

Abstract
Mice that globally overexpress the transcription factor Fos-related antigen-2 (Fra-2) develop extensive pulmonary fibrosis and pulmonary vascular remodeling. To determine if these phenotypes are a consequence of ectopic Fra-2 expression in vascular smooth muscle cells and myofibroblasts, we generated mice that overexpress Fra-2 specifically in these cell types (α-SMA-rtTA;tetO-Fra-2). Surprisingly, these mice did not develop vascular remodeling or pulmonary fibrosis but did develop a spontaneous emphysema-like phenotype characterized by alveolar enlargement. Secondary septa formation is an important step in the normal development of lung alveoli, and α-smooth muscle actin (SMA)-expressing fibroblasts (myofibroblasts) play a crucial role in this process. The mutant mice showed reduced numbers of secondary septa at postnatal day 7 and enlarged alveolae starting at postnatal day 12, suggesting impairment of secondary septa formation. Lineage tracing using α-SMA-rtTA mice crossed to a floxed TdTomato reporter revealed that embryonic expression of α-SMA Cre marked a population of cells that gave rise to nearly all alveolar myofibroblasts. Comprehensive transcriptome analyses (RNA sequencing) demonstrated that the overwhelming majority of genes whose expression was significantly altered by overexpression of Fra-2 in myofibroblasts encoded secreted proteins, components of the extracellular matrix (ECM), and cell adhesion-associated genes, including coordinate upregulation of pairs of integrins and their principal ECM ligands. In addition, primary myofibroblasts isolated from the mutant mice showed reduced migration capacity. These findings suggest that Fra-2 overexpression might impair myofibroblast functions crucial for secondary septation, such as myofibroblast migration across alveoli, by perturbing interactions between integrins and locally produced components of the ECM.
Keywords: Fos-related antigen-2, alveolarization, septation, myofibroblasts, lung development
secondary septa formation is critical for alveolarization of the saccular lung in normal lung development. During this process, budding secondary septa form new walls between alveoli that result in significantly increased alveolar surface area and, thereby, expansion of future gas exchange capabilities (8, 30, 33). Since human secondary alveolar septa formation occurs in the last trimester of gestation and extends into the postnatal period, premature infants have a high risk for impairment of lung alveolarization. One example is bronchopulmonary dysplasia (BPD), a chronic lung disease characterized by persistent respiratory dysfunction in extremely premature infants (21, 33). Because of advances in neonatal care, pathological changes associated with classical BPD (severe fibrosis, epithelial metaplasia, and smooth muscle hypertrophy in small airways) have become infrequent. In the new classification of BPD, however, the arrest of secondary alveolar septation and subsequent respiratory dysfunction remain a serious cause of morbidity, especially in very preterm infants born before 28 wk gestational age (1, 2, 4, 19).
Several previous studies revealed that two distinct alveolar fibroblast subsets, myofibroblasts and lipofibroblasts, play crucial roles in the formation of secondary septa (10, 40). Thus, proper differentiation of these two fibroblast subpopulations is important for normal alveolarization (24, 31, 34). Lipofibroblasts contain lipid droplets and localize in the central region of the alveolar septum. Lipofibroblasts serve as an important storage site for retinoic acid, which is crucial for development of new alveolar septa (27, 29). Myofibroblasts are identified during development as α-smooth muscle actin (SMA)-expressing alveolar interstitial cells. During the saccular phase that precedes alveolar septation, myofibroblast precursors exist on the wall of prospective terminal sacs; in the alveolar phase, they are present at the tips of secondary septa and are considered to regulate secondary septa formation (10). Platelet-derived growth factor subunit A (PDGFA) signaling is crucial for myofibroblast differentiation and migration. In PDGFA-null mice, myofibroblast precursors are absent from the primary septa, resulting in severe alveolarization defects (5, 20, 25). Myofibroblasts are thus thought to be crucial for alveolar septation, and dysregulation of myofibroblast function is assumed to underlie the pathogenesis of BPD (3, 20). However, the molecular pathways that regulate myofibroblast function during alveolar septation have not been fully elucidated.
Fos-related antigen-2 (Fra-2), a transcription factor and a member of the activator protein 1 family, modulates a variety of cell functions, including migration, proliferation, and apoptosis, by regulating specific gene expression (41, 42). Previously, we and others reported that Fra-2 transgenic mice developed severe lung structural alterations, including fibrosis, vascular remodeling, and inflammation (12, 35, 36). However, in Fra-2 transgenic mice, Fra-2 is globally and constitutively expressed, and it is difficult to determine the contributions of altered gene expression in specific cell types. To test the hypothesis that vascular remodeling and tissue fibrosis in Fra-2 transgenic mice are due to overexpression of Fra-2 in vascular smooth muscle cells and myofibroblasts, we generated mice with inducible overexpression of Fra-2 under control of the α-SMA promoter. Surprisingly, these mice did not develop pulmonary vascular remodeling or fibrosis but did develop a simplified alveolar architecture with fewer and larger alveoli. We further determined that the simplified alveolar architecture in smaFra-2 mice is due, at least in part, to a defect in alveolar secondary septa formation. Genome-wide expression analysis of purified myofibroblasts from developing lungs suggested that this effect might be explained by Fra-2-mediated modulation of extracellular matrix (ECM) production and cell adhesion.
MATERIALS AND METHODS
Mice.
tetO-Fra-2 mice were generated as previously described (16). α-SMA-rtTA, tetO-Cre, and PDGFRα-GFP mice were already well established (9, 15). Ai14 (Rosa26-LSL-TdTomato) mice were obtained from the Jackson Laboratory (26). Experimental mice used in this study were tetO-Fra-2 homozygous/α-SMA-rtTA-positive mice, and controls were tetO-Fra-2 homozygous/α-SMA-rtTA-negative mice and, in selected experiments, tetO-Fra-2 homozygous/α-SMA-rtTA-positive mice not fed doxycycline. The genotypes of these transgenic mice were determined by PCR analysis using tail DNA. All the mice were on a pure C57BL/6 background or were backcrossed at least six times to C57BL/6. Mice were maintained in specific-pathogen-free conditions in the Animal Barrier Facility of the University of California, San Francisco. All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco.
Histological evaluation of alveolar size.
Ten fields from 5-μm-thick lung sections obtained in a blinded manner were randomly selected and stained with hematoxylin-eosin for histological analyses, and the mean alveolar chord length was automatically calculated from ~4,000 readouts per lung using Scion Image software and macros provided by Caroline A. Owen (Harvard Medical School) (22). For secondary crest analyses, the number of >10-μm crests per field was counted, and the mean values from 10 fields per mouse were compared between smaFra-2 and control mice.
Lung function analyses.
Mice were anesthetized with ketamine (100 mg/kg), xylazine (10 mg/kg), and acepromazine (3 mg/kg) and paralyzed with pancuronium (0.1 mg/kg). Pulmonary compliance and elastance were determined using the flexiVent system (SCIREQ), as previously described (9). All analyses were performed in a blinded fashion.
Bronchoalveolar lavage fluid analysis.
