

Summary
MYOD-directed fibroblast trans-differentiation into skeletal muscle provides a unique model to investigate how one transcription factor (TF) reconfigures the three-dimensional chromatin architecture to control gene expression, which is otherwise achieved by the combinatorial activities of multiple TFs. Integrative analysis of genome-wide high-resolution chromatin interactions, MYOD and CTCF DNA-binding profile, and gene expression, revealed that MYOD directs extensive re-wiring of interactions involving cis-regulatory and structural genomic elements, including promoters, enhancers and insulated neighborhoods (INs). Re-configured INs were hot-spots of differential interactions, whereby MYOD binding to highly constrained sequences at IN boundaries and/or inside INs leads to alterations of promoter-enhancer interactions to repress cell-of-origin genes and to activate muscle-specific genes. Functional evidence shows that MYOD-directed re-configuration of chromatin interactions temporally preceded the effect on gene expression and was mediated by direct MYOD-DNA binding. These data illustrate a model whereby a single TF alters multi-loop hubs to drive somatic cell trans-differentiation.
Graphical Abstract
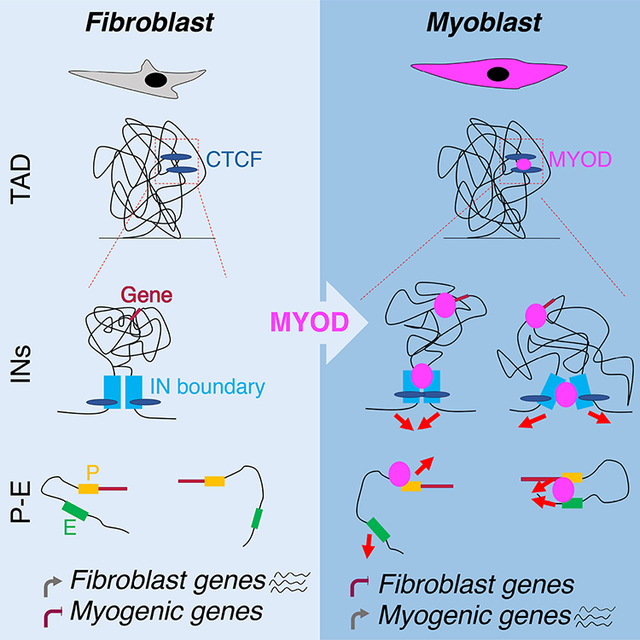
Dall’Agnese et al. finds that the myogenic master transcription factor MYOD drives significant re-wiring of the 3D chromatin architecture during somatic reprogramming toward transdifferentiation, in order to erase the cell-of-origin transcriptional program and activate skeletal myogenesis. MYOD-directed reconfiguration of chromatin interactions involve involving cis-regulatory and structural genomic elements and temporally precede transcriptional regulation of target genes.
Introduction
Growing evidence indicates the importance of the three-dimensional (3D) genome organization for the spatiotemporal regulation of gene expression (Andrey and Mundlos, 2017; Bonev and Cavalli, 2016; Bonev et al., 2017; Franke et al., 2016; Guerreiro et al., 2016; Hnisz et al., 2016a; Kragesteen et al., 2018; Lupianez et al., 2015; Noordermeer et al., 2014; Noordermeer et al., 2011; Ong and Corces, 2014; Palstra et al., 2003; Remeseiro et al., 2016; Rodriguez-Carballo et al., 2017; Schauer et al., 2017; Spielmann et al., 2018; Symmons et al., 2016). The genome is folded into a hierarchy of chromatin domains (Dekker and Mirny, 2016; Dixon et al., 2012; Dowen et al., 2014; Hnisz et al., 2016a; Nora et al., 2012; Phillips-Cremins et al., 2013; Rao et al., 2014; Sexton et al., 2012), which facilitate and constrain interactions between regulatory elements and genes. Among these chromatin domains, topologically associating domains (TADs) and insulated neighborhoods (INs) are structural units that are largely conserved across cell types (Beagan et al., 2016; Bonev et al., 2017; Chandra et al., 2015; Dixon et al., 2015; Ji et al., 2016; Jin et al., 2013; Krijger et al., 2016; Siersbaek et al., 2017). TADs consist of genomic regions that interact more frequently within the domain than with regions outside, and are separated by boundaries across which chromatin interactions are relatively scarce (Dixon et al., 2012; Nora et al., 2012). SubTADs within individual TADs might further provide hierarchical and nested topological organization (Phillips-Cremins et al., 2013; Schmitt et al., 2016b). TADs and subTADs are generally composed of and/or contain INs (Hnisz et al., 2016a), which are regions of the DNA that contain one or more genes and whose boundaries are co-bound by CTCF and cohesin and interact with each other (Dowen et al., 2014; Flavahan et al., 2016; Hnisz et al., 2016b; Ji et al., 2016; Narendra et al., 2015). INs constrain gene regulation within their boundaries, by harboring interactions between cis-regulatory elements, such as promoter-enhancer communication (Sun et al., 2019).
While higher genomic structures appear to be generally conserved, chromatin interactions within TADs, subTADs and INs, could rather be cell-type specific and dynamic (Bonev et al., 2017; Dixon et al., 2015; Hnisz et al., 2013; Hnisz et al., 2016a; Javierre et al., 2016; Ji et al., 2016; Phanstiel et al., 2017; Remeseiro et al., 2016; Siersbaek et al., 2017). Importantly, the role of transcription factors (TFs), and in particular cell type-specific “master” TFs (mTFs), in regulating these interactions at the genome-wide level has not been directly addressed yet. Previous studies have been mostly based on correlative analysis of 3D chromatin reorganization with DNA sequence motifs for multiple ubiquitous TFs (Phanstiel et al., 2017); DNA binding of neural (Bonev et al., 2017) or pluripotency TFs (Stadhouders et al., 2018); cMYC expression (Kieffer-Kwon et al., 2017). Furthermore, the ubiquitous TF YY1 has been shown to contribute to the formation of enhancer/promoter loops (Weintraub et al., 2017). Other studies investigated promoter-related chromatin loops formed by tissue-specific TFs at individual gene level (de Wit et al., 2013; Krijger et al., 2016). However, the causative role of a single tissue-specific TF in directing genome-wide rewiring of 3D chromatin organization during lineage commitment and differentiation has yet to be determined.
Somatic cell reprogramming into another somatic cell type (trans-differentiation) or toward pluripotency (induced pluripotency) by ectopic expression of TFs provides an experimental model to address the role of mTFs in re-wiring chromatin interactions to regulate gene expression during establishment of cell identity, stemness, lineage commitment and terminal differentiation. However, nuclear somatic cell reprograming almost invariably requires the combinatorial overexpression and activity of multiple mTFs (Caiazzo et al., 2011; Chronis et al., 2017; Ieda et al., 2010; Pang et al., 2011; Pfisterer et al., 2011; Qian et al., 2012; Schaub et al., 2018; Stadhouders et al., 2018; Takahashi et al., 2007; Tsunemoto et al., 2018; Vierbuchen et al., 2010; Wada et al., 2013), which complicates the interpretation of the relative contribution of each individual mTF to this process. A notable exception is provided by somatic cell trans-differentiation into skeletal muscle cells through the ectopic expression of one single mTF, MYOD, which is sufficient to reprogram virtually all somatic cells into skeletal muscles (Davis et al., 1987; Weintraub et al., 1989). MYOD-mediated trans-differentiation also permits the study of two separate and sequential stages of trans-differentiation: lineage commitment and terminal differentiation. Several distinctive features of MYOD, even among other myogenic bHLH factors (Conerly et al., 2016; Gerber et al., 1997), predict that MYOD possesses unique properties that enable epigenetic and transcriptional events necessary to coordinate repression of cell-of-origin gene expression and transcription of new lineage-specific genes, a complicated task that is otherwise carried out by the concerted action of multiple mTFs (Sartorelli and Puri, 2018). As such, MYOD-mediated somatic cell trans-differentiation into skeletal muscles provides a unique experimental system to investigate whether and how one single TF can re-wire 3D chromatin architecture to orchestrate activation and repression of gene expression during lineage commitment and terminal differentiation.
Genome-wide analysis of MYOD DNA binding revealed a pervasive binding through the genome; however, only a small percentage of MYOD binding sites are associated with regional gene expression (i.e. binding to proximal promoters of target genes) (Cao et al., 2010; Fong et al., 2012). Thus, the function of most MYOD binding sites remains unknown. Previous works reporting on MYOD interactions with architectural proteins, such as CTCF (Delgado-Olguin et al. 2011), and on MYOD-regulated chromatin interactions (Harada et al. 2015; Battistelli et al., 2014; Busanello et al., 2012), suggest that MYOD could regulate gene expression also by altering the 3D genome architecture.
Results
MYOD-driven myogenic conversion of primary human fibroblasts
To investigate the impact of MYOD on 3D chromatin architecture during skeletal myogenesis, we exploited the model of MYOD-directed reprogramming of fibroblasts into skeletal muscle (Davis et al., 1987; Weintraub et al., 1989). To this purpose, we introduced a tetracycline-inducible Myod1 transgene (MYOD) or vector control (EMPTY) in human primary IMR90 fibroblasts (Figure 1A). Upon doxycycline treatment in growth media (GM) for 24hrs, ~95% of cells transfected with Myod1 expressed Myod1, but not the skeletal muscle differentiation marker myosin heavy chain (MyHC) (Figure 1B and Figures S1A–C). At this stage, MYOD-expressing cells continued to proliferate (Figure S1D), indicating that Myod1 expression levels were compatible with proliferation and therefore the progenitor state. Following 72hrs in differentiation medium (DM), most MYOD-expressing cells (over 90%) differentiated into MyHC-expressing multinucleated myotubes (Figure 1B, Figures S1A–C). We further validated MYOD-mediated transdifferentiation of IMR90 at the trascriptome level by RNA-seq in two biological replicates. We identified 1,446 or 2,772 differentially expressed (DE) genes (see Methods) during MYOD-mediated commitment (MYOD GM vs EMPTY GM) or differentiation (MYOD DM vs MYOD GM), respectively. Gene ontology (GO) and upstream regulator prediction analysis of the DE genes showed that MYOD committed IMR90 fibroblasts toward the skeletal muscle lineage in GM by activating myogenic transcriptional networks and repressing pro-fibrotic and pro-inflammatory transcriptional networks (Figure S1E and Figure 1C) that are typically active in fibroblasts and antagonize skeletal myogenesis (Gerber et al., 1997; Liu et al., 2001; Loell and Lundberg, 2011; Puri and Sartorelli, 2000). The exposure to DM was required for the activation of the gene expression program of terminal muscle differentiation (Figure S1E). Activation or repression of these transcriptional networks was also observed when we compared RNA-seq data between EMPTY GM and primary human myotubes (hMTs, data from ENCODE), revealing that ~40% of the DE genes between MYOD DM and EMPTY GM were in common with the DE genes between hMTs and EMPTY GM (Figure S1F). Activation or repression of these transcriptional networks was validated by RT-qPCR (Figure S1G). Taken together, these results show that this system is highly suitable for investigating MYOD activity during myogenic commitment and differentiation.
Figure 1: MYOD regulation of gene expression in the linear sequence of the DNA. See alsoFigure S1.

A, Experimental design. IMR90 fibroblasts containing doxycycline-inducible MYOD or EMPTY vector were treated with doxycycline (dox) for 24hrs in growth media (GM) prior differentiation stimuli (DM) with doxycycline for 72hrs. Experiments were always performed at these time points, unless otherwise stated.
B, Representative immunofluorescence of IMR90 cells stained for MYOD (magenta) and MyHC antibody (green). Nuclei were stained with Hoechst (blue).
C, Transcriptional networks predicted to be altered by MYOD comparing MYOD GM vs EMPTY GM. For all predictions, p<0.001.
D, Percentage of MYOD peaks at promoters of differentially expressed (DE) genes (yellow) or not (grey).
E, Observed/expected ratio of MYOD binding at the genomic regions listed in the y axes, as described in Chronis et al, 2017 (see Methods).
The repression of the original transcriptional program is a feature shared with fibroblast reprogramming to induced-pluripotent stem cells (iPSCs). Analysis of mouse embryonic fibroblasts (MEFs) and MEF-derived iPSCs using available gene expression data (Chronis et al., 2017) revealed that OCT4, SOX2 and NANOG inhibited the same transcriptional networks repressed upon MYOD expression in IMR90 fibroblasts (Figure S1H). These results indicate that master TFs share the ability to coordinately activate and repress specific transcriptional programs during reprogramming, as previously suggested (Ciglar et al., 2014) and that MYOD can integrate multiple activities that are otherwise accounted for by the combinatorial activity of multiple TFs.
A small fraction of MYOD binding sites are associated with local transcription regulation
To study whether MYOD regulates gene expression by direct DNA binding, we performed ChIP-seq for MYOD in IMR90-derived myoblasts (MYOD GM) and myotubes (MYOD DM). We found that MYOD pervasively bound the genome (~50,000 and ~80,000 MYOD binding sites in MYOD GM and DM, respectively), with a large preference for the prototypical E-box motif CA(G/C)GTG (Figure S1I), as previously reported (Cao et al., 2010). Two examples are reported in Figure S1J. By integrating MYOD ChIP-Seq and RNA-Seq analyses, we found that only ~5% of MYOD binding sites were located at promoters of DE genes, both during myogenic commitment (GM) and differentiation (DM) (Figure 1D), in agreement with previous studies (Cao et al., 2010; Fong et al., 2012). Since only a small fraction of MYOD binding sites are associated to local transcription regulation, we investigated MYOD DNA binding distribution to cis-regulatory elements and insulators using publicly available H3K27ac and CTCF ChIP-seq datasets in IMR90, human myoblasts (hMBs) and myotubes (hMTs) (Consortium, 2012; Jin et al., 2013; Yue et al., 2014). We found enrichment of MYOD binding i) at promoters of DE genes (Figure 1E, see Methods), with no preference for promoters of up or down-regulated genes (Figure S1K), ii) at distal enhancers, and iii) at CTCF-binding sites in IMR90, hMBs and hMTs during both MYOD-mediated commitment (MYOD GM vs EMPTY GM) and differentiation (MYOD DM vs MYOD GM) (Figure 1E). These results suggest that MYOD might regulate transcription by binding distal regulatory and/or structural genomic elements.
MYOD DNA binding correlates with significant alterations in chromatin interactions
The enrichment of MYOD binding at cis-regulatory elements and at DNA elements bound by CTCF, an architectural protein implicated in chromatin looping (Hnisz et al., 2016a; Nora et al., 2017; Ong and Corces, 2014; Tsui et al., 2016), prompted us to investigate whether MYOD could regulate transcription by re-organizing interactions between functional and/or structural genomic elements.
