

Abstract
Recent research demonstrating that the glutamatergic modulator ketamine has rapid, robust and sustained antidepressant effects has been a turning point in drug discovery for depression. The recent FDA approval of esketamine for adults with treatment-resistant major depressive disorder (MDD) has further underscored the relevance of this agent in spurring investigation into novel and mechanistically distinct agents for use in depression. Over the past two decades, ketamine research has ushered in a new wave of studies seeking to not only identify its mechanism of action but also to examine the antidepressant potential of novel or repurposed agents. This article reviews the approaches that have proven particularly fruitful for the field of neuropsychiatry.
Keywords: Depression, glutamate, serotonin, ketamine, hallucinogens, novel antidepressant mechanisms
Introduction
The discovery of ketamine as a highly effective and rapid-acting treatment for major depression has been hailed as arguably the most significant development in psychiatry during the past few decades [1]. The paradigm-shifting nature of the rapid antidepressant response to ketamine in patients was a significant breakthrough in neuropsychopharmacology and a turning point in antidepressant research. Globally, depression remains a leading cause of distress and disability and a major contributor to the overall global burden of disease. Depression currently affects >300 million people worldwide and is the leading cause of suicide, with an annual related suicide rate reaching close to 800 000 people [2]. Currently available standard antidepressants, which are mostly monoaminergic-based, are effective for a large proportion of patients; however, a significant subset do not respond to these agents [3].
Ketamine was initially introduced into clinical practice in the 1960s as a safer alternative to the anesthetic phencyclidine (PCP) [4]. Although ketamine acts on diverse receptors and neurotransmitter systems throughout the brain, it has formally been classified as a noncompetitive N-methyl-d-aspartate (NMDA) antagonist [5]. Approved by the FDA in 1970, today ketamine is on the WHO list of essential medicines. It is one of the most commonly used anesthetics in human and veterinary medicine and is also used to treat a variety of pain conditions, including cancer pain, chronic pain and acute perioperative pain. In addition, ketamine is often given in emergency room, battlefield and intensive care settings for the management of acute behavioral agitation [6]. Although generally considered safe, in the USA ketamine is nevertheless classified as a Schedule III drug owing to its potential for physical and psychological abuse and dependence. Even at subanesthetic doses it produces transient dissociative and psychotomimetic effects that resemble the positive and negative symptoms of schizophrenia [7].
Recent research has shown that ketamine has considerable promise for treating a wide range of treatment-refractory neuropsychiatric disorders, including obsessive compulsive disorder (OCD), post-traumatic stress disorder (PTSD), bipolar disorder, suicide ideation, addiction and, most notably, treatment-resistant major depressive disorder (MDD). Although this research has taken place almost exclusively within the past two decades, evidence of ketamine’s neuropsychiatric effects appeared long before this. For example, ketamine was used throughout the 1970s in Mexico as part of psychedelic therapy sessions that combined traditional healing practices with psychoanalytic techniques [8]; and in Argentina as an adjunct to regression therapy [9]. In addition, Dr Edward Domino, who conducted ketamine’s initial anesthetic clinical trials in the 1960s, described several patients who abused ketamine and PCP to alleviate their depressive symptoms; Domino noted that the patients claimed that these drugs worked far better than their prescribed antidepressants [4]. Nevertheless, Domino and other researchers in the field viewed this behavior as bizarre and worried that it might potentially lead to unrestrained use of the drug in settings other than anesthesia, where unconsciousness prevented the active experience of its psychotomimetic effects [10].
The reasons behind the long gap between ketamine use as an anesthetic and research into its salutary antidepressant potential are not entirely clear. One possibility is that as its medicinal use grew its recreational use did as well, which undermined its psychiatric utility. Ketamine use as an anesthetic grew during the late 1960s and 1970s, a time when hallucinogenic and psychedelic drug [e.g., lysergic acid diethylamide (LSD), psilocybin] abuse was coincidentally also widespread. Although preliminary research demonstrated that these substances might have therapeutic potential, they were also considered dangerous and socially disruptive. As a result, there was little financial investment into research for psychiatric purposes [11]. In addition, ketamine was initially only administered intravenously (i.v.), making its use for other clinical indications less practical. With the advent of tricyclic antidepressants (TCAs), monoamine oxidase inhibitors (MAOIs) and, eventually, selective serotonin reuptake inhibitors (SSRIs), neuropsychopharmacology research shifted its focus to the role of monoaminergic neurotransmitters [12]. However, despite its undisputed value to the field, the monoamine hypothesis of depression cannot fully explain the heterogeneity of MDD [12]. Furthermore, it is not sufficient to treat the entire spectrum of MDD [13]. Indeed, by the 1990s, animal models began to implicate glutamate – one of the major excitatory neurotransmitters in the mammalian central nervous system (CNS) – as well as its ionotropic NMDA receptor in the etiology and treatment of mood disorders [14]. This paper will review ketamine’s role in revolutionizing drug discovery in depression, discuss recent investigations seeking to uncover novel and mechanistically distinct agents for use in depression, examine the antidepressant potential of novel or repurposed agents and explore how the quest to identify ketamine’s mechanism of action has spurred research. This article also reviews the approaches that have proven particularly fruitful for the field of neuropsychiatry.
Ketamine: historical overview of its antidepressant properties
Trullas and Skolnick [14] were among the first to examine the possible link between depression and glutamatergic system dysfunction. Because inescapable stress can lead to behavioral symptoms of depression as well as disrupted long-term potentiation in the hippocampus [15] – a process mediated by NMDA receptor activation [16] – Trullas and Skolnick reasoned that NMDA antagonists might have antidepressant properties. Indeed, preclinical data supported their hypothesis, and the tested drugs did in fact exert significant antidepressant effects comparable to those of known antidepressants. Over the next several years, the glutamate hypothesis of depression garnered further support as studies began to show that glutamate-modulating agents could alleviate depression-like behaviors in rodents [17,18]. Reduced NMDA function was also shown to correlate with long-term antidepressant treatment [19], implicating NMDA receptor modulation in the mechanisms underlying antidepressant efficacy. Despite this promising line of preclinical research, NMDA receptor antagonists were not examined as potential antidepressants in humans until the following decade.
Building on previous preclinical evidence [19], in 2000, Berman and colleagues discovered that ketamine exerted rapid, robust and relatively sustained antidepressant effects in depressed patients [20]. Using a randomized, placebo-controlled, crossover design, each patient received an i.v. infusion of 0.5 mg/kg of either ketamine or saline on the first test day. On the following test day, which took place at least 1 week later, treatments were switched. The authors found that ketamine exerted antidepressant effects that began within 4 h of the infusion, peaked at 72 h and persisted for 1–2 weeks post-infusion.
Despite the groundbreaking nature of the results, the paper nevertheless did not have the dramatic impact on the field that one would expect. The medical community could have viewed such rapid and robust antidepressant effects as a ‘fluke’ or, perhaps, researchers might not have wanted to test a drug that possessed abuse potential and psychotomimetic effects [21]. Nevertheless, once Zarate and colleagues successfully replicated the finding in an independent group of 18 patients with treatment-resistant MDD, interest in this line of research grew dramatically [22]. Since then, numerous placebo-controlled studies have shown that subanesthetic-dose ketamine exerts rapid, robust and relatively sustained antidepressant effects in individuals with treatment-resistant MDD and bipolar depression [23]. Ketamine has also been shown to have distinct and independent antisuicidal and anti-anhedonic effects in patients with mood disorders [24,25].
It should be noted here that exploration of ketamine’s antidepressant effects only began in earnest after the initial study’s findings were replicated in an independent, placebo-controlled trial. This valuable reminder of the importance of replicating initial positive results is particularly vital when considering that many initial neuroscientific studies have not been replicated, in part because traditional research in general and funding in particular tend to incentivize novel findings. For example, a study examining clinical trial results published between 2000 and 2002 in the five-highest-ranked psychiatry journals at the time found that, although highly cited, 48% of the initial findings were not replicated until 2013 [26]. In this context, successful repurposing of existing drugs – not just novel ones – similarly demands well-designed replication attempts.
