

Abstract
Background:
15-lipoxygenase 1 (15LO1) is expressed in airway epithelial cells in Type-2-High asthma, in association with eosinophilia. Chronic rhinosinusitis with nasal polyps (CRSwNP) is also associated with Type-2 inflammation and eosinophilia. CCL26/Eotaxin3 has been reported to be regulated by 15LO1 in lower airway epithelial cells. However, its relation to 15LO1 in CRSwNP or mechanisms for its activation are unclear.
Objective:
To evaluate 15LO1 and CCL26 in nasal epithelial cells (NECs) from CRSwNP subjects and health controls (HCs) and determine whether 15LO1 regulates CCL26 in NECs via ERK activation.
Methods:
15LO1, CCL26, and phospho-ERK were evaluated from CRSwNP and HCs NECs. 15LO1/CCL26 and CCL26/Cytokeratin5 were co-localized by immunofluorescence. IL-13 stimulated NECs were cultured at an air-liquid interface with or without ALOX15 DsiRNA transfection, a specific 15LO1 enzymatic inhibitor and two ERK inhibitors. 15LO1 and CCL26 mRNA and protein were analyzed by qRT-PCR, Western blot and ELISA.
Results:
15LO1 was increased in nasal polyp epithelial cells compared to middle turbinate epithelial cells from CRSwNP subjects and HCs. 15LO1 correlated with CCL26 expression and co-localized with CCL26 in basal cells of middle turbinate and nasal polyps from CRSwNP subjects. In primary NECs in vitro, IL-13 induced 15LO1 and CCL26 expression. 15LO1 knockdown and inhibition decreased IL-13-induced ERK phosphorylation and CCL26 expression. ERK inhibition (alone) similarly decreased IL-13-induced CCL26. Phospho-ERK was increased in NECs from CRSwNP subjects and positively correlated with both 15LO1 and CCL26.
Conclusions:
15LO1 is increased in nasal polyp epithelial cells and contributes to CCL26 expression through ERK activation. 15LO1 could be considered a novel therapeutic target for CRSwNP.
Keywords: 15-lipoxygenase 1, CCL26, epithelial cell, nasal polyps, chronic rhinosinusitis, ERK
Graphical Abstract
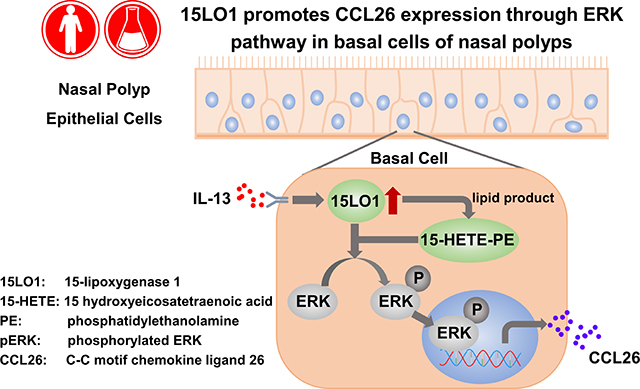
Capsule Summary:
15LO1 is increased in nasal polyp epithelial cells in association with CCL26. 15LO1 induced by the Type-2 cytokine IL-13 in nasal polyp epithelial cells contributes to CCL26 production through ERK pathway activation. Blocking 15LO1 expression could inhibit tissue eosinophilia in CRSwNP subjects.
Introduction
Chronic rhinosinusitis with nasal polyps (CRSwNP) is a complex heterogeneous disease which presents as an inflammatory outgrowth of sinonasal tissue.1–3 In Western populations, CRSwNP is often characterized by Type-2 (T2) inflammation with elevated levels of IL-13 and eosinophilia in the polyp tissue.1,4 T2 inflammation is considered critical to the development and maintenance of nasal polyps (NP), as recent T2-targeted therapies (both anti-IL-5 and anti-IL-4Ra) have decreased polyp size and, in some cases, improved clinical symptoms.5–10 Thus, understanding the pathways by which inflammatory cells, including eosinophils, accumulate in nasal polyp tissue could lead to improved therapies.
Eosinophil trafficking from the circulation into tissue is generally believed to be controlled by activation of IL-4Rα pathways.11 Engagement of IL-4Rα is well known to activate JAK/STAT pathways.12 However, IL-4/13 also activates MEK-ERK pathways, through increased 15-lipoxygenase 1 (15LO1) expression.13 Increases in 15LO1 initiates a binding interaction with phosphatidylethanolamine binding proteins-1 (PEBP1), also known as Raf kinase inhibitory protein. Under low 15LO1 conditions, PEBP1 preferentially binds Raf-1, inhibiting activation of ERK.14–16 Increases in 15LO1 displace Raf-1 from PEBP1 to activate ERK, and generate oxidized phospholipids.13
IL-4Rα activation increases expression of eosinophilic chemokines, including the eotaxin family (CCL11, 24 and 26).17–19 Previous reports consistently showed increased expression of CCL24 and CCL26 in NP tissues compared to healthy controls (HCs), while less consistent increases in CCL11.20–23 15LO1 has been reported to contribute to CCL26 expression in human lower airway epithelial cells (LAECs).13 In the upper airways, a recent RNA sequencing analysis of NPs reported 15LO1 as one of the top differentiating genes compared to HC tissues.24 Thus, we hypothesized that increases in 15LO1 in NPs would increase expression of CCL26 through MAP kinase/ERK related pathways, thereby contributing to the eosinophilia associated with nasal polyps. To address this hypothesis, fresh middle turbinate (MT) and NP epithelial cells from CRSwNP subjects and inferior turbinate (IT) and MT epithelial cells from HCs were analyzed for expression of 15LO1, CCL24, CCL26 and phosphorylated and total ERK. Human nasal epithelial cells (NECs) were also cultured in air-liquid interface (ALI) under IL-13 and control conditions. 15LO1 and CCL26 expression were measured and the role of 15LO1 and ERK activation to CCL26 expression determined by specific knockdown and inhibition of 15LO1 and ERK pathways. The results demonstrate that 15LO1 upregulation in NP epithelial cells potentially promotes CCL26 production and secretion through ERK pathway activation in patients with CRSwNP.
Methods
Study participants.
