

Abstract
Alcohol consumption exacerbates hepatitis C virus (HCV) pathogenesis and promotes disease progression, although the mechanisms are not quite clear. We have previously observed that acetaldehyde (Ach) continuously produced by the acetaldehyde-generating system (AGS), temporarily enhanced HCV RNA levels, followed by a decrease to normal or lower levels, which corresponded to apoptosis induction. Here, we studied whether Ach-induced apoptosis caused depletion of HCV-infected cells and what role apoptotic bodies (AB) play in HCV-alcohol crosstalk. In liver cells exposed to AGS, we observed the induction of miR-122 and miR-34a. As miR-34a has been associated with apoptotic signaling and miR-122 with HCV replication, these findings may suggest that cells with intensive viral replication undergo apoptosis. Furthermore, when AGS-induced apoptosis was blocked by a pan-caspase inhibitor, the expression of HCV RNA was not changed. AB from HCV-infected cells contained HCV core protein and the assembled HCV particle that infect intact hepatocytes, thereby promoting the spread of infection. In addition, AB are captured by macrophages to switch their cytokine profile to the proinflammatory one. Macrophages exposed to HCV+ AB expressed more IL-1β, IL-18, IL-6, and IL-10 mRNAs compared with those exposed to HCV− AB. The generation of AB from AGS-treated HCV-infected cells even enhanced the induction of aforementioned cytokines. We conclude that HCV and alcohol metabolites trigger the formation of AB containing HCV particles. The consequent spread of HCV to neighboring hepatocytes via infected AB, as well as the induction of liver inflammation by AB-mediated macrophage activation potentially exacerbate the HCV infection course by alcohol and worsen disease progression.
Keywords: HCV RNA, acetaldehyde, apoptosis, hepatocytes, macrophages
alcohol exposure exacerbates hepatitis C virus (HCV) infection severity, increases liver inflammation, and potentiates liver injury progression. However, the pathogenesis of HCV-alcohol interactions is not clear yet. The major difficulty in HCV-alcohol studies is related to the lack of adequate models that can recapitulate both viral replication and ethanol metabolism—two important features that potentiate liver injury progression in alcohol-abusing HCV patients. Human liver cell lines permissive for HCV (Huh7.5 or Huh 7.5.1) that are currently used for HCV-alcohol-related in vitro studies do not metabolize ethanol, while ethanol-metabolizing rodent liver cells cannot support HCV replication. Studies conducted with isolated human hepatocytes also have failed, since these cells require at least 4–5 days to become infected with HCV; however, by this time, hepatocytes lose the expression and functional activity of CYP2E1 and alcohol dehydrogenase (ADH), the main ethanol-metabolizing enzymes (47). In vivo models of HCV infection are also questionable, since small rodents cannot be infected with human HCV, and they only transgenically express HCV proteins, in the absence of replicating virus. The best option in this case is to use immunodeficient mice with humanized livers that are able to replicate the human virus, but to our best knowledge, while infection of the liver with HCV is widely reported (5, 26, 57), HCV-ethanol studies on this expensive and demanding model are very few (37).
Many investigators attribute the potentiating effects of ethanol to enhanced HCV replication. However, these reported studies were conducted on either ethanol-nonmetabolizing cells or cells that express only CYP2E1, but not ADH, suggesting that they did not generate appreciable levels of acetaldehyde (Ach) (36, 51, 62). While some studies claimed the role of ethanol-induced redox changes in promoting HCV replication (46), meta-analysis data summarizing the effects of ethanol on HCV RNA in HCV-infected alcoholic patients, revealed minimal contribution of alcohol in enhancing HCV levels (1).
Altered ethanol- or Ach-induced cellular signaling may worsen HCV progression by affecting the regulation of HCV RNA production, packaging, or dissemination. MicroRNAs have the potential to regulate a number of steps in HCV progression. For example, miR-122 has two seed-binding sites in the 5′UTR of the HCV RNA genome that are upstream of the internal ribosomal entry site and are necessary for viral replication (34). Reduced miR-122 binding decreases, while increased binding elevates HCV replication (3, 22). Alternatively, we have postulated a role for apoptosis in HCV propagation, and miR-34a is known to regulate apoptosis in hepatocytes (10).
Recently, we showed a biphasic effect of ethanol metabolites on HCV RNA expression in JFH1-infected Huh7.5-CYP2E1+ cells exposed to ethanol-containing acetaldehyde-generating system (AGS) that makes physiological levels of Ach. We observed an enhancement of HCV RNA levels by 24 h of AGS cell exposure, but HCV RNA returned to untreated or even lower levels at 48 h of exposure. Furthermore, similar kinetics for HCV core protein expression on lipid droplets in these cells was observed using immunofluorescent staining (17), confirming the HCV RNA data. In addition to the loss in HCV RNA at 48 h of AGS exposure, we observed the induction of apoptosis in HCV-infected cells (2). Thus, we hypothesized that HCV replication, induced by ethanol metabolism (Ach), sensitizes infected liver cells to apoptosis. We further hypothesized that the generated apoptotic bodies (AB) potentiate the spread of viral infection and contribute to the induction of liver injury by programming liver macrophages toward an inflammatory phenotype. Here, we provide evidence for this scenario that explains how ethanol exacerbates HCV pathogenesis.
MATERIALS AND METHODS
Reagents and Media
High-glucose DMEM and FBS were purchased from Invitrogen (Carlsbad, CA). Antibodies to the cleaved caspase-3 and PARP were obtained from Cell Signaling (Beverly, MA); Antibody to β-actin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell Lines and Treatment
CYP2E1+ Huh7.5 (RLW) cells were obtained and cultured as described before (17). They were infected with JFH1 virus at multiplicity of infection (MOI) = 0.1 (24) or left uninfected. The cells were exposed for 48 h to 50 mM ethanol (EtOH), yeast ADH (0.02 U/ml), and 2 mM nicotinamide adenine dinucleotide (NAD). This system designated as AGS was designed to produce Ach to mimic the effect of ethanol metabolism via ADH-mediated Ach release. In AGS, ethanol serves as a substate for yeast ADH-mediated enzymatic reaction utilizing NAD as a cofactor. We have previously shown that AGS generates a high level of Ach in a cell-free medium, but in the presence of RLW cells, a medium level of Ach measured by gas chromatography (GC) fluctuates between about 250 (at 1–4-h exposure) and 50 μM (at 18–48 h exposure) (for details, see Ref. 17). These levels of Ach correspond to the amount of Ach produced by ADH-expressing liver cells (14, 38). AGS provides sustained generation of Ach, which recapitulates the in vivo conditions observed upon ethanol metabolism in the liver. According to the results of lactate hydrogenase measurement, AGS is not toxic to RLW cells (17).