Tracheas were cannulated, and lungs were lavaged three times with 1 ml of PBS. Numbers of cells in lavage fluid were counted using a hemocytometer. The bronchoalveolar lavage fluid (BALF) was centrifuged (Cytospin) at 500 g for 5 min, and differential leukocyte counts were performed on a smear stained with Hema3 (Fisher Scientific, Pittsburgh, PA) (35).
Single-cell tissue dissociation for flow cytometry.
Tissue dissociation was conducted as described previously with minor modifications (35). Briefly, lung tissues were excised, minced with scissors, and digested in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen) containing liberase (0.13 IU/ml; Sigma-Aldrich, St. Louis, MO) at 37°C for 20 min. Single-cell suspensions prepared with a gentleMACS dissociator (Miltenyi Biotec) were passed through 100- and 40-μm cell strainers and suspended in PBS supplemented with 50% fetal bovine serum. After live/dead staining with 4′,6-diaminido-2-phenylindole (DAPI; Sigma-Aldrich), live single cells with reporters were sorted using a FACSAria (BD Biosciences).
Quantitative RT-PCR.
An RNeasy Plus kit (Qiagen, Venlo, The Netherlands) was used to isolate RNA from lung tissues or cells. cDNA was analyzed by SYBR Green RT-PCR with a thermocycler (model 7900HT, Applied Biosystems) and normalized to Gapdh expression. Primers are listed in Table 1.
Table 1.
Primers used for quantitative RT-PCR
| Primer |
|||
|---|---|---|---|
| Gene Abbreviation | Gene Definition | Forward | Reverse |
| Gapdh | Glyceride phosphate dehydrogenase | AGG TCG GTG TGA ACG GAT TTG | GGG GTC GTT GAT GGC AAC A |
| Acta2 | α2-Actin, smooth muscle | CCC AGA CAT CAG GGA GTA ATG G | TCT ATC GGA TAC TTC AGC GTC A |
| Tagln | Transgelin | CCA ACA AGG GTC CAT CCT ACG | ATC TGG GCG GCC TAC ATC A |
| Epcam | Epithelial cell adhesion molecule | CTG GCG TCT AAA TGC TTG GC | CCT TGT CGG TTC TTC GGA CTC |
| Pecam | Platelet and endothelial cell adhesion molecule | ACG CTG GTG CTC TAT GCA AG | TCA GTT GCT GCC CAT TCA TCA |
| Elastin | TTG CTG ATC CTC TTG CTC AAC | GCC CCT GGA TAA TAG ACT CCA C | |
| ADRP | Adipose differentiation-related protein | GAC CTT GTG TCC TCC GCT TAT | CAA CCG CAA TTT GTG GCT C |
| Thy1 | Thymus cell antigen 1 | TGC TCT CAG TCT TGC AGG TG | TGG ATG GAG TTA TCC TTG GTG TT |
| Pparg | Peroxisome proliferator-activated receptor-γ | GGA AGA CCA CTC GCA TTC CTT | GTA ATC AGC AAC CAT TGG GTC A |
| LPA1 | Lysophosphatidic acid receptor type 1 | AGC CAT GAA CGA ACA ACA GTG | CAT GAT GAA CAC GCA AAC AGT G |
| Fra-2-Flag | Fos-related antigen-2-Flag | TGG GGA CCA GTC ATC AGA C | TGT CGT CGT CGT CCT TGT AG |
| Col1a1 | Collagen I-α1 | GCT CCT CTT AGG GGC CAC T | CCA CGT CTC ACC ATT GGG G |
| Col3a1 | Collagen III-α1 | CTG TAA CAT GGA AAC TGG GGA AA | CCA TAG CTG AAC TGA AAA CCA CC |
| Col4a1 | Collagen IV-α1 | CTG GCA CAA AAG GGA CGA G | ACG TGG CCG AGA ATT TCA CC |
| Pdgfra | Platelet-derived growth factor receptor-α | AGA GTT ACA CGT TTG AGC TGT C | GTC CCT CCA CGG TAC TCC T |
| Itga5 | Integrin-α5 | CTT CTC CGT GGA GTT TTA CCG | GCT GTC AAA TTG AAT GGT GGT G |
| Itga8 | Integrin-α8 | TGG CTG GGA TTC CAA GAG GA | GTG CCC CGA CCA ATA TGT CA |
| Enpp2 | Ectonucleotide pyrophosphatase/phosphodiesterase 2 | TTT GCA CTA TGC CAA CAA TCG G | GGA GGC ACT TTA GTC CTG TAC TT |
| Lama1 | Laminin-α1 | CCT GGA CTT ACG GCA GGT C | AGG GTT TGA ACT TGA CGC CAT |
| Sftpc | Surfactant protein C | ATG GAG AGT CCA CCG GAT TAC | ACC ACG ATG AGA AGG CGT TTG |
| SCGB1A1 (CC10) | Secretoglobulin, family 1A, member 1 | ATG AAG ATC GCC ATC ACA ATC AC | GGA TGC CAC ATA ACC AGA CTC T |
| Pdpn | Podoplanin | ACC GTG CCA GTG TTG TTC TG | AGC ACC TGT GGT TGT TAT TTT GT |
| Aebp1 | Adipocyte enhancer-binding protein 1 | TTG GAA ACG CTG GAT CGG TTA | CTT GAC CTT GCC AGG CAT TT |
| Mmp2 | Matrix metalloproteinase 2 | CAA GTT CCC CGG CGA TGT C | TTC TGG TCA AGG TCA CCT GTC |
| Mmp9 | Matrix metalloproteinase 9 | CAA GTT CCC CGG CGA TGT C | CTG TCG GCT GTG GTT CAG T |
| Mmp12 | Matrix metalloproteinase 12 | CTG CTC CCA TGA ATG ACA GTG | AGT TGC TTC TAG CCC AAA GAA C |
| Npnt | Nephronectin | GAA GCC TCG GCC CTG TAA G | AGC ATG TAT CCG TTG AGA CAG TA |
| Dcn | Decorin | GTC ATC TTC GAG TGG TGC AGT | CAA GGT TGT GTC GGG TGG AAA |
| Igf1 | Insulin-like growth factor I | CTG GAC CAG AGA CCC TTT GC | GGA CGG GGA CTT CTG AGT CTT |
Preparation of cDNA library and RNA sequencing.
TdTomato-positive and DAPI-negative cells (α-SMA-expressing lineage cells) were sorted from α-SMA-rtTA;tetO-Cre;Ai14 reporter mice with (smaFra-2 mice, n = 4) or without (control mice, n = 4) the tetO-Fra-2 transgene at 7 days of age using a FACSAria, as described above. Cells were directly sorted into RLT Plus buffer (Qiagen) and stored at −80°C until RNA extraction. Total RNA was extracted from lysates using RNeasy Plus Micro kits (Qiagen) according to the manufacturer’s instructions. RNA was quantified using a photometer (model ND-1000, NanoDrop Technologies), and RNA quality was confirmed using a bioanalyzer (model 2100, Agilent Technologies, Palo Alto, CA). mRNA was isolated from 50 ng of total RNA using a Dynabeads mRNA purification kit (Ambion). The Ovation RNA-Seq System V2 (NuGEN Technologies, San Carlos, CA) was used for cDNA amplification from selected mRNA, and then cDNA libraries were prepared using the Nextera XT DNA library preparation kit (Illumina, San Diego, CA). Differentially expressed genes between control and smaFra-2 mice (false discovery rate-adjusted P < 0.05) were identified and analyzed using DAVID (Database for Annotation, Visualization, and Integrated Discovery) (17).
