

Abstract
Although osteoarthritis (OA) was historically referred to as the non-inflammatory arthritis, it is now considered a condition involving persistent low-grade inflammation and activation of innate inflammatory pathways. Synovitis increases the risk of OA onset and progression and involves the recruitment of monocytes, lymphocytes, and other leukocytes. In particular, macrophages are important mediators of synovial inflammatory activity and pathologic cartilage and bone responses that are characteristic of OA. Advances in understanding how damage-associated molecular patterns (DAMPs) trigger monocyte/macrophage recruitment and activation in joints provide opportunities for disease-modifying therapies. However, the complexity and plasticity of macrophage phenotypes that exist in vivo have thus far prevented the successful development of macrophage-targeted treatments. Current studies show that synovial macrophages are derived from distinct cellular lineages, which correspond to unique functional roles for maintaining joint homeostasis. An improved understanding of the etiology of synovial inflammation in specific OA-subtypes, such as with obesity or genetic risk, is a potential strategy for developing patient selection criteria for future precision therapies.
Keywords: Macrophages, Innate Immunity, Osteoarthritis, Synovitis
Introduction
The name osteoarthritis (OA) implies that it is an inflammatory disease. However, for many years the role of inflammation was contested, spawning the use of alternative names such as osteoarthrosis and degenerative joint disease (1). This controversy was based in part on the belief that the central pathological features of the disease were articular cartilage erosion and pathological bone growth, as seen by osteophytes and subchondral bone sclerosis. In addition, compared to “inflammatory joint diseases” such as rheumatoid arthritis, OA patients were characterized by lower levels of pro-inflammatory serum biomarkers and less remarkable synovitis (2,3). Questions continued as studies linked biomechanical factors to OA risk (4–7), and clinical trials testing anti-inflammatory therapies failed to modify OA progression (8–10). Yet, evidence connecting inflammation and OA remained (11,12) and eventually flourished with the advent of more sophisticated techniques to broadly interrogate cellular, molecular, and genetic factors associated with OA. These studies revealed persistent low-grade inflammation, and in particular activation of innate inflammatory pathways, as central mediators of OA pathogenesis (13–15).
The persistent low-grade activation of innate inflammatory pathways has led OA to be likened to a chronic wound (16). This concept is based on evidence of an aberrant wound-healing response in the OA joint (17). Classically, the wound-healing response involves a clotting reaction to stop bleeding, inflammation, cellular proliferation, and tissue remodeling, resulting in the resolution of inflammation and formation of a scar. The initial inflammatory response is triggered by danger/damage associated molecular patterns (DAMPs), which are endogenous molecules released into the extra-cellular space following tissue damage or cellular stress (18–20). DAMPs stimulate innate immunity by interacting with pattern-recognition receptors, including toll-like receptors (TLRs), expressed on sentinel tissue-resident immune cells and joint tissue stromal cells (21). The resulting cellular signaling cascade leads to the production of cytokines, chemokines, growth factors, and matrix proteases, which coordinate cellular proliferation and tissue remodeling responses. There is increasing evidence that tissues throughout the whole joint contribute to this wound-like response in an autocrine and paracrine-like manner (22,23). Notably, sustained protease-mediated breakdown of cartilage and meniscus tissue generates an ongoing source of DAMPs, which feed into a continuing cycle of inflammation and tissue destruction (18,20,24). Altered joint biomechanics may also contribute to the cycle of inflammation with the recognition that mechanically induced tissue damage and mechanotransduction stress signaling can trigger cytokine production and cell-derived DAMPs (25–27). These observations have helped to unite what previously seemed like distinct pathological mechanisms under the umbrella of inflammation. Many questions remain, however, about the specific cellular and molecular mediators that initiate the onset of disease and drive its progression (28,29). These topics have been discussed in numerous other recent reviews (30–35). In this review, we focus on evidence surrounding the role of synovial macrophages in OA pathophysiology.
Synovitis and Synovial Macrophages in OA
Inflammatory cell infiltration of the synovial membrane occurs to a lesser degree and is more heterogeneous in patients with OA compared to those with rheumatoid arthritis (3,36). Nevertheless, many features of inflammatory arthritis, including lymphoid follicles and perivascular fibrosis, occur in a portion of OA cases (3). Neovascularization and mild synovitis are also present in the joints of patients at risk for OA due to soft-tissue injuries (36). In a large prospective epidemiological study, joint effusion and synovitis were detected in approximately half of the study participants who had OA symptoms but no radiographic pathology (37), further indicating that synovitis is not restricted to late stages of disease. Moreover, baseline synovial thickening and synovitis were shown to increase the rate of OA progression and are associated with increased pain and dysfunction (38,39). Micro-array analysis of inflamed synovial tissue revealed a distinct transcriptional C-C family chemokine signature, suggesting that synovitis in patients at risk for OA involves the recruitment of monocytes, lymphocytes, and other leukocytes (39).
