahydronaphthalen-1-yl)-2-methylpropane-2-sulfinamide (18b).
17b (80 mg, 1.72 mmol) and (R)-(+)-2-methyl-2-propanesulfinamide (80 mg, 0.63 mmol) were dissolved in THF (0.7 mL) under an inert atmosphere. Ti(OEt)4 (0.3 mL, 1.3 mmol) was added and the mixture heated to reflux for 18 h. To the cooled slurry was added further THF (1 mL), aiding dissolution. The mixture was cooled to −60 °C (dry ice/IPA) and NaBH4 (50 mg, 1.3 mmol) was added portion-wise and stirred for 0.5 h, then warmed to 0 °C and upon complete reaction quenched by the drop-wise addition of MeOH until gas evolution ceased. To the solution was added EtOAc/brine (1:1) and the resulting slurry stirred vigorously for 1 h. The aqueous layer was separated and extracted with EtOAc (x2). Combined organics were dried (MgSO4) and concentrated in vacuo; purification by flash column chromatography (12 g, 0–2.5% MeOH in CH2Cl2) affording the product as a pale yellow solid (56 mg, 0.16 mmol, 50%). 1H NMR (400 MHz, CD3OD) δH 7.77 (s, 1H), 7.67 (d, 1H, J = 8.0 Hz), 7.28 (s, 1H) 7.12 (d, 1H, J = 8.0 Hz), 4.51 (d, 1H, J = 5.0 Hz) 4.14 (t, 2H, J = 7.2 Hz), 3.05 (t, 2H, J = 7.5 Hz), 2.89–2.61 (m, 2H), 2.65 (p, 2H, J = 7.5 Hz), 2.04–1.98 (m, 4H), 1.24 (s, 9H); LCMS (ESI): Rt = 0.85 min, m/z = 358 [M+H]+; > 95% (215, 254 nm).
(R)-6-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-2,3-dihydro-1H-inden-1-amine hydrochloride (19a).
18a (140 mg, 0.41 mmol) was dissolved in MeOH (3 mL) and HCl ([4.0 M] in dioxane, 0.31 mL, 1.2 mmol) was added drop-wise at r.t., and stirred for 1 h upon complete addition. The mixture was concentrated in vacuo, and dried in a vacuum oven to afford crude product as a pale yellow solid (110 mg, 97%), which was used without purification. LCMS (ESI): Rt = 0.09 min, m/z = 240 [M+H]+; > 98% (215, 254 nm).
(R)-7-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazol-2-yl)-1,2,3,4-tetrahydronaphthalen-1-amine hydrochloride (19b).
18b (50 mg, 0.14 mmol) was dissolved in MeOH (1 mL), and HCl (4 M in dioxane, 0.11 mL, 0.42 mmol) was added drop-wise. The mixture was stirred for 1 h, then concentrated under vacuum. The crude residue was used immediately without purification. LCMS (ESI): Rt = 0.12 min, m/z = 254 [M+H]+; > 95% (215, 254 nm).
Protein Expression and Purification.
Truncated WDR5 (Δ23, residues 22–334) was cloned into a pET vector with a 6xHis-SUMO tag fused at the N-terminus. The plasmids WDR5 was transformed into E. coli BL21 (DE3) cells. The overnight culture was used to start a 10 L fermentation (BioFlo 415, New Brunswick Scientific) grown at 37 °C. For NMR samples, uniformly 15N-labeled protein was produced in minimal M9 medium, where 15NH4Cl (Cambridge Isotope Laboratories) and D-glucose were used as sole nitrogen and carbon sources. When the cell density reached OD600 = 2.5, the temperature was lowered to 30 °C. The protein was expressed overnight with 1 mM isopropyl-β-D-thiogalactoside (IPTG). MLL1 peptide (WIN peptide Ac-ARTEVHLRKS-NH2) for NMR experiments was purchased from Genscript as HPLC purified synthetic polypeptide (>95% purity) and dissolved in DMSO for further use.
Cell pellets were dissolved in lysis buffer (1XPBS plus 300 mM NaCl, 20 mM imidazole, 5 mM BME, and 10 % glycerol), and broken by homogenization (APV-2000, APV). The lysate was cleared by centrifugation and filtering, and then applied to an affinity column (140 mL, ProBond, Invitrogen). Bound protein was eluted by an imidazole gradient. The His-SUMO-tag was removed by SUMO protease cleavage during dialysis and the subsequent subtractive second nickel-column. WDR5 protein was then purified by size-exclusion chromatography (HiLoad 26/60, Superdex 75, GE Healthcare) using NMR or crystallization buffer.