Immunohistochemistry and immunofluorescence.
Paraffin-embedded sections were deparaffinized, rehydrated, and heated in citrate buffer (10 mM, pH 6.0), as described previously (37). For frozen tissue sections, mouse lungs were inflated through the trachea and fixed in 4% paraformaldehyde overnight at 4°C. Thereafter, 4% paraformaldehyde was replaced with PBS containing 20% sucrose, and tissue was snap-frozen in OCT embedding compound (Tissue Tek) and stored at −80°C until use. Ten-micrometer-thick sections were cut using a cryostat microtome (Leica, Deerfield, IL), placed onto gelatin-coated slides, and air-dried. After the sections were blocked for 30 min at room temperature in blocking buffer (PBS containing 5% serum, 0.5% bovine serum albumin, and 0.1% Triton X-100), they were stained with the primary antibodies: Alexa 488-conjugated anti-α-SMA (clone 1A4, Sigma-Aldrich) (35), goat anti-Flag (Abcam, Cambridge, UK) (13), and rat anti-Fra-2 (clone REY146C) (16).
Migration assay.
The migratory capacity of lung myofibroblasts was evaluated using an Oris cell migration assay kit (fibronectin-coated) (Platypus Technologies, Madison, WI), as previously described with minor modification (38). Briefly, after isolation of TdTomato-positive and DAPI-negative cells from α-SMA-rtTA;tetO-Cre;Ai14 reporter mice, 1 × 105 cells in 100 μl of DMEM containing 10% fetal bovine serum were dispensed into each well of a 96-well plate containing a cell-seeding stopper. After an overnight incubation in a CO2 incubator, the stopper was removed and the wells were washed with PBS to remove nonadherent cells. Images of the TdTomato-positive cells were acquired using a confocal microscope (Carl Zeiss Microscopy, Jena, Germany) after a further 0, 12, 24, or 36 h of incubation. The area of TdTomato-positive cells in the central circle of each well was quantified using ImageJ version 1.51n. The percentage of the covered area was calculated by subtraction of the 0-h value from the value at each time point.
Western blotting.
Lung tissue samples were lysed in RIPA buffer [0.1% SDS, 0.5% deoxycholate, 1% NP-40, 150 mM NaCl, and 50 mM Tris (pH 8.0)] supplemented with protease and phosphatase inhibitors. Lysates containing equal amounts of protein were electrophoresed via SDS-PAGE and transferred to Immobilon-P membranes (Millipore, Billerica, MA). Membranes were probed using primary antibodies followed by peroxidase-conjugated secondary antibodies. The primary antibodies were GAPDH (Cell Signaling Technology, Danvers, MA) and Fra-2 (clone REY146C) (16). For densitometry, blots were analyzed using ImageJ software.
Statistical analysis.
Values are means ± SE. The significance of differences between two sample means was determined by two-tailed Student’s t-test. P < 0.05 was considered statistically significant. Statistical analyses were carried out using GraphPad Prism software.
RESULTS
Transgenic mice with Fra-2 overexpressed in α-SMA-expressing cells spontaneously display alveolar simplification.
To determine cell-specific roles of Fra-2 in cells that express α-SMA in vivo, we generated transgenic mice in which Fra-2 was specifically expressed in α-SMA-expressing cells in the presence of doxycycline by crossing α-SMA-rtTA mice with tetO-Fra-2 mice (smaFra-2 mice; Fig. 1A). To evaluate the contribution of α-SMA-expressing cells that could arise at any stage of normal lung development or the progression of vascular remodeling and fibrosis, Fra-2 transgenes were induced in the mutant mice by continuous doxycycline feeding to both mothers and pups starting from the embryonic stage (embryonic day 0) and continuing until they were euthanized at 15 wk of age (Fig. 1B). Fra-2 transgene expression (Fra-2-Flag fusion proteins) was specifically detected in lung airway and vascular smooth muscle cells (Fig. 1C), supporting the efficiency and cell specificity of this conditional transgenic mouse line. We did not detect fibrosis, vascular remodeling, or inflammation in the lungs of 15-wk-old mice (Fig. 1D), but alveoli were significantly larger in mutant lungs than in lungs of littermate controls (Fig. 1E). Physiological analyses revealed increased lung compliance and decreased lung elastance in the mutant mice (Fig. 1F), consistent with alveolar simplification. The phenotype was not apparent in the α-SMA-rtTA-positive/tetO-Fra-2-negative mice or in the α-SMA-rtTA-positive/tetO-Fra-2-positive mice without doxycycline feeding, further supporting the idea that Fra-2 overexpression in α-SMA-expressing cells is crucial for the phenotype.
Fig. 1.

Conditional Fos-related antigen-2 (Fra-2) overexpression in α-smooth muscle actin (SMA)-positive cells results in an emphysema-like phenotype. A: construct of α-SMA-rtTA;tetO-Fra-2 (smaFra-2) mice, in which Fra-2 is overexpressed specifically in α-SMA-expressing cells by doxycycline (Dox) administration. B: transgenic mice were continuously fed doxycycline to induce Fra-2 expression until they were euthanized at 15 wk of age. C: Fra-2-Flag fusion proteins were specifically detected in nuclei of airway and vascular smooth muscle cells. Scale bar = 25 μm. D: representative hematoxylin-eosin (H and E)-stained lung sections from a smaFra-2 (α-SMA-rtTA+/−;tetO-Fra-2+/+) mouse and its littermate control (α-SMA-rtTA−/−;tetO-Fra-2+/+) at 15 wk of age. smaFra-2 mice showed no apparent fibrotic changes, vascular remodeling, or alveolar inflammation, but alveolar size was increased. Scale bar = 100 μm. E: mean chord length was significantly increased in lungs of smaFra-2 mice (n = 5 mice in each group). F: lung compliance was significantly increased, while lung elastance was significantly decreased, in smaFra-2 mice (n = 5 mice in each group).
Alveolar simplification in the mutant mice is, at least partially, attributable to a defect in secondary septation.
Since enlarged alveolar spaces in smaFra-2 mice were apparent as early as postnatal day 25 (P25; Fig. 2A), we carefully examined the early stages of alveolar development. In normal mice, secondary septa form between P7 and P12, and all postnatal alveolarization is completed by P25 (8). Alveoli were larger at P12 and P25 in smaFra-2 mice than littermate controls, while alveolar size at P7 was similar between the two groups (Fig. 2, B and C). Furthermore, the number of secondary crests was significantly decreased at P7 in the smaFra-2 mouse lungs compared with controls (Fig. 2D), suggesting that impairment of the maturation of secondary septa in the mutant lungs is likely responsible for the alveolar enlargement we observed.
Fig. 2.