Macrophages are well established as key cellular mediators of innate immunity and tissue remodeling in the wound healing response following injury (40,41). Macrophages also appear to play similar roles in OA. For example, more than 10 years ago, Bondeson and colleagues showed that depletion of macrophages in OA synovial explants significantly reduced the production of numerous cytokines, including tumor necrosis factor (TNF), interleukin-1 (IL-1), IL-6, and IL-8 (14). Importantly, macrophage depletion and neutralization of macrophage-derived TNF and IL-1 also downregulated matrix metalloproteinase (MMP) production, thereby linking synovial macrophages to cartilage degradation (14). Blom et. al. confirmed a role for synovial macrophages in MMP-mediated cartilage degeneration in vivo, using the murine collagenase-induced model of OA and intra-articular injection of clodronate to deplete macrophages prior to inducing OA (42). The same investigators previously demonstrated a critical role for synovial macrophages in promoting osteophytosis in the model (43). The effect on pathologic bone formation in OA was attributed to a decrease in macrophage-derived growth factors that are typical of wound-healing responses, but are also important in chondrogenesis and bone formation (TGFβ, BMP-2 and BMP-4) (43,44). In follow-up studies, this group showed that the Collagenase model of OA used in these studies, in contrast to injury-induced models, has a robust synovial inflammatory component and is dependent on activation by the S100A8/9 family of DAMPs (45). This body of work has implicated macrophage-mediated inflammatory activity in pathologic cartilage and bone responses characteristic of OA, although it also suggests that the impact of inflammation in OA is complex and influenced by the inciting stimulus.
The focus on macrophages and OA has continued to build in recent years. In a revealing set of studies, Kraus and colleagues investigated the presence of activated macrophages in patients with knee OA (KL: 1–4) directly in vivo using etarfolatide (EC20) imaging, which selectively detects folate receptor expression on activated, but not resting, macrophages (46). They found that the quantity of activated macrophages in knee joints correlated with radiographic OA severity and symptoms (46,47). In addition, etarfolatide uptake positively correlated with OA symptoms in joint sites throughout the body (47). Kraus and colleagues also compared etarfolatide uptake to soluble macrophage markers in synovial fluid and serum (46). The markers CD14 and CD163 are shed when macrophages are activated by pro- and anti-inflammatory cytokines, respectively. Notably, both CD14 and CD163 were detected in synovial fluid and correlated with EC20-quantified knee macrophage abundance (46). These data suggest that macrophages are etiological factors in OA pain and that measurement of soluble macrophage markers may predict the risk of OA progression.
If macrophages indeed drive OA progression then strategies that target their recruitment and accumulation in the synovium may be one approach for a disease-modifying OA therapy. The C-C family chemokine receptors CCR2 and CCR5 are important mediators of monocyte/macrophage recruitment to sites of inflammation. In OA patients, synovial fluid levels of CCR2 ligands (i.e., CCL2, CCL7, and CCL8) but not CCR5 ligands (i.e., CCL5, CCL3) were increased compared to a cohort of control individuals with a knee injury but no OA (48). CCL2 was expressed in OA chondrocytes and OA synovial fibroblasts stimulated with cartilage-derived debris and the OA-associated alarmin S100A8 (48). CCR2-positive macrophages were also observed in OA synovium adjacent to damaged cartilage (48). Further studies in mouse models of post-traumatic knee OA showed that genetic or pharmacologic inhibition of CCL2/CCR2 signaling reduced OA pathology and pain-related behavior, although differences have been reported between investigators in the effectiveness of targeting CCL2/CCR2 and may be dependent in part on the timing of inhibition (48–51). Therapeutically targeting the recruitment and accumulation of monocytes/macrophages to OA joints is one of several strategies to modify macrophage-mediated inflammation and OA pathology.
Macrophage activation and polarization states are tightly controlled and vital for regulating effective wound healing (40). Activated macrophages have traditionally been defined as “M1” and “M2” polarization states based on in vitro stimulation models that drive pro-inflammatory or immuno-regulatory phenotypes, respectively (52). In OA synovium and cartilage, cytokines associated with M1 macrophages (e.g., TNF, IL-1β, IL-6) are well established for their role in stimulating pro-catabolic mediators, such as aggrecanases and MMPs. However, it has been difficult to assess the polarization state of synovial macrophages at earlier stages of disease or even prior to the onset of OA. At the end stage of OA, both M1- and M2-like macrophages are present in joint synovium and adjacent adipose tissue (53). Using a genetic approach, Zhang and colleagues generated mice with either M1- or M2-enhanced macrophage polarization to evaluate the positive and negative effect of polarization bias on OA risk (54). Mice were subjected to two models of knee OA, Collagenase and meniscal destabilization, to evaluate the effect of macrophage polarization bias on OA models with high and low levels of synovitis, respectively. As predicted, mice with a pro-inflammatory M1 bias developed greater synovial inflammation and cartilage pathology; whereas, mice with an M2 bias were protected, albeit at later time points only (54). When analyzed in detail, M1-enhanced macrophages were found to promote cartilage degradation and osteophyte development through the production of Rspo2, a cytokine previously identified as a Wnt signaling agonist (54).
The focus on synovial macrophages as drivers of OA symptoms and pathology has led to therapeutic strategies involving the removal of synovial macrophages to slow the progression of disease. These strategies presuppose that macrophages primarily modulate OA pathology through their pro-inflammatory and pro-catabolic actions. However, in compelling preclinical study by Wu and colleagues, the authors showed the depleting joint macrophages using macrophage Fas-induced apoptosis (MaFIA)-transgenic mice did not reduce OA pathology (55). Although macrophage depletion acutely reduced both M1 and M2 macrophages in the joints of mice following meniscal destabilization surgery, it increased the infiltration of CD3+ T cells and neutrophils into the injured joint, resulting in greater synovitis and systemic inflammation (55). Thus, given that macrophages regulate synovial immune cell homeostasis, these findings indicate that a more detailed understanding of the functional roles of macrophage subtypes will be required for therapies that target macrophage removal.