Primary Human Hepatocytes
Human hepatocytes were obtained from Triangle Research Labs (Research Triangle Park, NC). Human hepatocytes were attached to collagen-coated six-well plates (8×105 cells/well) and then infected with JFH1 (HCV genotype 2a) virus at MOI = 0.1 (total time of infection is 5 days). Infected cells were cultured in William's medium with supplements (insulin, holo-transferin, L-ascorbic acid, dexamethazone, selenium, and antibiotics) and 10% FBS for 3 days and then exposed to AGS for 24 and 48 h.
Primary Human Macrophages
Monocytes were obtained from healthy donor blood elutriation. For macrophage (Mph) differentiation, human monocytes were cultured for 0–8 days in DMEM culture media containing human serum and macrophage colony-stimulating factor.
RNA Isolation and Real-Time PCR
Reagents used for RNA isolation, cDNA synthesis and real-time PCR were from Life Technologies (Carlsbad, CA). The relative HCV RNA expression level in infected cells was quantified as described before (61). MicroRNAs were amplified using TaqMan microRNA reverse transcription kit (ThermoFisher, Grand Island, NY) from 100 ng of total RNA. Stem loop primers and hydrolysis probes for miR-34a (hsa-miR-34a-5p, no. PN4427975-000426), miR-122 (has-miR-122-5p, no. PN442795-002245), and control, Z30 RNA (no. PN4427975-001092) were also obtained from ThermoFisher. Relative expression was calculated as using DeltaCT Method as described previously (19).
Immunoblotting
Cell lysis and immunoblotting were performed as described previously (53).
Apoptotic Bodies Generation
Apoptosis was induced by ultraviolet (UV) light (0–100 mJ/cm2, 140 s) (6). After 24 h, the floating apoptotic bodies were collected from 10-cm plates by centrifugation at 1,500 rpm for 5 min and resuspended in DMEM.
HCV Infectivity of Apoptotic Bodies
Huh7.5 cells (2–3 × 104 per well) were plated on eight-well chamber slides and incubated overnight; then, they were intensively washed 3×, and AB generated from (6 ×104) JFH1-infected or noninfected Huh7.5 cells were applied to the wells. JFH1 virus was used as a positive control. Medium from the last wash of AB separated by centrifugation was used as a negative control. AB, JFH1 virus, or medium were incubated with intact cells for 72 h, and then cells were stained with antibody to HCV core (clone: C7-50, ThermoScientific, cat no. MA1-080, dilution 1:300) for 1 h at room temperature followed by incubation with secondary antibody [Alexa Fluor 594 goat-anti-mouse (1:500), Invitrogen] and DAPI for 1 h in the dark. The presence of HCV was visualized using a 10× lens in a LSM 710 confocal microscope (Carl Zeiss, Peabody, MA).
Characterization of Apoptotic Cells
Hoechst 33342 staining: cell aliquots (1–5 × 105) from the ultraviolet light-treated cells were cytospun onto glass slides, fixed in 4% paraformaldehyde in PBS, incubated with 7 μg/ml of Hoechst 33342 (Sigma) at 37°C for 10 min, washed, and sealed under VectaShield. Fluorescent Hoechst H33342-stained nuclei were observed under fluorescent microscopy.
Flow Cytometry Analysis
The percentage of apoptotic cells were determined using annexin-V/propidium iodide (PI) apoptosis detection kit (BD Biosciences, San Diego, CA), according to the instructions of the manufacturer, where Annexin V+/PI− cells showed early apoptosis and Annexin V+/PI+ cells showed late apoptosis. Fluorescence was detected by flow cytometry (BD LSR-II-Green).
Macrophage Treatment with AB
After differentiation of Mph for 8 days, on day 9, an equal number of AB from HCV-infected Huh7.5 cells or HCV-infected Huh7.5 cells pretreated with AGS were added to macrophages (Mph) for 2, 4, 8, and 24 h (Mph: AB ratio was 1:3). AGS-treated or untreated HCV-noninfected cells used for AB generation served as a negative control for the effects of virus/viral proteins on cytokine mRNA induction in Mph measured by RT-PCR, as described above.
ApoB protein was quantified by ELISA method (Mabtech ELISA kit; Mabtech, Cincinatti, OH) in medium from cultured cells exposed or not to AGS.
Statistical Analyses
Data from at least three independent experiments were expressed as means ± SE. Comparisons among multiple groups were determined by one-way ANOVA, using a Tukey post-hoc test. For comparisons between two groups, we used Student's t-test. A probability value of 0.05 or less was considered significant.
RESULTS
Kinetics of HCV RNA, miR-122, and miR-34a Expression Upon Exposure of Cells to AGS
As reported before, we observed an increase in HCV RNA in RLW cells at 4, 18, and 24 h of AGS treatment, which then returned to control (untreated) levels at 48 h (Fig. 1A). In some experiments, we observed even suppression of HCV RNA at this latter time point. These changes in HCV RNA level are accompanied by upregulation of miR-34a at 24 and 48 h and miR-122 at 48 h (Fig. 1, B and C). Since miR-122 increases HCV replication by targeting HCV RNA (28), decrease in HCV RNA at 48 h exposure to AGS (when miR 122 is still high) was intriguing. However, upregulation of proapoptotic miR-34a (29), observed at 24 h of AGS treatment that further increases at 48 h, suggested a substantial apoptosis in HCV-replicating cells that could lead to HCV RNA depletion to amounts comparable to AGS-pretreatment levels.
Fig. 1.