Subjects with CRSwNP were enrolled from Otolaryngology Clinics at UPMC Mercy Hospital. HCs were recruited as part of the Severe Asthma Research Program (SARP)-SPLUNC1 ancillary study, or from both the Otolaryngology Clinics at UPMC Mercy Hospital and patients undergoing skull based neurosurgery (typically for benign adenomas) at UPMC Presbyterian Hospital. All studies were approved by the University of Pittsburgh Institutional Review Board, and all participants provided informed consent. Tissue specimens included those obtained from the MT and NP at the time of surgery, as well as from epithelial scrapings of the IT. All subjects with CRSwNP met the clinical criteria for CRSwNP, as defined by the American Academy of Otolaryngology–Head and Neck Surgery Chronic Rhinosinusitis Task Force.25,26 Subjects with established immunodeficiency, coagulation disorders, fungal rhinosinusitis, or cystic fibrosis were excluded from the study. The presence of sinusitis or NP was confirmed by means of office endoscopy and maxillofacial CT scan. HCs did not have any history of inflammatory upper airway diseases.
Primary NECs Air-Liquid Interface (ALI) culture and siRNA transfection.
NECs were collected by epithelial scrapings of the IT, MT and NP. Primary NECs were cultured in ALI and DsiRNA transfection (using Lipofectamine RNAiMAX) performed when appropriate, as previously described.27 For details, see the online supplement.
15LO1 enzymatic inhibition with BLX2477.
IL-13 treated NECs in ALI were treated with 2 μM (diluted in DMSO) of a specific 15LO1 inhibitor (BLX2477, gift of HE Claesson, Karolinska, Stockholm, Sweden) for 3 days prior to harvest.28 DMSO was used as a control. For longer exposure times, the culture medium with BLX2477 was changed every 24 hours.27
Exogenous 15-HETE-PE stimulation.
Human airway epithelial cells (HAECs) were treated with 10 μM 15-HETE-PE (HPLC/MS collection dissolved in methanol) for 5 days. DMPE (Dimyristoyl-phosphoethanolamine) dissolved in methanol was applied as vehicle control.
Western Blotting (WB).
Cell lysates were run on 4–12% gradient SDS-PAGE gels under reducing conditions as previously described.29 Western blot was performed using primary antibodies against 15LO1 (1: 1000 dilution, Gift of Dr. Doug Conrad, University of California, San Diego, USA), CCL26 (1:100 dilution, R&D, USA), phospho-ERK (pERK, 1: 5000 dilution, Sigma, USA), total-ERK (tERK, 1: 5000 dilution, Sigma, USA), and GAPDH (1: 1000 dilution, Novus, USA). Densitometry analysis was performed using Image J Software. The densitometry of pERK was presented as a ratio over tERK and the densitometry of 15LO1 and CCL26 were presented as a ratio over GAPDH. For details, see the online supplement.
Real-time PCR.
qRT-PCR was performed using a TaqMan method and Applied Biosystems 7900 Sequence Detection System. GUSB was used for internal control. Relative mRNA expression levels were calculated using the ΔCt method.30 For details, see the online supplement.
Immunohistochemistry staining (IHC).
Tissue slides were deparaffinized and incubated with hyaluronidase for antigen retrieval. Slides were blocked with 5% donkey serum and incubated with primary antibodies of 15LO1 (1: 500 dilution). Following overnight incubation at 4 degrees, HRP-conjugated secondary antibody was added for 1 hour at room temperature, the slides developed with DAB counterstained with hematoxylin and imaged with an Olympus Provis Fluorescence microscope.31,32 For details, see the online supplement.
Immunofluorescent staining (IF) and confocal microscopy.
Tissue sections were blocked with 5% donkey serum and incubated with primary antibodies against 15LO1 (1: 200 dilution), CCL26 (1:100 dilution), and Cytokeratin-5 (CK5, 1: 200 dilution, Novus, USA). Cells were then incubated with Alexa 488 and Cy3 conjugated secondary antibodies, counterstained with DAPI and imaged using a Nikon A1R confocal microscope.33 For details, see the online supplement.
CCL26 ELISA.
CCL26 protein levels were measured using quantitative sandwich ELISA (R&D, Minneapolis, USA) according to the manufacture instructions.34
Statistical Analysis.
Statistical analysis was performed using JMP software (SAS Institute, Cary, NC). Data were analyzed for distribution. Normally distributed data were represented as means ± SEM, while non-normally distributed were represented as medians with 25–75% interquartile range (Ex vivo 15LO1 mRNA data, CCL24 mRNA data, CCL26 mRNA and protein data, pERK and tERK protein data). When multiple groups were compared, appropriate parametric or non-parametric ANOVA were utilized with Bonferroni or Mann-Whitney U testing applied to determine intergroup differences in the presence of an overall significant difference. Pearson Chi-square tests were used for categorical variables. Paired Student t tests were used when comparisons were made between paired conditions. Correlation analysis (rho) were performed by Spearman rank correlation. P values of less than 0.05 were considered statistically significant.
Results
Demographics (Table 1).
Table 1.
Clinical characteristics of subjects
| All Subjects | HC(IT)1 | HC(MT) | CRSwNP without Asthma |
CRSwNP with Asthma |
P value |
|---|---|---|---|---|---|
| N | 13 | 5 | 17 | 18 | |
| Age (mean±SD) | 41.9±12.7 | 57.5±12.3 | 51.6±18.0 | 56.6±11.7 | 0.043 |
| Sex (Male/Female) | 4/9 | 3/2 | 12/5 | 5/13 | 0.016 |
| Race (C/AA/Other)2 | 12/1/0 | 5/0/0 | 15/1/1 | 16/2/0 | 0.825 |
| Asthma Severity (1/2/3/4)3 | NA | NA | NA | 4/3/4/7 | NA |
| AERD(Y/N)4 | NA | NA | 0/17 | 6/12 | 0.009 |
| CS (Y/N)5 | NA | NA | 13/4 | 15/3 | 0.612 |
HC: health control; IT: inferior turbinate; MT: middle turbinate; CRSwNP: chronic rhinosinusitis with nasal polyps;
C=Caucasian; AA=African American
Asthma Severity: 0 = No asthma; 1 = Mild; 2 = Moderate; 3 = Severe; 4 = Not specified
AERD: aspirin-exacerbated respiratory disease
CS: perioperative corticosteroids
Fresh NECs were obtained from a total of 18 HCs and 35 CRSwNP subjects. Of the 35 CRSwNP, 18 had concomitant asthma and 6 reported concomitant aspirin-exacerbated respiratory disease (AERD). Of the 18 HCs, IT NECs were available from 13, while MT NECs were available from 5. Both MT and NP tissues were available from 13 of the 35 CRSwNP patients. Due to the limited cell numbers, not all cells were used for all experiments (specific “n” given for each experiment, see online supplement, Figure S1).
