

Abstract
Heme oxygenase (HO) activity is exhibited by inducible (HO-1) and constitutive (HO-2) proteins. HO-1 protects against ischemic and nephrotoxic acute kidney injury (AKI). We have previously demonstrated that HO-2 protects against heme protein-induced AKI. The present study examined whether HO-2 is protective in ischemic AKI. Renal ischemia was imposed on young and aged HO-2+/+ and HO-2−/− mice. On days 1 and 2 after renal ischemia, there were no significant differences in renal function between young male HO-2+/+ and HO-2−/− mice, between young female HO-2+/+ and HO-2−/− mice, or between aged female HO-2+/+ and HO-2−/− mice. However, in aged male mice, HO-2 deficiency worsened renal function on days 1 and 2 after ischemic AKI, and, on day 2 after ischemia, such deficiency augmented upregulation of injury-related genes and worsened histological injury. Renal HO activity was markedly decreased in unstressed aged male HO-2−/− mice and remained so after ischemia, despite exaggerated HO-1 induction in HO-2−/− mice after ischemia. Such exacerbation of deficiency of HO-2 protein and HO activity may reflect phosphorylated STAT3, as activation of this proinflammatory transcription factor was accentuated early after ischemia in aged male HO-2−/− mice. This exacerbation may not reflect impaired induction of nephroprotectant genes, since the induction of HO-1, sirtuin 1, and β-catenin was accentuated in aged male HO-2−/− mice after ischemia. We conclude that aged male mice are hypersensitive to ischemic AKI and that HO-2 mitigates such sensitivity. We speculate that this protective effect of HO-2 may be mediated, at least in part, by suppression of phosphorylated STAT3-dependent signaling.
Keywords: acute kidney injury, age, biology of sex differences, heme oxygenase-2, phospho-STAT3
INTRODUCTION
The inducible (HO-1) and constitutive (HO-2) isoforms of heme oxygenase (HO) catalyze the breakdown of heme and, in the process, generate carbon monoxide and bile pigments and procure proteins that regulate the cellular handling of iron (2, 11, 35, 36, 60). HO-1 is readily inducible by diverse insults, including those that are ischemic, hypoxic, nephrotoxic, and inflammatory in origin, and this HO isoform has received the bulk of attention in the study of renal and other forms of tissue injury. These studies have demonstrated the potent protective effects of HO-1 against this range of insults in the kidney and have ascribed such beneficial effects to any one or combination of actions of HO-1: 1) degradation of heme as this toxic metabolite is released from destabilized heme proteins in the injured kidney, 2) generation of the vasodilating and anti-inflammatory gas carbon monoxide, 3) production of antioxidant, anti-inflammatory bile pigments, and 4) elicitation of iron-binding and iron-transporting proteins that safeguard cellular homeostasis of iron (2, 11, 35, 36, 60).
HO-2 possesses not only all these actions of HO-1 but also a particular attribute that HO-1 clearly lacks: as a constitutive protein, HO-2 is expressed and fully functional in the healthy kidney, and, should the healthy kidney ever be exposed to ischemic or other insults, the potentially protective actions of HO-2 are ongoing, in place, and readily available. Inducible systems such as HO-1 obligate a temporal delay for such systems to be fully functional. In the case of HO-1, the delay reflects the time needed, at a minimum, for the following critical steps: 1) activation and engagement of signaling proteins and transcription factors that regulate HO-1 gene expression, 2) transcription of HO-1 mRNA, 3) translation of HO-1 protein and trafficking of the nascent protein to its microsomal sites, and 4) functional interaction with other molecules (e.g., NADPH, cytochrome P-450 reductase, and biliverdin reductase) needed for a fully functional HO-1 system. During the phase at which such essential preparatory steps occur, the protective actions of HO-1 will not be in place, thereby leaving imposed insults relatively unchecked and, accordingly, permitting accrual of functional and structural damage. This potential drawback does not apply to an already functional, constitutive system such as HO-2.
There are very few studies that have examined the functional significance of HO-2 in acute kidney injury (AKI). Prior studies from this laboratory demonstrated that HO-2 confers protective effects in heme protein-induced AKI (39) and models of vascular injury (28, 29). The present study examined the role of HO-2 as a putative protectant against ischemic AKI.
