

Abstract
The pathology of acute lung injury (ALI) involves inducible nitric oxide (NO) synthase (iNOS)-derived NO-induced apoptosis of pulmonary endothelial cells. In vitro, iNOS-derived NO production has been shown to depend on the uptake of l-arginine by the cationic amino acid transporters (CAT). To test the hypothesis that mice deficient in CAT-2 (slc7a2−/−on a C57BL/6 background) would be protected from hyperoxia-induced ALI, mice (slc7a2−/− or wild-type) were placed in >95% oxygen (hyperoxia) or 21% oxygen (control) for 60 h. In wild-type mice exposed to hyperoxia, the exhaled nitric oxide (exNO) was twofold greater than in wild-type mice exposed to normoxia (P < 0.005), whereas in slc7a2−/− mice there was no significant difference between exNO in animals exposed to hyperoxia or normoxia (P = 0.95). Hyperoxia-exposed wild-type mice had greater (P < 0.05) lung resistance and a lower (P < 0.05) lung compliance than did hyperoxia-exposed slc7a2−/− mice. The lung wet-to-dry weight ratio was greater (P < 0.005) in the hyperoxia-exposed wild-type mice than in hyperoxia-exposed slc7a2−/− mice. Neutrophil infiltration was lower (P < 0.05) in the hyperoxia-exposed slc7a2−/− mice than in the hyperoxia-exposed wild-type mice as measured by myeloperoxidase activity. The protein concentration in bronchoalveolar lavage fluid was lower (P < 0.001) in the hyperoxia-exposed slc7a2−/− mice than in similarly exposed wild-type mice. The percent of TUNEL-positive cells in the lung following hyperoxia exposure was significantly lower (P < 0.001) in the slc7a2−/− mice than in the wild-type mice. These results are consistent with our hypothesis that lack of CAT-2 protects mice from acute lung injury.
Keywords: acute lung injury, cationic amino acid transporters, inducible nitric oxide synthase, slc7a2 knockout mice
INTRODUCTION
Inflammation leading to acute lung injury (ALI) is involved in the pathogenesis of a number of lung diseases, including acute respiratory distress syndrome (ARDS) (22). ALI/ARDS are often associated with conventional intensive care therapies, including mechanical ventilation and supplemental oxygen therapy (19, 22). Inflammatory lung diseases are associated with increased nitric oxide (NO) production largely because of the induction of inducible NO synthase (iNOS). iNOS metabolizes l-arginine to l-citrulline and NO and produces relatively large quantities of NO compared with other isoforms of NOS, such as endothelial NOS (eNOS) or neuronal NOS (nNOS). It is important to remember that NO is a molecule that in most situations is beneficial to the organism, but NO can also have deleterious effects on the host organism particularly when NO is over-produced (15, 18). In ARDS/ALI, endothelial cell apoptosis drives the formation of pulmonary edema, which is the hallmark of the disease (22). We have previously found in cultured human pulmonary endothelial cells that iNOS-derived NO results in endothelial cell apoptosis (4). In animal models of ARDS, inhibition of iNOS using pharmacological inhibitors has been shown to have beneficial effects (7, 21, 24). No clinical studies of NOS inhibition have been done in patients with ARDS. However, two large multi-center studies of NOS inhibitors in patients with shock, one because of sepsis and one cardiogenic, were stopped early because of increased mortality in one (13) and to prespecified futility in the other (1).
These disappointing results in clinical trials studying NOS inhibitors in shock may well be due to a lack of sufficient iNOS specificity, that is to say that the NOS inhibitors also inhibit constitutive NO production from eNOS and nNOS (2, 8). We have previously shown in cultured human pulmonary microvascular endothelial cells that inflammation-induced iNOS-derived NO production can be significantly attenuated by inhibition of the cellular uptake of l-arginine (4). l-arginine is taken up by pulmonary endothelial cells predominantly by the cationic amino acid transporters (CAT) (4, 14, 16). There are two main CAT family members expressed in endothelial cells, CAT-1 encoded by the solute carrier family 7 member 1 gene (slc7a1) on chromosome 13 and CAT-2 encoded by the slc7a2 gene on chromosome 8. We have previously shown that CAT-2 mRNA can be upregulated in pulmonary arterial endothelial cells (16) and in neonatal pig lungs (3). Therefore, we hypothesized that mice deficient in the Slc7a2 gene would be protected from hyperoxia-induced ALI, and demonstrate less NO production and apoptosis, when compared with wild-type animals. We used slc7a2−/− mice on a C57BL/6 background, which have recently become commercially available, with C57BL/6 mice serving as the wild type. We employed a hyperoxia model of ALI to test our hypothesis. Lung production of NO was measured using exhaled NO (exNO). To assess ALI we used pulmonary function testing and measured lung wet-to-dry weight ratios, bronchoalveolar lavage fluid (BALF) protein concentrations, and lung myeloperoxidase (MPO) activity.
METHODS
Mice.
C57BL/6 (wild-type) and slc7a2 knockout (slc7a2−/−) mice were purchased from the Jackson Laboratories (Bar Harbor, ME). The C57BL/6 and slc7a2−/− mice were bred in-house. All mice were maintained at 24°C with a relative humidity between 30% and 70% on a 12 h light:12 h dark cycle. The mice were fed Harlan Teklad irradiated diet (Harlan Sprague-Dawley) ad libitum. All animals received humane care in accordance with the guidelines of the National Institutes of Health. The study protocol was approved by the Institutional Animal Care and Use Committee of the Research Institute at Nationwide Children’s Hospital.
Exposures.
Adult wild-type and slc7a2−/− mice (8 to 12 wk old; 17–28 g body wt) were placed in a hyperoxic chamber with >95% O2 for a period of 60 h. The mice were monitored twice a day. Age- and weight-matched C57BL/6 and slc7a2−/− mice were kept under normoxia in similar conditions to serve as room air controls. Approximately the same numbers of female (25) and male mice (29) were used in these studies.
Measurement of pulmonary function.