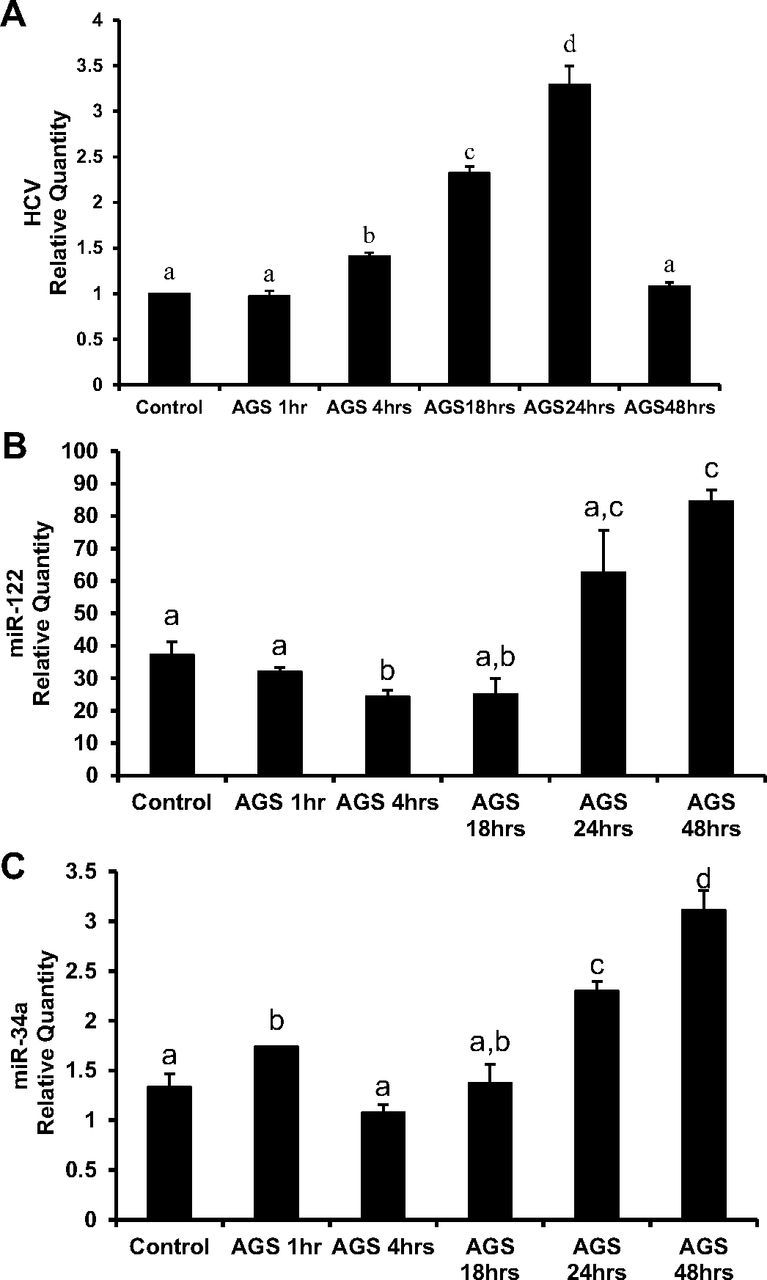
Effects of the acetaldehyde-generating system (AGS) on expression of hepatitis C virus (HCV) RNA and miRNAs. HCV-infected CYP2E1+ Huh7.5 (RLW) cells were treated with AGS for the indicated time. A: RT-PCR analysis of HCV RNA in RLW cells. GAPDH mRNA was used to normalize the gene of interest. B and C: quantitative PCR analysis of miR-122 and miR-34a. Z30 RNA was used to normalize the gene of interest. All data (representative results and quantification) were generated from three independent experiments and presented as means ± SE. Bars with different letters are significantly different at P ≤ 0.05.
AGS Suppresses Very Low-Density Lipoproteins Secretion
Since VLDLs serve as a vehicle for the secretion of intracellularly assembled HCV particle (21), to study whether the reduction in HCV RNA at 48 h is attributed to the effects of AGS on VLDL secretion, we measured ApoB levels in the medium of control and ethanol or AGS-treated RLW cells by ELISA. Ethanol in the absence of AGS did not affect ApoB release to the medium, while AGS suppressed ApoB secretion in infected and noninfected cells (Fig. 2, A and B), indicating the role of Ach in this process. Furthermore, cleared by centrifugation, supernatants from AGS-exposed infected cells were unable to infect intact Huh7.5 cells (infectivity assay, not shown).
Fig. 2.

Acetaldehyde (Ach) decreases ApoB levels in both HCV+ and HCV− RLW cell supernatants. A and B: ApoB levels were measured in Etoh and AGS-treated cell supernatants by ELISA. Data are generated from three independent experiments and presented as means ± SE. Bars with different letters are significantly different at P ≤ 0.05.
Apoptosis in HCV-Infected RLW Cells Exposed to AGS
We hypothesized that the decrease in HCV RNA at 48 h of AGS treatment was related to enhanced apoptosis in HCV+ cells exposed to AGS. Thus, we measured the kinetics of apoptosis induction (cleaved caspase 3 and PARP) upon EtOH or AGS treatment (Fig. 3). While ethanol induced no apoptosis in our CYP2E1+ RLW cells, AGS increased caspase 3 cleavage by seven-fold in HCV-infected cells and also activated PARP cleavage at 48 h (Fig. 3, A–C). Interestingly, even in the absence of HCV, AGS activated apoptosis, which was evident by a four-fold induction of caspase 3 cleavage by 48 h in RLW cells (not shown). These data indicate that either Ach alone or in a combination with CYP2E1-generated ethanol metabolites, but not CYP2E1-mediated ethanol metabolism, per se, enhanced apoptotic cell death and that the most potent effects of AGS were observed in HCV-presensitized cells. Furthermore, by 48 h of AGS treatment, there was an induction of lipid peroxidation in infected RLW cells, as evident from the accumulation of 4-HNE (Fig. 3D). Induction of miR-34a and/or increased production of 4-HNE could be the trigger for cells to undergo apoptosis.
Fig. 3.

Ach induces apoptosis in HCV-infected RLW cells. A and B: Pro-caspase-3, cleaved caspase-3, and β-actin [immunoblotting (IB) and quantification]. C: cleaved PARP and total PARP (IB). D: kinetics of 4-hydroxynonenal (4HNE) detection (IB). E: effect of pan-caspase inhibitor on HCV RNA. All data (representative results and quantification) were generated from three independent experiments and presented as means ± SE. Bars with different letters are significantly different at P ≤ 0.05.
To prove that HCV RNA reduction in AGS-treated RLW cells is apoptosis-related, we blocked apoptosis by treatment with a pan-caspase inhibitor, Z-VAD-FMK for the last 24 h of AGS exposure. While AGS treatment reduced HCV RNA, the reduction was abrogated by cotreatment with the pan-caspase inhibitor (Fig. 3E).
Apoptosis induction by combined HCV plus Ach exposure was attenuated by silencing miR-34a using a locked nucleic acid antagonist, LNA-34a. There was a 16% reduction in levels of cleaved caspase 3 in LNA-34a treatment compared with LNA controls (Fig. 4, A and B; see details of experiment in figure legends). These data are consistent with miR-34a-dependent induction of apoptosis by Ach in HCV+ cells.
Fig. 4.