MATERIALS AND METHODS
Experiments were performed as approved by the Institutional Animal Care and Use Committee of Mayo Clinic and were consistent with guidelines of the National Institutes of Health. As we have previously described, the present study involved mice with targeted disruption of the HO-2 gene (HO-2−/−) and littermate control (HO-2+/+) mice (16, 28, 29, 39). Colonies of HO-2+/+ and HO-2−/− mice were maintained by mating of HO-2+/− male and female mice. Experiments involving bilateral renal ischemia or sham ischemia were conducted on aged (76−86 wk old) and young (18−20 wk old) male and female HO-2+/+ and HO-2−/− mice, as described below. For each experiment involving male or female mice, available HO-2+/+ and HO-2−/− mice within each of these age ranges were randomized to groups, which were subjected to ischemia or sham ischemia. For each experiment, groups of HO-2+/+ and HO-2−/− mice were subjected to ischemia or sham ischemia simultaneously.
Renal ischemia-reperfusion model.
A murine model of bilateral renal ischemia was used as described in detail in our prior studies (25, 26, 37, 38, 50, 61). Briefly, mice were anesthetized with pentobarbital (50 mg/kg ip), the renal pedicles were gently dissected via a midline abdominal incision, and ischemia was induced with nontraumatic clamps (straight, 10-mm, 125-g pressure micro aneurysm clip, model no. RS5426, Roboz Surgical Instruments, Rockville, MD). In these experiments, mice were subjected to 15 or 20 min of ischemia. Sham procedures included the abdominal incision but excluded renal pedicle dissection and clamping. For all surgical procedures, buprenorphine (0.1 mg/kg sc) was administered for analgesia. Renal function was assessed daily for 2 sequential days after ischemia-reperfusion (IR) by measurement of plasma creatinine and blood urea nitrogen (BUN) levels using a Creatinine Analyzer 2 (Beckman Instruments, Fullerton, CA) and a commercially available kit (Pointe Scientific, Canton, MI), respectively.
Western blot analysis.
Protein expression was assessed by Western blot analysis, as described in our previous studies (29, 37, 39, 40). Briefly, extracted proteins were separated on 10% or 12% Tris·HCl gels (Bio-Rad) and transferred to polyvinylidene difluoride membranes. Overnight incubations with primary antibodies were performed for the following target and housekeeping proteins: β-catenin and β-actin (catalog nos. 610153 and 612656, respectively, BD Biosciences, San Jose, CA); sirtuin 1 (SIRT1), phosphorylated (p-)STAT3, GAPDH, p-p65, H2A histone family member X (H2AX), p-p38, high-mobility group protein B1 (HMGB1), p-ERK1/2, ETS1, and p-JNK (catalog nos. 8469, 9138, 2118, 3031, 7631, 4511, 6893, 4370, 14069, and 4668, respectively, Cell Signaling Technology, Danvers, MA); p16 and p21 (catalog nos. ab189034 and ab109199, respectively, Abcam, Cambridge, MA); and p53 (catalog no. MAB1355, R&D Systems, Minneapolis, MN). After incubation with horseradish peroxidase-conjugated secondary antibodies, bands were visualized using an enhanced chemiluminescence method.
Analysis of gene expression.
mRNA expression in renal tissues was measured by a two-step quantitative real-time RT-PCR method, as used in our previous studies (29, 37, 50). Total RNA was extracted by the TRIzol method (Invitrogen, Carlsbad, CA) and purified with an RNeasy Mini kit (Qiagen, Valencia, CA). cDNA, synthesized with a first-strand cDNA synthesis kit (Transcriptor, Roche Applied Science, Indianapolis, IN), was used in quantitative real-time PCR analysis performed on a real-time PCR system (Prism 7900HT, Applied Biosystems, Foster City, CA) using TaqMan gene expression assays (Applied Biosystems) for target and housekeeping genes. Expression of each target gene was normalized in reference to 18S rRNA expression.
Measurement of renal HO activity.
As described in our prior studies, HO activity was determined by measurement of the rate of bilirubin production from hemin (29, 41). Activity in renal tissues was assessed in microsomal preparations and expressed as picomoles of bilirubin produced per hour per milligram of microsomal protein, with the latter determined by the Lowry method.
Histological analysis.
Semiquantitative histological analysis was conducted on hematoxylin and eosin-stained sections of kidneys from HO-2+/+ and HO-2−/− mice on day 2 after IR (37). Necrosis was evaluated as described in our previous study by assessment of the percentage of tubules demonstrating any evidence of epithelial cell necrosis and the degree of extension of necrosis into the cortex, determined as the mean percentage of the distance from the corticomedullary junction to the surface of the kidney in which necrosis was present.