15LO1 expression is increased in NECs from subjects with CRSwNP and associated with CCL26 expression.
To determine whether 15LO1 is differentially expressed in NECs from IT, MT and NP, 15LO1 mRNA and protein expression were analyzed in NECs from these 3 anatomic locations across subjects with CRSwNP and HCs (MT and IT only). 15LO1 mRNA was increased in NP NECs compared to MT NECs from CRSwNP subjects and HCs, without difference between non-NP MT NECs from HCs vs CRSwNP (Figure 1A). 15LO1 protein was significantly higher in NP NECs from subjects with CRSwNP compared with HCs, as well as from MT NECs from CRSwNP patients (Figure 1B and 1C), including those with paired NECs (Figure 1D and 1E). There was no statistical difference in 15LO1 protein between MT cells from CRSwNP subjects and HCs (Figure 1C). To determine whether AERD has effect on 15LO1 expression, 15LO1 mRNA and protein were analyzed in NECs from CRSwNP with or without AERD subjects. There was no statistical difference of 15LO1 mRNA and protein in CRSwNP with or without AERD groups (Figure S2A and S2B in the Online Repository). To determine the localization of 15LO1 in turbinate and polyp tissues, 15LO1 IHC staining was performed. Overall 15LO1 positive cells were increased in MT and NP epithelial cells from subjects with CRSwNP, but particularly in NP epithelial cells, compared with MT from HCs. Although 15LO1 was mainly expressed in epithelial cells, it was also expressed in submucosal eosinophils and macrophages. (Figure 1F).
Figure 1.

15LO1 mRNA and protein are higher in fresh NECs of CRSwNP subjects compared to HCs. Total RNA and protein from freshly brushed NECs were harvested for RT-PCR and Western blot analysis. NP and MT tissues from subjects with CRSwNP and HCs were fixed for Immunohistochemistry studies. (A) 15LO1 mRNA is higher in NECs of NP from CRSwNP subjects compared to HC. (B) 15LO1 protein is higher in NECs of NP from CRSwNP compared to NECs of MT from HCs. (C) Densitometry analysis of 15LO1 protein from CRSwNP subjects and HCs by WB. (D) 15LO1 protein is higher in NECs of NP compared to MT from same CRSwNP subjects. (E) Densitometry analysis of the paired MT and NP 15LO1 protein. (F) Representative immunostaining photomicrograph showed 15LO1 positive staining is upregulated in NECs from CRSwNP subjects than MT of CRSwNP and HCs. Abbreviations: NECs=nasal epithelial cells, HC=healthy controls, NP=nasal polyps, MT=middle turbinate, IT=inferior turbinate, and CRSwNP=chronic rhinosinusitis with nasal polyps.
To determine whether 15LO1 expression was related to CCL24 and CCL26 expression in fresh NECs. CCL24 and CCL26 mRNA expression was measured in fresh IT, MT and NP NECs by qRT-PCR. CCL24 and CCL26 mRNA were increased in NECs from NP compared with MT tissues from HCs (Figure 2A and Figure S3A in the Online Repository). 15LO1 mRNA correlated with CCL26 mRNA (r=0.53, p=0.024; Figure 2B), but not with CCL24 (r=−0.14, NS; Figure S3B in the Online Repository). CCL26 protein in fresh NECs lysates (by ELISA) was increased in NP and MT NECs of CRSwNP compared with HCs (Figure 2C). CCL26 protein in IT or MT NECs was undetectable in HCs. Among the CRSwNP subjects, NEC lysate CCL26 protein (by ELISA) correlated with 15LO1 (by WB) (r=0.67, p=0.007; Figure 2D). To determine whether AERD has effect on CCL26 expression, CCL26 mRNA and protein were analyzed in NECs from CRSwNP with or without AERD subjects. There was no statistical difference of CCL26 mRNA and protein in CRSwNP with or without AERD groups (Figure S2C and S2D in the Online Repository).
Figure 2.

CCL26 mRNA and protein are higher in fresh NECs from CRSwNP compared to HCs and correlated with 15LO1 expression. Total RNA and protein from freshly brushed NECs were harvested for RT-PCR and ELISA analysis. (A) CCL26 mRNA is higher in NECs of NP from CRSwNP subjects compared to HCs. (B) CCL26 mRNA expression correlated with 15LO1 mRNA ex vivo. (C) CCL26 protein (by ELISA) is higher in NECs of MT and NP cell lysates from CRSwNP subjects, especially NP, compared to HCs. (D) CCL26 protein (by ELISA) expression correlates with 15LO1 protein expression (by WB) in NP cell lysates. Abbreviations: NECs=nasal epithelial cells, HC=healthy controls, NP=nasal polyps, MT=middle turbinate, and CRSwNP=chronic rhinosinusitis with nasal polyps.
15LO1 and CCL26 protein are colocalized in MT and NP tissues from CRSwNP ex vivo.
Similar to the immunohistochemistry staining, immunofluorescent staining of NP showed high 15LO1 and CCL26 expression in NP epithelial cells of CRSwNP patients, with lesser staining in MT NECs (Figure 3A), and minimal/no staining in MT tissue of HCs (Figure 3B). While 15LO1 was distributed widely throughout the epithelial layers of the nasal polyps, CCL26 was located primarily in basal NECs (Figure 3A). Confocal analysis confirmed colocalization of 15LO1 and CCL26, but primarily in basal NP NECs. The quantitation analysis result showed that 15LO1/CCL26 colocalization numbers increased in NPs and MT epithelial cells of CRSwNP subjects compared to HCs, particularly in NPs epithelial cells (Figure 3C).
Figure 3.

Immunostaining results showed that 15LO1/CCL26 and CCL26/cytokeratin-5 positive cell numbers increased in MT and NP tissues from CRSwNP subjects, especially in NP tissues, compared to HCs. NP and MT tissues from subjects with CRSwNP and HCs were fixed for IF analysis. (A) Representative immunostaining photomicrograph showed CCL26 positive cells are expressed in MT and NP epithelial cells of CRSwNP subjects. CCL26 colocalized with 15LO1 in MT (n=6) and NP (n=6) epithelial cells of CRSwNP. (B) Representative immunostaining photomicrograph showed no positive 15LO1/CCL26 double staining in MT (n=5) tissues of HCs. (C) Quantitation of 15LO1/CCL26 positive signals in NP and MT epithelial cells from CRSwNP and HCs. (D) Representative immunostaining photomicrograph showed CCL26 colocalized with cytokeratin-5 in MT (n=3) and NP (n=3) epithelial cells from CRSwNP subjects. (E) Quantitation of CCL26/CK5 positive signals in NP and MT tissues from CRSwNP and HCs (n=3). Abbreviations: HC=healthy controls, NP=nasal polyps, MT=middle turbinate, and CRSwNP=chronic rhinosinusitis with nasal polyps.