Secondary septation is impaired in lungs from smaFra-2 mice. A: gross appearance of hematoxylin-eosin-stained lung sections from a smaFra-2 mouse and its littermate control at postnatal day 25 (P25). Alveolar enlargement in smaFra-2 mice was already apparent at this age. Scale bar = 400 μm. B: hematoxylin-eosin-stained representative lung sections from smaFra-2 mice and their littermate controls at P7, P12, and P25. Scale bar = 100 μm. C: mean cord length was significantly increased in lungs of smaFra-2 mice at P12 and P25, but not P7 (n = 5 mice in each group). *P < 0.01, **P < 0.001. D: numbers of alveolar crests were significantly decreased in smaFra-2 lungs at P7 (n = 5 mice in each group). *P < 0.01. E: representative elastin Hart’s-stained lung sections from smaFra-2 and control mice at P7. Scale bar = 100 μm. F: no significant differences were observed in numbers of cells in bronchoalveolar lavage fluid (BALF) between control (n = 7) and smaFra-2 (n = 6) mice at P25. MΦ, macrophages; Lym, lymphocytes; Gra, granulocytes; NS, not significant. G: relative mRNA expression in whole lung lysates of control (n = 10) and smaFra-2 (n = 10) mice. See Table 1 for definitions of gene abbreviations.
In smaFra-2 mouse lungs at P7, there were no apparent pathological changes in elastin accumulation (elastin Hart’s staining; Fig. 2E). Furthermore, there were no apparent differences in the expression of cell markers for airway epithelium [secretoglobulin, family 1A, member 1 (CC10)] or alveolar epithelium type 1 [podoplanin (Pdpn)] or type 2 [surfactant protein C (Sftpc)] between the two groups. In addition, expression of matrix metalloproteinases (Mmp2, Mmp9, and Mmp12) and collagen (Col1a1, Col3a1, and Col4a1) in the total lungs at P7 was comparable between the two groups (Fig. 2G), and we did not observe morphological evidence of inflammation or fibrosis. The number of inflammatory cells in the alveolar area was also similar between the two groups at P25 (Fig. 2F). Myofibroblasts and lipofibroblasts are two major fibroblast subsets considered to be crucially involved in secondary septation. However, the proportion of lipid-containing cells evaluated by Oil-Red staining (data not shown) and whole lung gene expression for general markers of fibroblasts [platelet-derived growth factor receptor-α (Pdgfra)], myofibroblasts [α2-actin, smooth muscle (Acta2)], or lipofibroblasts [adipose differentiation-related protein (ADRP), thymus cell antigen 1 (Thy1), and peroxisome proliferator-activated receptor-γ (Pparg)] were not changed in the lungs of smaFra-2 mice (Fig. 2G).
Fra-2 is overexpressed in alveolar myofibroblasts during alveolarization in lungs of smaFra-2 mice.
We evaluated α-SMA and Fra-2 expression in developing lungs of wild-type and smaFra-2 mice. In parallel with secondary septa formation, α-SMA expression in the total mouse lungs was dramatically increased in both groups, especially between P4 and P9 (Fig. 3, A and B). These changes in α-SMA expression were similarly observed in smaFra-2 mice compared with control wild-type mice (Fig. 3B). As previously reported, α-SMA expression was well detected in alveolar interstitial cells of mouse lungs, especially at the tips of secondary septa (Fig. 3A, arrowheads), between P4 and P12, but not at P0 or P25 (Fig. 3A). In contrast, there were no apparent changes in Fra-2 expression during the postnatal alveolarization period. In accordance with the increase in α-SMA expression, Fra-2 transgene (Fra-2-Flag) was significantly upregulated shortly after birth and then gradually decreased (Fig. 3C). At P7, total Fra-2 protein expression was significantly increased in smaFra-2 lungs (Fig. 3D) and localized in the nuclei of myofibroblasts in the alveolar area (Fig. 3, E and F). In contrast, in wild-type lungs, Fra-2 protein expression was below the detection limit of our staining protocol.
Fig. 3.

Fra-2 is overexpressed in myofibroblasts in the alveolar region of lungs from smaFra-2 mice. A: representative α-SMA staining in lungs from wild-type mice at P0, P4, P7, P12, and P25. Arrowheads indicate myofibroblasts localizing at tips of secondary septa. A, airway; V, vessel. B: relative mRNA expression in whole lung lysates. Acta2 gene expression was significantly increased between P4 and P9 in wild-type (WT) and smaFra-2 mice (n = 2–4 in each group). C: no significant changes were observed in Fra-2 gene expression in whole lungs during postnatal lung development (n = 2–4 in each group). D: relative mRNA expression of Fra-2 transgene (Fra-2-Flag) in lungs from smaFra-2 mice (n = 2–4 in each group). E: Fra-2 expression was increased in whole lung lysates from smaFra-2 mice compared with littermate control mice at P7. Representative immunoblots and quantitative measurement by densitometry (n = 3 in each group) are shown. F: representative lung sections stained with antibody against Fra-2 from smaFra-2 and littermate control mice at P7. Fra-2 (red) was predominantly observed in nuclei of α-SMA-expressing myofibroblasts (green) in lungs from smaFra-2 mice. G: Fra-2 intensity in nuclei of alveolar myofibroblasts was calculated using ImageJ.
Characterization of progeny of α-SMA lineage-marked cells using α-SMA-rtTA;tetO-Cre reporter mice.
To determine and characterize α-SMA-expressing lineage cell populations in the postnatal lung, α-SMA-rtTA;tetO-Cre;Ai14 transgenic mice, in which TdTomato expression is specifically and permanently induced in α-SMA-expressing lineage cells, were employed (Fig. 4A). In the lungs of the reporter mice, TdTomato expression was localized in α-SMA-positive myofibroblasts, as well as in vascular and airway smooth muscle cells, at P7 (Fig. 4B), supporting the specificity of this reporter line. TdTomato-positive cells were observed in the alveolar area as early as P0, despite minimal detection of α-SMA protein by immunostaining at this time point (Fig. 4C, arrowheads), suggesting that using our reporter mice is likely more sensitive than immunostaining with the antibody available to us. This TdTomato-positive cell population expanded even when doxycycline induction was stopped at P0, with the number of TdTomato-positive cells in the lungs at P7 similar to that in lungs from mice continuously treated with doxycycline (Fig. 4D). These TdTomato-positive cells did not arise from airway or vascular smooth muscle cells (which are already well established before birth), because no TdTomato-positive cells were observed in the alveolar area at P7, when Cre induction was stopped 4 days before birth (Fig. 4E). These data suggest that this reporter line marked essentially all the precursors of myofibroblasts shortly after birth and that the reporter line should be suitable to trace myofibroblast lineage cells in postnatal lungs at least between P0 and P7.
Fig. 4.

Characterization of α-SMA-expressing lineage cells using α-SMA-rtTA;tetO-Cre reporter mice. A: construct of α-SMA-rtTA;tetO-Cre;Ai14flox (sma-Ai14) mice, in which TdTomato is induced specifically in α-SMA-expressing lineage cells in the presence of doxycycline. B–E: representative immunofluorescence of lungs from sma-Ai14 mice at P7 (B, D, and E) and P0 (C). Doxycycline was given starting from the early embryonic stage to P7 (B) and continuing to day 0 (C and D) or to embryonic day 16 (E). At P0, some alveolar cells express TdTomato with no evidence of α-SMA expression (arrowheads), while interstitial cells in alveolar ducts express both TdTomato and α-SMA (arrow). Proportion of TdTomato-positive cells in myofibroblasts is shown at right. A, airway; V, vessel. Scale bar = 100 μm.