Ach-induced apoptosis is attenuated by miR-34a antagonist in HCV-infected RLW cells. Immunoblotting of cleaved caspase-3 and β-actin (A) and quantification of Western blot (B). HCV-infected RLW cells were transfected using RNAiMAX transfection reagent (Life Technologies) with miR-34a antagonist, LNA-34a, an antisense-locked nucleic acid oligonucleotide specific for human miR-34a (MIMAT0000255) and LNA-negative control A. LNAs were obtained from Exiqon (Woburn, MA) and used at a final concentration of 20 nM. Twenty-four hours after transfection, cells were exposed to AGS for 48 h. Caspase-3 cleavage was detected by Western blot analysis. All data (representative results and quantification) were generated from three independent experiments. Data are presented as means ± SE. Bars with different letters are significantly different at P ≤ 0.05.
HCV RNA and Apoptosis in HCV-Infected Human AGS-Exposed Hepatocytes
To confirm that the changes observed in hepatoma cells upon AGS treatment are reproducible in human hepatocytes, we infected hepatocytes with JFH-1 virus for 72 h and then exposed to AGS for 24 and 48 h. HCV RNA was upregulated at 24 h of AGS exposure and returned to control levels by 48 h (Fig. 5A). Accordingly, by 48 h of AGS treatment, we observed cleavages of caspase 3 and PARP (Fig. 5, B–E).
Fig. 5.

Ach regulates HCV RNA levels and induces apoptosis in HCV-infected hepatocytes. A: HCV-RNA in hepatocytes treated with AGS for 24 h and 48 h, RT-PCR. B and C: immunoblotting of cleaved caspase-3, β-actin, and quantification. Human primary hepatocytes were infected with JFH-1 virus for 72 h and then treated with AGS for 24 h and 48 h. D and E: immunoblotting of Cleaved PARP, total PARP, and quantification. All data (representative results and quantification) were generated from three independent experiments. Data are presented as means ± SE. Bars with different letters are significantly different at P ≤ 0.05.
AB Contain HCV Proteins and Virus
To better characterize AB, we induced apoptosis in HCV-infected and noninfected RLW cells by exposure to UV light. Apoptosis was confirmed by Hoechst staining, as well as by flow cytometric analysis of propidium iodide (PI) and Annexin V-FITC staining (Fig. 6, A and B). This UV exposure generated a sufficient amount of AB of 80–90% purity that could be used for Western blot (WB) analysis and infectivity assay. HCV core protein was detected by WB analysis in AB from both control and AGS-treated infected cells (Fig. 7, A and B); the amount of HCV core protein was three-fold lower in live (attached) AGS-treated cells corroborating the observed reduction of HCV RNA at this time point (Fig. 7, C and D). Furthermore, washed AB (but not medium from the last wash) were able to infect intact Huh7.5 cells (Fig. 7E), indicating that AB contain not just viral proteins, but infection-competent virus.
Fig. 6.

Hoechst 33342 staining of normal and apoptotic cells. A: control cells show normal nuclear staining, while the cells undergoing apoptosis demonstrated apoptotic chromatin changes: blebbing, fragmentation, and condensation under a fluorescence microscope at ×20. Apoptotic cells showed typical morphological features as DNA condensation, fragmentation, and nuclear shrinkage in UV light-exposed cells. B: flow cytometric analysis of annexin V-FITC/PI double-staining: Apoptotic cells collected after UV light treatment were incubated with Annexin V-FITC and/or propidium iodide (PI) and analyzed by flow cytometry. Annexin V+/PI− cells are early apoptotic cells; Annexin V+/PI+ cells are late apoptotic.
Fig. 7.

Ach reduces HCV core protein levels in HCV-infected live cells, but not in apoptotic bodies. A and B: HCV core protein (IB and quantification) in AGS-treated AB. C and D: HCV core protein (IB and quantification) in AGS-treated live cells. β-actin was used as internal control. E: infectivity of AB (immunostaining of HCV core protein in RLW cells infected with AB obtained from HCV+ untreated and treated cells). All data were generated from three independent experiments and presented as means ± SE. Bars with different letters are significantly different at P ≤ 0.05.
HCV+ AB Program Macrophage Cytokine Profile
Human in vitro differentiated Mph (primary cells) were exposed to HCV+ and HCV− AB for 2, 4, 8, and 24 h at a 1:3 ratio. mRNAs were determined in Mph at all time points for the following cytokines: TNF-α, IL-6, IL-1β, IL-18, IL-10, IL-12, and TGF-β. The optimal time point that demonstrated the highest levels of mRNA induction after Mph exposure to AB was chosen for all cytokines. Since engulfment of HCV− AB may also affect cytokine profile of Mph, to elucidate the impact of HCV on activation of cytokine genes, we calculated the ratio between cytokine mRNA expression resulting from Mph exposure to HCV+ AB vs. HCV− AB. As appeared, HCV activated proinflammatory IL-1β, IL-18, IL-6, and anti-inflammatory IL-10 cytokines (Fig. 8A, black bars). Because posttranslational protein modifications induced by RLW cell exposure to AGS can affect antigenic characteristics of AB and, thereby, influence the cytokine profile of Mph after engulfment of such AB, we generated HCV+ and HCV− AB from either untreated (control) cells or cells exposed to AGS. We observed that the HCV+ AB generated from AGS-treated cells induced higher levels of IL-1β, IL-18, and IL-10, but not IL-6 mRNA compared with the HCV− AGS-treated AB (Fig. 8A, gray bars).
Fig. 8.