Ketamine: a paradigm-shifting antidepressant
Existing antidepressant treatments [MAOIs, TCAs, SSRIs and serotonin-norepinephrine reuptake inhibitors (SNRIs)] are monoaminergic-based treatments. Although they have been in use for decades and have helped many patients, a significant subset of MDD patients showed little to no therapeutic benefit in response to these agents. For instance, the NIMH-funded, community-based Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study of >4000 MDD patients found that, even after four unique medication trials, augmentation or switch, ~33% of the patients did not respond to standard monoaminergic-based treatments [3]. This finding underscores the substantive proportion of treatment-resistant MDD patients and highlights their ongoing risks for decompensation and suicide in the absence of few viable treatment options. Furthermore, all of the monoaminergic antidepressants exhibit a delayed onset of action, in most cases taking up to several weeks to exert their salutary effects. In this context, it should be noted that a single dose of ketamine boasts a superior response rate within a matter of hours [27,28].
Another limitation of currently available antidepressants is that their clinical effects take more time to reach their full therapeutic potential (for instance, the mean onset for paroxetine is 13 days) [29]. This is a substantial disadvantage during an acute depressive crisis. Furthermore, even when these agents do alleviate depressive symptoms, evidence regarding their ability to successfully reduce suicide ideation and behavior remains inconclusive [30]. By contrast, a single dose (0.5 mg/kg) of intravenous ketamine exerts rapid and profound antidepressant effects within hours to days of administration [31]. Ketamine also rapidly reduces suicidal ideation [24], an effect that appears to occur independently of its antidepressant properties [32]. Ketamine’s pan-therapeutic effects also include alleviating fatigue [33] and anhedonia [34] as well as improving sleep measures such as circadian rhythm and slow-wave activity in MDD patients [35]. These symptoms, which are typically seen across several psychiatric diagnoses, are inadequately treated by standard antidepressants.
Given these profound differences in its antidepressant efficacy, the fundamental paradigm shift that ketamine ushered in is the concept that rapid improvement in depressive symptoms – occurring within hours or days instead of weeks or months – can and should be the overall goal in developing novel therapeutics for depression. A faster, better and prolonged antidepressant response represents a major challenge in the development of new effective therapeutics for depression but is a public health goal, the impact of which cannot be underestimated, given its potential to prevent the deleterious neurobiological and psychosocial effects secondary to recurrent or unremitting depressive episodes. In this context, the necessary i.v. administration of ketamine is a hurdle for outpatient settings across many healthcare systems throughout the world. Thus, researchers also began to investigate alternative and less invasive routes of ketamine administration. Lapidus and colleagues demonstrated that intranasal ketamine had antidepressant effects and led to sufficiently high ketamine plasma concentrations [36]. This body of work ultimately led to the development of esketamine – the S(+) enantiomer of ketamine – for intranasal use, which preserves the anesthetic and dissociative properties of its parent compound and has higher NMDA-receptor-binding affinity than the R(+) ketamine enantiomer. In 2013 and 2016, respectively, esketamine was the first antidepressant ever granted ‘breakthrough therapy’ designation by the FDA for treatment-resistant depression. This led to positive Phase III intranasal esketamine studies [37,38] and, in March 2019, the FDA approved intranasal esketamine (SPRAVATO™) for adults with treatment-resistant depression [39].
In this context, a note of caution is warranted. Like most clinically effective drugs that bind at multiple targets, ketamine and its derivative esketamine exhibit a distinctive side-effect profile. Although generally well-tolerated at subanesthetic antidepressant doses, ketamine is nevertheless associated with significant transient side effects, including dissociative symptoms, floating, tachycardia, hypertension, increased irritability and anxiety, impaired vision and nausea. At anesthetic doses and with chronic abuse, cases of cystitis have also been reported [23]. Ketamine also has potential abuse liability by patients with substance use disorders. Nevertheless, the lifetime prevalence of ketamine abuse in those 12 and older, at least in the USA, is relatively low: 1.3% compared with 7.0% for ecstasy and 9.6% for LSD, and its misuse appears to be declining [40]. Nevertheless, the abuse potential of ketamine, as well as the potential iatrogenic induction of ketamine dependence, should warrant extra caution. Thus, its side-effect profile should be thoughtfully balanced against its unparalleled antidepressant efficacy for those with debilitating MDD as well as for patients with acute and chronic suicide risk associated with treatment-resistant MDD.
In the absence of long-term safety guidelines and a dearth of repeat-dose studies, thorough post-market observations of esketamine are being implemented to monitor not only its efficacy but also assess any potential abuse liability and long-term side effects. Indeed, such necessary observations were taken into account when the FDA approved ketamine under a restricted distribution system, which requires (among many other factors) special training for prescribers and post-dosing safety procedures (e.g., no driving). The successful implementation of esketamine could serve as a precedent to approve other scheduled substances or, conversely, spur the development of alternative agents that are better tolerated and have fewer psychotomimetic properties. In this regard, ketamine metabolites such as hydroxynorketamine (HNK, see below) that appear to be devoid of side effects in preclinical animal models of depression would be potentially viable candidates for clinical testing.
Exploring ketamine mechanisms of action: the importance of translational neuroscience
Basic neuroscientific research and animal models have also significantly contributed to and substantially informed the development of ketamine as a treatment for depression, even though the mechanisms leading to clinical antidepressant efficacy have only partially been elucidated [41,42]. The search to clarify the underlying mechanisms of action of ketamine is ongoing and crucial for future progress in the field, as well as for identifying new or repurposing old rapid-acting agents with similar mechanisms of action that have more-favorable side-effect profiles and prolonged therapeutic effects. Such research is also likely to provide valuable insights into the neurobiology of MDD, suicide and stress-related diseases.
Interest in ketamine’s distinctive mechanism of action has spurred interest in similar agents, including the possible repurposing of existing agents. Pharmacologically, a drug’s efficacy is determined by its dose-dependent affinity for particular targets. The pharmacological profile of ketamine and its underlying mechanism of action go beyond modulating glutamate neurotransmission and include direct and indirect high affinity (~2 μM) antagonistic binding properties at the NMDA receptor, as well as α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) throughput modulation [43,44]. Ketamine also exhibits weak agonism at the mu, delta and kappa opioid receptors. Interestingly, preliminary evidence also suggests that mu-opiate receptor antagonism can attenuate ketamine’s antidepressant effects [45], underscoring the mechanistic links between ketamine and the opiate system. Ketamine is used as an adjunctive agent for treating pain and in opioid-sparing anesthesia regimens, particularly in opioid-tolerant patients [46]. Additional mechanisms could also contribute to the antidepressant efficacy, including agonism at the dopamine (D2) receptor and antagonism at the M1–3 muscarinic receptors. Ketamine also inhibits the reuptake of serotonin, dopamine and norepinephrine [47], although a recent positron emission tomography (PET) study found no evidence that ketamine treatment alters serotonin transporter occupancy [48].
Evidence from chronic rodent stress animal models suggests that spontaneous NMDA receptor-mediated inhibition of gamma aminobutyric acid (GABA)-ergic interneurons leads to disinhibition of pyramidal neurons and enhanced glutamate release and enables burst glutamate firing [41]. At postsynaptic sites, ketamine transiently enhances glutamate release that activates AMPA receptors, thus increasing TrkB receptor stimulation, which in turn facilitates mammalian target of rapamycin complex (mTORC) signaling and enables translation and release of brain-derived neurotrophic factor (BDNF) [49]. In response, spine synapses are generated in crucial cortical regions affected by MDD such as the prefrontal cortex (PFC). Interestingly, a recent clinical trial using a forward translational approach demonstrated that the mTORC1 inhibitor rapamycin did not block the rapid antidepressant effects of ketamine in patients with MDD [50]. Instead, and in contrast to their hypothesis, the authors detected significant prolongation of ketamine’s antidepressant effects in patients pretreated with rapamycin versus placebo. In addition, the percentage of responders was higher after 2 weeks [50]. The mechanisms behind this counterintuitive finding are not clear but could involve rapamycin-mediated stabilization of synapse formation.