Statistical analysis.
Results are expressed as means ± SE and were considered statistically significant for P < 0.05. A Student's t-test was used for parametric data, and the Mann-Whitney U-test was used for nonparametric data.
RESULTS
Plasma creatinine and BUN in young male HO-2+/+ and HO-2−/− mice were not significantly different on days 1 and 2 after 15 min of renal IR or sham IR (Table 1). Similarly, these indexes were not significantly different in young female HO-2+/+ and HO-2−/− mice on days 1 and 2 after renal IR (Table 2).
Table 1.
Renal filtration markers in young male HO-2+/+ and HO-2−/− mice on days 1 and 2 after 15 min of IR or sham operation
| Sham |
IR |
|||
|---|---|---|---|---|
| HO-2+/+ (n = 5) | HO-2−/− (n = 5) | HO-2+/+ (n = 6) | HO-2−/− (n = 8) | |
| Blood urea nitrogen, mg/dl | ||||
| Day 1 | 19 ± 1 | 20 ± 1 | 39 ± 6 | 36 ± 5 |
| Day 2 | 23 ± 2 | 24 ± 1 | 35 ± 2 | 35 ± 4 |
| Plasma creatinine, mg/dl | ||||
| Day 1 | 0.20 ± 0.02 | 0.20 ± 0.02 | 0.38 ± 0.06 | 0.37 ± 0.03 |
| Day 2 | 0.19 ± 0.03 | 0.20 ± 0.02 | 0.28 ± 0.04 | 0.28 ± 0.01 |
Values are means ± SE; n, number of mice. HO, heme oxygenase; IR, ischemia-reperfusion.
Table 2.
Renal filtration markers in young female HO-2+/+ and HO-2−/− mice on days 1 and 2 after 15 min of ischemia
| HO-2+/+ (n = 6) | HO-2−/− (n = 6) | |
|---|---|---|
| Blood urea nitrogen, mg/dl | ||
| Day 1 | 21 ± 1 | 20 ± 1 |
| Day 2 | 19 ± 1 | 21 ± 1 |
| Plasma creatinine, mg/dl | ||
| Day 1 | 0.28 ± 0.02 | 0.29 ± 0.01 |
| Day 2 | 0.22 ± 0.01 | 0.22 ± 0.01 |
Values are means ± SE; n, number of mice. HO, heme oxygenase.
Significant differences were found, however, in aged male mice: aged male HO-2−/− mice exhibited an increased sensitivity to 15 min of IR, as reflected by significantly higher levels of plasma creatinine and BUN on days 1 and 2 after acute renal IR compared with aged male HO-2+/+ mice (Fig. 1). BUN and serum creatinine were not significantly different in aged male HO-2+/+ and HO-2−/− mice after sham IR (Table 3).
Fig. 1.
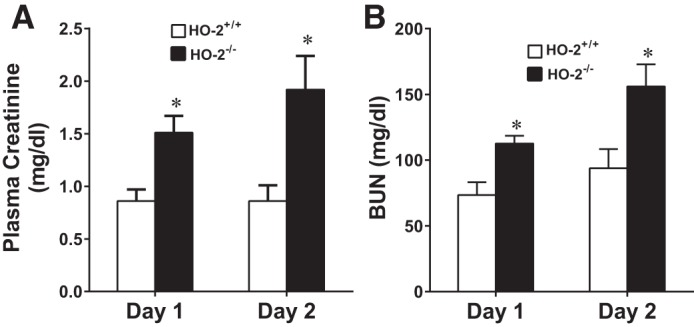
Renal filtration markers in aged male mice with targeted disruption of the heme oxygenase (HO)-2 gene (HO-2−/− mice) and littermate control (HO-2+/+) mice after acute renal ischemia [ischemia-reperfusion (IR)]. Plasma levels of creatinine (A) and blood urea nitrogen (BUN; B) were measured in aged male HO-2+/+ and HO-2−/− mice on days 1 and 2 after 15 min of IR. Values are means ± SE; n = 11 HO-2+/+ and 12 HO-2−/− mice. *P < 0.05 vs. HO-2+/+ mice on the same day.
Table 3.