NMR Experiments: Fragment Screening and Ki Determination.
NMR samples contained 2 mg/mL (~60 μM) 15N-labeled WDR5 in 25 mM phosphate buffer, pH = 6.0, 100 mM NaCl, and 1 mM DTT. The fragment library was screened as mixtures of 12 compounds (670 μM per compound), and the mixture hits were then deconvoluted. Nuclear magnetic resonance (NMR) screening was conducted using a Bruker Avance III 600 MHz NMR spectrometer equipped with a 5 mm single axis z-gradient cryoprobe and a Bruker Sample Jet sample changer. Two-dimensional, gradient-enhanced 1H-15N heteronuclear multiple-quantum coherence (SOFAST-HMQC) spectra were collected at 25 °C and used to track chemical shift perturbation upon fragment binding.36 Spectra were processed and visualized using Topspin (Bruker BioSpin).
Protein Crystallization, Data Collection, and Structure Refinement.
WDR5 was concentrated to 10 mg/mL (~300 μM) in the buffer of 20 mM HEPES, pH 7.0, 250 mM NaCl, and 5 mM DTT. Apo- and co-Crystals were obtained at 18 °C using the hanging drop method. The crystallization condition was 0.1 M Bis-Tris pH 6.0, 0.2 M ammonium acetate, 28% to 32% PEG3350. Soaking was also applied to some compounds using the apo-crystals. Crystals were flash frozen in liquid nitrogen directly. Diffraction data were collected on the Life Sciences Collaborative Access Team (LS-CAT) 21-ID-D and G beamlines at the Advanced Photon Source (APS), Argonne National Laboratory. Data were indexed, integrated, and scaled with HKL2000.37 Molecular replacement was achieved with Phaser44 as implemented in CCP4.4538 using a previously determined WDR5 structure (PDB: 3EG6). Refinement of the structural models was conducted with Phenix and included rounds of manual model building in COOT. All structure images were prepared with PyMOL.39 A summary of the final refinement statistics for F–1, 3h, N-Ac-ART-NH2, 4a, 4q, and 6b can be found in the Supporting Information.
FPA and TR-FRET Competition Assays.
Fluorescein isothiocyante (FITC) labeled MLL peptide (FITC-GSARAEVHLRKS) and 10mer-Thr-FAM (ARTEVHLRKS-(Ahx-Ahx)(Lys-(5-FAM)))6 were purchased from Genscript and used without additional purification. Anisotropy, fluorescence, and TR-FRET emissions were recorded on a BioTek Cytation 3 instrument.
FITC-MLL FPA peptide assay:
FITC-MLL peptide is used at 50 nM, while WDR5 (Δ23, residues 24–334) is added at the Kd value of the protein:peptide interaction (WDR5-WIN Kd = 2.5 μM). Stock compounds are dispensed in 384-well source plates as 30 mM solutions in DMSO. An Echo Liquid Handler distributes the compounds to the assay plate (384-well, black, flat-bottom; Greiner) in a 10-point, 3-fold dilution scheme in a final volume of 50 μL using a top concentration of 250 μM. Both the top concentration and the dilution scheme can be adjusted to fit the potency of the compounds to a lower Ki limit of ~1 μM. For the FITC-MLL assay, 2.5 μM WDR5 and 50 nM FITC-MLL peptide in assay buffer (1X Phosphate Buffered Saline, pH 6.0, 300 mM NaCl, 0.5mM TCEP, 0.1% CHAPS) is added to all compound-containing wells and to columns 2, 24 (negative control, 0% inhibition). To columns 1 and 23 (positive control, 100% inhibition) 2 μL of 50 nM FITC-MLL peptide alone in assay buffer is added. The assay performs with an average Z’ value of 0.5 and is tolerant to up to 5% DMSO. For compounds with an IC50 < 2.0 μM and Ki < 1 μM, the 10mer-Thr-FAM probe and FPA protocol described below can used for enhanced sensitivity.