Other alternative and mutually complementary NMDA-receptor-dependent mechanisms of ketamine include changing thalamo-prefrontal signaling via antagonism of the GluN2B subunit of NMDA receptors, preventing eEF2 phosphorylation, enhancing BDNF levels and promoting AMPA receptor trafficking at the synapse [42,51]. In addition, recent animal model studies found that ketamine rapidly silences NMDA-receptor-dependent firing bursts at the lateral habenula, thereby disinhibiting downstream reward centers [52], which could rapidly alleviate depressive symptoms. Epigenetic mechanisms such as stimulation of histone deacetylase 5 and subsequent stimulation of transcriptional activity could also play a part in rapid protein translation [53]. In addition, preclinical models have shown that ketamine and imipramine can reverse susceptibility-associated transcriptional changes as well as induce resilience-associated transcription in important brain areas such as the PFC [54]. Another recent study found that ketamine restores lost plasticity in the dendritic synapses of the medial PFC induced by chronic cortisol administration [55]. Intriguingly, ketamine facilitated microcircuit reconfigurations that appear to be crucial for restoring connectivity and synchrony in the PFC. This unique work adds intriguing evidence to the hypothesis that ketamine induces spine density formation and rapidly facilitates neuronal connectivity [56] (Figure 1). It should be noted that our knowledge of all these mechanisms has been informed by animal models and basic neuroscience research, underscoring the benefits of translating evidence from animal models to humans and vice versa.
Figure 1.
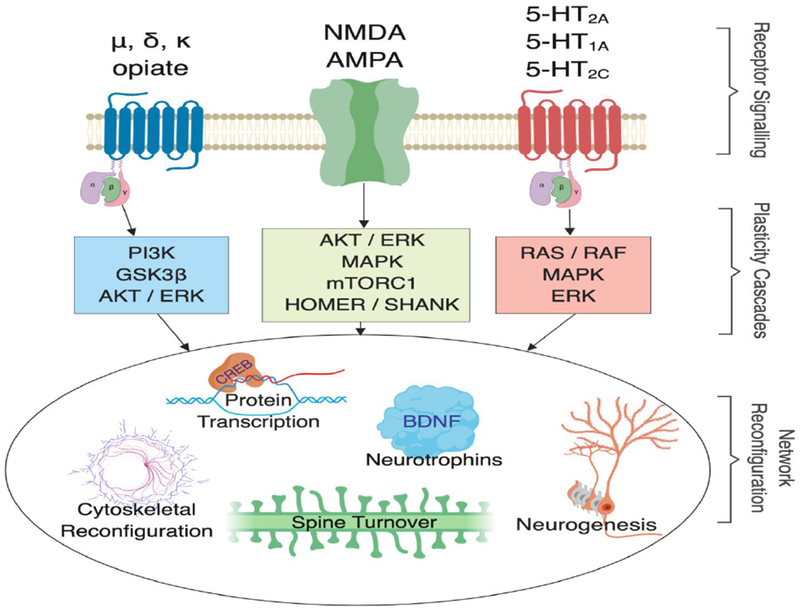
Shared antidepressant mechanisms in three different neurotransmitter systems. Opioid, glutamatergic and serotonergic receptors are linked to intracellular plasticity cascades that target shared neurobiological mechanisms of network reconfiguration. Notably, rapid-acting antidepressant effects occurring within days to 1 week have, so far, only been shown for glutamatergic and serotonergic drugs. Abbreviations: NMDA, N-methyl-d-aspartate receptor; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; 5-HT1/2A, serotonin-1A receptor, serotonin-2A receptor; 5-HT2C, serotonin-2C receptor; PI3K, phosphoinositide 3-kinase; GSK3β, glycogen synthase kinase 3 beta; AKT, protein kinase B; ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinase; mTORC1, mammalian target of rapamycin complex 1; HOMER, homer protein homolog 1; SHANK, SH3 and multiple ankyrin repeat domain 3; RAS/RAF, GTPase protein coupling calcium influx to forms of synaptic plasticity; BDNF, brain-derived neurotrophic factor; δ, delta; κ, kappa; μ, mu; CREB, cyclic adenosine monophosphate response-element-binding protein.
Investigating the potential of active ketamine pharmacometabolites and stereoisomers is another potential research avenue. Repurposing active metabolites and stereoisomers in neuropsychopharmacology is not new; relevant examples include desvenlafaxine (an active metabolite of venlafaxine) and escitalopram [an S(+) enantiomer of citalopram]. After administration, racemic (R,S)-ketamine undergoes demethylation via cytochrome P450 (CYP) liver enzymes CYP2B5 and CYP3A4, mostly to norketamine [57,58]. Norketamine is further metabolized to several HNK metabolites, of which (2R,6R;2S,6S)-HNK and (2S,6R;2R,6S)-HNK predominate in human plasma after ketamine administration, as well as a secondary metabolite, dehydronorketamine (DHNK) [43]. Interestingly, preclinical research recently demonstrated that HNK exerts antidepressant-like properties via indirect early and sustained activation of AMPA receptors probably resulting from a mechanism converging via mGlur2 receptor signaling [59–61]. Differential receptor engagement and side-effect profiles were noted between the S and R isoforms (NMDA vs AMPA receptors) [59]; notably, this finding that (2R,6R)-HNK has antidepressant-like effects that occur independently of NMDA-receptor inhibition was recently replicated by the same laboratory [62]. This research is particularly intriguing given that many of ketamine’s side effects appear to be related to NMDA-receptor-dependent inhibition. Studies investigating the potential antidepressant efficacy – and the side-effect profile – of (2R,6R)-HNK in humans will be a crucial next step; initial safety and tolerability studies are ongoing, and a Phase II clinical trial is planned.
Stimulating research into novel glutamatergic antidepressants
The success of ketamine shifted the field’s focus onto the glutamatergic system for developing novel and/or rapid-acting therapeutics and for investigating the underlying pathophysiology of MDD and other psychiatric disorders. This, in turn, led to advances in our conceptual understanding of depressive mechanisms, suggesting that the neurobiological events triggered by rapid-acting antidepressants might be deeply rooted in the rapid reconfiguration of limbic circuitries [41,42]. The most notable example of a successful rapid-acting agent is intranasal esketamine, the S(+) enantiomer of ketamine, which was FDA-approved in early 2019 for treatment-resistant MDD [39]. Several other rapid-acting glutamatergic agents have similarly shown promising preliminary results; in Table 1 we highlight a handful of examples based on their pharmacodynamic profile. For a more comprehensive overview of studies on novel glutamatergic antidepressants, we refer the interested reader to several recent review articles on the subject [63,64].
Table 1.
Example substances tested in humans with novel antidepressant mechanisms across three major pharmacological classes
| Compound | Pharmacodynamic mechanisms | Outcome parameter | Phase | N | Dosage | Placebo control | Results | Rapid-actinga | Refs |
|---|---|---|---|---|---|---|---|---|---|
| Glutamatergic modulators | |||||||||
| Ketamine | NMDA antagonist, μ- and κ-opioid agonist, D2 agonist, mACh antagonist, weak SERT, DAT, NET reuptake inhibition, 5-HT2 affinity | HAM-D | IV | 99 | 0.1,0.2,0.5 and 1.0 mg/kg | Midazolam 0.045 mg/kg | + | Y | [65] |
| Nitrous oxide | NMDA antagonist | HAM-D | II | 40 | 50% N2O/50% O2 for 1 h | 50% N/50% O2 | + | Y | [69] |
| Sarcosine | Glycine transporter-I inhibitor | HAM-D | II | 40 | 500 mg to 1500 mg | Citalopram 20 mg | + | N | [70] |
| GABAergic modulators | |||||||||
| Brexanolone (SAGE-547) | GABAAPAM, nACh and 5-HT3 NAM agonist at mPR | HAM-D | II | 21 | 30–90 μg/kg/h | Yes | + | Y | [77] |
| SAGE-217 | HAM-D | III | 89 | 30 mg | Yes | + | Y | [78] | |
| Opioids | |||||||||
| ALKS 5461 | Combination of a μ- and κ-opioid partial agonist and μ-opioid antagonist | MADRS | III | 790 | High-dose or low-dose (sublingual) | Yes | + | N | [85] |
| Buprenorphine | μ-receptor partial agonist, κ-opioid, δ-opioid antagonist | MADRS | III | 13 | 0.2 mg to 1.6 mg (sublingual) | No | + | N | [82] |
| Serotonergic hallucinogens | |||||||||
| Psilocybin | 5-HT2A, 5-HT2C, 5-HT2B agonist | QIDS | II | 12 | 10 mg and 25 mg | No | + | Y | [99] |
| DMT | 5-HT2A, 5-HT2C, 5-HT1A agonist | HAM-D | II | 35 | – | Yes | Ongoing | N/A | |
| LSD | 5-HT2A, 5-HT2C, 5-HT1A agonist | IDS | II | 60 | 100 or 200 μg | 25 μg LSD | Ongoing | N/A | |
Abbreviations: 5-HT1A, serotonin-1A receptor; 5-HT2C, serotonin-2C receptor; 5-HT2A, serotonin-2A receptor; 5-HT3, serotonin 3 receptor; δ, delta; D2, dopamine receptor 2; DAT, dopamine transporter; DMT, N-dimethyltryptamine; GABA, gamma aminobutyric acid; HAM-D, Hamilton Depression Rating Scale; IDS: Inventory of Depressive Symptomatology; κ kappa; LSD: lysergic acid diethylamide; μ: mu; mACh: muscarinic acetylcholine; MADRS, Montgomery–Asberg Depression Rating Scale; mPR, membrane progesterone receptor; mg, milligrams; μg, micrograms; NAM, negative allosteric modulator; NET, norepinephrine transporter; nACH, nicotinic acetylcholine receptor; NMDA, N-methyl-d-aspartate; PAM, positive allosteric modulator; QIDS, quick inventory of depressive symptomatology; SERT, serotonin transporter.