Mice were anesthetized with an intraperitoneal injection of ketamine (65 mg/kg, Hospira, Lake Forest, IL)/xylazine (10 mg/kg, LLOYD, Shenandoah, IA) to suppress spontaneous breathing. The trachea was cannulated via a tracheostomy and then connected to the FlexiVent system (SCIREQ, Montreal, QC, Canada) under the controlled mechanical ventilation mode with a tidal volume of 10 ml/kg at a frequency of 150 breaths/min and a positive end-expiratory pressure of 3 cmH2O. Total lung capacity perturbation was applied first to standardize volume history. Snapshot perturbation was then performed to measure compliance (CRS) and resistance (RRS) of the respiratory system (airways, lung, and chest wall). Pressure-volume loops were used to measure quasi-static compliance (Cst). All perturbations were repeated three times with acceptable measurements (coefficient of determination > 0.95), and the average was calculated for each individual subject.
Tissue preparation.
On the day of the study, the mice were euthanized with an intraperitoneal injection of ketamine (90 mg/kg)-xylazine (4.5 mg/kg). The right lungs were removed, weighed, and then dried in the oven at 60°C until the weight stabilized. The left lungs were removed, weighed, freeze-clamped in liquid nitrogen, and kept at −80°C for further analyses.
Real-time PCR.
mRNA was isolated from the frozen lungs using TRIzol (Invitrogen, Carlsbad, CA) following the manufacturer’s instructions. Real-time PCR for slc7a1, slc7a2, iNOS, COX-2, eNOS, and nNOS was performed as previously described (4, 5). Briefly, all RNA samples were treated with RNase-free DNase (Promega, Madison, WI) followed by GoScript reverse transcriptase (Promega), and then cDNAs were analyzed by real-time PCR using PowerUp SYBR Green Master Mix (Applied Biosystems, Austin, TX). Primers were ordered from Invitrogen using the following sequences, for mouse slc7a1-forward primer: 5′-TTC GGT TAT GGG ATC TGG CAC AGT-3′; reverse primer: 5′-TTT GCA CTG GTC CAA GTT GCT GTC-3′; for mouse slc7a2-forward primer: 5′-CCC GAG TAT CGT GGT GTC TT-3′; reverse primer: 5′-CTC TTG CGA CAC TGG ACG TA-3′; for mouse iNOS-forward primer: 5′-CTG CTG GTG GTG ACA AGC ACA TTT-3′; reverse primer: 5′-ATG TCA TGA GCA AAG GCG CAG AAC-3′; for mouse COX-2-forward primer: 5′-CCA CCT CTG CGA TGC TCT TC-3′; reverse primer 5′-CAT TCC CCA CGG TTT TGA CAT G-3′; for mouse eNOS-forward primer: 5′-ATG CCT ACA GCA TTG GTT GCA AGG-3′; reverse primer 5′-AAG CAT ATG AAG AGG GCA GCA GGA-3′; and for mouse nNOS-forward primer: 5′-TGG CTT CAA GAA ATT GGC AGA GGC-3′; reverse primer 5′-TGG CGT AGA GAA TGG TCG CTT TGA-3′. 18S was amplified using the forward primer (5′-CCA GAG CGA AAG CAT TTG CCA AGA-3′) and the reverse primer (5′-TCG GCA TCG TTT ATG GTC GGA ACT-3′). For each reaction, negative controls containing reaction mixture and primers without cDNA were performed to verify that primers and reaction mixtures were free of template contamination. Relative slc7a1, slc7a2, iNOS, COX-2, eNOS, and nNOS mRNA amounts were normalized to 18S expression using the ΔΔCT method. All samples were analyzed in duplicate. Data are shown as fold-change relative to normoxia-exposed wild-type mice.
BALF collection and analysis.
Following exposure mice were anesthetized and a tracheostomy tube was placed. The mouse lungs were then lavaged with 1 ml of PBS and massaged for 1 min before collection of the fluid. The procedure was repeated 2 more times and the combined fluid was centrifuged at 200 g for 5 min at 4°C. The supernatant was then collected and stored at −80°C for further analyses. The supernatant was analyzed for total protein content using the Bradford assay (Bio-Rad, Hercules, CA).
MPO activity assay.
The mouse lungs were homogenized in 4 times volume of ice-cold Dulbecco’s phosphate-buffered saline (pH 7.4) and then were centrifuged at 12,000 g for 15 min. The supernatants were collected and analyzed for total protein content using the Bradford assay. In a 96-well plate after adding the same amount of protein, Dulbecco’s phosphate-buffered saline, H2O2, and 10-acetyl-3, 7-dihydroxyphenoxazine (Sigma-Aldrich, St. Louis, MO) to each well the assays were performed immediately at 37°C. The fluorescence readings were acquired using excitation of 535 nm and emission of 590 nm for a total of 15 kinetic cycles with an interval of 20 s per cycle in a SpectraMax M2 Multimode Microplate Reader (Molecular Device, Sunnyvale, CA). Enzyme activity was defined as the slope of the fluorescence versus time plot for each well.
Cytokine assays.
The lungs were homogenized with 0.1 M K2HPO4 (pH 7.4) containing 5 mM EDTA, 0.1% Triton × −100, phosphatase inhibitors, and protease inhibitors. The tubes were allowed to sit for 30 min at 4°C and then centrifuged at 14,000 revolutions/min, 4°C, for 20 min. The supernatant was then transferred into a new tube and stored at −80°C. Cytokine levels were assayed in the lung lysates using ELISA; for TNF-α the mouse TNF-α ELISA ready-SET-go kit (cat. no. 88-7324-88) from eBioscience, Waltham, MA was used, and for IL-6 the mouse IL-6 ready-SET-go kit (cat. no. 88-7064-88) from eBioscience was used.
TUNEL assay.
A thoracotomy was done, and the lungs were fixed by placing a cannula in the trachea and instilling 10% neutral buffered formalin at 25 cmH2O pressure over 5 min. The trachea was tied, and the lungs were fixed overnight in 10% neutral buffered formalin. The next day the lungs were washed five times with PBS and stored in PBS at 4°C. The left lungs were then embedded in paraffin, and 5-µm sections were cut for each sample. Some sections were stained with hematoxylin and eosin (H&E) for assessment of lung histology. Other sections were stained for assessment of apoptosis using the Apo-BrdU in situ DNA fragment assay kit (BioVision, Milpitas, CA). Fifty images from four mice per group were scored for apoptosis for each condition.