To confirm basal cells as the source of CCL26 in MT and NP tissues from CRSwNP subjects, co-immunostaining for CCL26 with the basal cell marker cytokeratin-5 (CK5) was performed. CK5 positive cells were increased in MT and NP tissues of CRSwNP with MT of HCs (n=3). Confocal analysis confirmed colocalization of CCL26 and CK5 in MT and NP tissues of CRSwNP subjects (Figure 3D). The number of cells colocalized for CCL26 and CK5 was higher in NP and MT epithelial cells of CRSwNP subjects compared to HCs (Figure 3E).
IL-13 induces both 15LO1 and CCL26 expression In vitro.
To determine whether NECs respond to IL-13 in a similar manner as lower airway epithelial cells, ALI cultured NECs from HCs (3 IT and 2 MT) and NP (n=8) were treated with IL-13 (10 ng/ml) for 1 to 5 days. 15LO1 and CCL26 mRNA increased on Day 1 and were generally sustained for 5 days (Figure S4A and S4B in the Online Repository). IL-13 time dependently increased 15LO1 and CCL26 protein in cell lysates (Figure S4C–E in the online Repository). CCL26 in lower chamber supernatant also increased on Day1 and was sustained for 5 days (Figure S4F in the Online Repository). Day 5 was chosen for comparisons. 15LO1 and CCL26 mRNA both increased (Figure 4A and 4B) and strongly correlated (r=0.72, p=0.003; Figure 4C). To confirm these relationships, 15LO1 and CCL26 protein expression (both by WB) were also highly increased by IL-13 (Figure 4D) and strongly correlated (r=0.60, p=0.012; Figure 4E).
Figure 4.

IL-13 induces 15LO1 and CCL26 expression in ALI cultured NECs and 15LO1 positively correlates with CCL26 expression in vitro. ALI cultured primary NECs were stimulated with IL-13 for 5 days. Cells were harvested for RT-PCR and Western blot analysis. Lower supernatants were harvested for CCL26 levels by ELISA. IL-13 increased (A) 15LO1 mRNA expression, (B) CCL26 mRNA expression and (C) 15LO1 mRNA correlates with CCL26 mRNA expression in vitro. (D) IL-13 induces intracellular 15LO1 and CCL26 protein in cell lysates. (E) IL-13 induced 15LO1 protein correlates with CCL26 protein in vitro. Abbreviations: ALI=air-liquid interface, NECs=nasal epithelial cells.
ALOX15 (15LO1) DsiRNA knockdown and inhibition decrease IL-13 induced CCL26 expression, while exogenous 15-HETE-PE increases it.
To determine whether 15LO1 regulated the production and secretion of CCL26 in NECs, 15LO1 silencing was performed with Dicer siRNA transfection in ALI cultured NECs from HCs (2 IT and 1 MT) and CRSwNP subjects (3 NP) in the absence and presence of IL-13. 15LO1 knockdown (KD) decreased 15LO1 protein expression varied between 69–91% (median 78%; Figure S5 in the Online Repository). 15LO1 KD decreased IL-13 induced CCL26 mRNA (Figure 5A), intracellular (Figure 5B and 5D) and supernatant protein (Figure 5C).
Figure 5.

ALOX15 DsiRNA and 15LO1 specific inhibitor BLX2477 decreased IL-13 induced CCL26 expression. ALI cultured NECs were treated with ALOX15 DsiRNA for 5 days in the presence or absence of IL-13. ALI cultured NECs were treated with BLX2477 for 3 days in the presence of IL-13. Cells were harvested for RT-PCR and Western blot. Lower supernatants were harvested for CCL26 protein by ELISA. 15LO1 knockdown decreases (A) intracellular CCL26 mRNA and (B) intracellular CCL26 protein and (C) secreted CCL26 protein in lower supernatants. (D) Densitometry analysis of intracellular CCL26 protein in cell lysates. (E) The 15LO1 inhibitor decreases IL-13 induced CCL26 mRNA, (F) and CCL26 protein in cell lysates and (G) secreted CCL26 protein in lower supernatants. (H) Densitometry analysis of CCL26 protein in cell lysates. Abbreviations: ALI=air-liquid interface, NECs=nasal epithelial cells.
To confirm these results, IL-13 stimulated NECs [3 HC (IT) and 3 NP subjects] were also treated with the selective 15LO1 inhibitor BLX2477 (2 M) or control (DMSO) for 3 days in the presence of IL-13. BLX2477 significantly inhibited IL-13 induced CCL26 mRNA (Figure 5E), as well as intracellular (Figure 5F and 5H) and secreted (lower chamber) CCL26 protein (Figure 5G). Finally, Exogenous 15-HETE-PE, the stable oxidized phospholipid product of 15-lipoxygenase 1, induced CCL26 expression in HAECs (Figure S6 in the Online Repository).
Responses to IL-13, ALOX15 DsiRNA or 15LO1 inhibition in vitro did not differ by subject group (HCs vs CRSwNP) or cell source (IT vs MT vs NP).
15LO1 driven ERK activation regulates CCL26 expression.
Although IL-13 induced 15LO1 expression increases ERK activation and CCL26 expression, it is unclear whether specific ERK activation drives CCL26 expression. DsiRNA 15LO1 KD and 15LO1 inhibition (BLX2477) of IL-13 treated NECs decreased ERK1/2 phosphorylation, while total ERK1/2 did not change (Figure 6A–C).
Figure 6.

ALOX15 DsiRNA and 15LO1 inhibitor BLX2477 decreased pERK expression. Blocking ERK pathway inhibits CCL26 production in ALI cultured NECs. (A) ALOX15 DsiRNA decreased pERK but not tERK by WB. (B) The 15LO1 specific inhibitor decreased pERK but not tERK by WB. (C) ALOX15 DsiRNA and 15LO1 specific inhibitor BLX2477 decreased pERK/tERK ratio. (D) ERK inhibitor PD98059 decreased pERK and CCL26 protein by WB. (E) ERK inhibitor U0126 decreased pERK and CCL26 protein by WB. (F) ERK pathway inhibitors PD98059 and U0126 decreased CCL26 mRNA in ALI cultured NECs. (G) The ERK pathway inhibitor PD98059 and U0126 decreased secreted CCL26 in supernatants. Abbreviations: ALI=air-liquid interface, NECs=nasal epithelial cells, WB=western blot.