To study MYOD-mediated changes in chromatin structure during myogenic conversion, we conducted in situ Hi-C (Rao et al., 2014) in two biological replicates in EMPTY GM, MYOD GM and MYOD DM. We collectively detected 2.6 billion unique pairwise contacts (each map contained on average ~470M unique pairwise contacts). Our Hi-C libraries were of high quality (Table S1, see Methods) and were highly reproducible (Figure S2A–B, see Methods).
The genome is compartmentalized into TADs (Figure 2A) that we identified using Armatus (Filippova et al., 2014), and we called TAD boundaries as in Crane et al, 2015 (Crane et al., 2015) from Hi-C matrices binned at 40kb resolution, i.e., by partitioning the genome in 40kb bins. Although we detected MYOD DNA binding both inside TADs and at TAD boundaries, boundary location did not significantly differ during MYOD-mediated fibroblast conversion into myoblasts or myotubes (Figure 2B), since the percentage of overlap of TAD boundaries between samples (~90%) was similar to the percentage of overlap of TAD boundaries between biological replicates (~90%). This result is consistent with previous observations showing TAD conservation across cell types (Dixon et al., 2015; Schmitt et al., 2016a; Siersbaek et al., 2017). Interestingly, we observed a general pattern of co-regulation of genes within MYOD-bound TADs (Figure 2C), suggesting that MYOD could alter chromatin interactions between promoters, enhancers and insulators within TADs.
Figure 2: Profound alteration of chromatin contacts by MYOD during myogenic conversion. See alsoFigures S2 andS3.

A, Graphical representation of TADs and TAD boundaries.
B, Hi-C interaction pattern (red heat map) and TAD boundaries (light blue) in EMPTY GM, MYOD GM and MYOD DM, MYOD ChIP-seq in MYOD GM and MYOD DM (black).
C, Number of TADs with one or more DE genes. Black represents the TADs whose differentially expressed genes are all upregulated or all downregulated; gray represents the TADs containing upregulated genes and downregulated genes. LEFT: gene expression comparison between EMPTY GM and MYOD GM. TADs were identified in MYOD GM. RIGHT: gene expression comparison between MYOD GM and MYOD DM. TADs were identified in MYOD DM. pvalue represents the significant prevalence of TADs with two or more differentially expressed genes that were either all upregulated or all downregulated compared to TADs that have both upregulated genes and downregulated genes. pvalue was calculated using the two-sided exact binomial test.
D, Percentage of 4kb bins involved in at least one differential interaction only during MYOD-mediated commitment (magenta), only during MYOD-mediated differentiation (green), or at both stages (violet).
E, Number (N) of bins involved in altered chromatin interactions during MYOD-mediated commitment (magenta) or differentiation (green) observed or expected to be bound by MYOD. Expected bin number was calculated based on the number of bins bound by MYOD genome-wide. Chi-squared test was used for statistical analysis.
F, Percentage of DI bins bound by MYOD (red), DI bins directly interacting with MYOD-bound bins (orange), DI bins indirectly interacting (dark yellow), others (yellow) – see figure S3A for details
G, Left: number (N) of differential interactions, including all differential interactions (All) Promoters-all (interactions between promoters and any other genomic region), Enhancers-all (interactions between enhancers and any other genomic region), Promoter-enhancers and CTCF-bound regions during MYOD-mediated commitment (magenta) or differentiation (green). Right: percentage of the differential interactions described on the left that were bound by MYOD during myogenic commitment (magenta) or differentiation (green) (right). Dashed lines represent the percentage of all differential interactions bound by MYOD during commitment (red) or differentiation (blue).
To test this hypothesis, we analyzed the in situ Hi-C maps at 4kb resolution and identified differential intra-chromosomal interactions between bin-pairs using diffHic (Lun and Smyth, 2015). Around 14% of the genome differentially interacted in cis during MYOD-mediated myogenic commitment (MYOD GM vs EMPTY GM) and/or differentiation (MYOD DM vs MYOD GM) (Figure 2D). Examples of differential chromatin interactions are shown in Figure S2C and one illustrative example was validated by DNA FISH (Figure S2D), using 100kb probes centered around the 4kb bins that differentially interacted with each other.
By integrating the differential interactome maps with MYOD ChIP-seq profile, we found that the number of bins with altered chromatin interactions and bound by MYOD was significantly higher than expected by chance (chi-squared test, p<2.2×10−16, circular permutations, p=0, see Methods) during myogenic commitment or differentiation (Figure 2E).
To determine the extent to which the differential chromatin interactions were orchestrated by MYOD, we considered altered interactions directly bound as well as indirect events potentially generated by MYOD DNA binding, as illustrated in Figure S3A. According to this model the initial chromatin alterations are conceivably caused by MYOD binding to the DNA (Bin2, “MYOD-dependent and MYOD-bound”). MYOD binding to the DNA could promote the interaction with another bin that may be bound by MYOD (“MYOD-dependent and MYOD-bound”) or not (Bin3, “MYOD-dependent and directly interacting”). MYOD binding to the DNA could also dis-engage previously interacting bins (i.e., Bin1 and Bin2) thereby generating free bins (i.e., Bin1) available for new interactions with other bins (Bin?), either bound by MYOD (“MYOD-dependent and MYOD-bound”) or not (Bin?, “MYOD-dependent and indirectly interacting”). Moreover, some altered chromatin interactions can form independently on the initial chain of differential interactions triggered by MYOD DNA binding (others). These differential interactions could be mediated by other TFs, whose expression might be also regulated by MYOD (Figure S3A, co-operating TF).
When we applied this analysis to our experimental system, we found that about 50% of the differentially interacting (DI) bins could be dependent on MYOD binding to the DNA (Figure 2F). We observed that ~12% and ~18% of DI bins during MYOD-mediated commitment and differentiation, respectively, were “MYOD-dependent and MYOD-bound” bins; ~14% of the DI bins during both MYOD-mediated commitment and differentiation were “MYOD-dependent and directly interacting” bins; over 22% of the DI bins during both MYOD-mediated commitment and differentiation were “MYOD-dependent and indirectly interacting” bins (Figure 2F). Interestingly, directly interacting bins were enriched in binding motifs for TFs that are typically found at MYOD-bound promoters and/or enhancers, and reported to facilitate MYOD DNA binding (i.e., Pbx) (Berkes et al., 2004) and potentiate MYOD activation of target genes (i.e., MEF2) (Black et al., 1998; Dodou et al., 2003) (Figure S3B). Finally, ~50% of the DI bins detected in IMR90 upon MYOD expression do not appear to derive from the “domino effect” (illustrated in Figure S3A) of differential interactions triggered by direct MYOD-DNA binding. However, we note that bins involved in these interactions, as well as bins involved in MYOD-dependent indirect interactions, were enriched in motifs for endogenous TFs that were up- or down-regulated as a consequence of direct MYOD binding at their promoter (Figure S3C). This is consistent with a model whereby changes in chromatin topology during IMR90 trans-differentiation derive from initial MYOD DNA binding, with MYOD-regulated expression of TFs adding an additional layer of complexity to further expand the extent of 3D chromatin re-configuration through a cooperative action of TFs.
To determine the identity of the regulatory elements involved in the differential interactions (DIs) bound by MYOD, we divided the DIs into six categories: 1) all DIs (All), 2) DIs involving promoters (Promoter-all), 3) DIs involving enhancers (Enhancer-all), 4) DIs between promoters and enhancers (Promoter-enhancer), 5) DIs bound by CTCF (CTCF), 6) DIs between bins co-bound by CTCF (CTCF-CTCF) (Figure 2G). Interestingly, the percentage of Promoter-all DIs, Enhancer-all DIs, Promoter-enhancer DIs, CTCF DIs and CTCF-CTCF DIs that was bound by MYOD was higher than the percentage of all DIs bound by MYOD (Figure 2G), suggesting that MYOD could re-wire chromatin architecture at promoter, enhancers and insulators during fibroblast trans-differentiation into skeletal muscle.
MYOD DNA binding at regions showing differential interactions at gene promoters
We observed a significant enrichment in MYOD binding at altered chromatin interactions involving promoters (chi-squared p<2.2×10–16, Figure 3A) and at promoter-enhancer pairs (chi-squared p<2.2×10–16, Figure 3B). Of note, differential chromatin interactions anchored at promoters were more frequently associated to DE genes as compared to genomic regions not bound by MYOD during MYOD-mediated commitment or differentiation (chi-squared p<2.2×10−16, Figure 3C–D), suggesting that MYOD re-wires chromatin interactions to regulate transcription.
Figure 3: Characterization of MYOD-altered cis-regulatory interactions.

A, Number (N) of MYOD-bound bins involved in altered interactions between promoters and other genomic elements during myogenic commitment (magenta) or differentiation (green). The expected number of MYOD-bound bins was calculated based on MYOD-binding genome-wide (see methods).
B, Number (N) of MYOD-bound bins involved in altered enhancer-promoter interactions during commitment (magenta) or differentiation (green). The expected number of MYOD-bound bins was calculated based on MYOD-binding to enhancer or promoter (see methods).
C,D, Percentage (%) of MYOD-bound or unbound (dashed) differential interactions between promoters of DE genes and (C) other genomic elements or (D) enhancers.
E, Heatmap representing biological functions (using IPA) activated (orange) or inhibited (blue) based on DE genes, whose promoters are involved in MYOD-bound differential interactions during commitment or differentiation.
F, DE genes, whose promoters are involved in MYOD-bound differential interactions with enhancers during commitment or differentiation. Analysis performed using IPA.
G, Normalized contact heatmap at TNNT2 locus in EMPTY GM (top left) or MYOD GM (bottom right). The region in blue box corresponds to TNNT2 enhancer-promoter interaction. Enlargement of the interaction of interest in the corners.
H, From top to bottom: Magenta bars represent bins whose interaction increased during MYOD-mediated commitment determined by Hi-C. UCSC snapshots of: H3K27ac ChIP-seq in IMR90 (blue), hMB (violet), P indicates the TNNT2 promoter, E represents an enhancer, MYOD ChIP-seq in MYOD GM (magenta), RefSeq genes, black bars represent regions with increased H3K27ac levels in hMBs compared to IMR90. Close up representation of the enhancer region in the dashed blue box H3K27ac ChIP-seq in hMB (violet), and DpnII sites. Relative crosslinking frequencies (RCF) by in situ 3C using as view point MYOD peak at TNNT2 promoter (red eye) (n=3).
I, Relative enrichment of H3K27ac by ChIP-qPCR at TNNT2 promoter and enhancer, n=3. Data is represented as mean +/− SEM.
J, Relative mRNA expression of TNNT2 (n=3).
In A–D chi-squared test was used for statistical analysis, *** p<2.2×10–16
In H–J data is represented as mean + SEM. T-test was used for statistical analysis, * p<0.05, ** p<0.01.
GO analysis on the DE genes whose promoters were involved in MYOD-mediated differential chromatin interactions revealed that MYOD-bound differential interactions involving promoters were associated with cell proliferation and muscle contractility in GM (Figure 3E), consistent with MYOD ability to stimulate proliferation of myoblasts (Latella et al., 2017), while in DM MYOD-bound differential promoter interactions were associated with cell cycle arrest and terminal muscle differentiation (Figure 3E). GO analysis on the DE genes whose promoter was involved in differential MYOD-bound interactions with enhancers revealed activation of muscle specific genes and inhibition of anti-myogenic signaling (e.g. TGF-β) (Figure 3F).
A representative example of enhancer-promoter interactions increased by MYOD is illustrated by the interaction between TNNT2 promoter and a pre-existing putative enhancer, marked by H3K27ac, whose target gene was not previously known (Figure 3G and 3H). Upon Myod1 expression, MYOD bound the TNNT2 promoter (Figure 3H), and this binding correlated with a local increase of H3K27ac (Figure 3I) and with increased interaction between TNNT2 promoter and a pre-existing enhancer, as determined by Hi-C and validated by 3C analyses (Figure 3G and 3H bottom panel). Importantly, these events coincided with the upregulation of TNNT2 transcription (Figure 3J).
These results establish a functional link between MYOD-directed re-wiring of chromatin interactions among cis-regulatory elements and dynamic regulation of gene expression that enables fibroblast conversion into skeletal muscle cells, through a stepwise model of somatic cell reprogramming.
MYOD DNA binding at re-configured insulated neighborhoods
Changes in IN strength regulate chromatin interactions and expression of genes within INs during cell differentiation (Bonev et al., 2017); however, the molecular effectors of these events remain poorly understood. Since MYOD is known to physically and functionally interact with CTCF (Battistelli et al., 2014; Delgado-Olguin et al., 2011) (see also Figure 1E and Figure 2G), we postulated that MYOD could alter the INs present in fibroblasts by targeting CTCF-organized IN boundaries.
We defined altered INs as regions of DNA that contained at least one gene and whose boundaries were i) co-bound by CTCF in IMR90 and ii) showed differential interaction strength during MYOD-mediated commitment or differentiation (Figure 4A). Each altered IN was considered as a separate entity, regardless its inclusion within larger altered INs or the presence of smaller altered INs inside it.
Figure 4: MYOD alters insulated neighborhoods to regulate myogenesis. See alsoFigures S4 andS5 andTables S2 andS3.

A, Graphical representation of altered IN: black line represents the DNA, light-blue boxes represent IN boundaries, blue ovals represent CTCF, violet line represent gene, zig-zagged red lines represent altered interaction.
B, Percentage (%) and number (N) of differential interactions corresponding to altered IN boundary interactions.
C, Percentage (%) and number (N) of DIs with at least one bin that mapped inside altered INs during myogenic commitment or differentiation.
D, Percentage of DI bins genome-wide (GW) and distribution of percentages of DI bins inside altered INs.
E, Number (N) of IN boundaries which differentially interacted during myogenic commitment (magenta) or differentiation (green) that were observed (Obs) or expected (Exp) to be bound by MYOD. Expected number of MYOD-bound IN boundaries was calculated based on MYOD binding at bins containing CTCF genome-wide (see Methods). For statistical analysis Chi-squared test was used.
F, MYOD ChIP-seq signal over CTCF-summit +/−5kb at IN boundaries which differentially interacted during myogenic commitment or differentiation.
G, MYOD binding distribution at altered INs. For statistical analysis Chi-squared test was used.
H, Boxplots of the gene expression changes EMPTY GM vs MYOD GM of DE genes (p<0.01) in DI INs with strengthen interaction between boundaries (All); among these, DI INs not bound by MYOD at the boundaries (noMYOD), at only one boundary (oneSide), and at both boundaries (bothSides).
I, NGSplot of CTCF, MYOD and H3K27ac signal ChIP-seq from IMR90 (CTCF, H3K27ac EMPTY GM) and myoblast (MYOD, H3K27ac_myoblast). 167 regions
J, IPA-based GO analysis of the DE genes within MYOD-bound altered INs.