Isolation of α-SMA-expressing lineage cells from α-SMA-rtTA;tetO-Cre reporter mice.
Using α-SMA-rtTA;tetO-Cre reporter mice, we sorted the α-SMA-expressing lineage cells (TdTomato-positive and DAPI-negative) from singlet cells in dissociated lung tissues (Fig. 5A). Quantitative PCR analyses showed that the α-SMA-expressing lineage cells highly express myofibroblast and/or smooth muscle cell markers, including Acta2, transgelin (Tagln), and Pdgfra, compared with control cells or total lung cells (Fig. 5B). In contrast, the α-SMA-expressing lineage cells rarely express genes associated with epithelial cells, endothelial cells, or lipid-containing fibroblasts (Fig. 5B). Furthermore, these lineage cells did not express CD45, which is globally expressed in leukocytes (Fig. 5C). These results further confirm the specificity of this reporter line for myofibroblasts and smooth muscle cells.
Fig. 5.

Isolation of α-SMA-expressing lineage cells from α-SMA-rtTA;tetO-Cre reporter mice. A: live α-SMA-expressing lineage cells [TdTomato-positive and 4′,6-diaminido-2-phenylindole (DAPI)-negative] were sorted (SMA+) from sma-Ai14 mouse lungs at P7. TdTomato-negative cells were sorted as a negative control (Cont). B: relative mRNA expression in α-SMA-expressing lineage cells (SMA+), control cells (Cont), and total lung cells (Total) at P7 (n = 3 in each group). α-SMA-expressing lineage cells exclusively express mesenchymal markers (Acta2, Tagln, and Pdgfra) but rarely express epithelial (Epcam), endothelial (Pecam), or lipofibroblast (Adrp, Thy-1, and Pparg) markers. C: flow cytometry from dissociated lung cells at P7 revealed that α-SMA-expressing lineage cells rarely express the leukocyte marker CD45. D: representative images from lung sections of dual-reporter (PDGFRα-GFP;α-SMA-rtTA;tetO-Cre;Ai14) mice at P7. Significant overlap was observed in expression of TdTomato and GFP. Scale bar = 100 μm. E: representative flow cytometry of lung cells from dual-reporter (PDGFRα-GFP;α-SMA-rtTA;tetO-Cre;Ai14, top right), PDGFRα-GFP (top left), α-SMA-rtTA;tetO-Cre;Ai14 (bottom right), and double-negative (bottom left) mice at P7. Significant overlap was observed between PDGFRα-positive fibroblasts and α-SMA-expressing lineage cells (top right). F: relative mRNA expression changes in α-SMA-expressing lineage cells (n = 3–9 in each group). Fra-2 gene expression was decreased during P2 and P7.
It is well known that PDGFRα is predominantly expressed in lung fibroblasts and that PDGFA signaling plays a crucial role in secondary alveolar septation (5). In this context, PDGFRα-GFP reporter mice have been intensively used to trace the lung fibroblast population in studies of lung development (15, 28). To determine the contribution of PDGFRα-positive lung fibroblasts to the α-SMA-expressing lineage cell population, we generated dual-reporter mice, which carry both PDGFRα-GFP and α-SMA-TdTomato (PDGFRα-GFP;α-SMA-rtTA;tetO-Cre;Ai14). In the lungs of the dual-reporter mouse, significant overlap was observed between the two reporters: >90% of α-SMA-expressing lineage cells (TdTomato-positive) were PDGFRα-positive, suggesting similar populations of α-SMA- and PDGFRα-expressing cells in P7 lungs (Fig. 5, D and E). In these α-SMA-expressing lineage cells, Fra-2 gene expression was slightly decreased between P2 and P7 compared with P0 or P25 (Fig. 5F).
Comprehensive comparisons of transcriptome-identified critical gene expression changes in myofibroblasts of smaFra-2 mice.
To determine critical gene expression changes that might contribute to impaired secondary alveolar septation in the lungs of smaFra-2 mice, we performed RNA sequencing to compare mRNA expression in α-SMA-expressing cells from lungs of P7 smaFra-2 (n = 4) and control (n = 4) mice. A total of 4,3432 transcripts were analyzed; among these, 361 genes were differentially expressed (Fig. 6, A and B; GEO accession no. GSE94857). Functional annotation analyses using DAVID (17) showed that these differentially expressed genes were greatly enriched for transcripts encoding secreted and/or extracellular/cell surface molecules, especially components of the ECM or proteins involved in cell adhesion (Fig. 6C). In the genes categorized as secreted molecules, ectonucleotide pyrophosphatase/phosphodiesterase family member 2 (Enpp2, also known as autotaxin) was significantly decreased. Enpp2 is an enzyme that converts lysophosphatidylcholine (LPC) to lysophosphatidic acid (LPA), and LPA receptor type 1 (LPA1), one of the major LPA receptors through which LPA signaling is transmitted, was also downregulated in the α-SMA-expressing cells of smaFra-2 mice (Fig. 6D).
Fig. 6.

Differentially expressed genes in myofibroblasts of lungs from wild-type and smaFra-2 mice at P7. A: detected and differentially expressed genes in lung myofibroblasts from wild-type and smaFra-2 mice. B: MA plot showing log2 mean read counts on x-axis and log2 fold change on y-axis. Differentially expressed genes are shown in red. C: functional annotation analyses performed using DAVID. The top-20 gene categories are shown. D: relative mRNA expression (by real-time quantitative PCR) of Enpp2 and LPA1 (n = 6–9 in each group). *P < 0.05. E: heat map showing differentially expressed genes categorized as “extracellular matrix” by DAVID. F: quantitative PCR analyses revealed that several ECM genes and genes known to be associated with an active state of fibroblasts were upregulated (n = 6–8 in each group). G: heat map showing genes identified by DAVID as “cell adhesion”-associated genes. H: similar gene expression changes were observed in lung myofibroblasts of smaFra-2 mice at P4, but not P2 (n = 4–6 in each group).
The genes categorized as ECM molecules, summarized in Fig. 6E, include subsets of collagen, fibronectin, laminin, decorin, and nephronectin. Most of these were upregulated in the smaFra-2 group, suggesting that more ECM proteins are locally secreted by myofibroblasts in lungs of smaFra-2 mice than controls. In addition, we found upregulation of several genes that have been implicated in profibrotic activation of fibroblasts, including adipocyte enhancer-binding protein 1 (Aebp1, also known as aortic carboxypeptidase-like protein (Aclp)] (32, 39), insulin-like growth factor I (Igf1) (11, 18), and integrin-α8 (Itga8) (6, 23) (Fig. 6F). In addition, functional annotation analyses revealed upregulation of many cell adhesion-associated genes (Fig. 6G), including two integrin α-subunits (Itga5 and Itga8) and the genes encoding their specific ECM ligands [fibronectin 1 (Fn1) and nephronectin (Npnt)] (Fig. 6F), suggesting the possibility that Fra-2 overexpression enhances the adhesion of myofibroblasts. These gene expression changes in the myofibroblasts at P7 were similarly observed in smaFra-2 lungs at P4, but not P2 (Fig. 6H).