10mer-Thr-FAM peptide FPA assay:
Stock compounds are dispensed in 384-well source plates as before from 30 mM solutions in DMSO. An Echo Liquid Handler distributes the compounds in duplicate to the assay plate (384-well, black, flat-bottom; Greiner) in a 10-point, 3-fold dilution scheme in a final volume of 40 μL using a top concentration of 20–60 μM. 10mer-Thr-FAM peptide is used at 4 nM, while WDR5 is added at the Kd value of the protein:peptide interaction (WDR5–10mer-Thr Kd ~2–4 nM) using assay buffer (1X Phosphate Buffered Saline pH 6.0, 300 mM NaCl, 0.5mM TCEP, 0.1% CHAPS). To columns 1 and 23 (positive control, 100% inhibition) 10mer-Thr-FAM probe alone in assay buffer is added. The assay performs with an average Z’ value of 0.5 and is tolerant to up to 5% DMSO. In general, for compounds with an IC50 < 2 nM and a calculated Ki < 1 nM, the 10mer-Thr-FAM TR-FRET protocol described below can used for enhanced sub-nanomolar sensitivity.
10mer-Thr-FAM peptide TR-FRET assay:
LanthaScreen Elite Tb-anti His antibody (Tb-Ab) was purchased from Thermo-Fisher and used at 1 nM. 10mer-Thr-FAM peptide is used at 150 nM, while WDR5-His-SUMO tag protein is used at 2 nM. The working assay buffer composition was modified to pH 7.2 (1X Phosphate Buffered Saline, 300 mM NaCl, 0.5mM TCEP, 0.1% CHAPS). Stock compounds are dispensed to a white, flat-bottom OptiPlate plate (PerkinElmer) using an Echo Liquid Handler. A 10-point, 5 -fold dilution scheme with a top concentration of 5μM (0.003 nM low concentration) was used with a final volume of 20 μL. Both the top concentration and the dilution scheme can be adjusted to fit the anticipated potency of the compounds. Using the above probe concentration and assay conditions described, the calculated lower Ki limit is ~0.060 ± 0.020 nM. Positive control wells (0% displacement) consisted of 10mer-Thr-FAM probe and WDR5/terbium antibody mix occupying columns 2 and 24, while negative control wells (100% displacement) consisting of the protein/terbium antibody mix alone occupy columns 1 and 23. The assay performs with an average Z’ value of 0.7 and is tolerant to up to 5% DMSO.
For IC50 determination, plates are covered, shielded from light, and incubated for 1h at room temperature with rocking. For the FPA assay measurements anisotropy is measured an excitation wavelength of 480 nm, and emission of 535 nm. For TR-FRET assay measurement plates are excited at a wavelength of 340 nm, and emission wavelengths of 495 and 520 nm. The ratio of the 520/495 wavelengths are used to assess the degree of FRET signal and resulting peptide displacement. TR-FRET plate positive control wells include columns 2 and 24 containing 10mer-Thr-FAM peptide, His-SUMO-WDR5, and Tb-anti-His antibody to measure maximum signal from the FRET response. The change in anisotropy (FPA) or 520 / 495 emission ratio (TR-FRET) is used to calculate an IC50 (inhibitor concentration at which 50% of the bound peptide is displaced) by the fitting the inhibition data using XLFit software (Guilford, UK) to single-site binding model. This was converted into a binding inhibition/displacement constant (Ki) according the formula:40
where [I]50 is the concentration of the free inhibitor at 50% inhibition, [L]50 is the concentration of the free labeled ligand at 50% inhibition, [P]0 is the concentration of the free protein at 0% inhibition, and Kd pep represents the dissociation constant of the FITC-MLL or 10mer-Thr-FAM probe.
MLL1 Histone Methyl Transferase Inhibition.