K, Normalized contact heatmap for EMPTY GM (top left) and MYOD GM (bottom right) at ITGA7-RDH5 locus. Interaction under investigation is highlighted by blue boxes. Magnification of the blue boxes is shown in the corners.
L, From top to bottom: Magenta bars represent bins whose interaction increased during MYOD-mediated commitment. UCSC snapshots of: MYOD ChIP-seq in MYOD GM (magenta) and CTCF in IMR90 (blue), RefSeq genes from UCSC browser, DpnII sites (black). Close up representation of the region in the dashed red box. Relative crosslinking frequencies (RCF) by in situ 3C using as view point MYOD-CTCF peak at ITGA7 promoter (red eye) (n=3). 3C data is represented as mean + SEM. Relative mRNA expression of ITGA7 and RDH5 (n=3). Data is represented as mean +/− SEM. T-test was used for statistical analysis, * p<0.05, ** p<0.01, *** p<0.001.
We found that only ~2% (1,332 or 1,595) of altered interactions accounted for changes in interaction strength between IN boundaries during MYOD-mediated commitment or differentiation, respectively (Figure 4B). Interestingly, a large proportion (greater than 40%) of the altered genome-wide interactions, e.g. enhancer-promoter intreactions, involved DI bins within the altered INs and between bins located inside and outside altered INs (Figure 4C). The altered INs comprised a higher percentage of bins involved in differential interactions than expected by chance during both MYOD-mediated commitment and differentiation (Figure 4D). Thus, altered INs appear to be “hot-spots” of differential chromatin contacts during myogenic conversion of fibroblasts. This is consistent with the insulation effect of INs (Hnisz et al., 2016b; Lupianez et al., 2015; Nora et al., 2017; Sanborn et al., 2015; Schuijers et al., 2018; Sun et al., 2019).
By overlaying MYOD binding profile with the map of altered INs, we observed an enrichment of MYOD binding at IN boundaries whose interaction strength significantly changed during MYOD-mediated myogenic commitment or differentiation, as compared to the genome-wide binding of MYOD at CTCF-bound bins (chi-squared test p<2.2×10−16, Figure 4E, see Methods). Consistently, we detected an overlap between MYOD ChIP-seq signal and CTCF peak summits detected at changing IN boundaries (Figure 4F). Furthermore, MYOD-binding distribution at altered INs revealed that over 90% of altered INs during myogenic commitment or differentiation were bound by MYOD at the boundaries and/or inside INs (Figure 4G). These results suggest that MYOD alters IN strength and highly re-configures the chromatin interactions landscape at those INs.
We next set to analyze genetic determinants that could discriminate between MYOD-bound IN boundaries with increased or decreased interaction strength. DNA motif analysis indicated that MYOD-bound IN boundaries in both cases were enriched in CTCF- and MYOD-binding motifs, as expected, with no significant differences in nucleotide composition (Tables S2 and S3); however, while MYOD-bound IN boundaries with increased interaction strength were strongly enriched in AP1 (Jun/Fos dimers) motifs (Table S2), these motifs were notably absent in MYOD-bound IN boundaries with decreased interaction strength (Table S3). Conversely, MYOD-bound IN boundaries with decreased interaction strength were enriched in motifs for a collection of TFs that did not rank in the top 20 TF binding motifs found in MYOD-bound IN boundaries with increased interaction strength, with the notable exception of TCF 12 and 21, which encode potential bHLH heterodimerization partners of MYOD, and were common to both sets (Table S3). These results suggest that the presence of MYOD and other TFs at specidic loci may determine whether the interaction strength between IN boundaries is increased or decreased.
Given the transcriptional regulatory function of INs (Dowen et al., 2014; Flavahan et al., 2016; Hnisz et al., 2016b; Ji et al., 2016; Narendra et al., 2015; Sun et al., 2019) and our evidence of a significant clustering of differential interactions at altered INs (Figure 4D), we investigated whether a functional relationship exists between altered interaction strength of MYOD-bound IN boundaries and gene expression regulation within INs. Genes were considered within an IN when at least the promoter was inside the IN. We found that increased interaction strength of IN boundaries correlated with upregulation of genes within INs, especially when both IN boundaries were bound by MYOD compared to no MYOD binding or MYOD-binding at one IN boundary (Wilcoxon rank sum test p-value = 0.05 and 0.01, respectively, Figure 4H). Interestingly, these MYOD co-bound IN boundaries were also enriched in H3K27ac in the proximity of MYOD and CTCF binding sites, as compared to control IMR90 fibroblasts (Figure 4I). These results suggest that a functional relationship exists between MYOD binding, increased H3K27ac levels, transcription and increased IN boundary interactions.
GO analysis of the DE genes in INs altered by MYOD revealed inhibition of fibrosis (TGF-β1), activation of function of muscle and contractility of muscle (TNNT2, ACTC1) during MYOD-mediated commitment; activation of muscle differentiation and contractility during MYOD-mediated differentiation (Figures 4J).
An illustrative example of MYOD binding that correlated with increased IN boundary interaction and transcription upregulation is the ITGA7-RDH5 locus (Figures 4K–L). Upon ectopic expression, MYOD bound the promoter of ITGA7 and RDH5 at CTCF-bound elements in IMR90 (Figure 4L) and this coincided with increased interaction between the two CTCF-bound regions, as measured by Hi-C (Figures 4K–L) and validated by 3C (Figure 4L, bottom right). Importantly, these events correlated with an increased expression of both ITGA7 and RDH5 (Figure L, bottom left).
The TGF-β1 locus is an example of MYOD binding that correlated with multiple changes in chromatin interactions – e.g. decreased IN boundary interaction strength and disruption of interactions between regulatory elements – for transcriptional repression. TGF-β1 was downregulated by MYOD (Figure S1G and S4) and is contained within an IN whose boundaries were both bound by MYOD in GM and whose interaction intensity significantly decreased during MYOD-mediated commitment (Figure S4A–B). Furthermore, MYOD binding to TGF-β1 promoter coincided with increased interaction strength with a putative enhancer, whose H3K27ac levels were lower in hMB than in IMR90 (Figure S4B–C). Changes in interaction strength between IN boundaries as well as between the putative enhancer and TGF-β1 promoter were first observed by Hi-C and then validated by 3C (Figure S4A–C). TGF-β1 repression is therefore an example in which 3D chromatin reorganization occurs at multiple levels upon MYOD binding to IN boundaries as well as inside the IN that contains the TGF-β1 locus.
MYOD-bound differentially interacting elements are highly constraint and enriched in annotated pathogenic variants
To determine the biological significance of MYOD we performed genetic constraint analysis using context-dependent tolerance score (CDTS) (di Iulio et al., 2018), which is an estimate of sequence constraint and functional importance that is calculated as the absolute difference of the observed variation from the expected variation (di Iulio et al., 2018). This analysis revealed that differentially interacting bins (DI) were more constrained than the whole-genome (WG) (Figure S5A). Sequence constraint was even higher when considering DI bins either bound by CTCF (DI_CTCF) or MYOD (DI_MYOD) or co-bound by these two TFs (DI_MYOD_CTCF) (Figure S5A). Analysis of annotated pathogenic variants revealed that CTCF and/or MYOD-bound DI bins are enriched in single nucleotide variants associated with inflammatory and muscle diseases, with a notable preference for MYOD-bound DI bins (Figure S5B). Moreover, differentially interacting IN boundaries (Bd) were also significantly more constrained than CTCF-bound 4kb bins (WG_CTCF), DI bins (DI) and all bins (WG) (Figure S5C). The high constraint was further pronounced for differentially interacting IN boundaries bound by MYOD (Figure S5C). Of note, MYOD-anchored IN boundaries were enriched in annotated pathogenic variants, including inflammatory and skeletal muscle diseases (Figure S5D). The high level of constraint and the enrichment in disease-associated pathogenic variants observed at CTCF/MYOD-bound altered interactions indicate the biological relevance of MYOD-altered chromatin interactions.
These results also further emphasize the importance of studying the effects of mutations outside of the coding genome in altering the 3D chromatin architecture and interfering with transcriptional control as reviewed in (Spielmann and Mundlos, 2016).
MYOD expression is required for sustained MYOD re-wiring of chromatin interactions.
We investigated whether MYOD expression is required for the maintenance of MYOD effects on 3D chromatin interactions, once the myogenic commitment has been established. To address this question, we turned Myod1 expression on with doxycycline (ON) for 24hrs and then we decreased its expression by doxycycline withdrawal (OFF) for additional 48hrs, or maintained MYOD expression ON during the whole time (72hrs) (Figure 5A). Once verified the decreased expression of Myod1 after doxycycline withdrawal (Figure 5B–C), we investigated the effect of turning OFF Myod1 expression on MYOD-upregulated genes – TNNT2, ITGA7 and RDH5 – or repressed genes – TGF-β1.
Figure 5: MYOD expression is necessary for the maintenance of MYOD-regulated chromatin interactions. See alsoFigures S6 andS7.

A, Scheme of the experimental approach used for all experiment in Fig. 5, EMPTY or MYOD IMR90 were exposed to doxycycline for 24h in GM followed by additional 48h of with/out doxycycline (ON/ON, ON/OFF).
B, Relative mRNA expression of Myod1 compared to EMPTY ON/ON (n=3). Data is represented as mean +/− SEM.
C, Immunoblot analysis of the whole cell lysate. GAPDH is used as loading control
D, Relative mRNA expression of TNNT2 compared to EMPTY ON/ON (n=3). Data is represented as mean +/− SEM
E, ChIP-qPCR for MYOD at TNNT2 promoter relative to EMPTY ON/ON (n=3).
F, Relative crosslink frequency (RCF) values between MYOD peak at TNNT2 promoter (view point – red eye – see Fig. 3H) and the enhancer. Data is represented as mean + SEM (n=3).
G, Relative mRNA expression of ITGA7 and RDH5 compared to EMPTY ON/ON (n=3). Data is represented as mean +/− SEM
H, ChIPqPCR for CTCF and MYOD at regulatory elements in the locus relative to EMPTY ON/ON (n=3).
I, Relative crosslink frequency (RCF) values between CTCF MYOD peak at ITGA7 promoter (view point – red eye – see Fig. 4L) and the CTCF MYOD peak in RDH5. Data is represented as mean + SEM (n=3).
T-test was used for statistical analysis, * p<0.05, ** p<0.01, *** p<0.001
Upon MYOD induction in fibroblasts and its binding to TNNT2 promoter, we observed an increase in TNNT2 promoter-enhancer interaction together with an increase in TNNT2 expression (Figure 5D–F). Decreasing Myod1 expression at the commitment stage (GM) led to a reduction of MYOD binding to TNNT2 promoter, which coincided with a decrease in promoter-enhancer interaction strength to levels similar to those detected in control IMR90 fibroblasts, and a consensual reduction of TNNT2 expression (Figure 5D–F). We obtained similar results for ITGA7-RDH5 locus. Upon MYOD expression in fibroblasts, we observed increased ITGA7 and RDH5 expression, MYOD binding to CTCF-bound elements in ITGA7 and RDH5 promoters, increased CTCF binding at ITGA7 promoter and increased CTCF-CTCF interaction between ITGA7 and RDH5 promoters (Figure 5G–I). Decreasing Myod1 expression drastically reduced MYOD binding to CTCF-bound elements in ITGA7 and RDH5 promoters (Figure 5H), which was paralleled by reduction in the expression levels of ITGA7 and RDH5 (Figure 5G), decreased CTCF binding at ITGA7 promoter (Figure 5H), and reduced CTCF-CTCF interaction strength (Figure 5I). Finally, decreasing MYOD restored the original expression pattern of TGF-β1 (Figure S6A) and interactions (Figure S6B, C and E) as well as chromatin occupancy of MYOD and CTCF (Figure S6D and F) at the regulatory elements of TGF-β1 locus.
These results suggest that steady expression of MYOD is required for the maintenance of the 3D chromatin landscape at the stage of myogenic commitment. The reversible nature of MYOD-directed chromatin interactions to coordinate repression of fibrotic genes and activation of myogenic genes observed during lineage determination in our system could be implicated in the altered differentiation and gene expression observed in satellite cells upon acute loss of MYOD in vivo, as reported by Yamamoto et al (Yamamoto et al., 2018).
MYOD regulates chromatin interactions in mouse myoblasts.
To further validate the role of MYOD in regulating the 3D chromatin landscape within the physiological context of skeletal myogenesis, we analyzed, as a proof of concept, the Tnnt2 enhancer-promoter interaction in mouse C2C12 skeletal myoblasts. Upon siRNA-mediated silencing of Myod1 (Figure S7A), we observed a significant decrease in Myod1 and Tnnt2 expression (Figure S7B–C). We then used publicly available MYOD ChIP-seq dataset in C2C12 (Yue et al., 2014) and identified a MYOD peak in the murine Tnnt2 promoter (golden eye Figure S7D) that corresponds to the peak detected in MYOD-expressing IMR90 cells (shown in Figure 3H). By inspecting the sequence conservation between the human and murine genomes, we identified in myoblasts a DNA element that is conserved with the human TNNT2 enhancer region (shown in Figure 3H) (Figure S7D), suggesting that it could be a conserved Tnnt2 enhancer. Interestingly, we found that in myoblasts the MYOD-bound DNA element at Tnnt2 promoter interacted with the putative Tnnt2 enhancer by 3C; and the interaction strength between these two genomic regions dramatically decreased upon Myod1 silencing, (Figure S7D). These results extend to mouse skeletal muscle cells the notion that MYOD could regulate gene expression by re-organizing the 3D chromatin architecture.
MYOD rewires chromatin structure by direct DNA binding.
To investigate whether MYOD directly rewires chromatin interactions, and whether directly interplays with CTCF to alter INs, we employed a catalytically inactive Cas9 (dCas9) to block MYOD and/or CTCF binding at specific genomic loci. Briefly, we transfected IMR90 fibroblasts with dCas9-expressing vector and guide RNAs (gRNAs) that direct dCas9 to specific MYOD-bound DNA elements in the TNNT2 locus, to MYOD and CTCF co-bound DNA elements in ITGA7 locus or CTCF-bound elements in RDH5 locus (Figure 6A–C). We then monitored MYOD and CTCF DNA binding, gene expression and chromatin interaction changes after dCas9 blockade of MYOD and/or CTCF DNA binding at the target sites. When we targeted dCas9 to MYOD binding site at TNNT2 promoter, MYOD binding was decreased at TNNT2 promoter, but not at ITGA7-RDH5 locus (used as negative control) (Figure 6D), demonstrating the efficacy and specificity of this approach. We found that blocking MYOD binding at the TNNT2 promoter caused a decreased expression of TNNT2, while no effect on TNNT2 expression was observed by the same dCas9 in the absence of MYOD expression (EMPTY GM) (Figure 6E). MYOD-mediated TNNT2 promoter-enhancer interaction also decreased upon dCAS9-gRNA-mediated E-box targeting to TNNT2 promoter (Figure 6F). These results suggest a direct role of MYOD DNA binding in promoting TNNT2 promoter-enhancer interaction and TNNT2 expression.