Renal filtration markers in aged male HO-2+/+ and HO-2−/− mice on days 1 and 2 after sham operation
| HO-2+/+ (n = 4) | HO-2−/− (n = 4) | |
|---|---|---|
| Blood urea nitrogen, mg/dl | ||
| Day 1 | 16± 1 | 19 ± 1 |
| Day 2 | 21 ± 1 | 22 ± 1 |
| Plasma creatinine, mg/dl | ||
| Day 1 | 0.22 ± 0.03 | 0.22 ± 0.02 |
| Day 2 | 0.18 ± 0.02 | 0.22 ± 0.02 |
Values are means ± SE; n, number of mice. HO, heme oxygenase.
In aged male mice on day 2 after IR, HO-2 deficiency augmented the induction of injury-related genes, including acute inflammation-related genes [chemokine (C-C motif) ligand 2 (CCL2), IL-6, matrix metalloproteinase (MMP)-9, and plasminogen activator inhibitor 1 (PAI-1); Fig. 2], chronic inflammation-related genes [transforming growth factor (TGF)-β1, collagen type I (Col I), collagen type III (Col III), and collagen type IV (Col IV); Fig. 3], and other renal injury-related genes [platelet-derived growth factor (PDGF)-A, MMP-2, and p21; Fig. 4]. Histological analyses of the kidney showed that acute tubular necrosis was more severe in aged male HO-2−/− mice than in aged male HO-2+/+ mice after IR (Fig. 5). Semiquantitative assessment of the degree of necrosis resulting from 15 min of IR on day 2 demonstrated significantly more necrosis, both the percentage of necrotic tubules and degree of extension of necrosis into the cortex, in aged male HO-2−/− mice than in aged male HO-2+/+ mice (Fig. 6).
Fig. 2.

Renal expression of acute inflammation-related genes [chemokine (C-C motif) ligand 2 (CCL2), IL-6, matrix metalloproteinase (MMP)-9, and plasminogen activator inhibitor 1 (PAI-1)] in aged male mice with targeted disruption of the heme oxygenase (HO)-2 gene (HO-2−/− mice) and littermate control (HO-2+/+) mice on day 2 after acute (15 min) renal ischemia (IR). mRNA expression of CCL2 (A), IL-6 (B), MMP-9 (C), and PAI-1 (D) was determined by quantitative real-time RT-PCR. Values are means ± SE; n = 4 mice subjected to sham operation and n = 11 HO-2+/+ and 12 HO-2−/− mice subjected to IR. *P < 0.05 vs. HO-2+/+ mice subjected to IR.
Fig. 3.

Renal expression of chronic inflammation-related genes [transforming growth factor (TGF)-β1, collagen type I (Col I), collagen type III (Col III), and collagen type IV (Col IV)] in aged male mice with targeted disruption of the heme oxygenase (HO)-2 gene (HO-2−/− mice) and littermate control (HO-2+/+) mice on day 2 after acute (15 min) renal ischemia (IR). mRNA expression of TGF-β1 (A), Col I (B), Col III (C), and Col IV (D) was determined by quantitative real-time RT-PCR. Values are means ± SE; n = 4 mice subjected to sham surgery and n = 11 HO-2+/+ and 12 HO-2−/− mice subjected to IR. *P < 0.05 vs. HO-2+/+ mice subjected to IR.
Fig. 4.

Renal expression of injury-related genes [platelet-derived growth factor-A (PDGFA), matrix metalloproteinase (MMP)-2, p21, and p16] in aged male mice with targeted disruption of the heme oxygenase (HO)-2 gene (HO-2−/− mice) and littermate control (HO-2+/+) mice on day 2 after acute renal ischemia (IR). mRNA expression of PDGFA (A), MMP-2 (B), p21 (C), and p16 (D) was determined by quantitative real-time RT-PCR. Values are means ± SE; n = 4 mice subjected to sham surgery and n = 11 HO-2+/+ and 12 HO-2−/− mice subjected to IR. *P < 0.05 vs. HO-2+/+ mice subjected to IR.
Fig. 5.

Histological appearance of the kidney in aged male mice with targeted disruption of the heme oxygenase (HO)-2 gene (HO-2−/− mice; B and D) and littermate control (HO-2+/+) mice (A and C) on day 2 after acute (15 min) renal ischemia (IR). Lower-power ( ×100; A and B) and higher-power ( ×200; C and D) views are shown. Aged male HO-2−/− mice displayed more severe renal injury than aged male HO-2+/+ mice in response to IR, with a greater percentage of tubules exhibiting necrosis and with greater extension of necrosis into the cortex. No histological injury was observed in kidneys of aged male HO-2+/+ and HO-2−/− mice subjected to sham IR (not shown). Tissue sections were stained with hematoxylin and eosin. Scale bars = 250 μm (A and B) and 100 μm (C and D).