The cell-ECM adhesion strength is crucially implicated in cell migration. There is a bell-curve relationship between adhesion strength and migration speed, with low and very high adhesion strength leading to impaired cell migration. We therefore performed in vitro cell migration assays, tracking cells migrating onto fibronectin-coated dishes, since fibronectin is a ligand for both integrin-α5β1 and integrin-α8β1. We found a decrease in migration on fibronectin in α-SMA-expressing lineage cells from smaFra-2 compared with wild-type mouse lungs at P7 (Fig. 7, A and B). These findings suggest that the gene expression changes induced by Fra-2 overexpression critically affect the migratory function of myofibroblasts, possibly through increased cell-ECM adhesion strength, and that the impaired migration in the myofibroblasts could, at least partially, contribute to the secondary septation defect observed in the smaFra-2 mice.
Fig. 7.
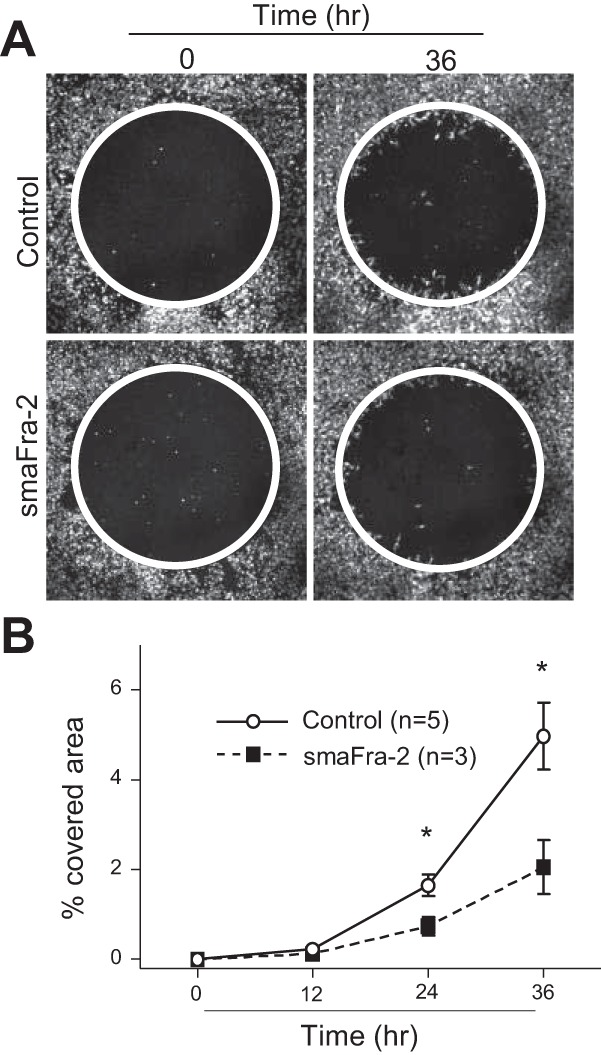
α-SMA-expressing cells isolated from smaFra-2 mouse lungs showed reduced migration on fibronectin. A: α-SMA-expressing (TdTomato-positive) cells were purified individually from lungs of control wild-type (n = 5) and smaFra-2 (n = 3) mice. Representative images of α-SMA-expressing cells (TdTomato-positive = white color) migrating on fibronectin are shown. Area of cells that had migrated into the cell-free zone (inside the circle) was assessed and is shown as proportion of the area within the circle (%covered area). B: migration on fibronectin was reduced in α-SMA-expressing cells in lungs from smaFra-2 compared with control wild-type mice. Values are means ± SE. *P < 0.05.
DISCUSSION
In this study we found that artificial Fra-2 expression in α-SMA-expressing lineage cells critically modulates secondary alveolar septation in developing murine lungs. The major alveolar cell type targeted by the tetracycline-inducible Fra-2 transgene in our study appears to be the alveolar myofibroblast, since the cells marked by Cre-mediated expression of a TdTomato reporter were nearly all PDGFRα-positive and were enriched at the tips of emerging secondary septae in P7 lungs. Although the transgene we used is also expressed in vascular and airway smooth muscle, we did not find evidence that developing airway or vascular smooth muscle cells contributed significantly to alveolar myofibroblasts. Induction of this transgene throughout embryonic development did not significantly target epithelial cells, endothelial cells, or lipofibroblasts, at least at P7 (Fig. 5B), suggesting that these cells diverge from developing myofibroblasts before the α-SMA gene is expressed. The defect in secondary septation we observed thus appears to be due to direct effects of Fra-2 on gene expression in alveolar myofibroblasts.
During normal postnatal lung development, Fra-2 mRNA expression was not significantly changed in the total lungs (Fig. 3C), whereas it was decreased in the myofibroblast lineage cells between P2 and P7 (Fig. 5F). In combination with our finding that artificial Fra-2 overexpression in the same cells resulted in a disturbance of normal alveolar septation, these results are consistent with the possibility that overexpression of Fra-2, directly or indirectly, impairs myofibroblast functions crucial for normal alveolar septation (e.g., migration capacity). We speculate that overexpression of Fra-2, or alteration in expression of the ECM and ECM responsive genes it regulates, could contribute to the pathogenesis of human lung developmental diseases.
Only a few molecules that regulate myofibroblast functions essential for secondary septa formation have been determined. PDGFRα is a well-known regulator of myofibroblasts, and its signaling is crucially implicated in proper myofibroblast differentiation, migration, and other functions, including correct assembly of elastin and formation of secondary septa. In lungs of smaFra-2 mice, however, no significant changes were observed in Pdgfra expression, elastin expression, or elastin accumulation. Our comprehensive transcriptome analyses showed that components of the ECM and cell adhesion-associated genes were among the major gene classes whose expression was significantly affected by Fra-2 overexpression. This finding is consistent with our previous report showing that Fra-2 is a positive regulator of matrix formation by virtue of its regulation of expression of several ECM molecules, including Col1a2 in bone cells (7). In particular, pairs of integrins (Itga5 and Itga8) and their principal ECM ligands (Fn and Npnt) are upregulated. It is well known that the strength of cell-ECM adhesion is a crucial factor to determine cell migration. At very low adhesion strength, migration is impaired. However, migration speed is optimal at intermediate adhesion strength and again reduced as the strength of adhesion is further increased. Our finding that migration on fibronectin was reduced in primary myofibroblasts isolated from the lungs of smaFra-2 mice compared with wild-type controls at P7 is consistent with the possibility that enhanced adhesion strength and reduced migration speed could contribute to the defect in secondary alveolar septation in these mice. However, in this study we did not directly prove that impaired cell myofibroblast migration was responsible for the in vivo septation defect we observed.
LPA is a bioactive phospholipid that is implicated in the survival, proliferation, differentiation, migration, adhesion, and morphology of a range of cell types during development (5). A recent study reported postnatal alveolarization impairment in LPA1-knockout mice similar to that observed in smaFra-2 mice (14). In smaFra-2 mouse lungs, Enpp2, an enzyme that converts lysophosphatidylcholine to LPA, and LPA1 were downregulated in myofibroblasts from smaFra-2 mice, suggesting that LPA signaling in the myofibroblasts and secondary septa was decreased. This downregulation of LPA signaling in the lungs of smaFra-2 mice could also contribute to the defect in secondary septation. Further studies are required to determine the relative contributions of the gene expression changes we observed to the lung development defect in smaFra-2 mice.