Figure 6: Direct MYOD binding is required for MYOD-directed changes in the 3D chromatin structure.

A, Scheme of the experimental approach used for all experiments in Fig. 6, EMPTY or MYOD IMR90 were transfected with plasmid encoding dCAS9 and specific gRNAs 36hours, then cells were treated with doxycycline for 24h in GM.
B, From top to bottom: TNNT2 locus - magenta bars represent bins whose interaction increased during MYOD-mediated commitment, UCSC genome browser snapshots of MYOD ChIP-seq in MYOD GM (magenta), H3K27ac ChIP-seq in hMB (violet), RefSeq genes, DpnII sites (black). Orange arrow indicates the region targeted by the gRNAs, which is MYOD and CTCF peak at TNNT2 promoter (TNNT2_M).
C, From top to bottom: ITGA7-RDH5 locus - magenta bars represent bins with increased interaction between during MYOD-mediated commitment, UCSC genome browser snapshots of MYOD ChIP-seq in MYOD GM (magenta), CTCF ChIP-seq in IMR90 (blue), RefSeq genes, DpnII sites (black). Red arrow indicates gRNAs targeting the MYOD-CTCF in the ITGA7 promoter (ITGA7_CM), green arrow indicates gRNAs targeting the CTCF in the RDH5 promoter (RDH5_C).
D, ChIP-qPCR for MYOD (left) or CTCF (right) at regulatory elements in ITGA7, RDH5 or TNNT2 loci. Data is represented as relative enrichment over MYOD expressing IMR90 transfected with CTRL gRNAs (n=3) Data is represented as mean + SEM.
E, Relative mRNA expression of TNNT2. Data is represented as mean +/− SEM
F, Close up representation of the enhancer region in the dashed blue box H3K27ac ChIP-seq in hMB (violet), and DpnII sites. Relative crosslinking frequencies (RCF) by in situ 3C using as view point MYOD peak at TNNT2 promoter (red eye, see Fig 3H) (n=3). 3C data is represented as mean + SEM.
G, Relative mRNA expression of ITGA7 and RDH5. Data is represented as mean +/− SEM
H, Close up representation of the enhancer region in the dashed red box MYOD ChIP-seq in MYOD GM (magenta), CTCF ChIP-seq in IMR90 (blue) and DpnII sites. Relative crosslinking frequencies (RCF) by in situ 3C using as view point CTCF-MYOD peak at ITGA7 promoter (red eye, see Fig 4L) (n=3). 3C data is represented as mean + SEM. T-test was used for statistical analysis, * p<0.05, ** p<0.01, *** p<0.001.
We next investigated the direct interplay between MYOD and CTCF in mediating IN boundary interaction between ITGA7 and RDH5 promoters (Figure 6C). Interestingly, we found that dCas9-mediated blockade of MYOD and CTCF DNA binding at ITGA7 gene decreased MYOD and CTCF binding at both ITGA7 and RDH5 promoters (Figure 6D), but not at a distal gene TNNT2. Likewise, dCas9-mediated blockade of CTCF DNA binding at RDH5 gene resulted in decreased binding for MYOD and CTCF at both ITGA7 and RDH5 promoters, but not at a distal gene TNNT2 (Figure 6D). Blocking CTCF and MYOD binding at ITGA7 promoter or blocking CTCF binding at RDH5 promoter invariably decreased MYOD-mediated CTCF-CTCF interactions and decreased ITGA7 and RDH5 expression (Figure 6G–H). These results show that CTCF and MYOD cooperate in recruiting each other at specific DNA elements, directly altering chromatin interactions that spatially regulate tissue-specific gene expression.
The absolute requirement of MYOD-DNA binding for changes in chromatin interactions in the above loci indicates a direct role of MYOD in re-configuring 3D chromatin architecture.
Relationship between MYOD-mediated chromatin interactions and transcription
It has been previously shown that transcription can be implicated in the formation of chromatin interactions (Isoda et al., 2017). We therefore investigated the dependency of MYOD-driven chromatin interactions on transcription in our system. We first performed a time-course experiment, in which we monitored the expression of Myod1, TNNT2 and ITGA7 and MYOD-driven interactions. We found that Myod1 expression and MYOD-mediated interactions preceded TNNT2 and ITGA7 upregulation (Figure 7A–D). We detected chromatin interactions already 3hrs after Myod1 induction (Figure 7C–D), while upregulation of TNNT2 and ITGA7 became apparent after 12hrs (Figure 7B). These results suggest that chromatin interactions can be dissociated temporally from the transcriptional regulation of their target genes. To address whether the interactions depend on active transcription, 6 hours after inducing Myod1 expression we inhibited transcription with the Polymerase II inhibitor Actinomycin D (ActD) for 30min. ActD treatment reduced the levels of GAPDH nascent RNA, but not of GAPDH mRNA, as compared to DMSO control, thus confirming that ActD effectively blocked transcription (Figure 7E). We then investigated the effect of ActD on MYOD-mediated chromatin interactions. Interestingly, we found that while ActD prevented MYOD-dependent enhancer-promoter interaction at TNNT2 locus (Figure 7F), it did not affect the MYOD-promoted CTCF-CTCF chromatin interaction at ITGA7-RDH5 locus (Figure 7G). These results indicate that, at least in some instances, MYOD-mediated chromatin interactions occur independently on active transcription.
Figure 7: MYOD loop formation and transcription.

A, Time course analysis of Myod1 expression in doxycycline-treated IMR90 cells. Representative immunofluorescence images of IMR90 cells stained for MYOD. The nuclei were stained with DAPI.
B, Relative expression of exogenous Myod1, endogenous TNNT2, ITGA7 (n=3). Data is represented as mean +/− SEM.
C,D, In situ 3C analysis of the TNNT2 (C), ITGA7 (D) loci at different time points of MYOD inductions. View point for TNNT2 locus is MYOD peak at TNNT2 promoter (Fig. 3H red eye). View point for ITGA7-RDH5 locus is MYOD and CTCF co-peak at ITGA7 promoter (Fig. 4L, red eye).
E, Relative expression of pre-GAPDH or GAPDH mRNA after treatment with 1μg/ml of Actinomycin D (ActD) or DMSO for 30 minutes at 37°C after 6 hours of Myod1 induction.
F,G, In situ 3C analysis of the TNNT2, ITGA7 loci after treatment with 1μg/ml of Actinomycin D (ActD) or DMSO for 30 minutes at 37°C after 6 hours of MYOD induction. 3C data is represented as mean + SEM. View point for TNNT2 locus is MYOD peak at TNNT2 promoter (Fig. 3H red eye). View point for ITGA7-RDH5 locus is MYOD and CTCF co-peak at ITGA7 promoter (Fig. 4L, red eye).
T-test was used for statistical analysis, * p<0.05, ** p<0.01, *** p<0.001. p-values have been calculated comparing 0hr vs 24hrs time point (blue), 3hrs vs 24hrs time point (green) in C and D and DMSO in F and G.
Discussion
Somatic cell nuclear reprogramming toward either trans-differentiation or pluripotency is a multi-step task that is typically achieved by the combinatorial activities of multiple TFs, consistent with a model whereby defined TFs complement each other activity, which is otherwise not sufficient to drive the entire program. Our data suggest that the unique property of MYOD to initiate a successful program of somatic cell trans-differentiation, upon its ectopic expression, relies on the ability to re-configure 3D chromatin architecture, via binding to its consensus DNA motifs – the myogenic E-boxes – at structural and cis-regulatory elements. In this regard, our data provide an initial model for TF-driven re-configuration of 3D chromatin architecture for somatic cell nuclear reprogramming, with the large majority of changes in chromatin interactions identified in fibroblasts upon the ectopic expression of MYOD being orchestrated by a single TF (e.g. MYOD), either directly or indirectly. This model (illustrated in Fig. S3A) posits that MYOD directs the processive re-configuration of the 3D chromatin interactions through an initial DNA binding that promotes changes in 3D chromatin interactions, which are further amplified by secondary events – i.e. expression of downstream TFs, which in turn promote additional chromatin interaction changes. This model is consistent with the current view of cooperative activity of TFs in nuclear reprogramming, which accounts for the expansion of the architectural repertoire of master TFs, such as MYOD.
Interestingly, a recent work that exploited the ectopic expression of just one of the pluripotency factors, KLF4, in mouse embryonic fibroblasts (MEFs), shows striking analogies with MYOD, including the ability to promote enhancer-promoter interactions enriched with H3K27ac that activate the expression of downstream genes (Di Giammartino D., 2018). However, KLF4 is unable to drive the entire somatic cell nuclear reprogramming toward pluripotency without the co-expression of other defined factors (i.e. NANOG, OCT4, MYC). We speculate that MYOD integrates multiple architectural and transcriptional properties into one TF, thereby providing a general paradigm for TF-directed re-wiring of chromatin interactions to instruct somatic cells toward a specific lineage.
Our data also show that MYOD-mediated changes in nuclear architecture temporally precede the changes in the expression of target genes, as also predicted bioinformatically (Liu et al., 2018) and shown previously for some genes (Ghavi-Helm et al., 2014). Moreover, while MYOD-mediated alteration of IN boundary interactions is independent on active transcription, at least some MYOD-mediated alterations of enhancer-promoter interactions appear to depend on active transcription. It is possible that short-lived RNAs may cooperate with MYOD in looping cis-regulatory elements, as recently proposed by Sartorelli and colleagues (Mousavi et al., 2013; Tsai et al., 2018).
MYOD-directed reconfiguration of chromatin interactions largely occurs at the subTAD level, by altering INs structure, via binding at CTCF-anchored boundaries, as well as by targeting interactions inside INs. Notably, the high constraint of sequences implicated in MYOD-directed genomic interactions in the human population and their enrichment in disease-associated single nucleotide variants indicate that TF-altered INs could be “hotspots” for the re-configuration of nuclear architecture during developmental and post-natal skeletal myogenesis. We also found a strong association between MYOD-mediated increased strength of IN boundaries, enrichment in H3K27ac and activation of genes within the INs. Moreover, the presence of AP1 motifs in proximity of MYOD/CTCF-bound IN boundaries correlates with the activation of genes within INs. Enrichment of AP1 binding sites flanking MYOD peaks has been consistently observed in ChIP-seq studies (Cao et al. 2010) and was anticipated by earlier studies (Bengal et al., 1992), suggesting that AP1 could be a genetic determinant of MYOD-directed control of local gene expression.
Another interesting aspect of MYOD-mediated somatic nuclear cell reprogramming concerns its ability to repress the expression of cell-of-origin genes by altering pre-existing chromatin interactions through binding to E-box sequences. This is well illustrated by the MYOD-mediated alterations of promoter-enhancer interactions at the insulated neighborhood that harbors the TGFβ locus. MYOD has been known since its discovery as a sequence-specific transcriptional activator (Weintraub et al., 1991), with no structural and functional features that can account for its ability to repress gene expression (Puri and Sartorelli 2000). Although transient interactions with co-repressors has been reported (Puri et al., 2001; Singh et al., 2015), this mechanism has been implicated in the temporal regulation of target gene activation, rather than the stable repression of other lineage genes. Thus, our data suggest that MYOD-mediated rewiring of chromatin interactions can account for its ability to stably repress gene expression, via direct DNA binding to E-box motifs.
Overall, our work revealed previously unappreciated features and mechanistic insights on alterations in 3D genome architecture by a single TF that allow significant changes in gene expression, leading to coordinated repression of cell of origin gene networks and activation of tissue-specific genes during somatic cell reprogramming. This significantly extend our knowledge on TF-mediated lineage activation and terminal differentiation (Heinz et al., 2010; Natoli, 2010; Spitz and Furlong, 2012). This knowledge can have a significant impact on our understanding of the regulation of developmental myogenesis and satellite cell biology at the molecular and epigenetic level.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, P.L.P (lpuri@sbpdiscovery.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human female lung primary fibroblasts isolated at 16 weeks gestation: IMR90 cells bought at doubling passage at freeze 7.74 (Coriell). Murin myoblast cell line: C2C12 (ATCC), strain C3H. IMR90 cells were grown in growth media (GM) consisting of EMEM (ATCC) supplemented with 10% FBS (Omega Scientific). Electroporation was performed in proliferating cells at passage 11–15. All other experiments were performed in proliferating cells at passage 23–28. Doubling passage is crucial for success of myogenic conversion. C2C12 cells (ATCC) were grown in growth media (GM) consisting of DMEM/High Glucose (HyClone) supplemented with 10% FBS.
METHOD DETAILS
Sequences.
Primers sequences for expression analysis, ChIP-qPCR and 3C experiments are provided in Supplementary Table 4, 5 and 6.
Antibodies and recombinant proteins.
The following primary antibodies were used in this study: rabbit polyclonal anti-MYOD (Santa Cruz, sc-760), mouse monoclonal anti-MYOD (Santa Cruz, sc-377460 and BD Bioscience, 554130), rabbit polyclonal anti-H3K27ac (Active Motif, 39135), mouse monoclonal anti-MyHC (DSHB, MF-20), mouse monoclonal anti-GAPDH (Abcam, ab9485), mouse monoclonal anti-beta Actin (Abcam, ab20272) and mouse monoclonal anti-TNNT2 (Abcam, ab10214). The secondary antibodies were anti-mouse IgG HRP conjugated (Thermo Fisher Scientific), goat anti-mouse IgG, Fc subclass 1 specific Cy3-conjugated (Jackson ImmunoResearch, 115-545-207) and goat anti-mouse IgG, Fc subclass 2b specific 488-conjugated (Jackson ImmunoResearch, 115-165-205). DpnII (R0543), T4 DNA Ligase (M0202L), Proteinase K (P8107) and BSA (B9000) were from NEB. Biotin-14-dATP from Life Technology (19524–016).
Cell Culture Experiments.
IMR90 cells (Coriell) were grown in growth media (GM) consisting of EMEM (ATCC) supplemented with 10% FBS (Omega Scientific). Electroporation was performed in proliferating cells at passage 11–15. All other experiments were performed in proliferating cells at passage 23–28. Doubling passage is crucial for success of myogenic conversion. C2C12 cells (ATCC) were grown in growth media (GM) consisting of DMEM/High Glucose (HyClone) supplemented with 10% FBS.
Myogenic conversion.
IMR90 cells were electroporated using the Neon Transfection System (Invitrogen, MPK5000, MPK10025) with helper plasmid and epB-Puro-TT containing or not murine MYOD cDNA. Cells were then selected with 2 ug/ml of puromycin dihydrochloride (MP Bio). When cells were 60% confluent, Myod1 was induced with 200 ng/ml doxycycline (Sigma) in GM for 24 hr and cells were collected for the GM point. When cells were 95–100% confluent, MYOD was induced with 200 ng/ml doxycycline (Sigma) in GM for 24 hr and then cells were differentiated in EMEM supplemented with 2% horse serum (Gibco), 1% ITS (Sigma), 200 ng/ml doxycycline for three days for the DM time point. Media with doxycycline was refreshed every 2 days.