Two studies published earlier this year bring new insight into macrophage subtypes in OA joints and their homeostatic role in synovial tissues. In the study by Wood et al. (56), flow cytometry was used to compare the population of immune cells in synovial tissue from OA patients undergoing total knee replacement and from inflammatory arthritis patients. The comparison showed that immune cells in OA patient synovium were overwhelmingly dominated by macrophages while T cells were dominant in the synovium of inflammatory arthritis patients (56). RNA sequencing of synovial tissue macrophages identified two distinct OA subgroups, one more similar to macrophages isolated from inflammatory arthritis synovium (i.e., proliferative, inflammatory-like OA subgroup) and one that was distinct and characterized by cartilage remodeling genes (i.e., classical OA subgroup) (56). An important observation was that neither subgroup aligned with an M1 or M2 phenotype. This points to the importance of detailed evaluations of macrophage phenotypes, and it also highlights the limitation of the M1/M2 paradigm to capture the complexity and plasticity of macrophage phenotypes that exist in vivo. Patient characteristics such as radiographic KL scores or serum C-reactive protein or erythrocyte sedimentation rate values were not predictive of the relative proportion of inflammatory and classical OA macrophages, suggesting that more specific biomarkers will be needed to identify patients with differing synovial macrophage subgroups.
Intriguingly, work by Culemann and colleagues showed that synovial macrophages are derived from distinct cellular lineages and that these differences are associated with different functional roles in the joint (57). Specifically, a population of CX3CR1+ tissue-resident macrophages were identified in mice that express tight junction proteins and create a barrier-forming population of macrophages that line the synovium (57). These “epithelial-like” macrophages are maintained through a local pool of proliferating CX3CR1− mononuclear cells in the synovial interstitium and are distinct from chemokine recruited monocyte-derived macrophages (57). The barrier-forming macrophage population was nearly gone in synovium from patients with inflammatory arthritis (57). Whether or not it is also impaired in the synovium of OA patients remains to be seen. Thus, understanding the origin and functional consequences of macrophage sub-populations will be vital for developing more specific macrophage-targeted OA-modifying therapies. Furthermore, developing biomarkers to assess the heterogeneity of macrophage sub-groups will likely be important for establishing patient selection criteria for macrophage therapies.
OA Risk Factors & Inflammatory Phenotypes: Targets for Future Therapies?
One of the most clinically significant risk factors for developing OA is obesity (58,59). Obesity increases OA risk in both knee and hand joints, although the greatest impact is on the knee where obesity doubles the lifetime risk of symptomatic OA compared to individuals with a body mass index (BMI) below 25 (60). Similarly, many components of the metabolic syndrome, such as central adiposity, dyslipidemia, hyperglycemia, and hypertension are associated with OA pathology and the risk of progression (31,61–63). Although the causal role of metabolic syndrome and its components in knee OA progression remain unclear (64,65), related factors such as high dietary fat consumption (66) and type 2 diabetes (67) are each associated with more rapidly progressing joint space narrowing in individuals with knee OA, even after adjusting for BMI. Given the strong causal relationship between metabolic dysfunction and pro-inflammatory macrophage activation that occurs with obesity (68,69), it seems likely that metabolic inflammation (“metaflammation”) also increases OA risk (31).
Chronic low-grade inflammation that occurs with obesity is due in part to the accumulation of pro-inflammatory macrophages in abdominal adipose tissue. This raised the question of whether a similar phenomenon occurs in the infra-patellar fat pad, thereby producing a local source of inflammation that increases OA risk. However, animal and clinical studies have largely shown that obesity does not increase the number of pro-inflammatory macrophages in the infra-patellar fat pad (70–73). Rather, obesity has a greater effect on the synovium where it causes synovial adipocyte hypertrophy, macrophage accumulation, fibrosis, and increased expression of TNF and TLR4 (73–76). In aged female mice fed a high-fat diet to induce obesity, genetic deletion of TLR4 prevented the development of knee OA, supporting a role for innate immune signaling via TLR4 in obesity-induced OA (77). A number of questions remain about which factors that are associated with obesity directly modulate synovial inflammation. Pre-clinical animal studies indicate that multiple factors could be involved, including synovial insulin resistance (74,78), dietary fatty acid composition (79–81), and gut microbiome composition (82,83). The complex and multi-factorial nature of these pro-inflammatory stimuli creates challenges for developing OA therapies for obese patients. Ongoing studies are seeking to establish better causal relationships between these factors and OA outcomes so that patient subsets might be identified for more precise therapies.
Genetic associations with OA also point to the importance of inflammatory mechanisms in OA. In 2014, polymorphisms in the IL-6 gene were found to be associated with radiographic hand OA in a British female twin cohort of Northern European ancestry, and in a Caucasian (Chuvash) population (84). That same year, an association between multi-joint (generalized) OA and a variant within the SMAD3 gene, a downstream mediator of TGFβ signaling, was reported (85). Although both TGFβ and IL-6 are products of synovial macrophages that have been implicated in driving OA-related joint pathology, the specific functional influence of these genetic polymorphisms on risk of OA remain to be described. More recently, two single-nucleotide polymorphisms (SNPs) within the Protease-activated receptor-2 (PAR-2) gene were associated with risk of knee OA in a Han Chinese cohort, and risk allele carriers expressed higher levels of IL-6 and IL-1β in synovial fluid (86). This receptor is activated by inflammatory proteases and plays an important role in promoting inflammatory signaling in a variety of cells. Genetic deficiency of PAR-2 in mice was shown to protect against injury-induced OA by two independent groups (87,88). The importance of TLRs in OA, which are highly expressed by monocyte/macrophage lineage cells and promote macrophage phenotypic differentiation, has also been mentioned earlier. Genetic studies in humans also support a role for these innate immune sensors in OA. TLR-related genetic associations have been reported in several populations. The T-1486C SNP in TLR9 was associated with knee OA in both a Chinese (89) and Turkish populations (90). Subsequently, two TLR3 SNPs were significantly associated with OA (91), and associations between OA and TLR7 and TLR8 SNPs were also found, but only in males. Taken together, these studies implicate genetic variation in inflammatory responses as a risk factor for OA. Whether they reveal important targets for therapy remains to be seen.