To determine whether this 15LO1 regulated ERK activation directly impacted CCL26 expression, ALI cultured primary NECs from HCs (3 IT) and CRSwNP subjects (6 NP) were treated with the ERK inhibitors, PD98059 and U0126 for 3 days in the presence of IL-13. Both inhibitors decreased ERK1/2 phosphorylation (Figure 6D and 6E), without affecting total-ERK1/2. ERK inhibition reduced both intracellular and secreted CCL26 mRNA (Figure 6F) and protein (Figure 6G), confirming that 15LO1 induced ERK phosphorylation regulates CCL26 expression.
Confirmation of in vitro results in ex vivo NECs.
Phosphorylation of ERK1/2 was compared between fresh NECs from HCs and CRSwNP subjects. pERK/tERK ratio was increased in NP NECs from CRSwNP subjects (Figure 7A and 7B) and pERK/tERK ratio correlated with 15LO1 by WB (r=0.63, p<0.001, Figure 7C). Supporting overall relationships, CCL26 (by ELISA) and pERK/tERK ratio (by WB) in NP and MT NECs from CRSwNP subjects also correlated (r=0.52, p=0.024, Figure 7D). pERK/tERK ratio was also analyzed in NPs and MT epithelial cells from CRSwNP subjects and pERK/tERK ratio is higher in NPs compared to MT NECs (Figure S7 in the Online Repository).
Figure 7.

Increased pERK correlates with 15LO1 and CCL26 in fresh NECs. (A) Phosphorylated ERK is increased in fresh NECs of NP compared to HCs. (B) Densitometry analysis of pERK/tERK in fresh NECs of CRSwNP and HCs. (C) pERK/tERK ratio correlates with 15LO1 protein in fresh NECs of CRSwNP and HCs. (D) pERK/tERK ratio correlates with CCL26 protein (by ELISA) in fresh NECs of CRSwNP. Abbreviations: NECs=nasal epithelial cells, HC=healthy controls, NP=nasal polyps, MT=middle turbinate, and CRSwNP=chronic rhinosinusitis with nasal polyps.
Discussion
15LO1, known to be upregulated in asthma and associated with Type-2 inflammation,31,35 is crucial for goblet cell differentiation and epithelial cell remodeling of relevance to asthma.27 More recently it has been identified as an enzyme essential for programmed cell death pathways, specifically ferroptosis, when bound to PEBP1.33 In this study, we expand the disease implications of 15LO1 to CRSwNP, an abnormality of the paranasal sinuses associated with epithelial cell dysfunction, mucus secretion, which commonly overlaps with comorbid asthma and T2/eosinophilic inflammation. The current data demonstrate 15LO1 is strongly upregulated in fresh NECs from CRSwNP patients, compared with HCs, and that it correlates with CCL26 expression. In vitro studies confirm that 15LO1 promotes expression and release of the eosinophilic chemokine CCL26 in NECs, which strongly relates to activation of MEK/ERK pathways. This increase in CCL26 ex vivo was seen primarily in basal cells of the nasal epithelium, a cell type believed to be dysregulated in nasal polyps,24 where the overall expression correlated with both 15LO1 and pERK. These results support a potential critical role for 15LO1 in CCL26 expression and subsequent tissue eosinophilia of relevance to the pathogenesis of CRSwNP.
CRSwNP is generally recognized as associated with Type-2 inflammatory responses.1–3,36 However, data supporting the association of T2 inflammation to 15LO1 in CRSwNP are more limited, with a previous study reported increased 15LO mRNA expression in all CRS patients with or without NP as compared with HCs.37 In the current study, we showed that 15LO1 mRNA and protein were significantly increased in NECs of both MT and NP from CRSwNP subjects as compared with HCs, with higher levels in NP. A recent single cell RNA sequencing study of NP showed that in non-polyp tissues 15LO1 was expressed mainly in ciliated cells, with less expression in apical, basal and glandular cells. In polyp cells, 15LO1 gene expression was seen at persistently high levels in ciliated cells but was also increased in basal, apical and glandular cells.24 These results are in line with our previous study of lower airway epithelial cells, and consistent with the current protein analysis, which shows 15LO1 expression in all subsets of NECs in MT and NP tissues of CRSwNP patients, although consistently in higher amount in NP cells.31
We previously reported that 15LO1 regulates IL-13 induced goblet cell differentiation and expression of the T2 “signature gene”, periostin.13,27 CCL26 is widely considered another T2 signature gene, primarily expressed by human airway epithelial cells.38 However, the specific epithelial cell type of origin for either periostin or CCL26 is unclear. CCL26 protein was reported to be increased in NP tissue lysates compared to control, but not in epithelial cell lysates, with the actual cell of origin undetermined.39,40 As noted earlier, the recent single cell RNA sequencing study reported ALOX15 to be one of the most differentially-expressed genes in nasal polyps, especially in basal cells.24 In that study, CCL26 mRNA expression was also primarily in basal cells with a significant increase in polyp compared to non-polyp basal cells.24 At the protein level, and in contrast to the RNA data, CCL26 was previously reported throughout the epithelial layer.41 In the present study, both CCL26 mRNA and protein were upregulated in NECs lysates of MT and NPs from CRSwNP subjects. However, consistent with the mRNA data, in ex vivo samples, CCL26 was largely colocalized with CK5, confirming their mRNA association at the protein level. Further, basal cells were enriched in NP, compared to MT tissue. While 15LO1 colocalized with CCL26 in some cells, 15LO1 was distributed more widely in a variety of NP (and MT) cell types. Despite this, there were strong correlations between all 3, supporting the ability of 15LO1 to induce CCL26 in vitro, and perhaps promote an increase or maintenance of the basal cell population as reported in NP.42–44
In contrast to the differential expression of 15LO1 and CCL26 in fresh NECs from fresh NP and MT cells, and as compared to HCs, these different cell types did not respond differently in vitro to stimulation with IL-13. These cell cultures have not yet been evaluated for the percentages of basal vs goblet vs ciliated cells, distributions which are likely to differ over time and culture conditions. Additional studies will determine whether cell culture conditions can be adjusted to model the conditions which limit basal cell differentiation to mature epithelial cells mimicking conditions suggested to occur in NP in vivo.24
We previously reported that IL-13-induced 15LO1 can catalyze either free or phosphatidylethanolamine (PE) conjugated arachidonic acid (AA) to 15-hydroperoxyeicosatetraenoic acid (15-HpETE) or 15-HpETE-PE. When 15LO1 binds to PEBP1, the enzyme preferentially oxidizes PE conjugated AA over free AA.13,33 Under IL-13/T2 conditions, high levels of 15LO1 displace the endogenous ERK inhibitor Raf-1 to allow ERK activation with 15LO1 KD decreasing IL-13 induced ERK pathway activation.13 However, release of Raf-1 from PEBP1 is not the only mechanism by which ERK pathways can be activated by 15LO1. Exogenous addition of PE conjugated 15 hydroxyeicosatetraenoic acid (15-HETE-PE), which is present in abundance when 15-HpETE-PE is metabolized by glutathione peroxidase (GPX)4 in HAECs, also activates ERK and, as reported in this study, subsequent CCL26 expression, even in the absence of 15LO1.13 The mechanism for this direct activation of ERK by the 15LO1 product is not known and could still be through release of Raf-1. Thus, it remains unclear whether release of Raf-1 or the increased generation of 15-HETE-PE (and perhaps 15 HpETE-PE) are driving increases in ERK activation. Finally, different mechanisms could be occurring in different cell types, perhaps explaining the different effects.