Based on our previous finding that partial blockade of TGF-β signaling in smooth muscle cells ameliorated vasculopathy in the lungs of Fra-2 transgenic mice, we expected that vascular smooth muscle cells might be the critical cells that respond to Fra-2 overexpression to cause the vascular phenotype. However, there was no apparent vascular remodeling, fibrosis, or inflammation in the lungs of smaFra-2 mice at 15 wk of age, when most of the global Fra-2 transgenic mice developed all these phenotypes. Notably, alveolar myofibroblasts in smaFra-2 mice overexpressed genes encoding ECM proteins, as well as genes implicated in fibroblast activation. However, even adult smaFra-2 mice showed no apparent fibrosis in their lungs. We found that the progeny of these α-SMA-expressing lineage cells (TdTomato-positive cells in α-SMA-rtTA;tetO-Cre;Ai14 mice) still survived in the alveolar region in adult mice, but they were only a fraction of PDGFRα-positive lung fibroblasts and did not express α-SMA (data not shown). Therefore, it seems likely that, in the transgenic system we used, Fra-2 was not significantly overexpressed in alveolar fibroblasts of uninjured adults.
In summary, we found that conditional Fra-2 expression in α-SMA-expressing cells impairs secondary alveolar septation, presumably by modulating expression of genes in myofibroblasts and/or their precursors. Comprehensive transcriptome analyses suggested that this effect might be explained by Fra-2-mediated modulation of ECM production and cell adhesion and/or by inhibition of LPA generation and signaling. Secondary septation is an important step in lung development, and its failure contributes to serious respiratory dysfunction, as in BPD. Our data suggest that Fra-2-mediated dysregulation of gene expression in myofibroblasts impairs secondary septation. Further evaluation of the impact of the gene expression changes we report here should provide clues to further elucidate the molecular mechanisms underlying this crucial step in normal lung development.
GRANTS
This work was supported by a grant from the Scleroderma Research Foundation (D. Sheppard).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.T., J.T.L., L.B., and D.S. conceived and designed research; K.T., T.T., and X.R. performed experiments; K.T. and T.T. analyzed data; K.T., J.T.L., T.T., and D.S. interpreted results of experiments; K.T. and T.T. prepared figures; K.T. and D.S. drafted manuscript; K.T., J.T.L., X.R., L.B., E.F.W., and D.S. edited and revised manuscript; K.T., J.T.L., T.T., X.R., L.B., E.F.W., and D.S. approved final version of manuscript.
REFERENCES
- 1.Abman SH, Bancalari E, Jobe A. The evolution of bronchopulmonary dysplasia after 50 years. Am J Respir Crit Care Med : 421–424, 2017. doi: 10.1164/rccm.201611-2386ED. [DOI] [PubMed] [Google Scholar]
- 2.Abman SH, Conway SJ. Developmental determinants and changing patterns of respiratory outcomes after preterm birth. Birth Defects Res A Clin Mol Teratol : 127–133, 2014. doi: 10.1002/bdra.23242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahlfeld SK, Conway SJ. Aberrant signaling pathways of the lung mesenchyme and their contributions to the pathogenesis of bronchopulmonary dysplasia. Birth Defects Res A Clin Mol Teratol : 3–15, 2012. doi: 10.1002/bdra.22869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhandari A, Bhandari V. Pitfalls, problems, and progress in bronchopulmonary dysplasia. Pediatrics : 1562–1573, 2009. doi: 10.1542/peds.2008-1962. [DOI] [PubMed] [Google Scholar]
- 5.Boström H, Willetts K, Pekny M, Levéen P, Lindahl P, Hedstrand H, Pekna M, Hellström M, Gebre-Medhin S, Schalling M, Nilsson M, Kurland S, Törnell J, Heath JK, Betsholtz C. PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell : 863–873, 1996. doi: 10.1016/S0092-8674(00)81270-2. [DOI] [PubMed] [Google Scholar]
- 6.Bouzeghrane F, Mercure C, Reudelhuber TL, Thibault G. α8β1-Integrin is upregulated in myofibroblasts of fibrotic and scarring myocardium. J Mol Cell Cardiol : 343–353, 2004. doi: 10.1016/j.yjmcc.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 7.Bozec A, Bakiri L, Jimenez M, Schinke T, Amling M, Wagner EF. Fra-2/AP-1 controls bone formation by regulating osteoblast differentiation and collagen production. J Cell Biol : 1093–1106, 2010. doi: 10.1083/jcb.201002111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burri PH. Structural aspects of postnatal lung development—alveolar formation and growth. Biol Neonate : 313–322, 2006. doi: 10.1159/000092868. [DOI] [PubMed] [Google Scholar]
- 9.Chen C, Kudo M, Rutaganira F, Takano H, Lee C, Atakilit A, Robinett KS, Uede T, Wolters PJ, Shokat KM, Huang X, Sheppard D. Integrin α9β1 in airway smooth muscle suppresses exaggerated airway narrowing. J Clin Invest : 2916–2927, 2012. doi: 10.1172/JCI60387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi CW. Lung interstitial cells during alveolarization. Korean J Pediatr : 979–984, 2010. doi: 10.3345/kjp.2010.53.12.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi JE, Lee SS, Sunde DA, Huizar I, Haugk KL, Thannickal VJ, Vittal R, Plymate SR, Schnapp LM. Insulin-like growth factor-I receptor blockade improves outcome in mouse model of lung injury. Am J Respir Crit Care Med : 212–219, 2009. doi: 10.1164/rccm.200802-228OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eferl R, Hasselblatt P, Rath M, Popper H, Zenz R, Komnenovic V, Idarraga MH, Kenner L, Wagner EF. Development of pulmonary fibrosis through a pathway involving the transcription factor Fra-2/AP-1. Proc Natl Acad Sci USA : 10525–10530, 2008. doi: 10.1073/pnas.0801414105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrando RE, Newton K, Chu F, Webster JD, French DM. Immunohistochemical detection of FLAG-tagged endogenous proteins in knock-in mice. J Histochem Cytochem : 244–255, 2015. doi: 10.1369/0022155414568101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Funke M, Knudsen L, Lagares D, Ebener S, Probst CK, Fontaine BA, Franklin A, Kellner M, Kühnel M, Matthieu S, Grothausmann R, Chun J, Roberts JD Jr, Ochs M, Tager AM. Lysophosphatidic acid signaling through the lysophosphatidic acid-1 receptor is required for alveolarization. Am J Respir Cell Mol Biol : 105–116, 2016. doi: 10.1165/rcmb.2015-0152OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamilton TG, Klinghoffer RA, Corrin PD, Soriano P. Evolutionary divergence of platelet-derived growth factor-α receptor signaling mechanisms. Mol Cell Biol : 4013–4025, 2003. doi: 10.1128/MCB.23.11.4013-4025.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hasenfuss SC, Bakiri L, Thomsen MK, Williams EG, Auwerx J, Wagner EF. Regulation of steatohepatitis and PPARγ signaling by distinct AP-1 dimers. Cell Metab : 84–95, 2014. doi: 10.1016/j.cmet.2013.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc : 44–57, 2009. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 18.Hung CF, Rohani MG, Lee SS, Chen P, Schnapp LM. Role of IGF-1 pathway in lung fibroblast activation. Respir Res : 102, 2013. doi: 10.1186/1465-9921-14-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jobe AH, Bancalari E. Bronchopulmonary dysplasia. Am J Respir Crit Care Med : 1723–1729, 2001. doi: 10.1164/ajrccm.163.7.2011060. [DOI] [PubMed] [Google Scholar]