Conclusion
Current evidence provides strong inference that innate inflammatory pathways are involved in the etiology of OA. However, the complexity of cell types involved, including heterogenous macrophages subtypes of distinct developmental origins (Figure 1), poses numerous challenges for developing strategies to resolve inflammation in the face of continuous DAMP activation. Strategies to resolve OA synovial inflammation may require a two-phase approach that combines inhibiting DAMP production and enhancing alternative macrophage activation, such as through exercise therapy, pre- or pro-biotic treatment, or increased consumption of anti-inflammatory ω–3 polyunsaturated fatty acids (PUFAs) (Figure 1). Indeed, metabolic approaches to tip the balance in favor of pro-resolving M2-like macrophages may be worth considering due to the distinct metabolic phenotypes between M1 and M2-like macrophages.
Figure 1: Complexity of Macrophage Phenotypes in the Osteoarthritic Synovial Membrane.
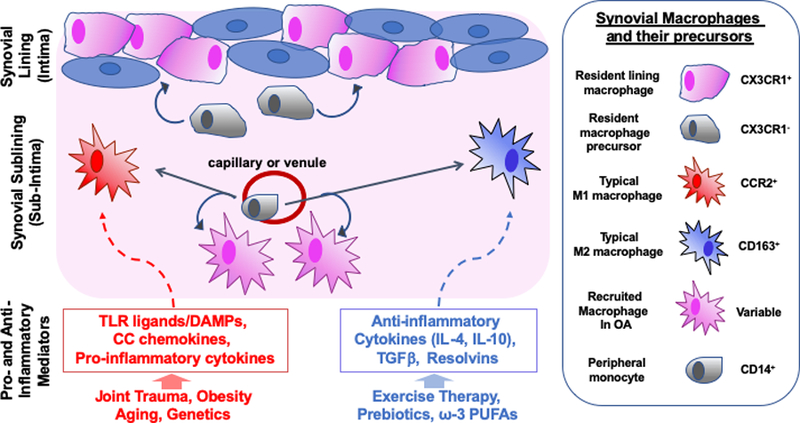
The synovial membrane of articular joints is typically separated into a lining layer (intima) composed of fibroblast-like synoviocytes (blue) and macrophage-like synoviocytes (pink), and a sublining layer (subintima) of vascularized loose connective tissue. Recent evidence suggests that the resident macrophages of the lining layer are derived from a distinct cellular lineage, express the fractalkine receptor (CX3CR1), and are repopulated by a proliferating pool of subintimal CX3CR1– precursors. In contrast, subintimal macrophages are largely derived from recruited monocytes from the periphery. In vitro, monocytes can differentiate into pro-inflammatory M1 macrophages in response to stimuli such as DAMPs, or into M2 anti-inflammatory or wound-healing macrophages in response to anti-inflammatory stimuli or resolvins. In OA, the majority of inflammatory cells in the synovium are macrophages, but recent studies suggest that these recruited macrophages do not clearly fit into M1 or M2 categories. This likely reflects a complex milieu of both pro- and anti-inflammatory stimuli in the OA joint. Whether the phenotypes and functions of the lining macrophages are similar to those of recruited macrophages is not yet clear, but their distinct phenotypic markers suggest a unique function. Attempts at “tipping the balance” of M1 and M2 stimuli have met with some limited success in preclinical models of OA. However, studies that have completely blocked macrophages in the joint have demonstrated that the anti-inflammatory functions of these cells may be critical in maintaining joint homeostasis. Thus, the complexity of synovial macrophage subtypes in the OA joint reflects functional variation important in both health and disease. This complexity and interactions with other immune and stromal cell types needs to be further explored to determine the most effective ways to target macrophage function or phentoypic modulation for an OA disease-modifying therapy.
Acknowledgements
TMG thanks Drs. Mary Beth Humphrey and Susan Kovats for many helpful discussions on innate inflammation and macrophage biology in osteoarthritis. TMG receives grant support from the National Institutes of Health (R01AG049058, R03AR066828), and CRS receives grant support from the Veteran’s Administration Rehabilitation Research and Development Program (RX001757–01). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or Veteran’s Administration.