Exhaled NO measurement.
exNO was measured as previously described (3, 9). The mice were anesthetized with ketamine (90 mg/kg)-xylazine (4.5 mg/kg) given by intraperitoneal injection, intubated, and mechanically ventilated using an NO-free room air-gas mixture. The gas mixture was supplied from a mylar balloon attached to the inhalation port of the ventilator. The animals were ventilated with a tidal volume of 0.2 ml and a respiratory rate of 150 breaths/minute. The exhaled gas was collected for 5 min using a mylar balloon attached to the ventilator exhaust port. The gas collected in the mylar balloon was analyzed within 5 min using a chemiluminescence NO analyzer (model 280i, Sievers, Boulder, CO). The analyzer was calibrated daily with authentic NO (1 parts per million in N2, Matheson, Chicago, IL) mixed with NO-free nitrogen using precision flow meters to obtain a standard curve with concentrations ranging from 0 to 500 parts per billion (vol/vol). The NO detection limit was 0.5 parts per billion. The exNO production (pmol/min) was calculated from the minute ventilation and the expired NO concentration.
RAW 264.7 cell culture.
The murine macrophage cell line RAW 264.7 (American Type Culture Collection, Manassas, VA) was cultured in Dulbecco’s modified Eagle’s medium (Mediatech, Manassas, VA) supplemented with 10% fetal bovine serum (Mediatech), 100 IU/ml penicillin, and 100 μg/ml streptomycin (Mediatech) at 37°C with 5% CO2.
Statistical analysis.
Two-way ANOVA was used to compare data among treatment and genotype groups, with genotype set as Factor A and exposure (room air or hyperoxia) set as Factor B. A Holm-Sidak post hoc test was used to identify groups that were different (SigmaPlot 12.5; Jandel Scientific, Carlsbad, CA). For the two-way ANOVA for the RAW experiments shown in Fig. 2, treatment with vehicle or LPS was set as Factor A, and room air or 85% oxygen was set as Factor B. Data that did not pass the normality test or equal variance test were log transformed. Data are plotted as box and whisker plots to demonstrate the variability within experimental groups better, where the box represents the 25th and 75th percentiles, and whiskers represent the 10th and 90th percentiles, the solid line in the box is the median (SigmaPlot 12.0; Jandel Scientific). Differences were considered significant when P < 0.05.
Fig. 2.

Hyperoxia and LPS increased slc7a1 mRNA expression in murine macrophages. RAW 264.7 cells were treated with LPS (0.5 µg/ml; n = 6 each exposure) or vehicle (n = 5 each exposure) and incubated in either 21% oxygen or 85% oxygen for 24 h. The cells were then harvested for determination of mRNA levels of slc7a1 and slc7a2. A: hyperoxia and LPS but neither stimulus alone resulted in increased slc7a1 mRNA levels. B: LPS resulted in substantially greater slc7a2 mRNA levels, and LPS and hyperoxia resulted in greater slc7a2 mRNA levels than LPS alone. *LPS different from vehicle same oxygen exposure, P < 0.001. #85% oxygen different from 21% oxygen same treatment, P < 0.001.
RESULTS
Hyperoxia increased slc7a1, eNOS, iNOS, and COX-2 mRNA expression.
We exposed wild-type and slc7a2−/− mice to either >95% O2 or 21% O2 for 60 h and then harvested mRNA from the lungs. Hyperoxic exposure caused an increase in slc7a1 mRNA expression in both the wild-type mice and in the slc7a2−/− mice (Fig. 1A). There was no difference in the slc7a1 mRNA expression between the wild-type and slc7a2−/− mice following exposure to hyperoxia. There was also no difference in slc7a2 mRNA levels in wild-type mice exposed to normoxia or hyperoxia (Fig. 1B). However, quantitative PCR results verified that the slc7a2 gene was knocked out in the slc7a2−/− mice (Fig. 1B). The mRNA levels of iNOS were greater following hyperoxic exposure than following normoxic exposure in both wild-type and slc7a2−/− mice (Fig. 1C). Interestingly, the mRNA levels of iNOS were greater in the slc7a2−/− mice than in the wild-type mice for both normoxia and hyperoxia exposures (Fig. 1C). Hyperoxia also caused a significant induction of COX-2 mRNA, but there was no difference between wild-type and slc7a2−/− mice in the hyperoxic COX-2 response (Fig. 1D). Similarly, hyperoxia caused a significant induction of eNOS mRNA that did not differ between genotypes (Fig. 1E), whereas neither hyperoxia nor genotype had any appreciable effect on nNOS mRNA levels (Fig. 1F).
Fig. 1.

Hyperoxia increased expression of slc7a1, iNOS, COX-2, and eNOS in the lung. Real-time PCR results for slc7a1 (A), slc7a2 (B), iNOS (C), cyclooxygenase 2 (COX-2) (D), eNOS (E) and nNOS (F) in mice exposed to either room air (n = 6) or hyperoxia (n = 6). *Hyperoxia different from room air, P < 0.005; #slc7a2−/− different from wild-type, P < 0.05. eNOS, endothelial NOS; iNOS, inducible NOS; NOS, nitric oxide synthase; nNOS, neuronal NOS.
The finding that slc7a1 mRNA was induced 10–12-fold in the lung following hyperoxia was a bit surprising to us, because our past work has shown slc7a1 induction with LPS to be of a smaller magnitude (4). Therefore, we studied slc7a1 and slc7a2 mRNA expression in RAW 264.7 cells treated with either vehicle or LPS (0.5 µg/ml) and then incubated in either room air or 85% oxygen for 24 h. The levels of slc7a1 mRNA were not significantly increased by treatment with LPS in room air and exposure to 90% oxygen alone did not significantly induce slc7a1 mRNA levels. However, both treatment with LPS and exposure to 85% oxygen resulted in a ~twofold induction of slc7a1 mRNA levels (Fig. 2A). On the other hand, slc7a2 mRNA levels were substantially induced following LPS treatment, and exposure to 85% oxygen resulted in significantly higher levels of slc7a2 mRNA following LPS treatment than did exposure to 21% oxygen following LPS treatment (Fig. 2B).
Hyperoxia-induced IL-6 expression in the lung.
Given the clear effect of LPS on the hyperoxia-induced slc7a1 and slc7a2 mRNA levels, we wondered if there was evidence of cytokine production following hyperoxic exposure in the lung. We chose to measure IL-6 and TNF-α levels in lung lysate as archetypical proinflammatory cytokines. We found that hyperoxia led to increased levels of IL-6 in the lung lysates and interestingly, that the levels in the lungs from the slc7a2−/− mice were significantly greater than those in the lungs from wild-type mice (Fig. 3A). There was no significant induction of TNF-α by hyperoxia in either genotype (Fig. 3B).