Rapid-acting was defined as response (−50% baseline scores) within 1 week of treatment.
Hypothetically, the affinity profile for all of ketamine’s targets could be more important than binding to one or two high-affinity ligands. That is, the antidepressant ‘sweet spot’ in terms of dosage for ketamine might result from the number of occupied NMDA receptors in combination with binding at other lower-affinity targets, triggering a series of events that lead to antidepressant efficacy. Interestingly, ketamine has dose-dependent neuropsychological effects even at subanesthetic doses, with antidepressant properties peaking at ~0.5–1.0 mg/kg [65]. At higher doses, ketamine’s anesthetic properties prevail. Theoretically, antidepressant efficacy at lower dosages might pertain for other drugs. For instance, antidepressant augmentation agents are mostly used at lower doses for drugs binding to the D2/3 or serotonin (5-hydroxytryptamine, 5-HT2 or 5-HT1) receptors, such as aripiprazole, lurasidone, quetiapine or olanzapine. In addition, animal studies co-administering ketamine in combination with 5-HT2A receptor or D2 receptor antagonists underscore the relevance of D2 and 5-HT2A receptors for ketamine’s pharmacological effects [66]. Notably, one study found that PCP and ketamine had more-similar affinities for D2 and 5-HT2 receptors than for NMDA receptors [67]. As a result, ketamine’s antidepressant properties might be mimicked by drugs with similar pharmacodynamic profiles.
As examples, we first briefly touch on mechanistically different agents with glutamatergic modulatory properties that showed antidepressant efficacy in clinical trials: nitrous oxide and sarcosine; as noted above, for a more extensive review of glutamatergic agents in depression, we refer the reader to several recent review articles [63,64]. Nitrous oxide has been used as an anesthetic for >150 years. Like ketamine, it exhibits NMDA receptor antagonism, has partial agonism for mu, kappa and delta opioid receptors, inhibits AMPA, kainite and gamma-aminobutyric acid receptors A and C (GABAA, GABAC), affects serotonin-3 receptors (5-HT3), and releases dopamine [68]. In a double-blind, placebo-controlled, crossover trial, depressive symptoms improved for participants receiving nitrous oxide within 2 h compared with those receiving placebo, an effect that remained significant at 1 day post-treatment [69]. Phase I and II trials are ongoing to determine optimal dose, safety and efficacy.
By contrast, the other agent, sarcosine (also known as N-methylglycine), is an amino acid that functions as a glycine transporter-1 inhibitor and has similarly shown promise in treating MDD. A 6-week, double-blind, randomized, citalopram-controlled trial in 20 MDD patients found that sarcosine possessed superior antidepressant properties compared with citalopram after 2 weeks [70]. In addition, researchers found that sarcosine was well-tolerated; no serious adverse events were reported. Notably, and in contrast to ketamine, sarcosine did not result in rapid-acting effects on the timescale of several days. Sarcosine has co-agonistic properties at the NMDA receptor and is an agonist at the inhibitory glycine receptor [71]. It also exhibits NMDA-enhancing properties, suggesting that AMPA-receptor-mediated or other downstream mechanisms might elicit antidepressant effects. NMDA receptor downregulation might also play a part; however, this is difficult to assess given the paucity of reliable PET radioligands.
Here, it should be noted that neither of these findings have yet been replicated, although such studies are ongoing. However, these two examples underscore that, with regard to drug development in particular, researchers have focused on developing or repurposing drugs with glutamatergic modulatory properties. Although some of these agents have shown antidepressant efficacy, many have not, and many others require further testing; for an excellent overview of the glutamatergic drug pipeline, see Wilkinson and Sanacora [72].
Pioneering research into novel GABAergic and opioidergic agents
Within 1 week of approving esketamine, the FDA also approved brexanolone (SAGE-547, market name Zulresso™) as the first drug specifically targeting postpartum depression (PPD) [73]; this agent is derived from allopregnanolone, an endogenous neuroactive steroid that acts as a positive allosteric modulator of GABAA receptors [74]. The drug was initially investigated for seizure disorder but later found to exhibit rapidacting and sustained antidepressant effects in PPD, a condition associated with the gradual rise and peak of progesterone by the end of the third trimester but rapid decline in progesterone levels in the aftermath of pregnancy. In addition to functioning as a positive allosteric modulator of the GABAA receptor, brexanolone exhibits affinity for nicotinic acetylcholine receptors, 5-HT3 receptors and membrane progesterone receptors, among others. How brexanolone exerts its rapid antidepressant effects is currently unclear but the compound is thought to bind to synaptic and extrasynaptic GABAA receptors, thus increasing receptor functionality [75]. Several double-blind, randomized, placebo-controlled trials demonstrated that brexanolone had potent and rapid antidepressant properties [76]. Two larger, Phase III trials conducted across 30 clinical research centers and psychiatric units across the USA found similarly positive results [77]. Unfortunately, the treatment was associated with four serious adverse events in a few patients (suicidal ideation, intentional overdose, syncope, altered state of consciousness), warranting close post-treatment follow-up.
SAGE-217 is a next-generation allosteric modulator with selectivity for synaptic and extrasynaptic GABAA receptors and a profile resembling that of brexanolone. However, instead of i.v. dosing, SAGE-217 is pharmacologically designed for once-daily oral dosing. SAGE-217 is currently being developed for MDD, PPD and certain other mood disorders. A recent Phase II trial of 89 MDD patients showed that the compound achieved its primary endpoint at 2 weeks post-randomization, but significant differences compared to placebo were found as early as day 2 [78]. Phase III trials are underway for MDD and PPD [79].
Interestingly, opioidergic drugs were used frequently as a treatment for melancholia through the 1950s, before safer, less addictive drugs (e.g., TCAs, MAOIs) became available [80]. Intrigued by the potential of nonaminergic antidepressant mechanisms, researchers have begun to re-evaluate the role of endogenous opioids in depression. For instance, buprenorphine (BUP), a drug currently used to treat opioid addiction and pain disorders, is being explored as a treatment for MDD. The compound has a wide variety of actions throughout the brain, including partial agonism at the mu opioid receptor and antagonism at the kappa and delta opioid receptors [81]; these are connected to intracellular signaling cascades that potentially mediate antidepressant effects (Figure 1). Several open-label studies of BUP in MDD have shown promising preliminary results [80,82], and a double-blind, randomized, placebo-controlled trial examining the effect of low-dose BUP on suicidal ideation similarly yielded positive results [83].
Several double-blind, randomized, placebo-controlled studies have also explored BUP in combination with samidorphan (SAM), a potent mu opioid antagonist that reduces the addictive potential of BUP; this drug combination is known as ALKS 5461. In one study, 32 MDD participants were randomly assigned to receive an 8:1 ratio of SAM to BUP, a 1:1 ratio of SAM to BUP or placebo. The researchers found that the 1:1 ratio most effectively blocked the opioid ‘high’ and, after 1 week of treatment, exerted a significant antidepressant effect [84]. The same research group subsequently conducted a larger trial of 142 MDD patients randomized to receive either BUP/SAM 8 mg/8 mg, BUP/SAM 2 mg/2 mg or placebo. After 4 weeks, both test groups showed improvement, although only the change in the 2 mg/2 mg group was statistically significant. Again, the treatments were well-tolerated and, importantly, neither group showed evidence of opiate withdrawal upon treatment discontinuation [85]. Two global, multicenter, placebo-controlled Phase III trials have provided additional support for the use of ALKS 5461 in depression. The first study, known as FORWARD-4 (n = 385), examined ALKS 5461 at doses of 2 mg/2 mg and 0.5 mg/0.5 mg; the second study, known as FORWARD-5 (n = 407), used doses of 2 mg/2 mg and 1 mg/1 mg. Although the pooled analysis of both studies revealed that the drug was superior to placebo in treating depression, only FORWARD-5 achieved the primary endpoint at 2 mg/2 mg. Again, ALKS 5461 was well-tolerated, with most adverse events classified as mild or moderate [86]. Nevertheless, the FDA stated that it will require more clinical data before the drug can be approved for MDD [87].