HMT inhibition activity was performed at Reaction Biology Corp. in a HotSpot, radioisotope based filter-binding format (assay catalog #HMT-15–105, MLL1 complex no extra WRAD2), using purified recombinant human MLL1 complex: MLL1 (aa 3745–3969, GenBank Accession No. NM_005933), WDR5 (aa 22–334, GenBank Accession No. NM_017588), RbBP5 (aa 1–538, GenBank Accession No. NM_005057), Ash2L (aa 2–534, GenBank Accession No. NM_001105214), DPY-30 (aa 1–99, GenBank Accession No. NM_0325742), all with N-terminal His tag were expressed in E. coli, and mixed molar ratio of 1:1:1:1:2, respectively, total MW = 212 kDa. Reaction buffer conditions: 50 mM Tris-HCl (pH 8.5), 5 mM MgCl2, 50 mM NaCl, 0.01% Brij35, 1 mM DTT, 1% DMSO. Purified oligonucleosomes were obtained from HeLa cells (primarily oligomers of 3–6 units, 600–1200 bp DNA), and S-adenosyl-L-[methyl-3H]methionine (SAM) was used as the methylation cofactor.31 S-Adenosylhomocysteine (SAH) was used as a positive control. Reaction conditions consisted of 7–10 nM of MLL1 complex, 0.05 mg/mL HeLa oligonucleosomes, and 1 μM of SAM substrate. Compounds were tested in duplicate using a 10-point CRC three-fold serial dilution from a 20 μM top concentration. See SI and www.reactionbiology.com for further details.
Cell-based anti-proliferative activity of compounds against MV4–11 leukemia cells.
MV4–11, cells were grown in IMDM media supplemented with 10% FBS and 1% penicillin/streptomycin. Compounds and a vehicle control (0.1% DMSO) were tested in a 7-day growth inhibition experiment using a 384-well plate (sterile Grainer 781091, clear bottom) format using an 11-point CRC with serial two-fold dilution starting from a 30 μM top concentration for test compounds. MV4–11 cells were counted and 28 μL at 3900 cells/mL were loaded in each well of 384-well plate (sterile Grainer 781091, clear bottom) and then 4 μL of series dilutions of compounds in media were added to each well (final volume was 32 μL per well). Cells were then incubated for 7 days at 37 °C with 5% CO2. Meanwhile, a control T = 0 plate was prepared to include 12 wells of each cell line containing the same number of cells in 32 μL media and tested with CellTiterGlo® right after as described below. This is the control assay for the experiment used to define the average viable cells at day zero. At the end of 7-day incubation, to each well 16 μL of CellTiterGlo® was added and the plates were centrifuged at 300 rpm for 1 minute while blocking light. The plates were allowed to sit for 15 min at room temperature and read on a BioTek luminescent instrument plate reader (Cytation 3). Growth inhibition at 50% (GI50) is calculated fitting the inhibition data to a 4-parameter logistical curve using XLFit software (Guilford, UK) and then applying the interpolated GI50 on the Y-axis. The average growth maximum is normalized to day zero (defined as 100%) and generated from the 7-day DMSO control wells using the relative luminescent units (LMU) by the following equation: % Growth Max = (100 * (RLU)) / (T=0 RLU). The GI50 for a given plate is defined as following: GI50 = ((%Growth Max – 100 / 2) + 100. The average GI50 values are reported based on biological replicate CRC’s and GI50 values.
Supplementary Material
Scheme 3.

Synthesis of 6a–6e.
(a) PdCl2(dppf)CH2Cl2, ArB(OH)2 or ArBpin, CH3CN, aq. K2CO3, 95 °C, 1–16 h; (b) (R)-(+)-2-methyl-2-propanesulfinamide, Ti(OEt)4 THF, reflux, 16 h; then (c) NaBH4, THF, −60 °C to rt (58–62%); (d) 4.0 M HCl, MeOH, rt (95–97%); (e) R2COCl, Et3N, CH2Cl2, rt, 2 h or R2CO2H, HATU, DMF, rt, 16–40 h.
ACKNOWLEDGMENT
The authors thank Dr. Mary Harner and Ms. Bethany Alicie for assessment of purchased analogs of fragment hits and for performing early FPA competition binding assays, and co-workers at the High-Throughput Screening Core facility at Vanderbilt University, TN, for compound management and Vanderbilt University Biomolecular NMR Facility for instrumentation. This facility receives support from an NIH SIG Grant (1S-10RR025677–01) and Vanderbilt University matching funds. The authors also thank contributing and supporting members of Leidos Biomedical Research at Frederick National Labs for Cancer Research and the NCI Chemical Biology Consortium. Use of the Advanced Photon Source, an Office of Science User Facility operated for the US Department of Energy Office of Science by the Argonne National Laboratory, was supported by US Department of Energy [Contract No. DE-AC02–06CH11357]. Use of the Life Sciences Collaborative Access Team Sector 21 was supported by the Michigan Economic Development Corporation and Michigan Technology Tri- Corridor Grant [grant number 085P1000817].