References
- 1.Dequeker J, Luyten FP. The history of osteoarthritis-osteoarthrosis. Ann Rheum Dis 2008;67:5–10. [DOI] [PubMed] [Google Scholar]
- 2.Farahat MN, Yanni G, Poston R, Panayi GS. Cytokine expression in synovial membranes of patients with rheumatoid arthritis and osteoarthritis. Ann Rheum Dis 1993;52:870–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Revell PA, Mayston V, Lalor P, Mapp P. The synovial membrane in osteoarthritis: a histological study including the characterisation of the cellular infiltrate present in inflammatory osteoarthritis using monoclonal antibodies. Ann Rheum Dis 1988;47:300–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Radin EL, Paul IL, Rose RM. ROLE OF MECHANICAL FACTORS IN PATHOGENESIS OF PRIMARY OSTEOARTHRITIS. The Lancet 1972;299:519–522. [DOI] [PubMed] [Google Scholar]
- 5.Radin EL. Protest the continuing common usage of the term osteoarthritis. Clinical Orthopaedics and Related Research 1990:311. [PubMed]
- 6.Sharma L, Song J, Felson DT, Cahue S, Shamiyeh E, Dunlop DD. The Role of Knee Alignment in Disease Progression and Functional Decline in Knee Osteoarthritis. JAMA 2001;286:188–195. [DOI] [PubMed] [Google Scholar]
- 7.Felson DT. Osteoarthritis as a disease of mechanics. Osteoarthritis Cartilage 2013;21:10–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Magnano MD, Chakravarty EF, Broudy C, Chung L, Kelman A, Hillygus J, et al. A pilot study of tumor necrosis factor inhibition in erosive/inflammatory osteoarthritis of the hands. J Rheumatol 2007;34:1323–1327. [PubMed] [Google Scholar]
- 9.Chevalier X, Goupille P, Beaulieu AD, Burch FX, Bensen WG, Conrozier T, et al. Intraarticular injection of anakinra in osteoarthritis of the knee: a multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum 2009;61:344–352. [DOI] [PubMed] [Google Scholar]
- 10.Verbruggen G, Wittoek R, Vander Cruyssen B, Elewaut D. Tumour necrosis factor blockade for the treatment of erosive osteoarthritis of the interphalangeal finger joints: A double blind, randomised trial on structure modification. Ann Rheum Dis 2012;71:891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huskisson EC, Dieppe PA, Tucker AK, Cannell LB. Another look at osteoarthritis. Ann Rheum Dis 1979;38:423–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pincus T. Clinical evidence for osteoarthritis as an inflammatory disease. Curr Rheumatol Rep 2001;3:524–534. [DOI] [PubMed] [Google Scholar]
- 13.Oehler S, Neureiter D, Meyer-Scholten C, Aigner T. Subtyping of osteoarthritic synoviopathy. Clin Exp Rheumatol 2002;20:633–640. [PubMed] [Google Scholar]
- 14.Bondeson J, Blom AB, Wainwright S, Hughes C, Caterson B, van den Berg WB. The role of synovial macrophages and macrophage-produced mediators in driving inflammatory and destructive responses in osteoarthritis. Arthritis Rheum 2010;62:647–657. [DOI] [PubMed] [Google Scholar]
- 15.Berenbaum F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!). Osteoarthritis Cartilage 2013;21:16–21. [DOI] [PubMed] [Google Scholar]
- 16.Scanzello CR, Plaas A, Crow MK. Innate immune system activation in osteoarthritis: is osteoarthritis a chronic wound? Curr Opin Rheumatol 2008;20:565–572. [DOI] [PubMed] [Google Scholar]
- 17.Orlowsky EW, Kraus VB. The role of innate immunity in osteoarthritis: when our first line of defense goes on the offensive. J Rheumatol 2015;42:363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu-Bryan R, Terkeltaub R. The growing array of innate inflammatory ignition switches in osteoarthritis. Arthritis Rheum 2012:n/a–n/a. [DOI] [PMC free article] [PubMed]
- 19.Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med 2009;15:774–780. [DOI] [PubMed] [Google Scholar]
- 20.Miller RE, Belmadani A, Ishihara S, Tran PB, Ren D, Miller RJ, et al. Damage-associated molecular patterns generated in osteoarthritis directly excite murine nociceptive neurons through Toll-like receptor 4. Arthritis Rheumatol 2015;67:2933–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gómez R, Villalvilla A, Largo R, Gualillo O, Herrero-Beaumont G. TLR4 signalling in osteoarthritis--finding targets for candidate DMOADs. Nat Rev Rheumatol 2015;11:159–170. [DOI] [PubMed] [Google Scholar]
- 22.Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum 2012;64:1697–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guermazi A, Roemer FW, Hayashi D, Crema MD, Niu J, Zhang Y, et al. Assessment of synovitis with contrast-enhanced MRI using a whole-joint semiquantitative scoring system in people with, or at high risk of, knee osteoarthritis: The MOST study. Ann Rheum Dis 2011;70:805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Lent PLEM, Blom AB, Schelbergen RFP, Slöetjes A, Lafeber FPJG, Lems WF, et al. Active involvement of alarmins S100A8 and S100A9 in the regulation of synovial activation and joint destruction during mouse and human osteoarthritis. Arthritis Rheum 2012;64:1466–1476. [DOI] [PubMed] [Google Scholar]
- 25.Guilak F, Fermor B, Keefe FJ, Kraus VB, Olson SA, Pisetsky DS, et al. The Role of Biomechanics and Inflammation in Cartilage Injury and Repair. Clinical Orthopaedics and Related Research 2004;423:17–26. [DOI] [PubMed] [Google Scholar]