Investigating controlled substances as novel and potentially rapid-acting antidepressants: serotonergic hallucinogens
One area that merits particular mention is the burgeoning exploration of serotonergic hallucinogens. In this context, ketamine’s success has not only shifted our understanding of rapid therapeutic response in depression it has also changed the way we look at existing psychopharmaceuticals more generally. Ketamine, which historically might have been written off by the psychiatric community as a hallucinogen with no psychiatric benefit [4], has led the field to reconsider what other scheduled or banned drugs might have been overlooked that could benefit psychiatric patients.
Before they were banned in the 1960s, serotonergic hallucinogens showed promise for treating a range of disorders, including anxiety, OCD, depression and alcoholism [88]; for instance, early papers described the therapeutic potential of LSD for major depression [89]. It should be noted that, by today’s standards, the methodology employed was suboptimal, because many early studies lacked a control group, failed to report adverse events, made no attempt to blind either party and/or used unvalidated outcome measures. However, in 1967, psychedelics were classified under Schedule I of the UN Convention of Drugs, effectively halting research. The reasons for this are complicated, and a lengthy discussion is beyond the scope of this paper. Briefly, by the 1960s, psychedelics had leaked into the community, leading to widespread misuse and reports of negative reactions. Furthermore, reports of these substances being administered covertly and unethically led to public outcry and dismay. Governments responded to these issues by banning the drugs and cutting research funding. However, the past three decades have led to a more tolerant view of these substances and their therapeutic potential, leading to a renewed interest in the field and better-quality studies [88,90].
Toward this end, a key question remains: are we missing potential existing drugs with antidepressant properties that would require dose adjustments in order to be repurposed? Possible examples include psychoactive drugs like psilocybin, LSD or 3,4-methylenedioxymethamphetamine (MDMA); despite their potential harmful side effects and abuse liability, it is hypothetically possible that their therapeutic pharmacological profile for treatment-resistant forms of MDD and other psychiatric disorders such as OCD and PTSD might be achieved by ‘microdosing’, echoing the subanesthetic ketamine doses needed to exert antidepressant effects. The present wave of psychedelic research started in the 1990s with several studies looking at the acute biopsychological effects of drugs like mescaline [91], dimethyltryptamine (DMT) [92] and psilocybin [93]. Through the late 1990s and 2000s, this area of study continued to grow as researchers began examining psychopharmacological and neuropsychological properties of the psychedelic state in healthy volunteers and conducted pharmacological neuroimaging trials [88]. Most of this research has looked at anxiety and depression, with some studies examining alcohol and substance dependence and OCD; for more information, we refer the interested reader to several recent reviews of the topic [11,88].
Of particular relevance to MDD, several open-label studies have examined the safety and feasibility of hallucinogens in MDD. An open-label trial of ayahuasca, a mixture of the South American rainforest liana Banisteriopsis caapi rich in the molecule DMT, found that this serotonergic receptor agonist was safe and well-tolerated in a group of six MDD patients. An ayahuasca mixture containing 96–160 mg DMT and 25.2–42.0 mg harmine (dose depending on bodyweight) led to significant symptom improvement within 1 day that remained at 3-week follow-up [94]. The same researchers subsequently replicated these findings in an open-label study of 17 MDD patients (11 of whom were newly enrolled) [95].
Studies have also examined the effect of open-label psilocybin – a potent 5-HT2 and 5-HT1 receptor partial agonist and one of the main hallucinogenic substances in ‘magic mushrooms’. An early, small, open-label study found that psilocybin significantly reduced depressive symptoms for up to 6 months post-treatment in a group of patients with advanced-stage cancer [96]. Building on this work, several double-blind, randomized, controlled trials found that psychedelics alleviated depression associated with end-stage cancer. Two of the largest come from Ross et al. [97] and Griffiths et al. [98], who examined the anxiolytic and antidepressant effects of a single dose of psilocybin in a group of 29 and 51 patients, respectively. Ross et al. randomly assigned participants to receive either 0.3 mg/kg of psilocybin with psychological support or 250 mg of a placebo (niacin) with psychological support. Crossover occurred 7 weeks after the first session. Griffiths and colleagues used a similar study design, but a low dose of psilocybin was used as the active control, and crossover occurred at 5 weeks. Both groups found that psilocybin was safe, well-tolerated and rapidly and significantly reduced anxiety and depressive symptoms; these improvements lasted through a 6- or 6.5-month follow-up.
Another study of 12 MDD patients used a safety dose of 10 mg of psilocybin followed by a treatment dose of 25 mg 7 days later, in conjunction with psychological support. Psilocybin was found to be safe and well-tolerated and to rapidly reduce depressive, anxiety and anhedonia symptoms within 1 week of administration; strikingly, these effects persisted at a 3-month follow-up [99]. The same researchers subsequently conducted a 6-month open-label trial of psilocybin in 20 patients with treatment-resistant MDD. They found that two doses (10 mg and 25 mg, 1 week apart) of psilocybin reduced depressive symptoms within 1 week for these patients, with improvements lasting through the 3- and 6-month assessment points [100].
To date, little controlled evidence exists for LSD, although at least one active-controlled study of 100 μg LSD and a low-dose active comparator is currently recruiting for MDD (University Hospital in Basel, Switzerland). A small 2015 study found that LSD-assisted psychotherapy was safe, well-tolerated and helped ten patients with life-threatening diseases deal with comorbid anxiety [101].
However, although these clinical trials with potent serotonergic agonists demonstrated tolerability, it is too early to conclude whether these drugs are safe to use in MDD. Toxicity and side-effect profiles after a single administration have been studied, but it is unclear whether rapid-acting effects might contribute to known psychological dependency. Nevertheless, taken together, the accumulating evidence regarding the therapeutic use of serotonergic hallucinogens to treat depression suggests that novel and potentially rapid-acting antidepressant mechanisms could be elicited by serotonin receptor agonists.
Supporting evidence for the rapid-acting antidepressant-like mechanisms of these agents being mediated by serotonergic receptors comes from animal models of stress [102] as well as from structural and functional changes in cortical neurons observed in vivo in rats [103]. Several common postsynaptic intracellular pathways that mediate plasticity as well as transcription factors such as cyclic adenosine monophosphate response-element-binding protein (CREB) are targeted by glutamatergic drugs that affect NMDA and AMPA receptors and serotonergic drugs [104] (Figure 1). To better understand the neurobiology of rapid antidepressant mechanisms, studies investigating the cellular and molecular underpinnings of differential onsets of action between slower-acting reuptake inhibitors and rapid-acting drugs are needed. It should also be noted here that investigations of cross-connectivity between monoaminergic, excitatory (glutamatergic) and/or inhibitory (GABAergic) neurotransmission could significantly enhance our understanding of novel and rapid-acting antidepressant mechanisms and the neurobiology of depression.
Concluding remarks and future perspectives
The FDA approval of esketamine for treatment-resistant MDD represents a major breakthrough in psychiatry. If ketamine or ketamine-like treatments continue to show promise for severe forms depression and other psychiatric diseases, these advances could improve quality of life for millions of patients who are not helped by currently available treatment options. Moreover, in light of growing safety data from esketamine trials, use of this agent could shift to earlier stages of the treatment algorithm, thereby optimizing treatment options, especially for those with acute depression and suicidal thoughts. Although ketamine’s potential efficacy for other neuropsychiatric disorders is still being evaluated, preliminary findings are promising. As noted above, initial evidence suggests that ketamine could also be clinically effective for the treatment of PTSD and OCD [105,106]; these disorders share several clinical and neurobiological characteristics with MDD and are currently treated with SSRIs. If these effects are confirmed, clinical applications beyond MDD could be on the horizon.