Funding Sources
This work was generously supported in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Chemical Biology Consortium Contract No. HHSN261200800001E (to S.W.F), grant CA200709 from the National Institutes of Health (to W.P.T), The Vanderbilt Ingram Cancer Center Support grant (NIH: CA68485), and startup funds to S.W.F. provided by Vanderbilt University. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
ABBREVIATIONS USED
- WDR5
WD repeat domain 5 protein
- MLL1
Mixed-Lineage Leukemia 1
- WIN
WDR5 interaction motif
- WRAD
WDR5-RbBP5-ASH2L-DPY30 complex
- HMQC
Heteronuclear Multiple Quantum Coherence
- FPA
fluorescence polarization anisotropy
- TR-FRET
time-resolved fluorescence energy transfer
Footnotes
Supporting Information. Further fragment screening hits and spectroscopic data for compounds, X-ray refinement statistics, representative assay curves. This material is available free of charge via the internet at http://pubs.acs.org.
Accession Codes. Atom coordinates and structure factors for WDR5-ligand complexes can be accessed in the PDB via the following accession codes: F-1 (6D9X), 3h (6DAI), 4a (6DAK), 4q (6DAR), 6b (6DAS).
REFERENCES
- 1.Smith TF; Gaitatzes C; Saxena K; Neer EJ The WD repeat: a common architecture for diverse functions. Trends Biochem Sci 1999, 24, 181–185. [DOI] [PubMed] [Google Scholar]
- 2.Stirnimann CU; Petsalaki E; Russell RB; Muller CW WD40 proteins propel cellular networks. Trends Biochem Sci 2010, 35, 565–574. [DOI] [PubMed] [Google Scholar]
- 3.Xu C; Min J Structure and function of WD40 domain proteins. Protein Cell 2011, 2, 202–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guarnaccia AD; Tansey WP Moonlighting with WDR5: A cellular multitasker. J Clin Med 2018, 7, 21; doi: 10.3390/jcm7020021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang C; Zhang F The Multifunctions of WD40 proteins in genome integrity and cell cycle progression. J Genomics 2015, 3, 40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karatas H; Townsend EC; Bernard D; Dou Y; Wang S Analysis of the binding of mixed lineage leukemia 1 (MLL1) and histone 3 peptides to WD repeat domain 5 (WDR5) for the design of inhibitors of the MLL1-WDR5 interaction. J Med Chem 2010, 53, 5179–5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun Y; Bell JL; Carter D; Gherardi S; Poulos RC; Milazzo G; Wong JW; Al-Awar R; Tee AE; Liu PY; Liu B; Atmadibrata B; Wong M; Trahair T; Zhao Q; Shohet JM; Haupt Y; Schulte JH; Brown PJ; Arrowsmith CH; Vedadi M; MacKenzie KL; Huttelmaier S; Perini G; Marshall GM; Braithwaite A; Liu T WDR5 supports an N-Myc transcriptional complex that drives a protumorigenic gene expression signature in neuroblastoma. Cancer Res 2015, 75, 5143–5154. [DOI] [PubMed] [Google Scholar]
- 8.Dai X; Guo W; Zhan C; Liu X; Bai Z; Yang Y WDR5 Expression is prognostic of breast cancer outcome. PLoS One 2015, 10, e0124964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen X; Xie W; Gu P; Cai Q; Wang B; Xie Y; Dong W; He W; Zhong G; Lin T; Huang J Upregulated WDR5 promotes proliferation, self-renewal and chemoresistance in bladder cancer via mediating H3K4 trimethylation. Sci Rep 2015, 5, 8293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carugo A; Genovese G; Seth S; Nezi L; Rose JL; Bossi D; Cicalese A; Shah PK; Viale A; Pettazzoni PF; Akdemir KC; Bristow CA; Robinson FS; Tepper J; Sanchez N; Gupta S; Estecio MR; Giuliani V; Dellino GI; Riva L; Yao W; Di Francesco ME; Green T; D’Alesio C; Corti D; Kang Y; Jones P; Wang H; Fleming JB; Maitra A; Pelicci PG; Chin L; DePinho RA; Lanfrancone L; Heffernan TP; Draetta GF In Vivo functional platform targeting patient-derived xenografts identifies WDR5-Myc association as a critical determinant of pancreatic cancer. Cell Rep 2016, 16, 133–147. [DOI] [PubMed] [Google Scholar]