MYOD Time-course.
When cells were 60% confluent, Myod1 was induced with 200 ng/ml doxycycline (Sigma) in GM and cells were collect for IF, RNA or 3C after 3, 6, 12 and 24 hrs.
Transcription inhibition.
When cells were 60% confluent, Myod1 was induced with 200 ng/ml doxycycline (Sigma) in GM for 6 hours and treated with 1μg/ml of Actinomycin D (Sigma) for 30 minutes at 37C. DMSO was used as vehicle control. Following the treatment cells were collected for gene expression and 3C analyses.
siRNA transfection.
C2C12 cells were transfected with 12.5 pmol of siScr (Dharmacon) or siMyod (Ambion) using Lipofectamine RNAiMAX (Life Technologies) according the manufacturer’s instructions. 48hrs post transfection media containing transfection mix has been replaced with 2 ml fresh GM media. Cells have been collected after additional 24hrs in culture.
Generation of gRNAs expressing plasmid.
gRNA plasmids have been generated according to Kabadi et al (2014)(Kabadi et al., 2014). Briefly oligos DNA, with the appropriate overhangs have been annealed and cloned into the appropriate donor plasmid and subsequently cloned into pLV hUbC-dCas9-T2A-GFP. phU6-gRNA; pmU6-gRNA; phH1-gRNA; p7SK-gRNA and pLV hUbC-dCas9-T2A-GFP are gift from Charles Gersbach (Addgene # 53187, 53187, 53186, 53189, 53191 respectively).
Plasmid Transfection.
IMR90 were grown in GM media until approximately 60–70% confluency and transfected with gRNA expressing vectors using Lipofectamine 2000 (Life Technologies) according to the manufacturer’s instructions. 36 hrs after transfections media was changed with GM media containing 200 ng/ml doxycycline to induce MYOD expression and cells were grown for additional 24hrs before being collected for in-situ 3C and RNA expression analysis.
Immunofluorescence.
Cells were fixed with 4% PFA in PBS, permeabilized with 0.5% TX100 and blocked with 5% BSA in PBS. Cells were stained with anti-MYOD (BD Bioscience, 554130) and anti-myosin heavy chain (DSHB, MF20) for 3 hrs or O/N at RT followed by anti-mouse IgG, Fc-subclass 2b 488 conjugate (Jackson ImmunoResearch) and anti-mouse IgG, Fc-subclass 1 Cy3 conjugated (Jackson ImmunoResearch) for 1hr at RT in the dark. Nuclei were then counterstained with 2 ug/ml Hoechst 33258 pentahydrate (bis-benzimide) (Life Technologies). Images were acquired with fluorescence microscope. Fields reported in figures are representative of all examined fields.
Western Blot.
Cells were lysed in RIPA Buffer (50 mM Tris-HCl, 0.1M NaCl, 0.5% sodium deoxycholate, 1% IGEPAL CA630, 0.5% SDS, 1mM EDTA) supplemented with 1mM PMSF (Sigma) and protease inhibitors cocktail (Roche). Protein concentration was measured by BCA Protein Assay Kit (Invitrogen). 5–20ug of proteins were run on a 4%–12% or 10% tris-glycine gel (Novex) and transferred to a 0.45 μm nitrocellulose membrane. Membrane was blocked with 2.5% skim milk (BD) in PBS-Tween (PBS with 0.1% Tween 20) for 1 hr at RT. Membrane was incubated with primary antibodies anti-MYOD (1:1000 BD Bioscience, 554130) and anti-myosin heavy chain (DSHB, MF20), anti-TNNT2 (1:1000 Abcam, ab10214) O/N at 4C or with anti-GAPDH (1:1000 Abcam, ab9485) anti-bACTIN (1:1000 Abcam ab20272) for 1hr at RT. After three washes in PBS-Tween, membrane was incubated O/N with anti-mouse IgG HRP (Thermo Fisher Scientific). For detection, ECL (Thermo Scientific, 32106) was used.
mRNA expression analysis.
Cells were lysed in Trizol (Ambion) and RNA was extracted following manufacture’s recommendation. RNA concentration was measured on Qubit (Invitrogen). 100–500 ng of RNA was reverse transcribed using QuantiTek Reverse Transcrition Kit (Qiagen). Real-time quantitative PCR (qPCR) was performed using Power SYBR Green Master Mix (Life Technologies) following manufacture’s indications. Expression was normalized to Gadph for IMR90 cells or b-actin for C2C12 using 2−ΔΔCt method.
DNA-FISH.
Cells were grown on glass coverslip and fixed with 4% PFA in PBS for 10min, washed three times in PBS for 5min and stored at 4C. Following permeabilization of cells with 0.5% TX100 for 10min at RT, cells were washed three times in PBS for 5min, incubated for 1min in 70% ethanol, for 1min in 85% ethanol and for 1min in 100% ethanol. After air-drying the coverslips, cells were incubated in 70% ethanol, 85% ethanol and then 100% ethanol for 1min at RT. Probe hybridization solution was made mixing 7μl of FISH Hybridization Buffer (Agilent G9400A), 1μl of FISH probes and 1μl of water. 10μl of mixture was added on a slide and coverslip was placed on top (cell-side toward the hybridization mixture). After sealing the coverslip with rubber cement, genomic DNA and probes were denatured at 78°C for 5 min and slides were incubated at 37C in the dark O/N. Coverslip was removed from slide and washed in warmed Wash buffer 1 (Agilent, G9401A) at 73°C for 2 min and in Wash Buffer 2 (Agilent, G9402A) for 1 min at RT. Air-dried slides were stained with Hoechst in PBS for 5min at RT. After washing the coverslips three times in PBS, they were mounted on slide using Vectashield and sealed with nail polish. Images were acquired using the RPI Spinning Disk confocal microscope with 100x objective using MetaMorph acquisition software and a Hammamatsu ORCA-ER CCD camera (W.M. Keck Microscopy Facility, MIT). DNA FISH probes were custom-made by Agilent and were centered around two 4kb bins that differentially interacted with each other. DNA FISH probe 1 design region: chr22:50111857–50212146. DNA FISH probe 1 design region: chr22:50537538–50636147.
DNA-FISH analysis.
For analysis of DNA FISH, custom Python scripts were written to process and analyze 3D image data gathered in both FISH channels. Nuclear stains were blurred with a median filter (k = 5 pixels), thresholded via the scikit-image.filters method with the triangle algorithm, and touching nuclei were separated by the watershed algorithm. FISH foci were either manually called with ImageJ or automatically called using the scipy ndimage package. For automatic detection, an intensity threshold (mean + 3*standard deviation) was applied to the FISH channel. The ndimage find_objects function was then used to call contiguous FISH foci in 3D. For manual calling, FISH foci were identified in maximum z-projections of the FISH channel, and the x and y coordinates were used as reference points to find the maximum signal in the z. Using the centroid of each FISH spot in both channels, the 3D Euclidean distance was calculated between spots in different channels for each nucleus. When multiple spots were identified in a given nucleus, the minimum distance between two spots was used.
RNA Sequencing and data analysis.
Cells were collected from the plate using trypsin, that was then inhibited by adding the media cells were in before trypsinization. Spike-in were added based on number of nuclei, but not used for the analysis. PolyA RNA-seq Libraries were prepared using TruSeq RNA Library Prep Kit (Illumina) and deep sequenced on the HiSeq2500 ~50 million reads per conditions. Read quality was determined using FASTQC. Reads were mapped to the female Homo sapiens hg19 genome using TopHat2.1.1 (Kim et al., 2013) using the following options: : -p 8 -g 1 – segment-length 17 -library-type fr-firststrand. Over 84% of the reads successfully mapped. HTSeq-0.6.1p1173 with -stranded=reverse option was used to assign mapped reads to Homo Sapiens GRCh37.75 genes. Differential expression analysis was performed using DESeq2 (Love et al., 2014). Genes were considered differentially expressed if p<0.05 and fold change was lower than 0.5 or higher than 2. For integrated analysis with ChIP-seq and Hi-C, we considered differentially expressed if p<0.05 and fold change was lower than 0.5 or higher than 2 and gene transcript per million was higher or equal to 1 in at least one of the conditions compared. Ingenuity pathway analysis (IPA®, QIAGEN Redwood City, www.qiagen.com/ingenuity) was used for gene ontology. Human skeletal myotubes RNASeq was taken from ENCODE database (SRR307932.sra and SRR307933.sra). Reads were trimmed to 50 bases using fastx_trimmer. Reads were mapped to the male Homo sapiens hg19 genome using TopHat2.1.1{Kim:2013eo} using the following options: -p 8 -g 1 --segment-length 17. HTSeq-0.6.1p1173 with – stranded=no option was used to assign mapped reads to Homo Sapiens GRCh37.75 genes. Differential analysis was performed as described above.
ChIP and ChIP-seq.
Cells were fixed in 1% formaldehyde (Sigma, F8775) in PBS for 15 min at RT. Formaldehyde was then quenched with 125mM Glycine for 5 min at RT. Cells were washed in PBS and harvested in PBS supplemented with 1mM PMSF and protease inhibitors. Dry cell pellet was stored at −80C. Nuclei were then extracted and then lysed in lysis buffer containing 50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 5 mM EDTA, pH 8.0, 0.5% SDS, 0.5% NP-40, 1 mM PMSF and a protease inhibitor. Chromatin was sheared with sonicator (ColeParmer, Misonix 3000) to an average DNA fragment length of 200–500bp. Chromatin was then diluted 5 times in lysis buffer without SDS. DNA amount was measured with the Qubit (Invitrogen Q32854). DNA was immunoprecipated with either rabbit anti-MYOD (Santa Cruz), or rabbit anti-H3K27ac (Active Motif) O/N at 4C. The immunocomplexes were captured with protein A magnetic beads (Life Technologies) for 3–4 hrs at 4C. After four washes with buffer containing 50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 5 mM EDTA, pH 8.0, 0.1% SDS, 1% NP-40, 0.5% sodium deoxycholate, one wash with a buffer containing 250 mM LiCl, 100 mM NaCl, 5 mM EDTA, pH 8.0, 1% NP-40, 1% sodium deoxycholate and two washes with TE buffer (10mM Tris-HCl pH=8, 1mM EDTA) chromatin was then eluted and decrossliked with 1% SDS in TE O/N at 65C 600RPM rotation. Also, the input is decrosslinked with 1% SDS in TE O/N at 65C 600RPM rotation. After 2 hrs digestion at 37C with 0.2 mg/ml proteinase K, DNA was extracted with phenol/chloroform and ethanol precipitated O/N at −20C. Prior to sequencing, DNA was then suspended in mQ water. The DNA was then analysed by qPCR calculating the amount of immunoprecipitated DNA relative to the input DNA (percentage of input). Library preparation and sequencing of immunoprecipitated and input DNA were performed as described http://bioinformatics-renlab.ucsd.edu/RenLabLibraryProtocolV1.pdf.
ChIP-seq analysis.
Read quality was determined using FASTQC. Reads were mapped using bowtie2–2.0.5/bowtie2 to the female Homo sapiens hg19 genomes with options: -- very-sensitive-local. Over 85% of the reads successfully mapped. Duplicate reads were removed using samtools1.3. Peaks were called using macs2 2.1.1.20160309 with qvalue=0.01, macs2 2.1.1.20160309 was also used for differential peak calling among samples. Reads were extended based on the fragment size predicted with macs2. Heatmap of ChIP-seq signal was generated using Seqminer. We also analyzed previously published ChIP-seq data. To compare H3K27ac levels between IMR90, hMB and hMT we started from the same number of reads. These data were analyzed following the same workflow as our MYOD ChIP-seq data. Motif analysis was performed using MEME Suite (Bailey et al., 2009), Jaspar (Mathelier et al., 2014) or HOMER (Heinz et al., 2010).
In situ Hi-C.
Hi-C was performed as previously described (Rao et al., 2014) with the following modifications. Cells were cross-linked with 2% formaldehyde in media. Formaldehyde was then quenched with 200mM of glycine for 5 min at RT. Cells were then washed in PBS and pelleted. Cell pellet was then saved at −80C. 2×106 cells were then lysed with lysis buffer (10mM Tris-HCl pH 8.0, 10mM NaCl, 0.2% Igepal CA630). Incubation of cells in 0.5% SDS in mQ at 62C for 10 min is followed by SDS quenching with TritonX-100 for 15 min at 37C. NEB3 buffer was added to reach 1X final concentration. DNA was then digested with DPNII O/N at 37C at 900RPM. Inactivation of DPNII was performed by incubating the samples at 62C for 20 min. Fill in of the digested end was performed by adding biotin-14-dATP (Life Technology, 19524–016), dCTP, dGTP, dTTP (Invitrogen) and Klenow (NEB, M0210). Mixture was incubated at 37C for 90 min 500RPM. Ligation was performed in 1.2ml by adding mQ water, T4 DNA ligase buffer to concentration 1X (NEB, B0202), 0.083% TritionX-100, 0.01mg/mL BSA, 2000U/uL T4 DNA Ligase (NEB, M0202) for 4 hr at RT with slow rotation. DNA is then ethanol precipitated, resuspended in 10mM Tris-HCl pH=8 and sheared using Covaris sonicator. Size selection of DNA (200–600bp) was performed using AmpureXP beads. DNA ends were then repaired and biotin removed from un-ligated samples by incubating the DNA at 37C for 30 min in 1X T4 DNA ligase buffer with 0.5mM dNTPs (Invitrogen), 50U T4 PNK (NEB, M0201), 12U T4 DNA Polymerase (NEB, M0203) and 5U Klenow. Biotin-labelled DNA was pulled down using Dynabeads My One T1 Streptavidin beads (Life Tech). Illumina Indexed adapter are then ligated with NEB DNA Quick Ligase (NEB, M2200). Beads were then washed and dissolved in 10mM Tris-HCl pH=8. KAPA qPCR assay was then performed to estimate concentration and cycle number needed for final PCR.
Hi-C analyses.