Fig. 3.

Hyperoxia increased IL-6 levels in lung lysates. Lung lysates (n = 5 in slc7a2−/− room air, and n = 6 in the other 3 groups) were assayed for IL-6 and TNF-α levels using ELISA. A: IL-6 levels were greater in lung lysates from hyperoxia exposed mice. Hyperoxic IL-6 levels were greater in lung lysates from slc7a2−/− mice than in lung lysates from wild-type mice. B: TNF-α levels were not different in lung lysates from any of the experimental groups. *Hyperoxia different from room air same genotype, P < 0.001. #slc7a2−/− different from wild-type same exposure, P < 0.005.
Slc7a2 knockout attenuated the hyperoxia-induced increase in exNO production.
To determine whether NO production is dependent on CAT-2-mediated l-arginine uptake, we measured exNO from the wild-type and slc7a2−/− mice following 60 h of exposure to either hyperoxia or room air. Wild-type mice exposed to hyperoxia had significantly greater exNO production than did wild-type mice exposed to normoxia (Fig. 4A). Although slc7a2−/− mice exposed to hyperoxia had levels of exNO similar to those seen in the slc7a2−/− mice exposed to normoxia (Fig. 4A). The wild-type mice exposed to hyperoxia had a twofold greater exNO production than wild-type mice exposed to room air (Fig. 4B) as perhaps expected with the induction of iNOS and eNOS in the lung. However, in the slc7a2−/− mice there was no difference in exNO production between the two exposures (Fig. 4B), despite the hyperoxia-induced iNOS and eNOS mRNA induction.
Fig. 4.

Hyperoxia increased exhaled nitric oxide (exNO) production from the lungs of wild-type mice but not from the lungs of slc7a2−/− mice. Mice were exposed to 60 h of either room air or >95% oxygen and then anesthetized, intubated, and mechanically ventilated using an NO-free room air gas mixture at a set minute ventilation (tidal volume of 0.2 ml with a respiratory rate of 150 breaths/minute). The exhaled gas was collected for 5 min and analyzed using a chemiluminescence NO analyzer. The exNO production rate (pmol/min) was calculated from the minute ventilation and the expired NO concentration. A: exNO production rate from lungs of wild-type or slc7a2−/− mice exposed to room air (n = 4 for wild-type and n = 5 for slc7a2−/−) or >95% O2 (n = 5 for wild-type and n = 6 for slc7a2−/−) for 60 h. *Wild-type hyperoxia different from wild-type room air, P < 0.005. B: exNO production from hyperoxia exposed mice normalized to that from room air exposed mice of the same genotype. There was an ~twofold increase in exNO production in wild-type mice exposed to >95% oxygen, whereas there was no increase in exNO production in slc7a2−/− mice exposed to >95% oxygen. *Different from wild-type, P < 0.001.
Knockout of slc7a2 in the mice prevented the hyperoxia-induced alterations in pulmonary function.
Following 60 h of exposure to either normoxia or hyperoxia the mice were anesthetized and connected to the FlexiVent system, and CRS, RRS, and Cst were measured. In the wild-type mice, exposure to hyperoxia resulted in a significantly lower CRS than did exposure to normoxia (Fig. 3A). Although in the slc7a2−/− mice there was no difference in CRS between the normoxic- and hyperoxic-exposed animals (Fig. 5A). Indeed, CRS in the hyperoxia-exposed wild-type mice was significantly (P < 0.001) lower than that seen in the hyperoxia-exposed slc7a2−/− mice (Fig. 5A). As expected, exposure to hyperoxia in the wild-type mice resulted in a significantly greater RRS than did exposure to normoxia (Fig. 5B). However, in the slc7a2−/− mice there was no difference in RRS between animals exposed to normoxia or hyperoxia (Fig. 5B). Indeed, the RRS in the hyperoxia-exposed wild-type mice was significantly (P < 0.05) greater than that in the hyperoxia-exposed slc7a2−/− mice (Fig. 5B). Similarly, Cst in the slc7a2−/− mice exposed to hyperoxia showed no difference from the room air wild-type mice but was significantly higher than that in the hyperoxia-exposed wild-type mice (Fig. 5C).
Fig. 5.

Hyperoxia exposure resulted in a decrease in the compliance in the respiratory system in the wild-type mice but not in the slc7a2−/− mice. slc7a2−/− mice had significantly lower respiratory system resistance than wild-type mice under hyperoxia exposure. A: respiratory system compliance in the wild-type and slc7a2−/− mice after exposure to either room air (n = 8 for each genotype) or >95% O2 (n = 19 for each genotype) for 60 h. *Different from the wild-type room air mice, P < 0.05; #slc7a2−/− hyperoxia different from wild-type hyperoxia, P < 0.005. B: respiratory system resistance in the wild-type and slc7a2−/− mice after exposure to either room air (n = 8 for each genotype) or >95% O2 (n = 19 for each genotype) for 60 h. *Different from the wild-type room air mice, P < 0.001; #Hyperoxic slc7a2−/− different from hyperoxic wild-type mice, P < 0.005. C: quasi-static compliance in the wild-type and slc7a2−/− mice after exposure to either room air (n = 8 for each genotype) or >95% oxygen (n = 19 for each genotype) for 60 h. In the wild-type mice hyperoxic exposure resulted in a lower quasi-static compliance than in normoxia, whereas in the slc7a2−/− mice there was no significant effect of hyperoxia on quasi-static compliance. *Hyperoxic wild-type mice different from the wild-type room air mice, P < 0.05; #Hyperoxic slc7a2−/− different from hyperoxic wild-type mice, P < 0.001.
Slc7a2 knockout prevented the hyperoxia-induced increase in lung edema in the mice.
H&E staining of the lungs demonstrated that hyperoxia exposure in the wild-type mice led to substantial perivascular edema (white arrows in Fig. 6A) and areas of alveolar consolidation (black arrows in Fig. 6A). However, in the slc7a2−/− mice there were little evidence of perivascular edema, and although there were some alveolar exudates there was little alveolar consolidation (Fig. 6A). The room air-exposed lungs from both genotypes showed no evidence of pulmonary edema or alveolar exudates (Fig. 6A). Furthermore and consistent with the H&E staining, wild-type mice exposed to >95% O2 displayed a substantial greater lung wet-to-dry weight ratio than did wild-type mice exposed to normoxia (Fig. 6B). Although in slc7a2−/− mice the lung wet-to-dry weight ratios were not different between those animals exposed to normoxia and those animals exposed to hyperoxia (Fig. 6B). Furthermore, the lung wet-to-dry weight ratio in the hyperoxia-exposed slc7a2−/− mice was significantly lower than that in the hyperoxia-exposed wild-type mice (Fig. 6B).