Although such findings certainly bring hope to the many individuals suffering from severe depression, the known and potentially unknown side effects associated with repeated ketamine administration require caution when moving forward. As an example, several case reports have noted ketamine-induced mania [107], although earlier studies from our laboratory found that a single ketamine infusion did not lead to mood switching in bipolar subjects [108]. In that regard, publication of post-marketing registries and observations of unwanted side effects – including ketamine dependence and switch-risk – are needed. Standardized, longitudinal databases could provide a solution to tackle many questions that are currently hard to answer in clinical trials.
Nevertheless, the success of ketamine has ushered in a new era in psychiatry, with new expectations regarding the speed of onset of antidepressant effects, and new mechanisms of action to explore. Several other novel glutamatergic antidepressants have similarly demonstrated preliminary potential for success [72]. Despite this progress, it is necessary to remain cautiously optimistic, because controlled, well-powered studies are needed to establish clinical efficacy for some of the agents discussed above, such as serotonergic hallucinogens. Furthermore, it is important to note that most of the research into the mechanism of action underlying rapid antidepressant response is still only in the early stages. Even for ketamine, by far the best studied of the novel agents, no unique mechanism of action has been revealed; rather, multiple potentially parallel-acting mechanisms appear to exist [42]. Yet, to develop future novel and/or rapid-acting antidepressant agents, research that identifies these multiple biological mechanisms is crucial.
In short, the discovery of ketamine’s antidepressant efficacy ushered in an era of paradigm-shifting research that raised the bar for developing the next generation of faster-acting and more-effective antidepressants. Clinical and preclinical ketamine research across the past two decades has ultimately paved the way toward ongoing work seeking to discover new approaches for preventing and treating this devastating illness.
Highlights:
The recent paradigm shift in drug development due to ketamine is described
A historical overview of novel and repurposed antidepressant drugs is provided
Mechanisms for ketamine and other novel agents with antidepressant effects are reviewed
The antidepressant properties of hallucinogenic drugs are assessed
Acknowledgments
Funding for this work was supported by the Intramural Research Program at the National Institute of Mental Health, National Institutes of Health (IRP-NIMH-NIH; ZIA MH002857), by a NARSAD Independent Investigator Award to Dr Zarate, and by a Brain and Behavior Mood Disorders Research Award to Dr Zarate. The NIMH had no further role in study design; in the collection, analysis or interpretation of data; in the writing of the report; or in the decision to submit the paper for publication. The authors thank the 7SE research unit and staff for support.
Biographies
Author biographies

Elia Acevedo-Diaz, MD, is a board-certified psychiatrist and clinical research fellow in the Experimental Therapeutics and Pathophysiology Branch (ETPB), National Institute of Mental Health (NIMH). Her research seeks to understand the mechanisms of action of novel fast-acting antidepressants and identify clinical markers of treatment response to guide clinical care for patients with treatment-resistant mood disorders.

Bashkim Kadriu, MD, is a board-certified psychiatrist and neuroscientist at the Experimental Therapeutics and Pathophysiology Branch (ETPB), National Institute of Mental Health (NIMH). His research interests include the neurobiological correlates of treatment-resistant mood disorders with a particular emphasis on discovering biosignatures that guide novel fast-acting antidepressant actions.

Christoph Kraus, MD, is a research psychiatrist conducting a post-doctoral visiting fellowship at the National Institute of Mental Health (NIMH). Dr Kraus received his training in clinical psychiatry at the Medical University of Vienna. His research seeks to establish biological correlates and predictors of antidepressant treatment by leveraging multimodal imaging methods.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Teaser: The discovery of the rapid antidepressant effects of ketamine was a turning point in drug discovery for depression; leading researchers explore several novel, mechanistically distinct and rapid-acting agents.
Conflicts of interest
Dr Zarate is listed as a co-inventor on a patent for the use of ketamine in major depression and suicidal ideation; as a co-inventor on a patent for the use of (2R,6R)-hydroxynorketamine, (S)-dehydronorketamine and other stereoisomeric dehydro and hydroxylated metabolites of (R,S)-ketamine metabolites in the treatment of depression and neuropathic pain; and as a co-inventor on a patent application for the use of (2R,6R)-hydroxynorketamine and (2S,6S)-hydroxynorketamine in the treatment of depression, anxiety, anhedonia, suicidal ideation and post-traumatic stress disorders. He has assigned his patent rights to the US government but will share a percentage of any royalties that might be received by the government. All other authors have no conflicts of interest to disclose, financial or otherwise.
References
- 1.Insel TR (2014) Director’s Blog: Ketamine Available at: http://www.nimh.nih.gov/about/director/2014/ketamine.shtml
- 2.World Health Organization (2018) Depression Fact Sheet. Available at: https://www.who.int/news-room/fact-sheets/detail/depression
- 3.Gaynes BN et al. (2009) What did STAR*D teach us? Results from a large-scale, practical, clinical trial for patients with depression. Psychiatr. Serv. 60, 1439–1445 [DOI] [PubMed] [Google Scholar]
- 4.Domino EF (2010) Taming the ketamine tiger. 1965. Anesthesiology 113, 678–684 [DOI] [PubMed] [Google Scholar]
- 5.Hirota K and Lambert DG (1996) Ketamine: its mechanism(s) of action and unusual clinical uses. Br. J. Anaesth. 77, 441–444 [DOI] [PubMed] [Google Scholar]
- 6.Riddell J et al. (2017) Ketamine as a first-line treatment for severely agitated emergency department patients. Am. J. Emerg. Med 35, 1000–1004 [DOI] [PubMed] [Google Scholar]
- 7.Krystal JH et al. (1994) Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry 51, 199–214 [DOI] [PubMed] [Google Scholar]
- 8.Kolp E et al. (2006) Ketamine enhanced psychotherapy: preliminary clinical observations on its effectiveness in treating alcoholism. The Humanistic Psychologist 34, 399–422 [Google Scholar]
- 9.Fontana A (1974) Terapia antidepressiva con Ci 581 (ketamine). Acta Psiquiat. Psicol. America Latina 4, 20–32 [PubMed] [Google Scholar]
- 10.Javitt DC and Zukin SR (1991) Recent advances in the phencyclidine model of schizophrenia. Am. J. Psychiatry 148, 1301–1308 [DOI] [PubMed] [Google Scholar]
- 11.Carhart-Harris RL and Goodwin GM (2017) The therapeutic potential of psychedelic drugs: past, present, and future. Neuropsychopharmacology 42, 2105–2113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirschfeld RM (2000) History and evolution of the monoamine hypothesis of depression. J. Clin. Psychiatry 61 (suppl. 6), 4–6 [PubMed] [Google Scholar]
- 13.Hillhouse TM and Porter JH (2015) A brief history of the development of antidepressant drugs: from monoamines to glutamate. Exp. Clin. Psychopharmacol 23, 1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trullas R and Skolnick P (1990) Functional antagonists at the NMDA receptor complex exhibit antidepressant actions. Eur. J. Pharmacol 185, 1–10 [DOI] [PubMed] [Google Scholar]
- 15.Shors TJ et al. (1989) Inescapable versus escapable shock modulates longterm potentiation in the rat hippocampus. Science 244, 224–226 [DOI] [PubMed] [Google Scholar]
- 16.Luscher C and Malenka RC (2012) NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol 4, pii:a005710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Layer RT et al. (1995) Antidepressant-like actions of the polyamine site NMDA antagonist, eliprodil (SL-82.0715). Pharmacol. Biochem. Behav 52, 621–627 [DOI] [PubMed] [Google Scholar]
- 18.Papp M and Moryl E (1994) Antidepressant activity of non-competitive and competitive NMDA receptor antagonists in a chronic mild stress model of depression. Eur. J. Pharmacol 263, 1–7 [DOI] [PubMed] [Google Scholar]
- 19.Skolnick P et al. (1996) Adaptation of /V-methyl-D-aspartate (NMDA) receptors following antidepressant treatment: implications for the pharmacotherapy of depression. Pharmacopsychiatry 29, 23–26 [DOI] [PubMed] [Google Scholar]
- 20.Berman RM et al. (2000) Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 47, 351–354 [DOI] [PubMed] [Google Scholar]
- 21.Blier P and Blier J (2016) Ketamine: clinical studies in treatment-resistant depressive disorders In Ketamine for Treatment-Resistant Depression: The First Decade of Progress (Mathew SJ. and Zarate CAJ., eds), pp. 31–42, Springer International Publishing [Google Scholar]