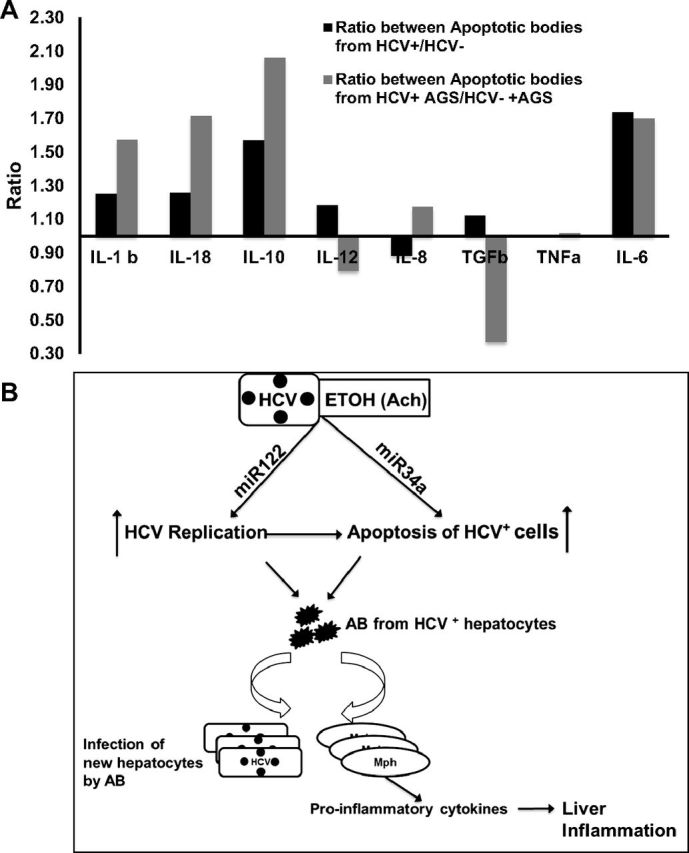
Induction of cytokine mRNA by AB in macrophages. A: RT-PCR analysis of cytokines. GAPDH was used to normalize the gene of interest. Results are presented as average ratio between cytokine mRNA obtained after macrophages (Mph) exposure to HCV+ over HCV− AB from control (black bars) or AGS-treated (gray bars) cells, results of three independent experiments. B: proposed mechanism of HCV pathogenesis exacerbation by alcohol metabolism (Ach).
DISCUSSION
Hepatocytes are primary sites of both HCV replication and ethanol metabolism. Ethanol is metabolized by major enzymes CYP2E1 and ADH. Metabolizing ethanol, CYP2E1 generates reactive oxygen species (ROS) and a small amount of Ach, while ADH is a main source of Ach. The RLW cells that we used in this study are Huh7.5 cells stably transfected with CYP2E1. They do not generate a measurable level of Ach, but they produce significant ROS levels when exposed to ethanol. Thus, to mimic ethanol metabolism by hepatocytes and to study the effects of Ach, we used AGS that provides enzymatic production of sustained and physiologically relevant amount of Ach. Here, when Huh7.5-CYP (RLW) cells were exposed to ethanol only, we observed no significant changes in HCV RNA, indicating that CYP2E1-mediated ethanol metabolism per se is not sufficient in regulating HCV RNA levels and requires sustained generation of Ach. One of the mechanisms by which ethanol exacerbates HCV pathogenesis is via an impaired methylation-dependent interferon signaling in hepatocytes that prevents protective antiviral interferon-stimulated gene activation (17). The role of methylation-dependent dysregulation of innate immunity by ethanol in HCV-infected liver cells has also been shown by others (54). The deficiency in innate immunity suggests that the level of HCV RNA should at least be doubled in alcohol-exposed liver cells. Instead, in previously published (17) and current studies, we observed the reduction of both HCV RNA and expression of HCV core protein in Huh7.5-CYP cells after extended exposure to Ach. These observations were contrary to the enhanced pathogenesis seen in HCV-infected alcoholics. The current study was undertaken to understand this paradigm.
There is no direct evidence that ethanol metabolites, and particularly AGS, suppress HCV replication. In contrast, AGS exposure upregulated miR-122, which usually enhances HCV replication (20, 45, 48) and has been shown to be elevated in alcoholic liver disease patients (49). In addition, here, AGS treatment increased miR-34a, known to be a proapoptotic (10, 11). Thus, miRNAs potentially affect two simultaneous events in response to Ach: the activation of HCV replication and apoptosis in infected liver cells. Indeed, our experiments demonstrated an increase in HCV RNA by 24 h of AGS exposure to cells followed by the decrease in HCV RNA due to apoptosis induced by 48 h, as revealed by caspase 3 and PARP cleavages. These apoptotic events became the most prominent at the highest levels of lipid peroxidation (determined by 4-HNE expression) activated by AGS in RLW cells. The crucial role of apoptosis in Ach-mediated depletion of HCV-infected cells is supported by the fact that Ach failed to reduce HCV RNA levels when apoptosis was blocked by pan-caspase inhibitor.
It has been reported before that HCV induces apoptosis of hepatocytes via BAX-mediated mitochondrion-dependent caspase 3 pathway (12). Moreover, other authors showed that HCV induces TRIAL-dependent apoptosis in HCV-infected liver cells (15, 27). Additionally, the ability of HCV to sensitize human hepatocytes to apoptosis has been demonstrated in the model of Scid-Alb-uPA mice with humanized livers (23).
HCV-induced apoptosis of hepatocytes was confirmed in primary cell cultures (39), and alcohol metabolism is known to induce apoptosis to potentiate the effects of HCV (8). Here, we also observed that HCV-infected human hepatocytes undergo apoptosis when exposed to AGS, which is accompanied by the decrease in HCV RNA in live cells. The mechanisms by which combined HCV and Ach treatment induces apoptosis may involve a number of pathways. Previous studies have shown that Ach can inhibit AKT activity, while Ach or ethanol results in proapoptotic posttranslational modifications to the transcription factor FoxO3 (30). The combination of ethanol, or its major metabolite acetaldehyde, plus HCV further dysregulates FoxO3 (55) and alters methylation reactions in cells (17), which usually are protective from apoptosis in the liver (25). Here, we additionally found that the combination increased expression of miR-34a, a known apoptosis inducer that targets silent information regulator 1 (SIRT1) expression and causes p53 acetylation (44, 52, 58, 60). Antagonism of miR-34a only partially reversed caspase 3 cleavage induced by HCV plus Ach, thereby demonstrating that this microRNA was responsible for part, but not all, of the apoptotic stimulation by these agents.
An induction of apoptosis in HCV-infected cells could be generally interpreted as a beneficial event that promotes elimination of infected cells. Thus, Ach-induced apoptosis in hepatocytes should provide protection from HCV infection. Instead, the clinical course of HCV infection in alcohol-consuming patients suggests that alcohol exposure promotes chronic persistence of the virus, considerable liver damage, and progression to hepatocellular carcinoma (HCC) (35, 36, 41, 43), indicating that the combination of HCV and alcohol is quite harmful.