- 26.Garcia M, Knight MM. Cyclic loading opens hemichannels to release ATP as part of a chondrocyte mechanotransduction pathway. J Orthop Res 2009:n/a–n/a. [DOI] [PubMed]
- 27.Buckwalter JA, Anderson DD, Brown TD, Tochigi Y, Martin JA. The Roles of Mechanical Stresses in the Pathogenesis of Osteoarthritis: Implications for Treatment of Joint Injuries. Cartilage 2013;0:194760351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scanzello CR. Role of low-grade inflammation in osteoarthritis. Curr Opin Rheumatol 2017;29:79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalaitzoglou E, Griffin TM, Humphrey MB. Innate Immune Responses and Osteoarthritis. Curr Rheumatol Rep 2017;19:45. [DOI] [PubMed] [Google Scholar]
- 30.Greene MA, Loeser RF. Aging-related inflammation in osteoarthritis. Osteoarthritis Cartilage 2015;23:1966–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berenbaum F, Griffin TM, Liu-Bryan R. Review: Metabolic Regulation of Inflammation in Osteoarthritis. Arthritis Rheumatol 2017;69:9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scanzello CR. Chemokines and inflammation in osteoarthritis: Insights from patients and animal models. J Orthop Res 2017;35:735–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang X, Hunter DJ, Jin X, Ding C. The importance of synovial inflammation in osteoarthritis: current evidence from imaging assessments and clinical trials. Osteoarthritis Cartilage 2018;26:165–174. [DOI] [PubMed] [Google Scholar]
- 34.Urban H, Little CB. The role of fat and inflammation in the pathogenesis and management of osteoarthritis. Rheumatology (Oxford) 2018;57:iv10–iv21. [DOI] [PubMed] [Google Scholar]
- 35.Woodell-May JE, Sommerfeld SD. The Role of Inflammation and the Immune System has in the Progression of Osteoarthritis. J Orthop Res 2019. [DOI] [PubMed]
- 36.Pessler F, Dai L, Diaz-Torne C, Gomez-Vaquero C, Paessler ME, Zheng D-H, et al. The synovitis of “non-inflammatory” orthopaedic arthropathies: a quantitative histological and immunohistochemical analysis. Ann Rheum Dis 2008;67:1184–1187. [DOI] [PubMed] [Google Scholar]
- 37.Roemer FW, Guermazi A, Felson DT, Niu J, Nevitt MC, Crema MD, et al. Presence of MRI-detected joint effusion and synovitis increases the risk of cartilage loss in knees without osteoarthritis at 30-month follow-up: the MOST study. Ann Rheum Dis 2011;70:1804–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roemer FW, Guermazi A, Collins JE, Losina E, Nevitt MC, Lynch JA, et al. Semi-quantitative MRI biomarkers of knee osteoarthritis progression in the FNIH biomarkers consortium cohort - Methodologic aspects and definition of change. BMC Musculoskelet Disord 2016;17:466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scanzello CR, McKeon B, Swaim BH, DiCarlo E, Asomugha EU, Kanda V, et al. Synovial inflammation in patients undergoing arthroscopic meniscectomy: molecular characterization and relationship to symptoms. Arthritis Rheum 2011;63:391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim SY, Nair MG. Macrophages in wound healing: activation and plasticity. Immunol Cell Biol 2019;97:258–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simkin J, Sammarco MC, Marrero L, Dawson LA, Yan M, Tucker C, et al. Macrophages are required to coordinate mouse digit tip regeneration. Development 2017;144:3907–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blom AB, van Lent PL, Libregts S, Holthuysen AE, van der Kraan PM, van Rooijen N, et al. Crucial role of macrophages in matrix metalloproteinase-mediated cartilage destruction during experimental osteoarthritis: Involvement of matrix metalloproteinase 3. Arthritis Rheum 2007;56:147–157. [DOI] [PubMed] [Google Scholar]
- 43.Blom AB, van Lent PLEM, Holthuysen AEM, van der Kraan PM, J Roth, van Rooijen N, et al. Synovial lining macrophages mediate osteophyte formation during experimental osteoarthritis. Osteoarthritis Cartilage 2004;12:627–635. [DOI] [PubMed] [Google Scholar]
- 44.van Lent PLEM, Blom AB, van der Kraan P, Holthuysen AEM, Vitters E, van Rooijen N, et al. Crucial role of synovial lining macrophages in the promotion of transforming growth factor beta-mediated osteophyte formation. Arthritis Rheum 2004;50:103–111. [DOI] [PubMed] [Google Scholar]
- 45.Schelbergen RF, Geven EJ, van den Bosch MHJ, Eriksson H, Leanderson T, Vogl T, et al. Prophylactic treatment with S100A9 inhibitor paquinimod reduces pathology in experimental collagenase-induced osteoarthritis. Ann Rheum Dis 2015;74:2254–2258. [DOI] [PubMed] [Google Scholar]