- 11.Tan X; Chen S; Wu J; Lin J; Pan C; Ying X; Pan Z; Qiu L; Liu R; Geng R; Huang W PI3K/AKT-mediated upregulation of WDR5 promotes colorectal cancer metastasis by directly targeting ZNF407. Cell Death Dis 2017, 8, e2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel A; Dharmarajan V; Cosgrove MS Structure of WDR5 bound to mixed lineage leukemia protein-1 peptide. J Biol Chem 2008, 283, 32158–32161. [DOI] [PubMed] [Google Scholar]
- 13.Patel A; Vought VE; Dharmarajan V; Cosgrove MS A conserved arginine-containing motif crucial for the assembly and enzymatic activity of the mixed lineage leukemia protein-1 core complex. J Biol Chem 2008, 283, 32162–32175. [DOI] [PubMed] [Google Scholar]
- 14.Song JJ; Kingston RE WDR5 interacts with mixed lineage leukemia (MLL) protein via the histone H3-binding pocket. J Biol Chem 2008, 283, 35258–35264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hess JL MLL: A histone methyltransferase disrupted in leukemia. Trends Mol. Med 2004, 10, 500–507. [DOI] [PubMed] [Google Scholar]
- 16.Meyer C; Hofmann J; Burmeister T; Gröger D; Park TS; Emerenciano M; Pombo de Oliveira M; Renneville A; Villarese P; Macintyre E; et al. The MLL recombinome of acute leukemias in 2013. Leukemia 2013, 27, 2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muntean AG; Hess JL The pathogenesis of mixed-lineage leukemia. Annu. Rev. Pathol. Mech. Dis 2012, 7 (1), 283–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Winters AC; Bernt KM MLL-Rearranged leukemias—An update on science and clinical approaches. Front. Pediatr 2017, 5, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thiel AT; Blessington P; Zou T; Feather D; Wu X; Yan J; Zhang H; Liu Z; Ernst P; Koretzky GA; et al. MLL-AF9-Induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer Cell 2010, 17 (2), 148–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Milne TA; Kim J; Wang GG; Stadler SC; Basrur V; Whitcomb SJ; Wang Z; Ruthenburg AJ; Elenitoba-Johnson KSJ; Roeder RG; et al. Multiple interactions recruit MLL1 and MLL1 fusion proteins to the HOXA9 locus in leukemogenesis. Mol. Cell 2010, 38 (6), 853–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Y; Anastassiadis K; Kranz A; Stewart AF; Arndt K; Waskow C; Yokoyama A; Jones K; Neff T; Lee Y; et al. MLL2, not MLL1, plays a major role in sustaining MLL-rearranged acute myeloid leukemia. Cancer Cell 2017, 31 (6), 755–770.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dias J; Van Nguyen N; Georgiev P; Gaub A; Brettschneider J; Cusack S; Kadlec J; Akhtar A Structural analysis of the KANSL1/WDR5/KANSL2 complex reveals that WDR5 is required for efficient assembly and chromatin targeting of the NSL complex. Genes Dev 2014, 28, 929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomas LR; Wang Q; Grieb BC; Phan J; Foshage AM; Sun Q; Olejniczak ET; Clark T; Dey S; Lorey S; Alicie B; Howard GC; Cawthon B; Ess KC; Eischen CM; Zhao Z; Fesik SW; Tansey WP Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC. Mol Cell 2015, 58, 440–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karatas H; Townsend EC; Cao F; Chen Y; Bernard D; Liu L; Lei M; Dou Y; Wang S High-affinity, small-molecule peptidomimetic inhibitors of MLL1/WDR5 protein-protein interaction. J Am Chem Soc 2013, 135, 669–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao F; Townsend EC; Karatas H; Xu J; Li L; Lee S; Liu L; Chen Y; Ouillette P; Zhu J; Hess JL; Atadja P; Lei M; Qin ZS; Malek S; Wang S; Dou Y Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol Cell 2014, 53, 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karatas H; Li Y; Liu L; Ji J; Lee S; Chen Y; Yang J; Huang L; Bernard D; Xu J; Townsend EC; Cao F; Ran X; Li X; Wen B; Sun D; Stuckey JA; Lei M; Dou Y; Wang S Discovery of a highly potent, cell-permeable macrocyclic peptidomimetic (MM-589) targeting the WD repeat domain 5 protein (WDR5)-mixed lineage leukemia (MLL) protein-protein interaction. J Med Chem 2017, 60, 4818–4839. [DOI] [PubMed] [Google Scholar]