Fig. 6.
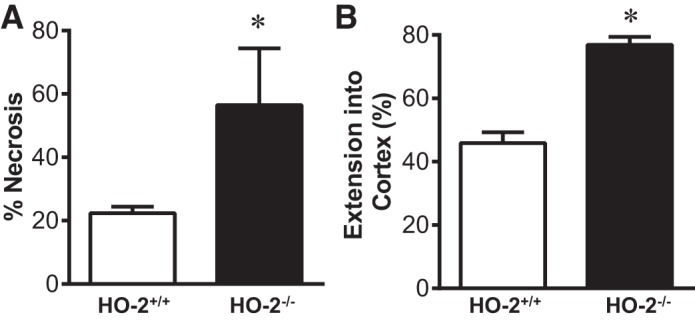
Semiquantitative assessment of necrosis in the kidney on day 2 after acute renal ischemia (IR) in aged male mice with targeted disruption of the heme oxygenase (HO)-2 gene (HO-2−/− mice) and littermate control (HO-2+/+) mice. A: percentage of tubules exhibiting tubular epithelial cell necrosis. B: extension of cortical necrosis, determined as the mean percentage of the distance from the corticomedullary junction to the surface of the kidney. Values are means ± SE; n = 11 HO-2+/+ and 12 HO-2−/− mice subjected to IR. *P < 0.05 vs. HO-2+/+ mice subjected to IR.
Aged female HO-2+/+ and HO-2−/− mice subjected to the same duration of IR (15 min) as aged male mice (compare with Fig. 1) showed no significant differences in BUN and plasma creatinine on days 1 and 2 after IR or sham IR (Table 4). To determine whether HO-2 deficiency or HO-2 sufficiency in aged female mice required greater renal ischemic stress to evince more severe renal functional impairment, experiments were undertaken with a longer period (20 min) of IR. Even after 20 min of IR, there were no significant differences in plasma creatinine and BUN in aged female HO-2+/+ and HO-2−/− mice on days 1 and 2 after IR (Table 5).
Table 4.
Renal filtration markers in aged female HO-2+/+ and HO-2−/− mice on days 1 and 2 after 15 min of IR or sham operation
| Sham |
IR |
|||
|---|---|---|---|---|
| HO-2+/+ (n = 4) | HO-2−/− (n = 4) | HO-2+/+ (n = 7) | HO-2−/− (n = 7) | |
| Blood urea nitrogen, mg/dl | ||||
| Day 1 | 12 ± 1 | 13 ± 1 | 46 ± 6 | 62 ± 12 |
| Day 2 | 18 ± 2 | 17 ± 2 | 30 ± 3 | 52 ± 15 |
| Plasma creatinine, mg/dl | ||||
| Day 1 | 0.20 ± 0.03 | 0.20 ± 0.03 | 0.45 ± 0.04 | 0.56 ± 0.12 |
| Day 2 | 0.18 ± 0.02 | 0.20 ± 0.03 | 0.30 ± 0.02 | 0.32 ± 0.05 |
Values are means ± SE; n, number of mice. HO, heme oxygenase; IR, ischemia-reperfusion.
Table 5.
Renal filtration markers in aged female HO-2+/+ and HO-2−/− mice on days 1 and 2 after 20 min of ischemia
| HO-2+/+ (n = 7) | HO-2−/− (n = 7) | |
|---|---|---|
| Blood urea nitrogen, mg/dl | ||
| Day 1 | 70 ± 7 | 77 ± 16 |
| Day 2 | 67 ± 19 | 73 ± 23 |
| Plasma creatinine, mg/dl | ||
| Day 1 | 0.73 ± 0.07 | 0.85 ± 0.14 |
| Day 2 | 0.66 ± 0.10 | 0.69 ± 0.15 |
Values are means ± SE; n, number of mice. HO, heme oxygenase.
To determine the basis for the heightened sensitivity of aged male HO-2−/− mice to renal IR, we examined the HO system in aged male HO-2+/+ and HO-2−/− mice under basal unstressed conditions and after IR. Unstressed aged male HO-2−/− mice showed comparable expression of HO-1 mRNA and markedly reduced HO activity; indeed, in intact unstressed aged male kidneys, the bulk of HO activity originated from the HO-2 isoform (Fig. 7), a finding that has not yet been reported in the literature.