We observed that AB generated from HCV+ cells not only express HCV core protein, but are able to infect intact hepatocytes. Since the canonical pathway of assembled HCV particle release from HCV-containing cells is through VLDL secretion, and VLDL secretion was suppressed by Ach, HCV-containing AB might be an alternative route for infecting neighboring cells in alcohol consumers. To our knowledge, this role of Ach-induced AB in HCV spread and possible chronic persistence of infection has not been reported before. Importantly, in our previous study, we did see longer HCV persistence (without appreciable effect of ethanol feeding on HCV RNA level) in ethanol-fed Scid-Alb-uPA- mice transplanted with human hepatocytes (humanized mice) compared with the same animals on control diet (37). It is not known which cell receptors could be involved in the process of infecting naïve hepatocytes with virus-containing AB. One candidate is the hepatocyte asialoglycoprotein receptor (ASGP-R), previously identified as a mediator of AB uptake (33). However, since the uptake is markedly impaired by ethanol (56) and this impairment can be more visible in certain liver zones, such as in perivenule vs. portal cells (7), it is possible that the in vivo spread of infection takes place in areas less affected by ethanol, where the function of ASGP-R is preserved. We cannot exclude that other receptors (different from ASGP-R) are also involved in AB-hepatocyte interactions. As an option, AB may infect hepatocytes via the receptors for HCV entry or those used for HCV+ exosome uptake (42). The detailed mechanism by which AB generated from HCV-infected cells infect surrounding intact liver cells and the receptor involvement is currently being investigated in our laboratory.
Infection of intact hepatocytes is not the only way that Ach-generated HCV+ AB contributes to liver injury. AB also may induce inflammation by programming liver macrophages (Kupffer cells) toward a proinflammatory phenotype. This, in turn, would promote the uptake of virus by hepatocytes (16) and finally contribute to cirrhosis/HCC development (32). The regulation of a cytokine profile of dendritic cells phagocytosed HCV+ AB was reported before (59). By testing a panel of cytokine mRNAs induced by exposure of macrophages to AB (AB phagocytosis), we found that AB made from HCV+ cells induced higher levels of IL-1β, IL-18, IL-6, and IL-10 than HCV− AB. The toleragenic ability of macrophages to secrete IL-10 in response to HCV core and nonstructural NS3 protein is known to involve the activation of the Toll-like receptor 2 (TLR2) (9), while triggering some other TLRs by viral host factors and alcohol may induce inflammation (13, 50). Interestingly, in our study, when AB were generated from HCV+ AGS-treated 4-HNE highly expressing cells, the activation of IL-1β, IL-18, and IL-10 was even higher and corresponded to the reported ability of alcohol to induce inflammasome formation (40). We cannot exclude that the adduction of Ach-exposed liver cells by lipid peroxidation products (like 4-HNE) enhances antigenic properties of AB generated from these cells, which, in turn, could further induce proinflammatory cytokine activation when phagocytosed by liver macrophages.
Thus, the possible scenario of alcohol-mediated progression of HCV infection may be summarized as the following (Fig. 8B): ethanol metabolism (Ach) upregulates miR-122 and miR-34a, which, on one hand, activate replication of HCV and on the other hand, promote apoptosis of heavily infected cells. Apoptotic bodies (AB) containing replication-competent virus infect neighboring intact hepatocytes and also program macrophages toward a proinflammatory phenotype; this response is further activated by ethanol-induced lipid peroxidation products that may coexist in AB along with HCV. Similarly, promotion of fibrosis could occur when the AB generated by the ethanol exposure of HCV-infected ethanol-metabolizing cells are engulfed by stellate cells (4, 18, 31).
We conclude that the combination of HCV and alcohol metabolites generate AB formation from infected hepatocytes. The consequent spread of HCV to neighboring hepatocytes via AB as a source of infection, as well as induction of liver inflammation by AB-mediated macrophage activation, potentially contributes to exacerbation of HCV infection course by alcohol and the disease progression.
GRANTS
This work was supported by Merit Review BX001673 from the Department of Veterans Affairs, Office of Research and Development (Biomedical Laboratory Research and Development).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.G. and N.A.O. conception and design of research; M.G., S.K.N., and J.Z. performed experiments; M.G., S.K.N., J.Z., and N.A.O. analyzed data; M.G., S.K.N., J.L.M., L.I.P., and N.A.O. interpreted results of experiments; M.G. prepared figures; M.G., K.K.K., and N.A.O. drafted manuscript; J.L.M., L.I.P., B.L.M., K.K.K., D.J.T., and N.A.O. edited and revised manuscript; D.J.T. and N.A.O. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. C. Rice for the Huh7.5 cells, Dr. T. Wakita for the JFH1 virus, and Dr. Raghubendra Sing for helping with flow cytometry analysis.
REFERENCES
- 1.Anand BS, Thornby J. Alcohol has no effect on hepatitis C virus replication: a meta-analysis. Gut : 1468–1472, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aruin AS, Kanekar N, Lee YJ, Ganesan M. Enhancement of anticipatory postural adjustments in older adults as a result of a single session of ball throwing exercise. Exp Brain Res : 649–655, 2015. [DOI] [PubMed] [Google Scholar]
- 3.Bala S, Marcos M, Szabo G. Emerging role of microRNAs in liver diseases. World J Gastroenterol : 5633–5640, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bataller R, Sancho-Bru P, Gines P, Lora JM, Al-Garawi A, Sole M, Colmenero J, Nicolas JM, Jimenez W, Weich N, Gutierrez-Ramos JC, Arroyo V, Rodes J. Activated human hepatic stellate cells express the renin-angiotensin system and synthesize angiotensin II. Gastroenterology : 117–125, 2003. [DOI] [PubMed] [Google Scholar]
- 5.Calattini S, Fusil F, Mancip J, Dao Thi VL, Granier C, Gadot N, Scoazec JY, Zeisel MB, Baumert TF, Lavillette D, Dreux M, Cosset FL. Functional and biochemical characterization of hepatitis C virus (HCV) particles produced in a humanized liver mouse model. J Biol Chem : 23,173–23,187, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Canbay A, Feldstein AE, Higuchi H, Werneburg N, Grambihler A, Bronk SF, Gores GJ. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology : 1188–1198, 2003. [DOI] [PubMed] [Google Scholar]