- 27.Grebien F; Vedadi M; Getlik M; Giambruno R; Grover A; Avellino R; Skucha A; Vittori S; Kuznetsova E; Smil D; Barsyte-Lovejoy D; Li F; Poda G; Schapira M; Wu H; Dong A; Senisterra G; Stukalov A; Huber KVM; Schonegger A; Marcellus R; Bilban M; Bock C; Brown PJ; Zuber J; Bennett KL; Al-Awar R; Delwel R; Nerlov C; Arrowsmith CH; Superti-Furga G Pharmacological targeting of the WDR5-MLL interaction in C/EBPα N-terminal leukemia. Nat Chem Biol 2015, 11, 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Getlik M; Smil D; Zepeda-Velazquez C; Bolshan Y; Poda G; Wu H; Dong A; Kuznetsova E; Marcellus R; Senisterra G; Dombrovski L; Hajian T; Kiyota T; Schapira M; Arrowsmith CH; Brown PJ; Vedadi M; Al-Awar R Structure-based optimization of a small molecule antagonist of the interaction between WD repeat-containing protein 5 (WDR5) and mixed-lineage leukemia 1 (MLL1). J Med Chem 2016, 59, 2478–2496. [DOI] [PubMed] [Google Scholar]
- 29.Li D-D; Chen W-L; Wang Z-H; Xie Y-Y; Xu X-L; Jiang Z-Y; Zhang X-J; You Q-D; Guo X-K High-affinity small molecular blockers of mixed lineage leukemia 1 (MLL1)-WDR5 interaction inhibit MLL1 complex H3K4 methyltransferase activity. Eur. J. Med. Chem 2016, 124, 480–489. [DOI] [PubMed] [Google Scholar]
- 30.See reference 14 for description of adopted WIN-site subpocket nomenclature based upon the ARA core sequence.
- 31.Ferry JJ; Smith RF; Denney N; Walsh CP; McCauley L; Qian J; Ma H; Horiuchi KY; Howitz KT Development and use of assay conditions suited to screening for and profiling of SET-domain-targeted inhibitors of the MLL/SET1 family of lysine methyltransferases. Assay Drug Dev Technol 2015, 13, 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Performed at Walter Reed Army Institute of Research, www.wrair.army.mil
- 33.6,7-Dihydro-5H-pyrrolo[1,2-a]imidazoles has a calculated pKb = 6.6
- 34.Borg G; Cogan DA; Ellman JA One-pot asymmetric synthesis of tert-butanesulfinyl-protected amines from ketones by the in situ reduction of tert-butanesulfinyl ketimines. Tetrahedron Letters 1999, 40, 6709–6712. [Google Scholar]
- 35.Colyer JT; Andersen NG; Tedrow JS; Soukup TS; Faul MM Reversal of diastereofacial selectivity in hydride reductions of N-tert-butanesulfinyl imines. J Org Chem 2006, 71, 6859–6862. [DOI] [PubMed] [Google Scholar]
- 36.Schanda P; Kupce E; Brutscher B SOFAST-HMQC experiments for recording two-dimensional heteronuclear correlation spectra of proteins within a few seconds. J Biomol NMR 2005, 33, 199–211. [DOI] [PubMed] [Google Scholar]
- 37.Otwinowski Z; Minor W Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Pt A 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 38.Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AGW; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS Overview of the CCP4 suite and current developments. Acta Crystallographica Section D-Biological Crystallography 2011, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adams PD; Grosse-Kunstleve RW; Hung LW; Ioerger TR; McCoy AJ; Moriarty NW; Read RJ; Sacchettini JC; Sauter NK; Terwilliger TC PHENIX: building new software for automated crystallographic structure determination. Acta Crystallographica Section D-Biological Crystallography 2002, 58, 1948–1954. [DOI] [PubMed] [Google Scholar]
- 40.Wang ZX; Jiang RF A novel two-site binding equation presented in terms of the total ligand concentration. FEBS Lett 1996, 392, 245–249. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.