We did not address the relationship of CCL26 expression to polyp eosinophil numbers. However, a previous study reported IL-13 and CCL26 expression were correlated with each other and with NP tissue eosinophilia.40 Interestingly, in lower airway epithelial cells, CCL26 protein is not measurable in fresh cell lysates, and the relation of CCL26 mRNA to sputum eosinophils is modest.38 Of substantial difference, CCL26 protein was readily detectable in fresh NP and MT epithelial cells.38 The reasons for these differences are unknown, although CCL26 is degraded by the mast cell enzyme tryptase, found in abundance in asthmatic lower airway epithelial cell brushings.45 This may imply fewer mast cells in polyp epithelial layers, although this requires further study.
Finally a very recent genome-wide association study of patients with NPs across several cohorts consistently identified a single nucleotide polymorphism (SNP) in the ALOX15 gene associated with >65% reduction in the risk of nasal polyps.46 This coding SNP causes a loss of function amino acid change in the 15LO1 protein rendering it inactive. rs34210653 was associated with the highest odds ratios for the relation to nasal polyps of any gene identified. Intriguingly, potential SNPs in the 15 HpETE-PE metabolizing gene, GPX4, were also identified among the genes associated with both eosinophilia and NPs.
Limitations of this study include the small sample size. However, MT cells from the same patients allowed us to use a matched pair approach, increasing the robustness of the comparisons. We also chose to focus specifically on epithelial cells and did not evaluate whole NP tissues as done in similar studies. While this enhanced our ability to detect specific epithelial changes, it did not allow us to study the other cell types (including eosinophils of interest to the relationship to both 15LO1 and CCL26 expression). We also did not include NECs from patients with chronic rhinosinusitis without nasal polyps (CRSsNP) in this study. Compared with nasal polyps, these tissues are characterized by less Type-2 inflammation and possibly increased Type-1 inflammation, situations where 15LO1 is likely to contribute less.40
In conclusion, this study identifies high levels of 15LO1 in NECs of nasal polyps, associated with activation of ERK and increased CCL26 expression. These studies add mechanistic insights to the identification of 15LO1 as a key differentiating factor both at the tissue mRNA and most recently at the SNP level in NPs.46 Together these studies strongly support 15LO1 as a potential therapeutic target with the potential to inhibit eosinophil recruitment, mucus generation and potentially epithelial remodeling in patients with CRSwNP.
Supplementary Material
Key Messages:
15LO1 is highly expressed in nasal polyp epithelial cells from CRSwNP subjects compared to HCs and in nasal polyps, correlates with CCL26 expression.
IL-13 induced 15LO1 promotes intracellular and secreted CCL26 expression through ERK pathway activation.
Acknowledgments:
This work was supported by National Institute of Health (NIH) (HL069174, HL064937, AI40600 and P01 AI106684), National Heart, Lung and Blood Institute (NHLBI) (U10 HL109152, and HL125128), generous donation from Dellenback Family, National Natural Science Foundation of China (81870700, 81502339), Shanghai Science and Technology Committee grant (16ZR1426100) and Shanghai Jiaotong University medical school grant (BXJ201637).
Disclosure of potential conflict of interest: ZP. Li, M. Z, YH. Deng, JM. Zhao, XX. Zhou, J. B. Trudeau and S. E. Wenzel’s institution has received funding from the National Institute of Health (NIH), National Heart, Lung and Blood Institute (NHLBI) and donation from Dellenback Family. HW. Chu and Peter. Di’s institution has received funding from the NIH and NHLBI. ZP. Li, M. Z, WT. Zhang, SK. Yin and Z. Liu’s institution has received funding from National Natural Science Foundation of China. ZP. Li and WT. Zhang’s institution has received funding from Shanghai Science and Technology Committee. The rest of the authors declare that they have no relevant conflicts of interest.