- 22.Zarate CA Jr, et al. (2006) A randomized trial of an /V-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry 63, 856–864 [DOI] [PubMed] [Google Scholar]
- 23.Kraus C et al. (2017) Administration of ketamine for unipolar and bipolar depression. Int. J. Psychiatry Clin. Pract 21,2–12 [DOI] [PubMed] [Google Scholar]
- 24.Wilkinson ST et al. (2018) The effect of a single dose of intravenous ketamine on suicidal ideation: a systematic review and individual participant data metaanalysis. Am. J. Psychiatry 175, 150–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lally N et al. (2014) Anti-anhedonic effect of ketamine and its neural correlates in treatment-resistant bipolar depression. Transl. Psychiatry 4, e469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tajika A et al. (2015) Replication and contradiction of highly cited research papers in psychiatry: 10-year follow-up. Br. J. Psychiatry 207, 357–362 [DOI] [PubMed] [Google Scholar]
- 27.aan het Rot M et al. (2012) Ketamine for depression: where do we go from here. Biol. Psychiatry 72, 537–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Phelps LE et al. (2009) Family history of alcohol dependence and initial antidepressant response to an /V-methyl-D-aspartate antagonist. Biol. Psychiatry 65, 181–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katz MM et al. (2004) Onset and early behavioral effects of pharmacologically different antidepressants and placebo in depression. Neuropsychopharmacology 29, 566–579 [DOI] [PubMed] [Google Scholar]
- 30.Leon AC et al. (2014) Risk of suicidal behavior with antidepressants in bipolar and unipolar disorders. J. Clin. Psychiatry 75, 720–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murrough JW et al. (2013) Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am. J. Psychiatry 170, 1134–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ballard ED, lonescu DF, Vande Voort JL, Niciu MJ, Richards EM, Luckenbaugh DA et al. (2014) Improvement in suicidal ideation after ketamine infusion: relationship to reductions in depression and anxiety. J. Psychiatr. Res 58, 161–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saligan LN et al. (2016) An assessment of the anti-fatigue effects of ketamine from a double-blind, placebo-controlled, crossover study in bipolar disorder. J. Affect. Disord 194, 115–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lally N et al. (2015) Neural correlates of change in major depressive disorder anhedonia following open-label ketamine. J. Psychopharmacol 29, 596–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duncan WC et al. (2017) Motor-activity markers of circadian timekeeping are related to ketamine’s rapid antidepressant properties. Biol. Psychiatry 82, 361–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lapidus KA et al. (2014) A randomized controlled trial of intranasal ketamine in major depressive disorder. Biol. Psychiatry 76, 970–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Daly EJ et al. (2018) Efficacy and safety of intranasal esketamine adjunctive to oral antidepressant therapy in treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry 75, 139–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Canuso CM et al. (2018) Efficacy and safety of intranasal esketamine for the rapid reduction of symptoms of depression and suicidality in patients at imminent risk for suicide: results of a double-blind, randomized, placebo-controlled study. Am. J. Psychiatry 175, 620–630 [DOI] [PubMed] [Google Scholar]
- 39.US Food & Drug Administration (2019) FDA approves new nasal spray medication for treatment-resistant depression. Available at: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm632761.htm
- 40.US Food and Drug Administration (2017) FDA Briefing Document. Available at: https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/PsychopharmacologicDrugsAdvisoryCommittee/UCM630970.pdf
- 41.Duman RS et al. (2016) Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat. Med 22, 238–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kadriu B et al. (2019) Glutamatergic neurotransmission: pathway to developing novel rapid-acting antidepressant treatments. Int. J. Neuropsychopharmacol 22, 119–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zanos P et al. (2018) Ketamine and ketamine metabolite pharmacology: insights into therapeutic mechanisms. Pharmacol. Rev 70, 621–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maeng S et al. (2008) Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol. Psychiatry 63, 349–352 [DOI] [PubMed] [Google Scholar]
- 45.Williams NR et al. (2018) Attenuation of antidepressant effects of ketamine by opioid receptor antagonism. Am. J. Psychiatry 175, 1205–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gabriel RA et al. (2019) State of the art opioid-sparing strategies for postoperative pain in adult surgical patients. Expert Opin. Pharmacother 20, 949–961 [DOI] [PubMed] [Google Scholar]
- 47.Kohrs R and Durieux ME (1998) Ketamine: teaching an old drug new tricks. Anesth. Analg 87, 1186–1193 [DOI] [PubMed] [Google Scholar]
- 48.Spies M et al. (2018) Assessment of ketamine binding of the serotonin transporter in humans with positron emission tomography. Int. J. Neuropsychopharmacol 21, 145–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li N et al. (2010) mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329, 959–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abdallah CG et al. (2018) Rapamycin, an immunosuppressant and mTORCI inhibitor, triples the antidepressant response rate of ketamine at 2 weeks following treatment: a double-blind, placebo-controlled, cross-over, randomized clinical trial. bioRxiv doi: 10.1101/500959 [DOI] [Google Scholar]
- 51.Miller OH. et al. (2014) GluN2B-containing NMDA receptors regulate depression-like behavior and are critical for the rapid antidepressant actions of ketamine. Elite 3, e03581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang Y et al. (2018) Ketamine blocks bursting in the lateral habenula to rapidly relieve depression. Nature 554, 317–322 [DOI] [PubMed] [Google Scholar]
- 53.Choi M et al. (2015) Ketamine produces antidepressant-like effects through phosphorylation-dependent nuclear export of histone deacetylase 5 (HDAC5) in rats. Proc. Natl. Acad. Sci. U. S. A 112, 15755–15760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bagot RC et al. (2017) Ketamine and imipramine reverse transcriptional signatures of susceptibility and induce resilience-specific gene expression profiles. Biol. Psychiatry 81,285–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moda-Sava RN et al. (2019) Sustained rescue of prefrontal circuit dysfunction by antidepressant-induced spine formation. Science 364, pii:eaat8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beyeler A (2019) Do antidepressants restore lost synapses? Science 364, 129–130 [DOI] [PubMed] [Google Scholar]
- 57.Moaddel R et al. (2016) Subchronic administration of (R,S)-ketamine induces ketamine ring hydroxylation in Wistar rats. J. Pharm. Biomed. Anal 127, 3–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moaddel R et al. (2010) A parallel chiral-achiral liquid chromatographic method for the determination of the stereoisomers of ketamine and ketamine metabolites in the plasma and urine of patients with complex regional pain syndrome. Talanta 82, 1892–1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zanos P et al. (2016) NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 533, 481–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zanos P et al. (2018) Convergent mechanisms underlying rapid antidepressant action. CNS Drugs 32, 197–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zanos P et al. (2019) (2R,6R)-hydroxynorketamine exerts mGlu2 receptor-dependent antidepressant actions. Proc. Natl. Acad. Sci. U. S. A 116, 6441–6450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lumsden EW et al. (2019) Antidepressant-relevant concentrations of the ketamine metabolite (2R,6R)-hydroxynorketamine do not block NMDA receptor function. Proc. Natl. Acad. Sci. U. S. A 116, 5160–5169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Garay RP et al. (2017) Investigational drugs in recent clinical trials for treatment-resistant depression. Expert Rev. Neurother 17, 593–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Henter ID et al. (2018) Glutamatergic modulators in depression. Harv. Rev. Psychiatry 26, 307–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fava M et al. (2018) Double-blind, placebo-controlled, dose-ranging trial of intravenous ketamine as adjunctive therapy in treatment-resistant depression (TRD). Mol. Psychiatry doi: 10.1038/s41380-018-0256-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Amargos-Bosch M et al. (2006) Clozapine and olanzapine, but not haloperidol, suppress serotonin efflux in the medial prefrontal cortex elicited by phencyclidine and ketamine. Int. J. Neuropsychopharmacol 9, 565–573 [DOI] [PubMed] [Google Scholar]