- 7.Casey CA, Kragskow SL, Sorrell MF, Tuma DJ. Zonal differences in ethanol-induced impairments in receptor-mediated endocytosis of asialoglycoproteins in isolated rat hepatocytes. Hepatology : 260–266, 1991. [PubMed] [Google Scholar]
- 8.Chakraborty JB, Oakley F, Walsh MJ. Mechanisms and biomarkers of apoptosis in liver disease and fibrosis. Int J Hepatol : 648915, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang S, Dolganiuc A, Szabo G. Toll-like receptors 1 and 6 are involved in TLR2-mediated macrophage activation by hepatitis C virus core and NS3 proteins. J Leukoc Biol : 479–487, 2007. [DOI] [PubMed] [Google Scholar]
- 10.Chen H, Wang J, Hu B, Wu X, Chen Y, Li R, Yuan W. MiR-34a promotes Fas-mediated cartilage endplate chondrocyte apoptosis by targeting Bcl-2. Mol Cell Biochem : 21–30, 2015. [DOI] [PubMed] [Google Scholar]
- 11.Chen Q, Li L, Tu Y, Zheng LL, Liu W, Zuo XY, He YM, Zhang SY, Zhu W, Cao JP, Cui FM, Hou J. MiR-34a regulates apoptosis in liver cells by targeting the KLF4 gene. Cell Mol Biol Lett : 52–64, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng L, Adachi T, Kitayama K, Bungyoku Y, Kitazawa S, Ishido S, Shoji I, Hotta H. Hepatitis C virus infection induces apoptosis through a Bax-triggered, mitochondrion-mediated, caspase 3-dependent pathway. J Virol : 10,375–10,385, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dolganiuc A, Norkina O, Kodys K, Catalano D, Bakis G, Marshall C, Mandrekar P, Szabo G. Viral and host factors induce macrophage activation and loss of Toll-like receptor tolerance in chronic HCV infection. Gastroenterology : 1627–1636, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donohue TM, Osna NA, Clemens DL. Recombinant Hep G2 cells that express alcohol dehydrogenase and cytochrome P450 2E1 as a model of ethanol-elicited cytotoxicity. Int J Biochem Cell Biol : 92–101, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Fischer R, Baumert T, Blum HE. Hepatitis C virus infection and apoptosis. World J Gastroenterol : 4865–4872, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fletcher NF, Sutaria R, Jo J, Barnes A, Blahova M, Meredith LW, Cosset FL, Curbishley SM, Adams DH, Bertoletti A, McKeating JA. Activated macrophages promote hepatitis C virus entry in a tumor necrosis factor-dependent manner. Hepatology : 1320–1330, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ganesan M, Zhang J, Bronich T, Poluektova LI, Donohue TM Jr, Tuma DJ, Kharbanda KK, Osna NA. Acetaldehyde accelerates HCV-induced impairment of innate immunity by suppressing methylation reactions in liver cells. Am J Physiol Gastrointest Liver Physiol : G566–G577, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gieseler RK, Marquitan G, Schlattjan M, Sowa JP, Bechmann LP, Timm J, Roggendorf M, Gerken G, Friedman SL, Canbay A. Hepatocyte apoptotic bodies encasing nonstructural HCV proteins amplify hepatic stellate cell activation: implications for chronic hepatitis C. J Viral Hepat : 760–767, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gupta S, Read DE, Deepti A, Cawley K, Gupta A, Oommen D, Verfaillie T, Matus S, Smith MA, Mott JL, Agostinis P, Hetz C, Samali A. Perk-dependent repression of miR-106b-25 cluster is required for ER stress-induced apoptosis. Cell Death Dis : e333, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hou W, Bukong TN, Kodys K, Szabo G. Alcohol facilitates HCV RNA replication via up-regulation of miR-122 expression and inhibition of cyclin G1 in human hepatoma cells. Alcohol Clin Exp Res : 599–608, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang H, Sun F, Owen DM, Li W, Chen Y, Gale M Jr, Ye J. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc Natl Acad Sci USA : 5848–5853, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Israelow B, Mullokandov G, Agudo J, Sourisseau M, Bashir A, Maldonado AY, Dar AC, Brown BD, Evans MJ. Hepatitis C virus genetics affects miR-122 requirements and response to miR-122 inhibitors. Nat Commun : 5408, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joyce MA, Walters KA, Lamb SE, Yeh MM, Zhu LF, Kneteman N, Doyle JS, Katze MG, Tyrrell DL. HCV induces oxidative and ER stress, and sensitizes infected cells to apoptosis in SCID/Alb-uPA mice. PLoS Pathog : e1000291, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kato T, Date T, Murayama A, Morikawa K, Akazawa D, Wakita T. Cell culture and infection system for hepatitis C virus. Nat Protoc : 2334–2339, 2006. [DOI] [PubMed] [Google Scholar]
- 25.Kharbanda KK. Alcoholic liver disease and methionine metabolism. Sem Liver Dis : 155–165, 2009. [DOI] [PubMed] [Google Scholar]
- 26.Kosaka K, Hiraga N, Imamura M, Yoshimi S, Murakami E, Nakahara T, Honda Y, Ono A, Kawaoka T, Tsuge M, Abe H, Hayes CN, Miki D, Aikata H, Ochi H, Ishida Y, Tateno C, Yoshizato K, Sasaki T, Chayama K. A novel TK-NOG based humanized mouse model for the study of HBV and HCV infections. Biochem Biophys Res Commun : 230–235, 2013. [DOI] [PubMed] [Google Scholar]
- 27.Lan L, Gorke S, Rau SJ, Zeisel MB, Hildt E, Himmelsbach K, Carvajal-Yepes M, Huber R, Wakita T, Schmitt-Graeff A, Royer C, Blum HE, Fischer R, Baumert TF. Hepatitis C virus infection sensitizes human hepatocytes to TRAIL-induced apoptosis in a caspase 9-dependent manner. J Immunol : 4926–4935, 2008. [DOI] [PubMed] [Google Scholar]
- 28.Li H, Jiang JD, Peng ZG. MicroRNA-mediated interactions between host and hepatitis C virus. World J Gastroenterol : 1487–1496, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin Q, Mao Y, Song Y, Huang D. MicroRNA34a induces apoptosis in PC12 cells by reducing B cell lymphoma 2 and sirtuin1 expression. Mol Med Rep : 5709–5714, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma H, Li J, Gao F, Ren J. Aldehyde dehydrogenase 2 ameliorates acute cardiac toxicity of ethanol: role of protein phosphatase and forkhead transcription factor. J Am Coll Cardiol : 2187–2196, 2009. [DOI] [PubMed] [Google Scholar]
- 31.Malhi H, Guicciardi ME, Gores GJ. Hepatocyte death: a clear and present danger. Physiol Rev : 1165–1194, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinez-Esparza M, Tristan-Manzano M, Ruiz-Alcaraz AJ, Garcia-Penarrubia P. Inflammatory status in human hepatic cirrhosis. World J Gastroenterol : 11,522–11,541, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McVicker BL, Tuma DJ, Kubik JA, Hindemith AM, Baldwin CR, Casey CA. The effect of ethanol on asialoglycoprotein receptor-mediated phagocytosis of apoptotic cells by rat hepatocytes. Hepatology : 1478–1487, 2002. [DOI] [PubMed] [Google Scholar]