Abbreviations:
- 15LO1
15-Lipoxygenase 1
- NP
nasal polyps
- CRSwNP
chronic rhinosinusitis with nasal polyps
- CRSsNP
chronic rhinosinusitis without nasal polyps
- NECs
nasal epithelial cells
- HCs
healthy controls
- MT
middle turbinate
- IT
inferior turbinate
- CCL11
C-C motif chemokine ligand 11
- CCL24
C-C motif chemokine ligand 24
- CCL26
C-C motif chemokine ligand 26
- CK5
cytokeratin 5
- ERK
extracellular-signal-regulated kinases
- ALI
air-liquid interface
- siRNA
short interfering RNA
- ALOX15
gene name for 15LO1
- IHC
immunohistochemistry
- IF
immunofluorescence
- SEM
standard error of the mean
- PEBP1
phosphatidyl ethanolamine binding protein 1
- 15-HpETE
15-hydroperoxyeicosaetetranoic acid
- 15-HETE
15 hydroxyeicosatetraenoic acid
- PE
phosphatidylethanolamine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schleimer RP. Immunopathogenesis of Chronic Rhinosinusitis and Nasal Polyposis. Annu Rev Pathol Mech Dis. 2017;12:331–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stevens WW, Schleimer RP, Kern RC. Chronic Rhinosinusitis with Nasal Polyps. J Allergy Clin Immunol Pract. 2016;4:565–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hulse KE, Stevens WW, Tan BK, Schleimer RP. Pathogenesis of nasal polyposis. Clin Exp Allergy. 2015;45:328–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Bruaene N, Pérez-Novo CA, Basinski TM, Van Zele T, Holtappels G, De Ruyck N, et al. T-cell regulation in chronic paranasal sinus disease. J Allergy Clin Immunol. 2008;121:16–8. [DOI] [PubMed] [Google Scholar]
- 5.Kolbeck R, Kozhich A, Koike M, Peng L, Andersson CK, Damschroder MM, et al. MEDI-563, a humanized anti-IL-5 receptor α mAb with enhanced antibody-dependent cell-mediated cytotoxicity function. J Allergy Clin Immunol. 2010;125:1344–53. [DOI] [PubMed] [Google Scholar]
- 6.Gevaert P, Lang-Loidolt D, Lackner A, Stammberger H, Staudinger H, Van Zele T, et al. Nasal IL-5 levels determine the response to anti-IL-5 treatment in patients with nasal polyps. J Allergy Clin Immunol. 2006;118:1133–41. [DOI] [PubMed] [Google Scholar]
- 7.Bachert C, Zhang N, Holtappels G, De Lobel L, Van Cauwenberge P, Liu S, et al. Presence of IL-5 protein and IgE antibodies to staphylococcal enterotoxins in nasal polyps is associated with comorbid asthma. J Allergy Clin Immunol. 2010;126:962–68. [DOI] [PubMed] [Google Scholar]
- 8.Bachert C, Mannent L, Naclerio RM, Mullol J, Ferguson BJ, Gevaert P, et al. Effect of subcutaneous dupilumab on nasal polyp burden in patients with chronic sinusitis and nasal polyposis: A randomized clinical trial. JAMA - J Am Med Assoc. 2016;315:469–79. [DOI] [PubMed] [Google Scholar]
- 9.Bachert C, Sousa AR, Lund VJ, Scadding GK, Gevaert P, Gilbert J, et al. Reduced need for surgery in severe nasal polyposis with mepolizumab : Randomized trial. J Allergy Clin Immunol. 140:1024–1031.e14. [DOI] [PubMed] [Google Scholar]
- 10.Gevaert P, Bruaene N Van, Cattaert T, Steen K Van. Mepolizumab , a humanized anti – IL-5 mAb , as a treatment option for severe nasal polyposis. J Allergy Clin Immunol. 128:989–995.e8. [DOI] [PubMed] [Google Scholar]
- 11.Spencer LA, Melo RCN, Perez SAC, Bafford SP, Dvorak AM, Weller PF. Cytokine receptor-mediated trafficking of preformed IL-4 in eosinophils identifies an innate immune mechanism of cytokine secretion. Proc Natl Acad Sci. 2006;103:3333–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wenzel SE, Trudeau JB, Barnes S, Zhou X, Cundall M, Westcott JY, et al. TGF- and IL-13 Synergistically Increase Eotaxin-1 Production in Human Airway Fibroblasts. J Immunol. 2002;169:4613–9. [DOI] [PubMed] [Google Scholar]
- 13.Zhao J, O’Donnell VB, Balzar S, St. Croix CM, Trudeau JB, Wenzel SE. 15-Lipoxygenase 1 interacts with phosphatidylethanolamine-binding protein to regulate MAPK signaling in human airway epithelial cells. Proc Natl Acad Sci. 2011;108:14246–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keller ET, Fu Z, Brennan M. The role of Raf kinase inhibitor protein (RKIP) in health and disease. In: Biochemical Pharmacology. 2004. p. 1049–53. [DOI] [PubMed] [Google Scholar]
- 15.Trakul N, Rosner MR. Modulation of the MAP kinase signaling cascade by Raf kinase inhibitory protein. Cell Research. 2005. p. 19–23. [DOI] [PubMed] [Google Scholar]
- 16.Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, et al. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature. 1999;401:173–7. [DOI] [PubMed] [Google Scholar]
- 17.Abonyo BO, Alexander MS, Heiman AS. Autoregulation of CCL26 synthesis and secretion in A549 cells: a possible mechanism by which alveolar epithelial cells modulate airway inflammation. Am J Physiol Cell Mol Physiol. 2005;289:L478–88. [DOI] [PubMed] [Google Scholar]
- 18.Sun Q, Yang X, Zhong B, Jiao F, Li C, Li D, et al. Upregulated Protein Arginine Methyltransferase 1 by IL-4 Increases Eotaxin-1 Expression in Airway Epithelial Cells and Participates in Antigen-Induced Pulmonary Inflammation in Rats. J Immunol. 2012;188:3506–12. [DOI] [PubMed] [Google Scholar]
- 19.van Wetering S, Zuyderduyn S, Ninaber DK, van Sterkenburg MAJA, Rabe KF, Hiemstra PS. Epithelial differentiation is a determinant in the production of eotaxin-2 and −3 by bronchial epithelial cells in response to IL-4 and IL-13. Mol Immunol. 2007;44:803–11. [DOI] [PubMed] [Google Scholar]
- 20.Olze H, Förster U, Zuberbier T, Morawietz L, Luger EO. Eosinophilic nasal polyps are a rich source of eotaxin, eotaxin-2 and eotaxin-3. Rhinology. 2006;44:145–50. [PubMed] [Google Scholar]
- 21.Cho DY, Nayak JV., Bravo DT, Le W, Nguyen A, Edward JA, et al. Expression of dual oxidases and secreted cytokines in chronic rhinosinusitis. Int Forum Allergy Rhinol. 2013;3:376–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bartels J, Maune S, Meyer JE, Kulke R, Schlüter C, Röwert J, et al. Increased eotaxin-mRNA expression in non-atopic and atopic nasal polyps: Comparison to RANTES and MCP-3 expression. Rhinology. 1997;35:171–4. [PubMed] [Google Scholar]