Read quality was determined using FASTQC. For interaction matrix, HiCPro-v2.7.7 was used for read mapping, detection of valid ligation products, quality control, and sparse chromosomal interaction maps(Servant et al., 2015) using the following settings: BOWTIE2_GLOBAL_OPTIONS = --very-sensitive -L 30 --score-min L,−0.6,−0.2 --end-to-end -reorder, BOWTIE2_LOCAL_OPTIONS = --very-sensitive -L 20 --score-min L,−0.6,−0.2 --end-to-end -reorder, REFERENCE_GENOME = hg19_XX, LIGATION_SITE = GATCGATC, MIN_FRAG_SIZE = 100, MAX_FRAG_SIZE = 100000, MIN_INSERT_SIZE = 100, MAX_INSERT_SIZE = 600, MAX_ITER = 100, FILTER_LOW_COUNT_PERC = 0.02, FILTER_HIGH_COUNT_PERC = 0, EPS = 0.1. Quality of the libraries based on percentage of mapped reads (for both ends), percentage of reported pairs (removal of unmapped pairs, multiple pairs alignments, low quality pairs, not reported pairs and pairs with singleton - % considering the total number of reads), percentage of valid putative interaction pairs (removal of dangling ends, fragments with no restriction site, self-circles etc - % considering the number of reported pairs), percentage of unique read pairs (removal of duplicates - % considering the valid putative interaction pairs), number of unique read pairs, percentage of cis-read pairs, number of long-range cis-read pairs, percentage of trans read pairs was determined using HiCPro-v2.7.7 (Servant et al., 2015). HiTC was used to transform sparse matrices to NxN matrices(Servant et al., 2012). For Hi-C library quality analysis presented in Table S1 please refer to (Servant et al., 2015). Hi-C data reproducibility between replicates per chromosome was calculated in two ways a) as previously described by Dixon et al, 2012 (Dixon et al., 2012) and b) using HiC-spector (Yan et al., 2017). For the first method, the set of all possible intra-chromosomal interactions for two replicates were correlated by comparing each point in interaction matrix at 4kb resolution from one replicate with the same point from the second replicate. We restricted the correlation to a maximum distance between points of 2Mb (500 bins), since Hi-C data is skewed toward proximal interactions (Dixon et al., 2012). We used the cor function of R (version 3.2.3) to calculate the Pearson correlation between the two vectors. For the second method, we used the python script HiC-spector (Yan et al., 2017) on 4kb raw intra-chromosomal triple sparse format matrices. Hi-C heatmap in Figure 2B and Figure S4 were generated using HiCPlotter (Akdemir and Chin, 2015).
TAD calling:
TADs were called on NxN ICE-normalized matrices using Armatus (Filippova et al., 2014) (v2.1), with gamma-max (-g) set to 0.3, resolution (-r) set to 40kb and the remaining parameters left as default. We called TADs at various resolutions, but we report data on TADs called at 40kb resolution, because 40kb was the highest resolution that gave us reproducible TAD calls between biological replicates.
Boundaries calling:
TAD boundaries were called following a previously described method (Crane et al., 2015) using a window size of 400kb, which was selected based on the reproducibility across biological replicates. We chose the smallest window size that would give high (>=90%) reproducibility. For co-regulation analysis, TADs were called at 40kb resolution using 75M reads, all other analyses were performed with TADs called using all the number of reads from deep sequencing.
Differential interaction calling:
differential interactions were called with diffHic (Lun and Smyth, 2015) v1.4.2, in R (v3.3.1), on raw matrices at 4kb resolution, with two biological replicates for each condition. Triplet sparse format matrices generated by HiCPro were converted to InteractionSet objects with the GInteractions function from the InteractionSet package (v1.0.4) and then organized in a counts matrix with the InteractionSet function. Differential analysis was performed for each chromosome separately, considering only intra-chromosomal bin pairs where the average logCPM > 1 across samples. Data were normalized using Loess normalization, applied separately to regions near (<=2L) and far from the Hi-C matrix diagonal. InteractionSet was converted to a DGEList object to be used as input for EdgeR (Robinson et al., 2010) v3.14.0. Once calculated data dispersion with the estimateDisp and glmQLFit functions, differential analysis is performed using a Quasi-Likelihood F-Test and Benjamini-Hochberg FDR correction. Requirement for significant differential interaction: fold change lower than 0.5 or higher than 2 and p-value lower than 0.05. All differential interactions falling on the diagonal were excluded. For fold change distribution analysis, we converted all fold changes between 0 and 1 as −1/(Fold change).
Data integration analysis.
To integrate data from RNA-seq, ChIP-seq and Hi-C experiments, we used bedtools 2.26.0(Quinlan and Hall, 2010) and R packages. Promoters were defined based on the location of the TSS of Homo sapiens GRCh37.75 genes (+/− 4kb from annotated TSS). Enhancers were defined as DNA regions containing H3K27ac that did not overlap with promoters.
Circular permutations.
Chromosome-bound circular permutations test described in Cabrera et al., 2012(Cabrera et al., 2012) was applied to evaluate if the observed overlap between Hi-C differentially interacting bins found in the different comparisons (i.e., EMPTY GM vs. MYOD GM and MYOD GM vs. MYOD DM) and MYOD ChIP-seq peaks was significantly enriched compared to the expected overlap. For each permutation, differentially interacting bins obtained from the comparison in analysis were shifted of a randomly generated number of bins (comprised between 1 and the maximum number of bins of the smallest chromosome in analysis, chr21), for a total of 10,000 permutations. When the shift exceeded the end of the chromosome, the permutation continued from the chromosome start, thus regarding chromosomes as circularized. Randomly generated genomic intervals were then overlapped with MYOD ChIP-seq peaks found in MYOD GM and MYOD DM, respectively. The number of permuted datasets, n, having a number of bins overlapping MYOD peaks greater than or equal to the observed number were noted and used to estimate approximate p-values (n/10,000) for enrichment.
Gene expression analysis inside differential INs.
For Figure S5A, the list of INs whose boundaries show a higher interaction strength between boundaries in MYOD GM as compared to EMPTY GM, and the list of MYOD ChIP-seq peaks in MYOD GM, were overlapped with bedtools (intersectbed, with parameters -wa and -u) in order to partition the differential INs in three categories: i) INs whose boundaries are not bound by MYOD, ii) INs where only one boundary is bound by MYOD and iii) INs where MYOD binds both boundaries. These sub-lists, together with the initial list of all INs whose boundaries interaction is strengthen upon MYOD expression, were overlapped with the list of human promoters (NB. genes were considered within an IN when at least the promoter overlapped the IN). Promoters were defined based on the location of the TSS of Homo sapiens GRCh37.75 genes (+/− 4kb from annotated TSS). Genes were subsequently filtered in order to retain only those with p-val<0.01 from the RNA-seq differential analysis (EMPTY GM vs MYOD GM comparison) and mapping exclusively to only one of the subgroups, in order to exclude the potential confounding effects due to the presence of nested INs. The bed intervals of the INs whose boundary interaction strength increases upon MYOD expression and that are bound by MYOD at both boundaries were used to plot the ChIP-seq signal of CTCF, MYOD and H3K27 acetylation (the latter both in EMPTY GM and in human myoblasts) with the NGSplot R s, with parameters -G hg19 -R bed -L 2500 -MW 7 -YAS 0,0.7 (Shen et al., 2014).
Calculations of “expected number” of events for each figure panel.
For Figure 1E, observed/expected ratios were calculated as in Chronis et al., 2017 (Chronis et al., 2017), as the ratio between the observed and the expected overlap for each feature based on their sizes and the size of the hg19 human genome:
where F is the number of base pairs annotated for the feature F (e.g. MYOD peaks), S is the size of the feature S (e.g. promoters of differentially expressed genes) and G is the length of the human genome.
For Figure 2E, expected frequency of MYOD binding at differentially interacting bins (fex) = (total number of 4kb bins bound by MYOD)/(total number of 4kb bins genome-wide) Expected number of bins that differentially interacted that are bound by MYOD = fex* (number of 4kb bins that differentially interacted).
For Figure 3A, expected frequency of MYOD binding at differentially interacting bins (fex) = (total number of 4kb bins bound by MYOD)/(total number of 4kb bins genome-wide) Expected number of bins that differentially interacted that are bound by MYOD = fex* (number of 4kb bins that differentially interacted, in this case one of the differentially interacting partner bin has to be in a promoter).
For Figure 3B, expected frequency of MYOD binding at differentially interacting bins (fex) = (number of 4kb bins that coincide with promoters or enhancers that are bound by MYOD)/(number of 4kb bins genome-wide that coincide with promoters or enhancers) Expected number of bins that differentially interacted that are bound by MYOD = fex* (number of differentially interacting 4kb bins involved in differential enhancer-promoter interaction).
For Figure 4E, expected frequency of MYOD binding at differentially interacting bins co-bound by CTCF (fex) = (number of 4kb bins that are co-bound by MYOD and CTCF)/(number of 4kb bins that are bound by CTCF) Expected number of bins that differentially interacted that are bound by MYOD = fex* (number of 4kb bins that differentially interacted with each other and that are both bound by CTCF)
To determine statistical significance between observed and expected, chi-squared test was performed using R version 3.2.3, function chisq.test().
Genetic constraint analysis.
The hg19 human reference genome was downloaded from ftp://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/ and split into 4kb bins, which corresponds to the experimental resolution. The bins were further classified into separate categories, using bedops (v2.4.30)(Neph et al., 2012), depending on whether they contained MYOD and/or CTCF peaks, were present in differentially interacting regions or boundaries. Of note, the categories are not mutually exclusive. The names and number of bins per categories are the following: DI (N=71,501 in EMPTY_GMvsMYOD_GM and N=74,327 in MYOD_GMvsMYOD_DM); DI_CTCF (N=7,414 in EMPTY_GMvsMYOD_GM and N=7,327 in MYOD_GMvsMYOD_DM); DI_MYOD (N=9,096 in EMPTY_GMvsMYOD_GM and N= 13,206 in MYOD_GMvsMYOD_DM); DI_MYOD_CTCF (N=2,666 in EMPTY_GMvsMYOD_GM and N=3,447 in MYOD_GMvsMYOD_DM); WG (N=759,086); WG_CTCF (N=47,625); WG_MYOD (N=44,741 MYOD_GM and N=68,989 MYOD_DM); Bd (N=2,241 in EMPTY_GMvsMYOD_GM and N=2,596 in MYOD_GMvsMYOD_DM); Bd_MYOD (N=933 in EMPTY_GMvsMYOD_GM and N=1,373 in MYOD_GMvsMYOD_DM). The mean CDTS value (di Iulio et al., 2018) of every 4kb bin was extracted using bedops and the cumulative distribution function of all genomic bins in a given category were plotted using R version 3.4.3. The CDTS used was computed with whole genome sequencing data obtained from the gnomAD dataset (N=15,496). The explanation is provided here: http://www.hliopendata.com/noncoding/Pipeline/README_compute_CDTS_fromPublic Dataset.txt
The difference in the cumulative distribution function of different categories was assessed using two-sample Kolmogorov-Smirnov test.
Pathogenic variants distribution analysis.
Variants from HGMD (Stenson et al., 2003) were filtered to retain only “High DM” flagged variants. The pathogenic variants were categorized into three non-mutually exclusive groups (All, Skeletal Muscle and Inflammation). All (N=154,503) – encompasses all pathogenic variants. Skeletal Muscle (N=5,888) – encompasses variants retrieved with the following key word extraction using grep: > grep “[Mm]usc\|[Mm]yopath\|[Mm]yogen\|[Mm]yasten” and grep -v “[Cc]ardio\|[Ss]tatin\|[Cc]ardiac\|[Cc]orne\|[Ss]mooth\|[Aa]drenoleukodystrophy”. Inflammation (N=2,737) – includes variants retrieved with the following key word extraction using grep: > grep “[Ii]nflam\|itis” and grep -v “British_Columbia\|Pseudohermaphroditism\|[Hh]epatitis [ABC]”. The fraction of genomic bins containing at least one pathogenic variant was then extracted. The difference in the fraction of bins with pathogenic variant of different categories was assessed using one sided Fisher’s Exact Test, with the following assumptions of expected fraction of variants per category: WG < DI; WG < WG_CTCF; WG < Bd; WG < Bd_MYOD; WG_CTCF < Bd; DI < Bd.
In situ 3C.
3C was performed as described for the in-situ Hi-C omitting the biotinylation step. Following Ethanol precipitation DNA is resuspended in 10mM Tris-HCl pH=8 and diluted to 25ng/μl. 1μl of the diluted DNA is then analysed by qPCR. Primers were designed using Primer3 and blasted to hg19 genome to ensure specificity for the fragment analyzed. For positive control, equimolar amount of BAC DNA containing the locus of interested were digested and ligated in the same way of sample DNA.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were carried out using Excel or R. Data are represented as Mean +/− SEM or Mean + SEM as described in the figure legend. We used various statistical tests depending on the statistical question: Student’s t test, or Two-way ANOVA was used for statistical analysis, corrected for multiple testing (Tukey), or chi-squared test, or two-sided exact binomial test, or One sided Fisher’s Exact Test, or Two-sample Kolmogorov-Smirnov test, or Wilcoxon rank sum test, or circular permutation. For differential gene expression, DESeq2 was used, for differential ChIP-seq peaks, MACS2 was used. The method used is stated in the legend and/or main text and/or STAR Method section of the relative figure.
DATA AND CODE AVAILABILITY
Sequencing data have been deposited in the GEO database under the accession number GEO: GSE98530. This work also used previous sequencing data deposited in the GEO database: GEO: GSM935404, GSM733762, GSM733783, GSM1055818, GSM915188, GSM733666, GSM733755, GSM915165, GSE128527, GSM2417196, GSM2417197, GSM2417198, GSM2417204, GSM2417203 (Chronis et al., 2017; Consortium, 2012; Jin et al., 2013; Sala et al., 2019; Yue et al., 2014). Codes generated for this study have been deposited to Mendeley.
DNA FISH analysis:
DOI: http://dx.doi.org/10.17632/tvb7yvpr4s.1#file-b6ac28c9-29d2-48ae-9658-ecf953457b98 and http://dx.doi.org/10.17632/tvb7yvpr4s.1#file-01a31ffd-8040-41cc-9b0e-98d9a4ac9c88
Circular permutation:
DOI: http://dx.doi.org/10.17632/rvzzpsh6vg.2#file-96dfb6d0-d6e3-4a42-ac13-a78fb2346b27
Supplementary Material
Table S1. Related to STAR Methods. Hi-C library quality.