- 67.Kapur S and Seeman P (2002) NMDA receptor antagonists ketamine and PCP have direct effects on the dopamine D(2) and serotonin 5-HT(2)receptors-implications for models of schizophrenia. Mol. Psychiatry 7, 837–844 [DOI] [PubMed] [Google Scholar]
- 68.Zarate CAJ and Machado-Vieira R (2015) Potential pathways involved in the rapid antidepressant effects of nitrous oxide. Biol. Psychiatry 78, 2–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nagele P et al. (2015) Nitrous oxide for treatment-resistant major depression: a proof-of-concept trial. Biol. Psychiatry 78, 10–18 [DOI] [PubMed] [Google Scholar]
- 70.Huang CC et al. (2013) Inhibition of glycine transporter-l as a novel mechanism for the treatment of depression. Biol. Psychiatry 74, 734–741 [DOI] [PubMed] [Google Scholar]
- 71.Zhang HX et al. (2009) The glycine transport inhibitor sarcosine is an inhibitory glycine receptor agonist. Neuropharmacology 57, 734–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wilkinson ST and Sanacora G (2019) A new generation of antidepressants: an update on the pharmaceutical pipeline for novel and rapid-acting therapeutics in mood disorders based on glutamate/GABA neurotransmitter systems. Drug Discov. Today 24, 606–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.FDA (2019) FDA approves first treatment for post-partum depression. Available at: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm633919.htm
- 74.Majewska MD et al. (1986) Steroid hormone metabolites are barbiturate-like modulators of the GABA receptor. Science 232, 1004–1007 [DOI] [PubMed] [Google Scholar]
- 75.Frieder A et al. (2019) Pharmacotherapy of postpartum depression: current approaches and novel drug development. CNS Drugs 33, 265–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kanes S et al. (2017) Brexanolone (SAGE-547 injection) in post-partum depression: a randomised controlled trial. Lancet 390, 480–489 [DOI] [PubMed] [Google Scholar]
- 77.Meltzer-Brody S et al. (2018) Brexanolone injection in post-partum depression: two multicentre, double-blind, randomised, placebo-controlled, Phase 3 trials. Lancet 392, 1058–1070 [DOI] [PubMed] [Google Scholar]
- 78.Gunduz-Bruce H et al. (2019) A pivotal study of the oral GABA-A receptor positive allosteric modulator SAGE-217 in major depressive disorder: analysis of HAM-D total score, subscales and individual items. Presented at: American Society of Clinical Psychopharmacology Annual Meeting (May 28–31) [Google Scholar]
- 79.Therapeutics Sage (2018) Sage announces pivotal Phase 3 trial status for SAGE-217 in major depressive disorder and postpartum depression based on FDA breakthrough therapy meeting. Available at: https://investor.sagerx.com/news-releases/news-release-details/sage-announces-pivotal-phase-3-trial-status-sage-217-major
- 80.Bodkin JA et al. (1995) Buprenorphine treatment of refractory depression. J. Clin. Psychopharmacol 15, 49–57 [DOI] [PubMed] [Google Scholar]
- 81.Lutfy K and Cowan A (2004) Buprenorphine: a unique drug with complex pharmacology. Curr. Neuropharmacol 2, 395–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Karp JF et al. (2014) Safety, tolerability, and clinical effect of low-dose buprenorphine for treatment-resistant depression in midlife and older adults. J. Clin. Psychiatry 75, e785–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yovell Y et al. (2016) Ultra-low-dose buprenorphine as a time-limited treatment for severe suicidal ideation: a randomized controlled trial. Am. J. Psychiatry 173, 491–498 [DOI] [PubMed] [Google Scholar]
- 84.Ehrich E et al. (2015) Evaluation of opioid modulation in major depressive disorder. Neuropsychopharmacology 40, 1448–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fava M et al. (2016) Opioid modulation with buprenorphine/samidorphan as adjunctive treatment for inadequate response to antidepressants: a randomized double-blind placebo-controlled trial. Am. J. Psychiatry 173, 499–508 [DOI] [PubMed] [Google Scholar]
- 86.Fava M et al. (2018) Opioid system modulation with buprenorphine/samidorphan combination for major depressive disorder: two randomized controlled studies. Mol. Psychiatry doi: 10.1038/s41380-018-0284-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.FierceBiotech (2019) FDA rejects Alkermes’ depression drug ALKS 5461. Available at: https://www.fiercebiotech.com/biotech/fda-rejects-alkermes-depression-drug-alks-5461
- 88.Rucker JJH et al. (2018) Psychiatry & the psychedelic drugs. Past, present & future. Neuropharmacology 142, 200–218 [DOI] [PubMed] [Google Scholar]
- 89.Savage C (1952) Lysergic acid diethylamide (LSD-25): a clinical-psychological study. Am. J. Psychiatry 108, 896–900 [DOI] [PubMed] [Google Scholar]
- 90.Sessa B (2005) Can psychedelics have a role in psychiatry once again? Br. J. Psychiatry 186, 457–458 [DOI] [PubMed] [Google Scholar]
- 91.Hermle L et al. (1992) Mescaline-induced psychopathological, neuropsychological, and neurometabolic effects in normal subjects: experimental psychosis as a tool for psychiatric research. Biol. Psychiatry 32, 976–991 [DOI] [PubMed] [Google Scholar]
- 92.Strassman RJ et al. (1994) Dose-response study of Λ/,/V-dimethyltryptamine in humans. II. Subjective effects and preliminary results of a new rating scale. Arch. Gen. Psychiatry 51, 98–108 [DOI] [PubMed] [Google Scholar]
- 93.Vollenweider FX et al. (1997) Positron emission tomography and fluorodeoxyglucose studies of metabolic hyperfrontality and psychopathology in the psilocybin model of psychosis. Neuropsychopharmacology 16, 357–372 [DOI] [PubMed] [Google Scholar]
- 94.Osorio Fde L et al. (2015) Antidepressant effects of a single dose of ayahuasca in patients with recurrent depression: a preliminary report. Braz. J. Psychiatry 37, 13–20 [DOI] [PubMed] [Google Scholar]
- 95.Sanches RF et al. (2016) Antidepressant effects of a single dose of ayahuasca in patients with recurrent depression: a SPECT study. J. Clin. Psychopharmacol 36, 77–81 [DOI] [PubMed] [Google Scholar]
- 96.Grob CS et al. (2011) Pilot study of psilocybin treatment for anxiety in patients with advanced-stage cancer. Arch. Gen. Psychiatry 68, 71–78 [DOI] [PubMed] [Google Scholar]
- 97.Ross S et al. (2016) Rapid and sustained symptom reduction following psilocybin treatment for anxiety and depression in patients with life-threatening cancer: a randomized controlled trial. J. Psychopharmacol 30, 1165–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Griffiths RR et al. (2016) Psilocybin produces substantial and sustained decreases in depression and anxiety in patients with life-threatening cancer: a randomized double-blind trial. J. Psychopharmacol 30, 1181–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Carhart-Harris RL et al. (2016) Psilocybin with psychological support for treatment-resistant depression: an open-label feasibility study. Lancet Psychiatry 3, 619–627 [DOI] [PubMed] [Google Scholar]
- 100.Carhart-Harris RL et al. (2018) Psilocybin with psychological support for treatment-resistant depression: six-month follow-up. Psychopharmacology 235, 399–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gasser P et al. (2015) LSD-assisted psychotherapy for anxiety associated with a life-threatening disease: a qualitative study of acute and sustained subjective effects. J. Psychopharmacol 29, 57–68 [DOI] [PubMed] [Google Scholar]
- 102.Opal MD et al. (2013) Serotonin 2C receptor antagonists induce fast-onset antidepressant effects. Mol. Psychiatry 19, 1106–1114 [DOI] [PubMed] [Google Scholar]
- 103.Ly C et al. (2018) Psychedelics promote structural and functional neural plasticity. Cell Rep. 23, 3170–3182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kraus C et al. (2017) Serotonin and neuroplasticity – links between molecular, functional and structural pathophysiology in depression. Neurosci. Biobehav. Rev 77, 317–326 [DOI] [PubMed] [Google Scholar]
- 105.Feder A et al. (2014) Efficacy of intravenous ketamine for treatment of chronic posttraumatic stress disorder: a randomized clinical trial. JAMA Psychiatry 71, 681–688 [DOI] [PubMed] [Google Scholar]
- 106.Rodriguez CI et al. (2013) Randomized controlled crossover trial of ketamine in obsessive-compulsive disorder: proof-of-concept. Neuropsychopharmacology 38, 2475–2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Allen ND et al. (2019) A report of affective switching associated with ketamine: the case of ketamine-induced mania is not closed. Bipolar Disord. 21, 176–178 [DOI] [PubMed] [Google Scholar]
- 108.Niciu MJ et al. (2013) Subanesthetic dose ketamine does not induce an affective switch in three independent samples of treatment-resistant major depression. Biol. Psychiatry 74, 23–24 [DOI] [PMC free article] [PubMed] [Google Scholar]