- 23.Van Zele T, Claeys S, Gevaert P, Van Maele G, Holtappels G, Van Cauwenberge P, et al. Differentiation of chronic sinus diseases by measurement of inflammatory mediators. Allergy Eur J Allergy Clin Immunol. 2006;61:1280–9. [DOI] [PubMed] [Google Scholar]
- 24.Ordovas-Montanes J, Dwyer DF, Nyquist SK, Buchheit KM, Vukovic M, Deb C, et al. Allergic inflammatory memory in human respiratory epithelial progenitor cells. Nature. 2018;560:649–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meltzer EO, Hamilos DL, Hadley JA, Lanza DC, Marple BF, Nicklas RA, et al. Rhinosinusitis: Establishing definitions for clinical research and patient care. J Allergy Clin Immunol. 2004;114:155–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pearlman AN, Conley DB. Review of current guidelines related to the diagnosis and treatment of rhinosinusitis. Vol. 16, Current Opinion in Otolaryngology and Head and Neck Surgery. 2008. p. 226–30. [DOI] [PubMed] [Google Scholar]
- 27.Zhao J, Minami Y, Etling E, Coleman JM, Lauder SN, Tyrrell V, et al. Preferential generation of 15-HETE-PE induced by IL-13 regulates goblet cell differentiation in human airway epithelial cells. Am J Respir Cell Mol Biol. 2017;57:692–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pelcman B, Sanin A, Nilsson P, No K, Schaal W, Öhrman S, et al. 3-Substituted pyrazoles and 4-substituted triazoles as inhibitors of human 15-lipoxygenase-1. Bioorganic Med Chem Lett. 2015;25:3024–9. [DOI] [PubMed] [Google Scholar]
- 29.Albano GD, Zhao J, Etling EB, Park SY, Hu H, Trudeau JB, et al. IL-13 desensitizes β2-adrenergic receptors in human airway epithelial cells through a 15-lipoxygenase/G protein receptor kinase 2 mechanism. J Allergy Clin Immunol. 2015;135:1144–1153.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xie M, Mustovich AT, Jiang Y, Trudeau JB, Ray A, Ray P, et al. IL-27 and type 2 immunity in asthmatic patients: Association with severity, CXCL9, and signal transducer and activator of transcription signaling. J Allergy Clin Immunol. 2015;135:386–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chu HW, Balzar S, Westcott JY, Trudeau JB, Sun Y, Conrad DJ, et al. Expression and activation of 15-lipoxygenase pathway in severe asthma: Relationship to eosinophilic phenotype and collagen deposition. Clin Exp Allergy. 2002;32:1558–65. [DOI] [PubMed] [Google Scholar]
- 32.Li Z, Wang H, Liu L. Interleukin-25 enhances allergic inflammation through p38MAPK and NF-κB Pathways in mouse models of allergic rhinitis. Iran J Allergy, Asthma Immunol. 2014;13:412–9. [PubMed] [Google Scholar]
- 33.Wenzel SE, Tyurina YY, Zhao J, Croix CM St, Dar HH, Mao G, et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell. 2017;171:628–641.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trudeau J, Hu H, Chibana K, Chu HW, Westcott JY, Wenzel SE. Selective downregulation of prostaglandin E 2-related pathways by the T H2 cytokine IL-13. J Allergy Clin Immunol. 2006;117:1446–54. [DOI] [PubMed] [Google Scholar]
- 35.Zhao J, Maskrey B, Balzar S, Chibana K, Mustovich A, Hu H, et al. Interleukin-13-induced MUC5AC is regulated by 15-lipoxygenase 1 pathway in human bronchial epithelial cells. Am J Respir Crit Care Med. 2009;179:782–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bachert C, Zhang L, Gevaert P. Current and future treatment options for adult chronic rhinosinusitis: Focus on nasal polyposis. J Allergy Clin Immunol. 2015;136:1431–40. [DOI] [PubMed] [Google Scholar]
- 37.Pérez-Novo CA, Watelet JB, Claeys C, Van Cauwenberge P, Bachert C. Prostaglandin, leukotriene, and lipoxin balance in chronic rhinosinusitis with and without nasal polyposis. J Allergy Clin Immunol. 2005;115:1189–96. [DOI] [PubMed] [Google Scholar]
- 38.Coleman JM, Naik C, Holguin F, Ray A, Ray P, Trudeau JB, et al. Epithelial eotaxin-2 and eotaxin-3 expression: Relation to asthma severity, luminal eosinophilia and age at onset. Thorax. 2012;67:1061–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yao T, Kojima Y, Koyanagi A, Yokoi H, Saito T, Kawano K, et al. Eotaxin-1, −2, and −3 immunoreactivity and protein concentration in the nasal polyps of eosinophilic chronic rhinosinusitis patients. Laryngoscope. 2009;119:1053–9. [DOI] [PubMed] [Google Scholar]
- 40.Min JY, Ocampo CJ, Stevens WW, Price CPE, Thompson CF, Homma T, et al. Proton pump inhibitors decrease eotaxin-3/CCL26 expression in patients with chronic rhinosinusitis with nasal polyps: Possible role of the nongastric H,K-ATPase. J Allergy Clin Immunol. 2017;139:130–141.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tian P, Ou H, Wu F, Ma Y, Liu X, Chen Q, et al. Interleukin-4 induced posttranscriptional gene regulation of CCL26 by the RNA-binding protein HuR in primary human nasal polyp-derived epithelial cells. Int Forum Allergy Rhinol. 2018;00:1–11. [DOI] [PubMed] [Google Scholar]
- 42.Yu XM, Li CW, Chao SS, Li YY, Yan Y, Zhao XN, et al. Reduced growth and proliferation dynamics of nasal epithelial stem/progenitor cells in nasal polyps in vitro. Sci Rep. 2014;4:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li YY, Li CW, Chao SS, Yu FG, Yu XM, Liu J, et al. Impairment of cilia architecture and ciliogenesis in hyperplastic nasal epithelium from nasal polyps. J Allergy Clin Immunol. 2014;134:1282–92. [DOI] [PubMed] [Google Scholar]
- 44.Li CW, Shi L, Zhang KK, Li TY, Lin Z Bin, Lim MK, et al. Role of p63/p73 in epithelial remodeling and their response to steroid treatment in nasal polyposis. J Allergy Clin Immunol. 2011;127:765–72. [DOI] [PubMed] [Google Scholar]
- 45.Balzar S, Fajt ML, Comhair SAA, Erzurum SC, Bleecker E, Busse WW, et al. Mast cell phenotype, location, and activation in severe asthma: Data from the Severe Asthma Research Program. Am J Respir Crit Care Med. 2011;183:299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kristjansson RP, Benonisdottir S, Davidsson OB, Oddsson A, Tragante V, Sigurdsson JK, et al. A loss-of-function variant in ALOX15 protects against nasal polyps and chronic rhinosinusitis. Nat Genet. 2019;51:267–76. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.