This table reports the number and percentage of mapped reads, percentage of valid putative interaction pairs, percentage and number of unique read pairs, percentage and number of cis- or trans- read pairs.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal MYOD | Santa Cruz | M-318 |
| Rabbit polyclonal H3K27Ac | Active Motif | |
| Mouse monoclonal MYOD | BD Bioscience | 554130 |
| Mouse monoclonal MyHC | DSHB | MF20 |
| Goat anti-mouse IgG, Fc subclass 1 specific | Jackson Immuno | NC0469362 |
| Goat anti-mouse IgG, Fc subclass 2b specific | Jackson Immuno | NC0266980 |
| Mouse monoclonal GAPDH | Abcam | ab9485 |
| Anti-bActin-HRP | Abcam | ab20272 |
| Anti-human TNNT2 | Abcam | ab10214 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Doxycycline | Sigma-Aldrich | D3072–1 ML |
| Puromycin dihydrochloride | MP Biomedicals | ICN10055210 |
| ITS | Sigma-Aldrich | I2146 |
| Hoechst 33258 | Life Technologies | H3569 |
| PMSF | Sigma-Aldrich | 93482 |
| Protease inhibitors | Roche | 11697498001 |
| DpnII | NEB | R0543 |
| T4 DNA Ligase | NEB | M0202L |
| Proteinase K | NEB | P8107 |
| BSA | NEB | B9000 |
| biotin-14-dATP | Life Technology | 19524–016 |
| Critical Commercial Assays | ||
| Neon Transfection System | Invitrogen | MPK5000, MPK10025 |
| BCA Protein Assay Kit | Invitrogen | 23235 |
| TruSeq Stranded mRNA Library Prep Kit set A | Illumina | RS-122–2101 |
| Deposited Data | ||
| RNA-seq, ChIP-seq, Hi-C | This paper | GEO: GSE98530 |
| Other ChIP-seq data | (Consortium, 2012; Jin et al., 2013; Sala et al., 2019; Yue et al., 2014) | GEO: GSM935404, GSM733762, GSM733783, GSM1055818, GSM915188, GSM733666, GSM733755, GSM915165, GSE128527 |
| Other RNA-seq data | (Chronis et al., 2017) | GEO: GSM2417196, GSM2417197, GSM2417198, GSM2417204, GSM2417203 |
| Experimental Models: Cell Lines | ||
| IMR90 | Coriell | I90–83 |
| C2C12 | ATCC | CRL-1772 |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| PCR primers - see list at the end of STAR Methods | This paper | N/A |
| siScr | Dharmacon | D-001210-01-05 |
| siMyod | Ambion | s232596 |
| Recombinant DNA | ||
| Helper plasmid | Provided by Dr. Alessandro Rosa | N/A |
| epB-Puro-TT plasmid EMPTY | Provided by Dr. Alessandro Rosa | N/A |
| epB-Puro-TT plasmid EMPTY mouse Myod1 cDNA | Provided by Dr. Alessandro Rosa | N/A |
| Software and Algorithms | ||
| TopHat2.1.1 | (Kim et al., 2013) | https://ccb.ihu.edu/software/tophat/index.shtml |
| HTSeq-0.6.1 | (Anders et al., 2015) | https://htseq.readthedocs.io/en/release0.11.1/ |
| DESeq2 | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Ingenuity Pathway Analysis | Qiagen | https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/ |
| Bowtie2–2.0.5/bowtie2 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools1.3 | (Li et al., 2009) | http://samtools.sourceforge.net/ |
| Macs2v2.1.1 | (Zhang et al., 2008) | http://liulab.dfci.harvard.edu/MACS/ |
| Bedtoolsv2.26.0 | (Quinlan and Hall, 2010) | http://bedtools.readthedocs.io/en/latest/ |
| HiCPro-v2.7.7 | (Servant et al., 2015) | https://github.com/nservant/HiC-Pro |
| HiTC | (Servant et al., 2012) | https://bioconductor.org/packages/release/bioc/html/HiTC.html |
| HiCPlotter | (Akdemir and Chin, 2015) | https://github.com/kcakdemir/HiCPlotter |
| Armatus | (Filippova et al., 2014) | https://www.cs.cmu.edu/~ckingsf/software/armatus/ |
| DiffHic | (Lun and Smyth, 2015) | https://bioconductor.org/packages/release/bioc/html/diffHic.html |
| Jaspar | (Mathelier et al., 2014) | http://jaspar.genereg.net/ |
| MEME | (Bailey et al., 2009) | http://meme-suite.org/tools/meme-chip |
| HOMER | (Heinz et al., 2010) | http://homer.ucsd.edu/homer/ |
| DNA FISH analysis | This paper | DOI: http://dx.doi.org/10.17632/tvb7yvpr4s.1#file-b6ac28c9-29d2-48ae-9658-ecf953457b98 DOI: http://dx.doi.org/10.17632/tvb7yvpr4s.1#file-01a31ffd-8040-41cc-9b0e-98d9a4ac9c88 |
| Circular permutation | This paper | DOI: http://dx.doi.org/10.17632/rvzzpsh6vg.2#file-96dfb6d0-d6e3-4a42-ac13-a78fb2346b27 |
| Other | ||
| Olympus IX71 microscope | ||
| RPI spinning disk microscope | ||
Highlights.
MYOD drives the re-wiring of chromatin interactions during transdifferentiation
MYOD alters chromatin interactions between cis-regulatory elements
MYOD re-wires insulated neighborhoods, hotspots of differential interactions
MYOD re-wiring of chromatin interactions temporally precedes transcriptional changes
Acknowledgements
The authors would like to give special thanks to Dr. Alessandro Rosa for sharing Helper and epB-Puro-TT plasmids, Dr. David Huhta for helping with the computer cluster, Dr. Nicolas Servant for helping with HiC-Pro, Samantha Kuan and Bin Li for operatiing the sequencing instruments and data processing, Wendy Salmon and the W.M. Keck Microscopy Facility for miscoscopy support, Amy Cortez for helping with cell cycle analysis, Dr. Michael Walker for statistics guidance, Dr. Jonathan Henninger for DNA FISH analysis, Dr. Brian Abraham for helpful discussion on imaging analysis, the members of Puri’s laboratory for helpful discussion. We are also grateful to Dr. Richard Young for his helpful suggestions on the manuscript and Tong Lee for helpful discussions. This work was supported by: NIAMS R01 AR056712; R01AR052779; AR061303 to PLP; Epigen Project Progetto Bandiera Epigenomica to PLP and SB; Ellison Medical Foundation AG-NS-0843–11; AFAR G16294 AD. Ludwig Institute for Cancer Research and a pilot project from the SAN DIEGO MUSCLE RESEARCH CENTER (SDMRC) to BR. YL is funded by a postdoctoral fellowship from the Human Frontier Program. PLP dedicates this work to his unforgettable colleague, teacher and friend Elisabetta “Betta” De Marzio, who will reamain for ever in his heart.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
B.R. is a co-founder of Arima Genomics, Inc. A.S. is an employee and stockholder at Arima Genomics, Inc. They did not influence the scientific outcome of this work. The remaining authors declare no competing interests.
References
- Akdemir KC, and Chin L (2015). HiCPlotter integrates genomic data with interaction matrices. Genome Biol 16, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrey G, and Mundlos S (2017). The three-dimensional genome: regulating gene expression during pluripotency and development. Development 144, 3646–3658. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, and Noble WS (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37, W202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battistelli C, Busanello A, and Maione R (2014). Functional interplay between MyoD and CTCF in regulating long-range chromatin interactions during differentiation. J Cell Sci 127, 3757–3767. [DOI] [PubMed] [Google Scholar]
- Beagan JA, Gilgenast TG, Kim J, Plona Z, Norton HK, Hu G, Hsu SC, Shields EJ, Lyu X, Apostolou E, et al. (2016). Local Genome Topology Can Exhibit an Incompletely Rewired 3D-Folding State during Somatic Cell Reprogramming. Cell Stem Cell 18, 611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengal E, Ransone L, Scharfmann R, Dwarki VJ, Tapscott SJ, Weintraub H, and Verma IM (1992). Functional antagonism between c-Jun and MyoD proteins: a direct physical association. Cell 68, 507–519. [DOI] [PubMed] [Google Scholar]
- Berkes CA, Bergstrom DA, Penn BH, Seaver KJ, Knoepfler PS, and Tapscott SJ (2004). Pbx marks genes for activation by MyoD indicating a role for a homeodomain protein in establishing myogenic potential. Mol Cell 14, 465–477. [DOI] [PubMed] [Google Scholar]
- Black BL, Molkentin JD, and Olson EN (1998). Multiple roles for the MyoD basic region in transmission of transcriptional activation signals and interaction with MEF2. Mol Cell Biol 18, 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonev B, and Cavalli G (2016). Organization and function of the 3D genome. Nat Rev Genet 17, 661–678. [DOI] [PubMed] [Google Scholar]
- Bonev B, Mendelson Cohen N, Szabo Q, Fritsch L, Papadopoulos GL, Lubling Y, Xu X, Lv X, Hugnot JP, Tanay A, et al. (2017). Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell 171, 557–572 e524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera CP, Navarro P, Huffman JE, Wright AF, Hayward C, Campbell H, Wilson JF, Rudan I, Hastie ND, Vitart V, et al. (2012). Uncovering networks from genome-wide association studies via circular genomic permutation. G3 (Bethesda) 2, 1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caiazzo M, Dell’Anno MT, Dvoretskova E, Lazarevic D, Taverna S, Leo D, Sotnikova TD, Menegon A, Roncaglia P, Colciago G, et al. (2011). Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature 476, 224–227. [DOI] [PubMed] [Google Scholar]
- Cao Y, Yao Z, Sarkar D, Lawrence M, Sanchez GJ, Parker MH, MacQuarrie KL, Davison J, Morgan MT, Ruzzo WL, et al. (2010). Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev Cell 18, 662–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra T, Ewels PA, Schoenfelder S, Furlan-Magaril M, Wingett SW, Kirschner K, Thuret JY, Andrews S, Fraser P, and Reik W (2015). Global reorganization of the nuclear landscape in senescent cells. Cell Rep 10, 471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chronis C, Fiziev P, Papp B, Butz S, Bonora G, Sabri S, Ernst J, and Plath K (2017). Cooperative Binding of Transcription Factors Orchestrates Reprogramming. Cell 168, 442–459 e420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciglar L, Girardot C, Wilczynski B, Braun M, and Furlong EE (2014). Coordinated repression and activation of two transcriptional programs stabilizes cell fate during myogenesis. Development 141, 2633–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conerly ML, Yao Z, Zhong JW, Groudine M, and Tapscott SJ (2016). Distinct Activities of Myf5 and MyoD Indicate Separate Roles in Skeletal Muscle Lineage Specification and Differentiation. Dev Cell 36, 375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium EP (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane E, Bian Q, McCord RP, Lajoie BR, Wheeler BS, Ralston EJ, Uzawa S, Dekker J, and Meyer BJ (2015). Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature 523, 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RL, Weintraub H, and Lassar AB (1987). Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 51, 987–1000. [DOI] [PubMed] [Google Scholar]
- de Wit E, Bouwman BA, Zhu Y, Klous P, Splinter E, Verstegen MJ, Krijger PH, Festuccia N, Nora EP, Welling M, et al. (2013). The pluripotent genome in three dimensions is shaped around pluripotency factors. Nature 501, 227–231. [DOI] [PubMed] [Google Scholar]
- Dekker J, and Mirny L (2016). The 3D Genome as Moderator of Chromosomal Communication. Cell 164, 1110–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado-Olguin P, Brand-Arzamendi K, Scott IC, Jungblut B, Stainier DY, Bruneau BG, and Recillas-Targa F (2011). CTCF promotes muscle differentiation by modulating the activity of myogenic regulatory factors. J Biol Chem 286, 12483–12494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giammartino D, L. Y, Kloetgen A, Polyzos A, Kim D, Stadtfeld M, Tsirigos A, Apostolou E (2018). KLF4 binding during reprogramming is involved in 3D architectural rewiring and transcriptional regulation of enhancer hubs. Biorxiv. [Google Scholar]
- di Iulio J, Bartha I, Wong EHM, Yu HC, Lavrenko V, Yang D, Jung I, Hicks MA, Shah N, Kirkness EF, et al. (2018). The human noncoding genome defined by genetic diversity. Nat Genet 50, 333–337. [DOI] [PubMed] [Google Scholar]
- Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, et al. (2015). Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodou E, Xu SM, and Black BL (2003). mef2c is activated directly by myogenic basic helix-loop-helix proteins during skeletal muscle development in vivo. Mech Dev 120, 1021–1032. [DOI] [PubMed] [Google Scholar]
- Dowen JM, Fan ZP, Hnisz D, Ren G, Abraham BJ, Zhang LN, Weintraub AS, Schuijers J, Lee TI, Zhao K, et al. (2014). Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 159, 374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippova D, Patro R, Duggal G, and Kingsford C (2014). Identification of alternative topological domains in chromatin. Algorithms Mol Biol 9, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suva ML, and Bernstein BE (2016). Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong AP, Yao Z, Zhong JW, Cao Y, Ruzzo WL, Gentleman RC, and Tapscott SJ (2012). Genetic and epigenetic determinants of neurogenesis and myogenesis. Dev Cell 22, 721–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke M, Ibrahim DM, Andrey G, Schwarzer W, Heinrich V, Schopflin R, Kraft K, Kempfer R, Jerkovic I, Chan WL, et al. (2016). Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 538, 265–269. [DOI] [PubMed] [Google Scholar]
- Gerber AN, Klesert TR, Bergstrom DA, and Tapscott SJ (1997). Two domains of MyoD mediate transcriptional activation of genes in repressive chromatin: a mechanism for lineage determination in myogenesis. Genes Dev 11, 436–450. [DOI] [PubMed] [Google Scholar]
- Ghavi-Helm Y, Klein FA, Pakozdi T, Ciglar L, Noordermeer D, Huber W, and Furlong EE (2014). Enhancer loops appear stable during development and are associated with paused polymerase. Nature 512, 96–100. [DOI] [PubMed] [Google Scholar]