- 46.Daghestani HN, Pieper CF, Kraus VB. Soluble macrophage biomarkers indicate inflammatory phenotypes in patients with knee osteoarthritis. Arthritis Rheumatol 2015;67:956–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kraus VB, McDaniel G, Huebner JL, Stabler TV, Pieper CF, Shipes SW, et al. Direct in vivo evidence of activated macrophages in human osteoarthritis. Osteoarthritis Cartilage 2016;24:1613–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raghu H, Lepus CM, Wang Q, Wong HH, Lingampalli N, Oliviero F, et al. CCL2/CCR2, but not CCL5/CCR5, mediates monocyte recruitment, inflammation and cartilage destruction in osteoarthritis. Ann Rheum Dis 2017;76:914–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miller RE, Tran PB, Das R, Ghoreishi-Haack N, Ren D, Miller RJ, et al. CCR2 chemokine receptor signaling mediates pain in experimental osteoarthritis. Proc Natl Acad Sci U S A 2012;109:20602–20607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miotla Zarebska J, Chanalaris A, Driscoll C, Burleigh A, Miller RE, Malfait AM, et al. CCL2 and CCR2 regulate pain-related behaviour and early gene expression in post-traumatic murine osteoarthritis but contribute little to chondropathy. Osteoarthritis Cartilage 2017;25:406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller RE, Malfait AM. Can we target CCR2 to treat osteoarthritis? The trick is in the timing! Osteoarthritis Cartilage 2017;25:799–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 2014;41:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Clockaerts S, Bastiaansen-Jenniskens YM, Feijt C, De Clerck L, Verhaar JAN, Zuurmond A-M, et al. Cytokine production by infrapatellar fat pad can be stimulated by interleukin 1β and inhibited by peroxisome proliferator activated receptor α agonist. Ann Rheum Dis 2012;71:1012–18. [DOI] [PubMed] [Google Scholar]
- 54.Zhang H, Lin C, Zeng C, Wang Z, Wang H, Lu J, et al. Synovial macrophage M1 polarisation exacerbates experimental osteoarthritis partially through R-spondin-2. Ann Rheum Dis 2018;77:1524–1534. [DOI] [PubMed] [Google Scholar]
- 55.Wu C-L, McNeill J, Goon K, Little D, Kimmerling K, Huebner J, et al. Conditional Macrophage Depletion Increases Inflammation and Does Not Inhibit the Development of Osteoarthritis in Obese Macrophage Fas-Induced Apoptosis-Transgenic Mice. Arthritis Rheumatol 2017;69:1772–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wood MJ, Leckenby A, Reynolds G, Spiering R, Pratt AG, Rankin KS, et al. Macrophage proliferation distinguishes 2 subgroups of knee osteoarthritis patients. JCI Insight 2019;4:869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Culemann S, Grüneboom A, Nicolás-Ávila JÁ, Weidner D, Lämmle KF, Rothe T, et al. Locally renewing resident synovial macrophages provide a protective barrier for the joint. Nature 2019;572:670–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Felson DT, Zhang Y, Hannan MT, Naimark A, Weissman B, Aliabadi P, et al. Risk factors for incident radiographic knee osteoarthritis in the elderly. The framingham study. Arthritis Rheum 1997;40:728–733. [DOI] [PubMed] [Google Scholar]
- 59.Muthuri SG, Hui M, Doherty M, Zhang W. What if we prevent obesity? Risk reduction in knee osteoarthritis estimated through a meta-analysis of observational studies. Arthritis Care Res (Hoboken) 2011;63:982–990. [DOI] [PubMed] [Google Scholar]
- 60.Murphy L, Schwartz TA, Helmick CG, Renner JB, Tudor G, Koch G, et al. Lifetime risk of symptomatic knee osteoarthritis. Arthritis Rheum 2008;59:1207–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sowers M, Karvonen-Gutierrez CA, Palmieri-Smith R, Jacobson JA, Jiang Y, Ashton-Miller JA. Knee osteoarthritis in obese women with cardiometabolic clustering. Arthritis Rheum 2009;61:1328–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhuo Q, Yang W, Chen J, Wang Y. Metabolic syndrome meets osteoarthritis. Nat Rev Rheumatol 2012;8:729–737. [DOI] [PubMed] [Google Scholar]
- 63.Katz JD, Agrawal S, Velasquez M. Getting to the heart of the matter: osteoarthritis takes its place as part of the metabolic syndrome. Current Opinion in Rheumatology 2010;22:512–519. [DOI] [PubMed] [Google Scholar]
- 64.Niu J, Clancy M, Aliabadi P, Vasan R, Felson DT. Metabolic Syndrome, Its Components, and Knee Osteoarthritis: The Framingham Osteoarthritis Study. Arthritis Rheumatol 2017;69:1194–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Appleton CT, Hawker GA, Hill CL, Pope JE. Editorial: “Weighing in” on the Framingham Osteoarthritis Study: Measuring Biomechanical and Metabolic Contributions to Osteoarthritis. Arthritis Rheumatol 2017;69:1127–1130. [DOI] [PubMed] [Google Scholar]
- 66.Lu B, Driban JB, Xu C, Lapane KL, McAlindon TE, Eaton CB. Dietary Fat and Progression of Knee Osteoarthritis Dietary Fat Intake and Radiographic Progression of Knee Osteoarthritis: Data from the Osteoarthritis Initiative. Arthritis Care Res (Hoboken) 2016:1–23. [DOI] [PMC free article] [PubMed]
- 67.Eymard F, Parsons C, Edwards MH, Petit-Dop F, Reginster JY, Bruyère O, et al. Diabetes is a risk factor for knee osteoarthritis progression. Osteoarthritis Cartilage 2015;23:851–859. [DOI] [PubMed] [Google Scholar]
- 68.Glass CK, Olefsky JM. Inflammation and Lipid Signaling in the Etiology of Insulin Resistance. Cell Metabolism 2012;15:635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Johnson AR, Milner JJ, Makowski L. The inflammation highway: metabolism accelerates inflammatory traffic in obesity. Immunol Rev 2012;249:218–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Klein-Wieringa IR, Kloppenburg M, Bastiaansen-Jenniskens YM, Yusuf E, Kwekkeboom JC, El-Bannoudi H, et al. The infrapatellar fat pad of patients with osteoarthritis has an inflammatory phenotype. Ann Rheum Dis 2011;70:851–857. [DOI] [PubMed] [Google Scholar]