- 34.Mortimer SA, Doudna JA. Unconventional miR-122 binding stabilizes the HCV genome by forming a trimolecular RNA structure. Nucleic Acids Res : 4230–4240, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mueller S, Millonig G, Seitz HK. Alcoholic liver disease and hepatitis C: a frequently underestimated combination. World J Gastroenterol : 3462–3471, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Osna NA, Ganesan M, Kharbanda KK. Hepatitis C, innate immunity and alcohol: friends or foes? Biomolecules : 76–94, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Osna NA, Kharbanda KK, Sun Y, Simpson RL, Poluektova LE, Ganesan M, Wisecarver JL, Mercer DF. Ethanol affects hepatitis C pathogenesis: Humanized SCID Alb-uPA mouse model. Biochem Biophys Res Commun : 773–776, 2014. [DOI] [PubMed] [Google Scholar]
- 38.Osna NA, White RL, Thiele GM, Donohue TM Jr. Ethanol metabolism alters major histocompatibility complex class I-restricted antigen presentation in liver cells. Hepatology : 1308–1315, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ozaras R, Tahan V, Ozbay G, Ozturk R, Yenice N, Celikel CA, Midilli K, Gucin Z, Fincanci M, Tozun N, Senturk H, Osme A, Tabak F, Mert A. Hepatic apoptotic markers are not predictors for the virological response to interferon-based therapy in chronic hepatitis C patients. Eur J Gastroenterol Hepatol : 1057–1062, 2015. [DOI] [PubMed] [Google Scholar]
- 40.Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, Barrieau M, Min SY, Kurt-Jones EA, Szabo G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest : 3476–3489, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Punzalan CS, Bukong TN, Szabo G. Alcoholic hepatitis and HCV interactions in the modulation of liver disease. J Viral Hepat : 769–776, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramakrishnaiah V, Thumann C, Fofana I, Habersetzer F, Pan Q, de Ruiter PE, Willemsen R, Demmers JA, Stalin Raj V, Jenster G, Kwekkeboom J, Tilanus HW, Haagmans BL, Baumert TF, van der Laan LJ. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc Natl Acad Sci USA : 13,109–13,113, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Randall G, Panis M, Cooper JD, Tellinghuisen TL, Sukhodolets KE, Pfeffer S, Landthaler M, Landgraf P, Kan S, Lindenbach BD, Chien M, Weir DB, Russo JJ, Ju J, Brownstein MJ, Sheridan R, Sander C, Zavolan M, Tuschl T, Rice CM. Cellular cofactors affecting hepatitis C virus infection and replication. Proc Natl Acad Sci USA : 12,884–12,889, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, Bentwich Z, Oren M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell : 731–743, 2007. [DOI] [PubMed] [Google Scholar]
- 45.Sedano CD, Sarnow P. Interaction of host cell microRNAs with the HCV RNA genome during infection of liver cells. Semin Liver Dis : 75–80, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seronello S, Ito C, Wakita T, Choi J. Ethanol enhances hepatitis C virus replication through lipid metabolism and elevated NADH/NAD+. J Biol Chem : 845–854, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Severgnini M, Sherman J, Sehgal A, Jayaprakash NK, Aubin J, Wang G, Zhang L, Peng CG, Yucius K, Butler J, Fitzgerald K. A rapid two-step method for isolation of functional primary mouse hepatocytes: cell characterization and asialoglycoprotein receptor-based assay development. Cytotechnology : 187–195, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shrivastava S, Mukherjee A, Ray RB. Hepatitis C virus infection, microRNA and liver disease progression. World J Hepatol : 479–486, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Szabo G, Bala S. MicroRNAs in liver disease. Nat Rev Gastroenterol Hepatol : 542–552, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Szabo G, Mandrekar P, Dolganiuc A. Innate immune response and hepatic inflammation. Semin Liver Dis : 339–350, 2007. [DOI] [PubMed] [Google Scholar]
- 51.Szabo G, Saha B, Bukong TN. Alcohol and HCV: implications for liver cancer. Adv Exp Med Biol : 197–216, 2015. [DOI] [PubMed] [Google Scholar]
- 52.Tarasov V, Jung P, Verdoodt B, Lodygin D, Epanchintsev A, Menssen A, Meister G, Hermeking H. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle : 1586–1593, 2007. [DOI] [PubMed] [Google Scholar]
- 53.Thomes PG, Osna NA, Davis JS, Donohue TM Jr. Cellular steatosis in ethanol oxidizing-HepG2 cells is partially controlled by the transcription factor, early growth response-1. Int J Biochem Cell Biol : 454–463, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tikhanovich I, Kuravi S, Artigues A, Villar MT, Dorko K, Nawabi A, Roberts B, Weinman SA. Dynamic arginine methylation of tumor necrosis factor (TNF) receptor-associated factor 6 regulates Toll-like receptor signaling. J Biol Chem : 22,236–22,249, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tikhanovich I, Kuravi S, Campbell RV, Kharbanda KK, Artigues A, Villar MT, Weinman SA. Regulation of FOXO3 by phosphorylation and methylation in hepatitis C virus infection and alcohol exposure. Hepatology : 58–70, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tworek BL, Tuma DJ, Casey CA. Decreased binding of asialoglycoproteins to hepatocytes from ethanol-fed rats. Consequence of both impaired synthesis and inactivation of the asialoglycoprotein receptor. J Biol Chem : 2531–2538, 1996. [DOI] [PubMed] [Google Scholar]
- 57.von Schaewen M, Ding Q, Ploss A. Visualizing hepatitis C virus infection in humanized mice. J Immunol Methods : 50–59, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Welch C, Chen Y, Stallings RL. MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene : 5017–5022, 2007. [DOI] [PubMed] [Google Scholar]
- 59.Wertheimer AM, Polyak SJ, Leistikow R, Rosen HR. Engulfment of apoptotic cells expressing HCV proteins leads to differential chemokine expression and STAT signaling in human dendritic cells. Hepatology : 1422–1432, 2007. [DOI] [PubMed] [Google Scholar]
- 60.Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci USA : 13,421–13,426, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang J, Garrison JC, Poluektova LY, Bronich TK, Osna NA. Liver-targeted antiviral peptide nanocomplexes as potential anti-HCV therapeutics. Biomaterials : 37–47, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang T, Li Y, Lai JP, Douglas SD, Metzger DS, O'Brien CP, Ho WZ. Alcohol potentiates hepatitis C virus replicon expression. Hepatology : 57–65, 2003. [DOI] [PubMed] [Google Scholar]