Fig. 6.

Exposure to hyperoxia caused pulmonary edema in wild-type animals. A: representative H&E stains at ×20 magnification from wild-type and slc7a2−/− mice exposed to room air or >95% oxygen for 60 h. The white arrows demonstrate perivascular edema, and the black arrows demonstrate complete alveolar consolidation, which were only seen in the wild-type animals exposed to >95% oxygen. B: lung wet-to-dry weight ratio was only significantly increased in wild-type mice exposed to hyperoxia. After 60 h of exposure to either room air (n = 4 for wild-type and n = 6 for slc7a2−/−) or >95% oxygen (n = 10 for each genotype) the right lungs were removed, weighed (wet weight), and then dried in the oven at 60°C until the weight stabilized (dry weight). The wet-to-dry weight ratios were then calculated. *Wild-type hyperoxia different from wild-type room air, P < 0.001; #slc7a2−/− hyperoxia different from wild-type hyperoxia, P < 0.001.
Slc7a2 knockout attenuated the hyperoxia-induced alveolar protein leak and lung neutrophil accumulation.
We measured the protein concentration of the BALF from the mice. The protein concentration of BALF was substantially greater in the wild-type mice exposed to hyperoxia than in wild-type mice exposed to room air (Fig. 7A). In the slc7a2−/− mice, although the protein concentration of BALF was significantly greater in the hyperoxia-exposed animals than in the normoxia-exposed animals, the protein concentration in BALF was significantly lower in the hyperoxia-exposed slc7a2−/− mice than in the hyperoxia-exposed wild-type mice (Fig. 7A).
Fig. 7.
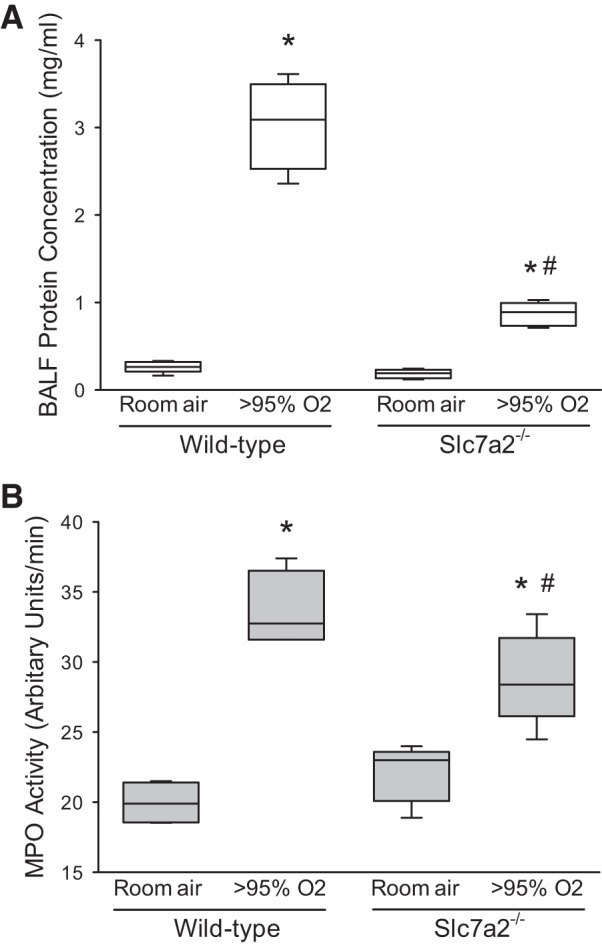
Hyperoxic exposure increased alveolar protein concentration and neutrophil accumulation in the lung, and deficiency of slc7a2 attenuated these hyperoxia-induced changes. A: hyperoxic exposure increased bronchoalveolar lavage fluid (BALF) protein concentration, whereas hyperoxic slc7a2−/− mice had significantly lower BALF protein concentration than did hyperoxic wild-type mice. Mice had catheters placed in the trachea and were given 1 ml of PBS, massaged for 1 min, and then the fluid was collected. The procedure was repeated 2 more times and the combined fluid was centrifuged at 200 g for 5 min at 4°C, and the supernatant was analyzed for total protein content. n = 5 for wild-type room air, slc7a2−/− room air, and slc7a2−/− hyperoxia, and n = 4 for wild-type hyperoxia. *Different from room air same genotype, P < 0.001, #slc7a2−/− hyperoxia different from wild-type hyperoxia, P < 0.001. B: hyperoxia exposure caused an increase in myeloperoxidase (MPO) activity in both wild-type and slc7a2−/− mice. MPO activity was measured in lung tissue homogenates from lungs of mice exposed to either normoxia (n = 4 for both genotypes) or >95% oxygen (n = 5 for both genotypes) for 60 h. *Hyperoxia different from room air same genotype, P < 0.001, #slc7a2−/− hyperoxia different from wild-type hyperoxia, P < 0.05.
To evaluate neutrophil infiltration in the mouse lungs, we measured MPO activity from the lungs. MPO activity was increased in the lungs from animals exposed to hyperoxia in both wild-type and slc7a2−/− mice (Fig. 7B). However, MPO activity was significantly lower in the hyperoxia-exposed slc7a2−/− mice than in the hyperoxia-exposed wild-type mice (Fig. 7B).
Slc7a2 knockout inhibited hyperoxia-induced apoptosis in the mouse lungs.
To determine apoptosis in the mouse lungs, we measured internucleosomal DNA fragmentation by labeling DNA breaks with TUNEL using immunohistochemistry. In the lungs of wild-type mice, hyperoxic exposure led to significantly greater numbers of TUNEL-positive cells than in normoxic-exposed wild-type animals (Fig. 8). However, in the slc7a2−/− mice there was no significant difference in the percent TUNEL-positive cells between those exposed to normoxia and those exposed to hyperoxia (Fig. 8). The percent TUNEL-positive cells was significantly lower in the hyperoxia-exposed slc7a2−/− mice than in the hyperoxia-exposed wild-type mice (Fig. 8).