- 71.de Jong AJ, Klein-Wieringa IR, Andersen SN, Kwekkeboom JC, Herb-van Toorn L, de Lange-Brokaar BJE, et al. Lack of high BMI-related features in adipocytes and inflammatory cells in the infrapatellar fat pad (IFP). Arthritis research & therapy 2017;19:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Barboza E, Hudson J, Chang W-P, Kovats S, Towner RA, Silasi-Mansat R, et al. Profibrotic Infrapatellar Fat Pad Remodeling Without M1 Macrophage Polarization Precedes Knee Osteoarthritis in Mice With Diet-Induced Obesity. Arthritis & Rheumatology 2017;69:1221–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Harasymowicz NS, Clement ND, Azfer A, Burnett R, Salter DM, Simpson AHWR. Regional Differences Between Perisynovial and Infrapatellar Adipose Tissue Depots and Their Response to Class II and Class III Obesity in Patients With Osteoarthritis. Arthritis Rheumatol 2017;69:1396–1406. [DOI] [PubMed] [Google Scholar]
- 74.Hamada D, Maynard R, Schott E, Drinkwater CJ, Ketz JP, Kates SL, et al. Suppressive Effects of Insulin on Tumor Necrosis Factor-Dependent Early Osteoarthritic Changes Associated With Obesity and Type 2 Diabetes Mellitus. Arthritis Rheumatol 2016;68:1392–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eymard F, Pigenet A, Citadelle D, Tordjman J, Foucher L, Rose C, et al. Knee and hip intra-Articular adipose tissues (IAATs) compared with autologous subcutaneous adipose tissue: A specific phenotype for a central player in osteoarthritis. Ann Rheum Dis 2017;76:1142–1148. [DOI] [PubMed] [Google Scholar]
- 76.Ioan-Facsinay A, Kloppenburg M. Osteoarthritis: Inflammation and fibrosis in adipose tissue of osteoarthritic joints. Nat Rev Rheumatol 2017;13:325–326. [DOI] [PubMed] [Google Scholar]
- 77.Kalaitzoglou E, Lopes EBP, Fu Y, Herron JC, Flaming JM, Donovan EL, et al. TLR4 Promotes and DAP12 Limits Obesity‐Induced Osteoarthritis in Aged Female Mice. JBMR Plus 2018;133:635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Griffin TM, Huffman KM. Editorial: Insulin Resistance: Releasing the Brakes on Synovial Inflammation and Osteoarthritis? Arthritis Rheumatol 2016;68:1330–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wu C-L, Jain D, McNeill JN, Little D, Anderson JA, Huebner JL, et al. Dietary fatty acid content regulates wound repair and the pathogenesis of osteoarthritis following joint injury. Ann Rheum Dis 2015;74:2076–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Donovan EL, Lopes EBP, Batushansky A, Kinter M, Griffin TM. Independent effects of dietary fat and sucrose content on chondrocyte metabolism and osteoarthritis pathology in mice. Dis Model Mech 2018;11:dmm034827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sekar S, Shafie SR, Prasadam I, Crawford R, Panchal SK, Brown L, et al. Saturated fatty acids induce development of both metabolic syndrome and osteoarthritis in rats. Sci Rep 2017;7:46457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Collins KH, Paul HA, Reimer RA, Seerattan RA, Hart DA, Herzog W. Relationship between inflammation, the gut microbiota, and metabolic osteoarthritis development: studies in a rat model. Osteoarthritis Cartilage 2015;23:1989–1998. [DOI] [PubMed] [Google Scholar]
- 83.Schott EM, Farnsworth CW, Grier A, Lillis JA, Soniwala S, Dadourian GH, et al. Targeting the gut microbiome to treat the osteoarthritis of obesity. JCI Insight 2018;3:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Blumenfeld O, Williams FMK, Valdes A, Hart DJ, Malkin I, Spector TD, et al. Association of interleukin-6 gene polymorphisms with hand osteoarthritis and hand osteoporosis. Cytokine 2014;69:94–101. [DOI] [PubMed] [Google Scholar]
- 85.Aref-Eshghi E, Zhang Y, Hart D, Valdes AM, Furey A, Martin G, et al. SMAD3 is associated with the total burden of radiographic osteoarthritis: The chingford study. PLoS ONE 2014;9:e97786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Han S-L, Zhang Y-J, Zhou M, Luan C, Wang P, Zhai L. Association of PAR-2 Gene Polymorphisms with the Inflammatory Response and Susceptibility to Knee Osteoarthritis in the Chinese Han Population. Genet Test Mol Biomarkers 2019;23:84–90. [DOI] [PubMed] [Google Scholar]
- 87.Huesa C, Ortiz AC, Dunning L, McGavin L, Bennett L, McIntosh K, et al. Proteinase-activated receptor 2 modulates OA-related pain, cartilage and bone pathology. Ann Rheum Dis 2016;75:1989–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jackson MT, Moradi B, Zaki S, Smith MM, McCracken S, Smith SM, et al. Depletion of protease-activated receptor 2 but not protease-activated receptor 1 may confer protection against osteoarthritis in mice through extracartilaginous mechanisms. Arthritis Rheumatol 2014;66:3337–3348. [DOI] [PubMed] [Google Scholar]
- 89.Su S-L, Yang H-Y, Lee C-H, Huang G-S, Salter DM, Lee H-S. The (−1486T/C) promoter polymorphism of the TLR-9 gene is associated with end-stage knee osteoarthritis in a Chinese population. J Orthop Res 2012;30:9–14. [DOI] [PubMed] [Google Scholar]
- 90.Balbaloglu O, Sabah Ozcan S, Korkmaz M, Yılmaz N. Promoter polymorphism (T-1486C) of TLR-9 gene is associated with knee osteoarthritis in a Turkish population. J Orthop Res 2017;35:2484–2489. [DOI] [PubMed] [Google Scholar]
- 91.Yang H-Y, Lee H-S, Lee C-H, Fang W-H, Chen H-C, Salter DM, et al. Association of a functional polymorphism in the promoter region of TLR-3 with osteoarthritis: a two-stage case-control study. J Orthop Res 2013;31:680–685. [DOI] [PubMed] [Google Scholar]