- 20.Kim N, Vu TH. Parabronchial smooth muscle cells and alveolar myofibroblasts in lung development. Birth Defects Res C Embryo Today : 80–89, 2006. doi: 10.1002/bdrc.20062. [DOI] [PubMed] [Google Scholar]
- 21.Kinsella JP, Greenough A, Abman SH. Bronchopulmonary dysplasia. Lancet : 1421–1431, 2006. doi: 10.1016/S0140-6736(06)68615-7. [DOI] [PubMed] [Google Scholar]
- 22.Laucho-Contreras ME, Taylor KL, Mahadeva R, Boukedes SS, Owen CA. Automated measurement of pulmonary emphysema and small airway remodeling in cigarette smoke-exposed mice. J Vis Exp 95: 52236, 2015. doi: 10.3791/52236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levine D, Rockey DC, Milner TA, Breuss JM, Fallon JT, Schnapp LM. Expression of the integrin α8β1 during pulmonary and hepatic fibrosis. Am J Pathol : 1927–1935, 2000. doi: 10.1016/S0002-9440(10)65066-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li A, Ma S, Smith SM, Lee MK, Fischer A, Borok Z, Bellusci S, Li C, Minoo P. Mesodermal ALK5 controls lung myofibroblast versus lipofibroblast cell fate. BMC Biol : 19, 2016. doi: 10.1186/s12915-016-0242-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lindahl P, Karlsson L, Hellström M, Gebre-Medhin S, Willetts K, Heath JK, Betsholtz C. Alveogenesis failure in PDGF-A-deficient mice is coupled to lack of distal spreading of alveolar smooth muscle cell progenitors during lung development. Development : 3943–3953, 1997. [DOI] [PubMed] [Google Scholar]
- 26.Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci : 133–140, 2010. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Massaro D, Massaro GD. Lung development, lung function, and retinoids. N Engl J Med : 1829–1831, 2010. doi: 10.1056/NEJMe1002366. [DOI] [PubMed] [Google Scholar]
- 28.McGowan SE, McCoy DM. Regulation of fibroblast lipid storage and myofibroblast phenotypes during alveolar septation in mice. Am J Physiol Lung Cell Mol Physiol : L618–L631, 2014. doi: 10.1152/ajplung.00144.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McGowan SE, Torday JS. The pulmonary lipofibroblast (lipid interstitial cell) and its contributions to alveolar development. Annu Rev Physiol : 43–62, 1997. doi: 10.1146/annurev.physiol.59.1.43. [DOI] [PubMed] [Google Scholar]
- 30.Mund SI, Stampanoni M, Schittny JC. Developmental alveolarization of the mouse lung. Dev Dyn : 2108–2116, 2008. doi: 10.1002/dvdy.21633. [DOI] [PubMed] [Google Scholar]
- 31.Rehan VK, Sakurai R, Wang Y, Santos J, Huynh K, Torday JS. Reversal of nicotine-induced alveolar lipofibroblast-to-myofibroblast transdifferentiation by stimulants of parathyroid hormone-related protein signaling. Lung : 151–159, 2007. doi: 10.1007/s00408-007-9007-0. [DOI] [PubMed] [Google Scholar]
- 32.Schissel SL, Dunsmore SE, Liu X, Shine RW, Perrella MA, Layne MD. Aortic carboxypeptidase-like protein is expressed in fibrotic human lung and its absence protects against bleomycin-induced lung fibrosis. Am J Pathol : 818–828, 2009. doi: 10.2353/ajpath.2009.080856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Silva DM, Nardiello C, Pozarska A, Morty RE. Recent advances in the mechanisms of lung alveolarization and the pathogenesis of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol : L1239–L1272, 2015. doi: 10.1152/ajplung.00268.2015. [DOI] [PubMed] [Google Scholar]
- 34.Torday JS, Torres E, Rehan VK. The role of fibroblast transdifferentiation in lung epithelial cell proliferation, differentiation, and repair in vitro. Pediatr Pathol Mol Med : 189–207, 2003. doi: 10.1080/pdp.22.3.189.207. [DOI] [PubMed] [Google Scholar]
- 35.Tsujino K, Reed NI, Atakilit A, Ren X, Sheppard D. Transforming growth factor-β plays divergent roles in modulating vascular remodeling, inflammation, and pulmonary fibrosis in a murine model of scleroderma. Am J Physiol Lung Cell Mol Physiol : L22–L31, 2017. doi: 10.1152/ajplung.00428.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsujino K, Sheppard D. Critical appraisal of the utility and limitations of animal models of scleroderma. Curr Rheumatol Rep : 4, 2016. doi: 10.1007/s11926-015-0553-9. [DOI] [PubMed] [Google Scholar]
- 37.Tsujino K, Takeda Y, Arai T, Shintani Y, Inagaki R, Saiga H, Iwasaki T, Tetsumoto S, Jin Y, Ihara S, Minami T, Suzuki M, Nagatomo I, Inoue K, Kida H, Kijima T, Ito M, Kitaichi M, Inoue Y, Tachibana I, Takeda K, Okumura M, Hemler ME, Kumanogoh A. Tetraspanin CD151 protects against pulmonary fibrosis by maintaining epithelial integrity. Am J Respir Crit Care Med : 170–180, 2012. doi: 10.1164/rccm.201201-0117OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsukui T, Ueha S, Shichino S, Inagaki Y, Matsushima K. Intratracheal cell transfer demonstrates the profibrotic potential of resident fibroblasts in pulmonary fibrosis. Am J Pathol : 2939–2948, 2015. doi: 10.1016/j.ajpath.2015.07.022. [DOI] [PubMed] [Google Scholar]
- 39.Tumelty KE, Smith BD, Nugent MA, Layne MD. Aortic carboxypeptidase-like protein (ACLP) enhances lung myofibroblast differentiation through transforming growth factor β receptor-dependent and -independent pathways. J Biol Chem : 2526–2536, 2014. doi: 10.1074/jbc.M113.502617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vaccaro C, Brody JS. Ultrastructure of developing alveoli. I. The role of the interstitial fibroblast. Anat Rec : 467–479, 1978. doi: 10.1002/ar.1091920402. [DOI] [PubMed] [Google Scholar]
- 41.Ye N, Ding Y, Wild C, Shen Q, Zhou J. Small molecule inhibitors targeting activator protein 1 (AP-1). J Med Chem : 6930–6948, 2014. doi: 10.1021/jm5004733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zenz R, Eferl R, Scheinecker C, Redlich K, Smolen J, Schonthaler HB, Kenner L, Tschachler E, Wagner EF. Activator protein 1 (Fos/Jun) functions in inflammatory bone and skin disease. Arthritis Res Ther : 201, 2008. doi: 10.1186/ar2338. [DOI] [PMC free article] [PubMed] [Google Scholar]