Fig. 8.

The percent of TUNEL-positive cells was significantly greater in hyperoxia only in the wild-type animals. Animals were exposed to either room air or >95% oxygen for 60 h, the lungs were then fixed, embedded in paraffin and 5 µm-sections were cut for each sample. The slides were assayed for internucleosomal DNA fragmentation by labeling DNA breaks with TUNEL using immunohistochemistry. Fifty images from four animals were scored for each condition. Representative TUNEL stain images (A); calculated percent TUNEL-positives cells (B). *Hyperoxia different from the room air same genotype, P < 0.001; #Hyperoxic slc7a2−/− different from hyperoxic wild-type, P < 0.001.
DISCUSSION
The main findings of this study in slc7a2−/− mice were that 1) hyperoxia led to greater iNOS mRNA expression, 2) lack of slc7a2 prevented the hyperoxia-induced increase in exNO production from the mice, 3) lack of slc7a2 attenuated the hyperoxia-induced increase in lung permeability and neutrophil infiltration, 4) lack of slc7a2 significantly suppressed apoptosis in the lungs induced by hyperoxia, and 5) lack of slc7a2 prevented the hyperoxia-induced decrease in compliance and increase in resistance in the lungs. Taken together, these findings support our hypothesis that mice deficient in the slc7a2 gene are protected from hyperoxia-induced ALI. Furthermore, the lower exNO in the slc7a2−/− mice supports the concept that the absence of slc7a2 results in lower lung NO production in hyperoxia despite greater levels of iNOS in the lungs. The attenuated NO production in the slc7a2−/− mice results in reduced hyperoxia-induced lung injury by attenuation of NO-induced apoptosis (Fig. 9). These results are consistent with a central role of CAT-2 in maintaining the hyperoxia-induced augmented NO production in the lungs.
Fig. 9.

Model summarizing findings and demonstrating a critical role for cationic amino acid transporter (CAT)-2-mediated l-arginine uptake in the development of acute lung injury (ALI). Hyperoxia upregulates iNOS and endothetlial NOS expression resulting in l-arginine-dependent augmented NO production, which leads to cellular damage and apoptosis resulting in inflammation, edema, and neutrophil accumulation culminating in ALI. CAT-2 (slc7a2) is necessary to maintain the augmented NO production leading to ALI, such that deficiency of CAT-2 protects the lung from the damaging effects of the augmented NO production by attenuating NO production. iNOS, inducible NOS; NO, nitric oxide; NOS, NO synthase.
Deletion of slc7a2 (CAT-2) in the lungs of these mice did not result in compensatory upregulation of slc7a1 (CAT-1). It is interesting that both genotypes exhibited an induction of slc7a1 mRNA following hyperoxic exposure. We have previously found a more modest upregulation of slc7a1 mRNA following LPS/TNF-α treatment in human pulmonary microvascular endothelial cells (4). It may be that the combination of hyperoxia and proinflammatory cytokine release seen in the hyperoxia exposed lungs resulted in the augmented induction of slc7a1. We did find that it took both LPS and hyperoxia in RAW cells to get induction of slc7a1. However, further studies are needed to elucidate the exact mechanisms underlying slc7a1 induction in the lung following exposure to hyperoxia. It is also likely that different cell types in the lung respond differently to exposure to hyperoxia, and it is the sum of these differing cellular responses that yields the overall lung response to hyperoxic injury (22). Although studies elucidating exactly which cell types are central to the development of hyperoxic lung injury are outside the scope of the current studies, our previous data in endothelial cells (4, 5, 16) and the experiments on RAW cells presented herein would suggest that both endothelial cells and macrophages response to hyperoxia-induced inflammation. It has long been known that endothelial cell apoptosis is a key early step in the pulmonary edema formation, neutrophil infiltration, and development of alveolar exudates that characterize ALI (19, 22). However, a key point in this study is that mice globally deficient in slc7a2 have significantly lower NO production and apoptosis in the lung following hyperoxia exposure than do wild-type mice following hyperoxia despite the induction of slc7a1. This supports a key role for slc7a2 in the hyperoxia-induced augmented lung NO production seen in wild-type mice exposed to hyperoxia. It is reasonable to speculate that endothelial cells deficient in slc7a2 will be protected from hyperoxia-induced apoptosis; however further studies are needed to determine if preventing CAT-2-mediated l-arginine uptake specifically in endothelial cells will have the same effect on attenuating hyperoxia-induced lung injury as described herein for global deficiency of slc7a2.
Although the slc7a2-deficient mice did have greater levels of iNOS in normoxia than did wild-type mice, the levels of exNO production were similar to normoxic wild-type mice. This suggests a role for CAT-2-mediated l-arginine uptake in iNOS-mediated NO production during normoxia, particularly considering that the levels of eNOS and nNOS mRNA were similar between the genotypes in normoxia. Both wild-type and slc7a2-deficient mice responded to hyperoxia with an increase in iNOS mRNA expression, although the levels of iNOS mRNA were greater in the slc7a2−/− hyperoxic mice than in the wild-type hyperoxic mice. In the hyperoxic slc7a2−/− deficient mice, exNO production was actually lower than in hyperoxic wild-type mice despite greater levels of iNOS. Although the wild-type mice had significantly greater exNO production in hyperoxia than in normoxia, the slc7a2−/− mice did not show any differences in exhaled NO production based on exposure, despite substantially greater levels of iNOS following hyperoxia exposure. In the original study of slc7a2−/− mice, Nicholson et al. (17) demonstrated that macrophages from Slc7a2−/− mice had substantially lower cytokine-induced NO production than did macrophages from wild-type mice, despite similar levels of iNOS protein and activity. In the same study, Nicholson et al. (17) also demonstrated that the macrophages had substantially lower l-arginine uptake than did wild-type macrophages. Similar results were reported in macrophages by Yeramian et al. (23). We have previously shown in isolated perfused lungs from LPS-treated neonatal pigs that inhibition of l-arginine uptake using the competitive inhibitor l-lysine decreased exhaled NO production (3). Furthermore, we reported that in isolated perfused mouse lungs the addition of l-arginine to the perfusate resulted in a significant increase in exhaled NO production (9). Taken together with our data, these findings are consistent with the concept that iNOS-mediated NO production depends on CAT-2-mediated l-arginine uptake following hyperoxia-induced lung injury.