- Guerreiro I, Gitto S, Novoa A, Codourey J, Nguyen Huynh TH, Gonzalez F, Milinkovitch MC, Mallo M, and Duboule D (2016). Reorganisation of Hoxd regulatory landscapes during the evolution of a snake-like body plan. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, and Young RA (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Day DS, and Young RA (2016a). Insulated Neighborhoods: Structural and Functional Units of Mammalian Gene Control. Cell 167, 1188–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Weintraub AS, Day DS, Valton AL, Bak RO, Li CH, Goldmann J, Lajoie BR, Fan ZP, Sigova AA, et al. (2016b). Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 351, 1454–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieda M, Fu JD, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, and Srivastava D (2010). Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142, 375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isoda T, Moore AJ, He Z, Chandra V, Aida M, Denholtz M, Piet van Hamburg J, Fisch KM, Chang AN, Fahl SP, et al. (2017). Non-coding Transcription Instructs Chromatin Folding and Compartmentalization to Dictate Enhancer-Promoter Communication and T Cell Fate. Cell 171, 103–119 e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javierre BM, Burren OS, Wilder SP, Kreuzhuber R, Hill SM, Sewitz S, Cairns J, Wingett SW, Varnai C, Thiecke MJ, et al. (2016). Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters. Cell 167, 1369–1384 e1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X, Dadon DB, Powell BE, Fan ZP, Borges-Rivera D, Shachar S, Weintraub AS, Hnisz D, Pegoraro G, Lee TI, et al. (2016). 3D Chromosome Regulatory Landscape of Human Pluripotent Cells. Cell Stem Cell 18, 262–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin F, Li Y, Dixon JR, Selvaraj S, Ye Z, Lee AY, Yen CA, Schmitt AD, Espinoza CA, and Ren B (2013). A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature 503, 290–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabadi AM, Ousterout DG, Hilton IB, and Gersbach CA (2014). Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Res 42, e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer-Kwon KR, Nimura K, Rao SSP, Xu J, Jung S, Pekowska A, Dose M, Stevens E, Mathe E, Dong P, et al. (2017). Myc Regulates Chromatin Decompaction and Nuclear Architecture during B Cell Activation. Mol Cell 67, 566–578 e510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kragesteen BK, Spielmann M, Paliou C, Heinrich V, Schopflin R, Esposito A, Annunziatella C, Bianco S, Chiariello AM, Jerkovic I, et al. (2018). Dynamic 3D chromatin architecture contributes to enhancer specificity and limb morphogenesis. Nat Genet 50, 1463–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijger PH, Di Stefano B, de Wit E, Limone F, van Oevelen C, de Laat W, and Graf T (2016). Cell-of-Origin-Specific 3D Genome Structure Acquired during Somatic Cell Reprogramming. Cell Stem Cell 18, 597–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latella L, Dall’Agnese A, Boscolo FS, Nardoni C, Cosentino M, Lahm A, Sacco A, and Puri PL (2017). DNA damage signaling mediates the functional antagonism between replicative senescence and terminal muscle differentiation. Genes Dev 31, 648–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Black BL, and Derynck R (2001). TGF-beta inhibits muscle differentiation through functional repression of myogenic transcription factors by Smad3. Genes Dev 15, 2950–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Chen H, Ronquist S, Seaman L, Ceglia N, Meixner W, Chen PY, Higgins G, Baldi P, Smale S, et al. (2018). Genome Architecture Mediates Transcriptional Control of Human Myogenic Reprogramming. iScience 6, 232–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loell I, and Lundberg IE (2011). Can muscle regeneration fail in chronic inflammation: a weakness in inflammatory myopathies? J Intern Med 269, 243–257. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun AT, and Smyth GK (2015). diffHic: a Bioconductor package to detect differential genomic interactions in Hi-C data. BMC Bioinformatics 16, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupianez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, et al. (2015). Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161, 1012–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathelier A, Zhao X, Zhang AW, Parcy F, Worsley-Hunt R, Arenillas DJ, Buchman S, Chen CY, Chou A, Ienasescu H, et al. (2014). JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res 42, D142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousavi K, Zare H, Dell’orso S, Grontved L, Gutierrez-Cruz G, Derfoul A, Hager GL, and Sartorelli V (2013). eRNAs promote transcription by establishing chromatin accessibility at defined genomic loci. Mol Cell 51, 606–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra V, Rocha PP, An D, Raviram R, Skok JA, Mazzoni EO, and Reinberg D (2015). CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science 347, 1017–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natoli G (2010). Maintaining cell identity through global control of genomic organization. Immunity 33, 12–24. [DOI] [PubMed] [Google Scholar]
- Neph S, Kuehn MS, Reynolds AP, Haugen E, Thurman RE, Johnson AK, Rynes E, Maurano MT, Vierstra J, Thomas S, et al. (2012). BEDOPS: high-performance genomic feature operations. Bioinformatics 28, 1919–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noordermeer D, Leleu M, Schorderet P, Joye E, Chabaud F, and Duboule D (2014). Temporal dynamics and developmental memory of 3D chromatin architecture at Hox gene loci. Elife 3, e02557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noordermeer D, Leleu M, Splinter E, Rougemont J, De Laat W, and Duboule D (2011). The dynamic architecture of Hox gene clusters. Science 334, 222–225. [DOI] [PubMed] [Google Scholar]
- Nora EP, Goloborodko A, Valton AL, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, and Bruneau BG (2017). Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 169, 930–944 e922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. (2012). Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong CT, and Corces VG (2014). CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet 15, 234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palstra RJ, Tolhuis B, Splinter E, Nijmeijer R, Grosveld F, and de Laat W (2003). The beta-globin nuclear compartment in development and erythroid differentiation. Nat Genet 35, 190–194. [DOI] [PubMed] [Google Scholar]
- Pang ZP, Yang N, Vierbuchen T, Ostermeier A, Fuentes DR, Yang TQ, Citri A, Sebastiano V, Marro S, Sudhof TC, et al. (2011). Induction of human neuronal cells by defined transcription factors. Nature 476, 220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfisterer U, Kirkeby A, Torper O, Wood J, Nelander J, Dufour A, Bjorklund A, Lindvall O, Jakobsson J, and Parmar M (2011). Direct conversion of human fibroblasts to dopaminergic neurons. Proc Natl Acad Sci U S A 108, 10343–10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phanstiel DH, Van Bortle K, Spacek D, Hess GT, Shamim MS, Machol I, Love MI, Aiden EL, Bassik MC, and Snyder MP (2017). Static and Dynamic DNA Loops form AP-1-Bound Activation Hubs during Macrophage Development. Mol Cell 67, 1037–1048 e1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips-Cremins JE, Sauria ME, Sanyal A, Gerasimova TI, Lajoie BR, Bell JS, Ong CT, Hookway TA, Guo C, Sun Y, et al. (2013). Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 153, 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri PL, Iezzi S, Stiegler P, Chen TT, Schiltz RL, Muscat GE, Giordano A, Kedes L, Wang JY, and Sartorelli V (2001). Class I histone deacetylases sequentially interact with MyoD and pRb during skeletal myogenesis. Mol Cell 8, 885–897. [DOI] [PubMed] [Google Scholar]
- Puri PL, and Sartorelli V (2000). Regulation of muscle regulatory factors by DNA-binding, interacting proteins, and post-transcriptional modifications. J Cell Physiol 185, 155–173. [DOI] [PubMed] [Google Scholar]
- Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, Conway SJ, Fu JD, and Srivastava D (2012). In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 485, 593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remeseiro S, Hornblad A, and Spitz F (2016). Gene regulation during development in the light of topologically associating domains. Wiley Interdiscip Rev Dev Biol 5, 169–185. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Carballo E, Lopez-Delisle L, Zhan Y, Fabre PJ, Beccari L, El-Idrissi I, Huynh THN, Ozadam H, Dekker J, and Duboule D (2017). The HoxD cluster is a dynamic and resilient TAD boundary controlling the segregation of antagonistic regulatory landscapes. Genes Dev 31, 2264–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala D, Cunningham TJ, Stec MJ, Etxaniz U, Nicoletti C, Dall’Agnese A, Puri PL, Duester G, Latella L, and Sacco A (2019). The Stat3-Fam3a axis promotes muscle stem cell myogenic lineage progression by inducing mitochondrial respiration. Nat Commun 10, 1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanborn AL, Rao SS, Huang SC, Durand NC, Huntley MH, Jewett AI, Bochkov ID, Chinnappan D, Cutkosky A, Li J, et al. (2015). Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci U S A 112, E6456–6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartorelli V, and Puri PL (2018). Shaping Gene Expression by Landscaping Chromatin Architecture: Lessons from a Master. Mol Cell 71, 375–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub JR, Huppert KA, Kurial SNT, Hsu BY, Cast AE, Donnelly B, Karns RA, Chen F, Rezvani M, Luu HY, et al. (2018). De novo formation of the biliary system by TGFbeta-mediated hepatocyte transdifferentiation. Nature 557, 247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer T, Ghavi-Helm Y, Sexton T, Albig C, Regnard C, Cavalli G, Furlong EE, and Becker PB (2017). Chromosome topology guides the Drosophila Dosage Compensation Complex for target gene activation. EMBO Rep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt AD, Hu M, Jung I, Xu Z, Qiu Y, Tan CL, Li Y, Lin S, Lin Y, Barr CL, et al. (2016a). A Compendium of Chromatin Contact Maps Reveals Spatially Active Regions in the Human Genome. Cell Rep 17, 2042–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt AD, Hu M, and Ren B (2016b). Genome-wide mapping and analysis of chromosome architecture. Nat Rev Mol Cell Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuijers J, Manteiga JC, Weintraub AS, Day DS, Zamudio AV, Hnisz D, Lee TI, and Young RA (2018). Transcriptional Dysregulation of MYC Reveals Common Enhancer-Docking Mechanism. Cell Rep 23, 349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant N, Lajoie BR, Nora EP, Giorgetti L, Chen CJ, Heard E, Dekker J, and Barillot E (2012). HiTC: exploration of high-throughput ‘C’ experiments. Bioinformatics 28, 2843–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant N, Varoquaux N, Lajoie BR, Viara E, Chen CJ, Vert JP, Heard E, Dekker J, and Barillot E (2015). HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol 16, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, and Cavalli G (2012). Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 148, 458–472. [DOI] [PubMed] [Google Scholar]
- Siersbaek R, Madsen JGS, Javierre BM, Nielsen R, Bagge EK, Cairns J, Wingett SW, Traynor S, Spivakov M, Fraser P, et al. (2017). Dynamic Rewiring of Promoter-Anchored Chromatin Loops during Adipocyte Differentiation. Mol Cell 66, 420–435 e425. [DOI] [PubMed] [Google Scholar]
- Singh K, Cassano M, Planet E, Sebastian S, Jang SM, Sohi G, Faralli H, Choi J, Youn HD, Dilworth FJ, et al. (2015). A KAP1 phosphorylation switch controls MyoD function during skeletal muscle differentiation. Genes Dev 29, 513–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielmann M, Lupianez DG, and Mundlos S (2018). Structural variation in the 3D genome. Nat Rev Genet 19, 453–467. [DOI] [PubMed] [Google Scholar]
- Spielmann M, and Mundlos S (2016). Looking beyond the genes: the role of non-coding variants in human disease. Hum Mol Genet 25, R157–R165. [DOI] [PubMed] [Google Scholar]
- Spitz F, and Furlong EE (2012). Transcription factors: from enhancer binding to developmental control. Nat Rev Genet 13, 613–626. [DOI] [PubMed] [Google Scholar]
- Stadhouders R, Vidal E, Serra F, Di Stefano B, Le Dily F, Quilez J, Gomez A, Collombet S, Berenguer C, Cuartero Y, et al. (2018). Transcription factors orchestrate dynamic interplay between genome topology and gene regulation during cell reprogramming. Nat Genet 50, 238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, and Cooper DN (2003). Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat 21, 577–581. [DOI] [PubMed] [Google Scholar]
- Sun F, Chronis C, Kronenberg M, Chen XF, Su T, Lay FD, Plath K, Kurdistani SK, and Carey MF (2019). Promoter-Enhancer Communication Occurs Primarily within Insulated Neighborhoods. Mol Cell 73, 250–263 e255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symmons O, Pan L, Remeseiro S, Aktas T, Klein F, Huber W, and Spitz F (2016). The Shh Topological Domain Facilitates the Action of Remote Enhancers by Reducing the Effects of Genomic Distances. Dev Cell 39, 529–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, and Yamanaka S (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. [DOI] [PubMed] [Google Scholar]
- Tsai PF, Dell’Orso S, Rodriguez J, Vivanco KO, Ko KD, Jiang K, Juan AH, Sarshad AA, Vian L, Tran M, et al. (2018). A Muscle-Specific Enhancer RNA Mediates Cohesin Recruitment and Regulates Transcription In trans. Mol Cell 71, 129–141 e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui S, Wang J, Wang L, Dai W, and Lu L (2016). CTCF-Mediated and Pax6-Associated Gene Expression in Corneal Epithelial Cell-Specific Differentiation. PLoS One 11, e0162071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunemoto R, Lee S, Szucs A, Chubukov P, Sokolova I, Blanchard JW, Eade KT, Bruggemann J, Wu C, Torkamani A, et al. (2018). Diverse reprogramming codes for neuronal identity. Nature 557, 375–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, and Wernig M (2010). Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada R, Muraoka N, Inagawa K, Yamakawa H, Miyamoto K, Sadahiro T, Umei T, Kaneda R, Suzuki T, Kamiya K, et al. (2013). Induction of human cardiomyocyte-like cells from fibroblasts by defined factors. Proc Natl Acad Sci U S A 110, 12667–12672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub AS, Li CH, Zamudio AV, Sigova AA, Hannett NM, Day DS, Abraham BJ, Cohen MA, Nabet B, Buckley DL, et al. (2017). YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 171, 1573–1588 e1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub H, Davis R, Tapscott S, Thayer M, Krause M, Benezra R, Blackwell TK, Turner D, Rupp R, Hollenberg S, et al. (1991). The myoD gene family: nodal point during specification of the muscle cell lineage. Science 251, 761–766. [DOI] [PubMed] [Google Scholar]
- Weintraub H, Tapscott SJ, Davis RL, Thayer MJ, Adam MA, Lassar AB, and Miller AD (1989). Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines by forced expression of MyoD. Proc Natl Acad Sci U S A 86, 5434–5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Legendre NP, Biswas AA, Lawton A, Yamamoto S, Tajbakhsh S, Kardon G, and Goldhamer DJ (2018). Loss of MyoD and Myf5 in Skeletal Muscle Stem Cells Results in Altered Myogenic Programming and Failed Regeneration. Stem Cell Reports 10, 956–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan KK, Yardimci GG, Yan C, Noble WS, and Gerstein M (2017). HiC-spector: a matrix library for spectral and reproducibility analysis of Hi-C contact maps. Bioinformatics 33, 2199–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, Sandstrom R, Ma Z, Davis C, Pope BD, et al. (2014). A comparative encyclopedia of DNA elements in the mouse genome. Nature 515, 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Related to STAR Methods. Hi-C library quality.
This table reports the number and percentage of mapped reads, percentage of valid putative interaction pairs, percentage and number of unique read pairs, percentage and number of cis- or trans- read pairs.
Data Availability Statement
Sequencing data have been deposited in the GEO database under the accession number GEO: GSE98530. This work also used previous sequencing data deposited in the GEO database: GEO: GSM935404, GSM733762, GSM733783, GSM1055818, GSM915188, GSM733666, GSM733755, GSM915165, GSE128527, GSM2417196, GSM2417197, GSM2417198, GSM2417204, GSM2417203 (Chronis et al., 2017; Consortium, 2012; Jin et al., 2013; Sala et al., 2019; Yue et al., 2014). Codes generated for this study have been deposited to Mendeley.
DNA FISH analysis:
DOI: http://dx.doi.org/10.17632/tvb7yvpr4s.1#file-b6ac28c9-29d2-48ae-9658-ecf953457b98 and http://dx.doi.org/10.17632/tvb7yvpr4s.1#file-01a31ffd-8040-41cc-9b0e-98d9a4ac9c88
Circular permutation:
DOI: http://dx.doi.org/10.17632/rvzzpsh6vg.2#file-96dfb6d0-d6e3-4a42-ac13-a78fb2346b27