A hallmark of oxygen-induced lung injury is increased lung permeability and neutrophil infiltration (6). We found that the slc7a2−/− mice exposed to hyperoxia had lower lung wet weights and lower BALF protein levels than did similarly exposed wild-type mice. Furthermore, the slc7a2−/− mice had less MPO activity in their lungs following hyperoxic exposure than did wild-type mice following hyperoxic exposure. Hyperoxia also induces apoptosis of cells, particularly endothelial cells, and this endothelial apoptosis contributes to the increased lung permeability and neutrophil infiltration (11). We found that the percent of TUNEL-positive cells in the lungs of hyperoxic slc7a2−/− mice was significantly lower than in the hyperoxic wild-type mice. Taken together our findings suggest that the slc7a2−/− mice were protected from the development of ALI following hyperoxia exposure because of a decrease in NO production resulting in less endothelial apoptosis, which was associated with the maintenance of the endothelial barrier and attenuation of the hyperoxia-induced lung edema and neutrophil infiltration. This concept is consistent with findings of Li et al. (12) where they showed that hyperoxia-exposed mice treated with IL-10 had lower levels of iNOS protein, lower BALF levels of NO, less MPO activity, less lung permeability, and lower levels of TUNEL-positive cells than did vehicle-treated hyperoxia-exposed mice. Similarly, Kondrikov et al. (11) reported that when treated with a peptide that inhibits NO production, mice exposed to hyperoxia had lower lung wet weights, less lung microvascular leakage, and less endothelial cell apoptosis than vehicle-treated mice. Another example is the report from Klings et al. (10) where mice deficient in γ-glutamyl transferase had greater iNOS expression, more NO production, greater lung wet weights, and more apoptosis than did wild-type mice. Furthermore, the γ-glutamyl transferase deficient mice had less apoptosis if treated with the iNOS inhibitor l-NAME (10). However, hyperoxia-induced ALI is a complex process that likely involves myriad molecular and cellular responses; taken together with our results, these findings are consistent with the notion that attenuating or preventing NO production during hyperoxic exposure protects against the development of ALI at least in part by attenuating the endothelial apoptotic response to hyperoxia and thereby decreasing lung edema and neutrophil infiltration.
We also used pulmonary mechanics measurements as a physiologic indicator of hyperoxia-induced ALI, and we found that pulmonary mechanics were better in the slc7a2−/− mice exposed to hyperoxia than in the wild-type mice exposed to hyperoxia. Specifically, CRS and Cst were higher, whereas RRS was lower in the hyperoxia-exposed slc7a2−/− mice than in wild-type mice. This finding is consistent with a study by Schwingshackl et al. (20) wherein 24 h of exposure to >95% oxygen resulted in decreased Cst. We found that the changes in CRS and RRS were associated with the changes in lung wet-to-dry weights. Specifically, we found that when lung wet-to-dry weight increased lung compliance decreased. This association suggests that pulmonary edema is responsible for the decreased lung CRS and RRS seen in the wild-type hyperoxia-exposed mice. The notion that pulmonary edema is a component of the ALI caused by hyperoxic exposure has been shown repeatedly in mice in the past (6, 7, 10–12, 20). Thus, deficiency of slc7a2 seemed to prevent the hyperoxia-induced pulmonary edema and thereby maintained essentially normal CRS and RRS values even after 60 h in hyperoxia.
In summary, we found that mice deficient in the slc7a2 gene, which encodes for CAT-2, were protected from the development of hyperoxia-induced ALI. We found that lung NO production was lower in the hyperoxic slc7a2−/− mice than in the hyperoxic wild-type mice, consistent with the notion that l-arginine uptake is necessary for iNOS-derived lung NO production following hyperoxic exposure. Furthermore, the lungs from the slc7a2−/− mice had less cell death as the result of apoptosis, lower BALF protein concentrations, less lung water, and fewer activated neutrophils than did the lungs from wild-type mice. We conclude that CAT-2-mediated l-arginine uptake is necessary for the hyperoxia-induced increase in lung NO production that leads to endothelial cell apoptosis and ALI. We speculate that CAT-2 may be a potential therapeutic target in ALI, wherein inhibition of CAT-2 activity may prevent or attenuate the physiological signs of ALI.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Y.L. and L.D.N. conceived and designed research; Y.J. performed experiments; Y.J., Y.L., and L.D.N. analyzed data; Y.J., Y.L., and L.D.N. interpreted results of experiments; Y.J. and L.D.N. prepared figures; L.D.N. drafted manuscript; Y.J., Y.L., and L.D.N. edited and revised manuscript; Y.J., Y.L., and L.D.N. approved final version of manuscript.
REFERENCES
- 1.Alexander JH, Reynolds HR, Stebbins AL, Dzavik V, Harrington RA, Van de Werf F, Hochman JS; The TRIUMPH Investigators . Effect of tilarginine acetate in patients with acute myocardial infarction and cardiogenic shock: the TRIUMPH randomized controlled trial. JAMA 297: 1657–1666, 2007. doi: 10.1001/jama.297.15.joc70035. [DOI] [PubMed] [Google Scholar]
- 2.Bailey A, Pope TW, Moore SA, Campbell CL. The tragedy of TRIUMPH for nitric oxide synthesis inhibition in cardiogenic shock: where do we go from here? Am J Cardiovasc Drugs 7: 337–345, 2007. doi: 10.2165/00129784-200707050-00003. [DOI] [PubMed] [Google Scholar]
- 3.Carter BW JR, Chicoine LG, Nelin LD. L-lysine decreases nitric oxide production and increases vascular resistance in lungs isolated from lipopolysaccharide-treated neonatal pigs. Pediatr Res 55: 979–987, 2004. doi: 10.1203/01.pdr.0000127722.55965.b3. [DOI] [PubMed] [Google Scholar]
- 4.Chicoine LG, Stenger MR, Cui H, Calvert A, Evans RJ, English BK, Liu Y, Nelin LD. Nitric oxide suppression of cellular proliferation depends on cationic amino acid transporter activity in cytokine-stimulated pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol 300: L596–L604, 2011. [Erratum in Am J Physiol Lung Cell Mol Physiol 301: L135, 2011]. doi: 10.1152/ajplung.00029.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cui H, Chen B, Chicoine LG, Nelin LD. Overexpression of cationic amino acid transporter-1 increases nitric oxide production in hypoxic human pulmonary microvascular endothelial cells. Clin Exp Pharmacol Physiol 38: 796–803, 2011. doi: 10.1111/j.1440-1681.2011.05609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galam L, Rajan A, Failla A, Soundararajan R, Lockey RF, Kolliputi N. Deletion of P2X7 attenuates hyperoxia-induced acute lung injury via inflammasome suppression. Am J Physiol Lung Cell Mol Physiol 310: L572–L581, 2016. doi: 10.1152/ajplung.00417.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garat C, Jayr C, Eddahibi S, Laffon M, Meignan M, Adnot S. Effects of inhaled nitric oxide or inhibition of endogenous nitric oxide formation on hyperoxic lung injury. Am J Respir Crit Care Med 155: 1957–1964, 1997. doi: 10.1164/ajrccm.155.6.9196102. [DOI] [PubMed] [Google Scholar]
- 8.Howes LG, Brillante DG. Expert opinion on tilarginine in the treatment of shock. Expert Opin Investig Drugs 17: 1573–1580, 2008. doi: 10.1517/13543784.17.10.1573. [DOI] [PubMed] [Google Scholar]
- 9.Jin Y, Chen B, Calvert TJ, Chicoine LG, Liu Y, Nelin LD. Chronic hypoxia decreases arterial and venous compliance in isolated perfused rat lungs: an effect that is reversed by exogenous arginine. Am J Physiol Heart Circ Physiol 304: H195–H205, 2013. doi: 10.1152/ajpheart.00188.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klings ES, Lowry MH, Li G, Jean JC, Fernandez BO, Garcia-Saura MF, Feelisch M, Joyce-Brady M. Hyperoxia-induced lung injury in gamma-glutamyl transferase deficiency is associated with alterations in nitrosative and nitrative stress. Am J Pathol 175: 2309–2318, 2009. doi: 10.2353/ajpath.2009.081017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kondrikov D, Gross C, Black SM, Su Y. Novel peptide for attenuation of hyperoxia-induced disruption of lung endothelial barrier and pulmonary edema via modulating peroxynitrite formation. J Biol Chem 289: 33355–33363, 2014. doi: 10.1074/jbc.M114.585356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li HD, Zhang QX, Mao Z, Xu XJ, Li NY, Zhang H. Exogenous interleukin-10 attenuates hyperoxia-induced acute lung injury in mice. Exp Physiol 100: 331–340, 2015. doi: 10.1113/expphysiol.2014.083337. [DOI] [PubMed] [Google Scholar]
- 13.López A, Lorente JA, Steingrub J, Bakker J, McLuckie A, Willatts S, Brockway M, Anzueto A, Holzapfel L, Breen D, Silverman MS, Takala J, Donaldson J, Arneson C, Grove G, Grossman S, Grover R. Multiple-center, randomized, placebo-controlled, double-blind study of the nitric oxide synthase inhibitor 546C88: effect on survival in patients with septic shock. Crit Care Med 32: 21–30, 2004. doi: 10.1097/01.CCM.0000105581.01815.C6. [DOI] [PubMed] [Google Scholar]
- 14.McDonald KK, Rouhani R, Handlogten ME, Block ER, Griffith OW, Allison RD, Kilberg MS. Inhibition of endothelial cell amino acid transport System y+ by arginine analogs that inhibit nitric oxide synthase. Biochim Biophys Acta 1324: 133–141, 1997. doi: 10.1016/S0005-2736(96)00226-X. [DOI] [PubMed] [Google Scholar]
- 15.Mocellin S, Bronte V, Nitti D. Nitric oxide, a double edged sword in cancer biology: searching for therapeutic opportunities. Med Res Rev 27: 317–352, 2007. doi: 10.1002/med.20092. [DOI] [PubMed] [Google Scholar]
- 16.Nelin LD, Nash HE, Chicoine LG. Cytokine treatment increases arginine metabolism and uptake in bovine pulmonary arterial endothelial cells. Am J Physiol Lung Cell Mol Physiol 281: L1232–L1239, 2001. doi: 10.1152/ajplung.2001.281.5.L1232. [DOI] [PubMed] [Google Scholar]
- 17.Nicholson B, Manner CK, Kleeman J, MacLeod CL. Sustained nitric oxide production in macrophages requires the arginine transporter CAT2. J Biol Chem 276: 15881–15885, 2001. doi: 10.1074/jbc.M010030200. [DOI] [PubMed] [Google Scholar]
- 18.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev 87: 315–424, 2007. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raghavendran K, Pryhuber GS, Chess PR, Davidson BA, Knight PR, Notter RH. Pharmacotherapy of acute lung injury and acute respiratory distress syndrome. Curr Med Chem 15: 1911–1924, 2008. doi: 10.2174/092986708785132942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwingshackl A, Teng B, Makena P, Ghosh M, Sinclair SE, Luellen C, Balasz L, Rovnaghi C, Bryan RM, Lloyd EE, Fitzpatrick E, Saravia JS, Cormier SA, Waters CM. Deficiency of the two-pore-domain potassium channel TREK-1 promotes hyperoxia-induced lung injury. Crit Care Med 42: e692–e701, 2014. doi: 10.1097/CCM.0000000000000603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su CF, Yang FL, Chen HI. Inhibition of inducible nitric oxide synthase attenuates acute endotoxin-induced lung injury in rats. Clin Exp Pharmacol Physiol 34: 339–346, 2007. doi: 10.1111/j.1440-1681.2007.04553.x. [DOI] [PubMed] [Google Scholar]
- 22.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 342: 1334–1349, 2000. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 23.Yeramian A, Martin L, Serrat N, Arpa L, Soler C, Bertran J, McLeod C, Palacín M, Modolell M, Lloberas J, Celada A. Arginine transport via cationic amino acid transporter 2 plays a critical regulatory role in classical or alternative activation of macrophages. J Immunol 176: 5918–5924, 2006. doi: 10.4049/jimmunol.176.10.5918. [DOI] [PubMed] [Google Scholar]
- 24.Yuba T, Nagata K, Yamada T, Osugi S, Kuwahara H, Iwasaki Y, Handa O, Naito Y, Fushiki S, Yoshikawa T, Marunaka Y. A novel potent inhibitor of inducible nitric oxide synthase, ONO-1714, reduces hyperoxic lung injury in mice. Respir Med 101: 793–799, 2007. doi: 10.1016/j.rmed.2006.08.001. [DOI] [PubMed] [Google Scholar]