Fig. 7.
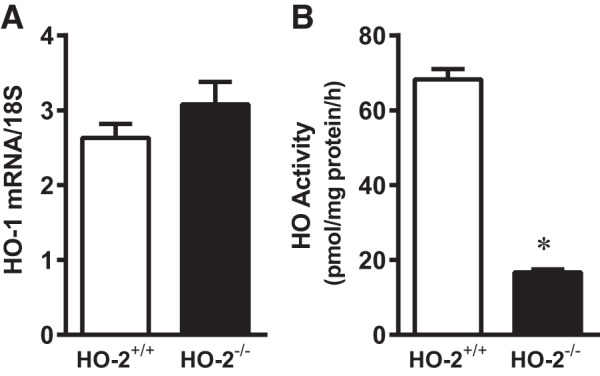
Basal expression of heme oxygenase (HO)-1 mRNA (A) and HO activity (B) in unstressed kidneys of aged male mice with targeted disruption of the HO-2 gene (HO-2−/− mice) and littermate control (HO-2+/+) mice. HO-1 mRNA was determined by quantitative real-time RT-PCR (A), and microsomal HO activity was measured in the intact unstressed kidney (B). Values are means ± SE; n = 8 for each group. *P < 0.05 vs. HO-2+/+ mice.
The HO system was then assessed in aged male HO-2+/+ and HO-2−/− mice after renal IR: we selected a time point relatively early (6 h) after IR, such that significant renal injury had not yet accrued (as occurred on day 2). As shown in Fig. 8A, HO activity was markedly decreased in HO-2−/− mice compared with HO-2+/+ mice 6 h after both IR and sham IR. The diminished HO activity in the kidney in HO-2−/− mice compared with HO-2+/+ mice persisted after IR, although, in response to IR, an exaggerated induction of HO-1 mRNA occurred in HO-2−/− mice (Fig. 8B).
Fig. 8.
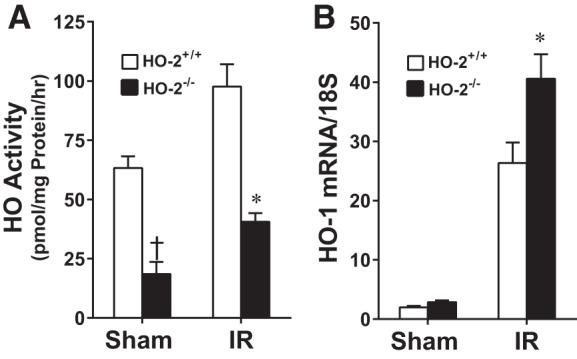
Renal microsomal heme oxygenase (HO) activity (A) and renal HO-1 mRNA expression (B) in aged male mice with targeted disruption of the HO-2 gene (HO-2−/− mice) and littermate control (HO-2+/+) mice 6 h after acute renal ischemia (IR). Values are means ± SE; n = 4 mice subjected to sham surgery and n = 9 mice subjected to IR in A and n = 4 mice subjected to sham surgery and n = 7 HO-2+/+ and 8 HO-2−/− mice subjected to IR in B. *P < 0.05 vs. HO-2+/+ mice subjected to IR; †P < 0.05 vs. HO-2+/+ mice subjected to sham surgery.
Our prior study (61) of renal IR demonstrated that when HO activity is deficient because of genetic deficiency of HO-1, the proinflammatory signaling molecule p-STAT3 is upregulated; such upregulation of p-STAT3 has been implicated in AKI (61). We thus examined p-STAT3 expression in aged male mice 6 h after IR. We observed that the decreased HO activity in aged male HO-2−/− mice was accompanied by an exaggerated upregulation of p-STAT3 protein at this early time point (Fig. 9).
Fig. 9.

Renal phosphorylated (p-)STAT3 expression in aged male mice with targeted disruption of the heme oxygenase (HO)-2 gene (HO-2−/− mice) and littermate control (HO-2+/+) mice 6 h after acute renal ischemia (IR). Renal expression of p-STAT3 protein was determined by Western blot analysis. Values are means ± SE; n = 4 mice subjected to sham surgery and n = 7 HO-2+/+ and 8 HO-2−/− mice subjected to IR. *P < 0.05 vs. HO-2+/+ mice subjected to IR.
At 6 h after renal IR, we also examined expression of factors that may account for the augmented renal injury observed in aged male HO-2−/− mice. At this time point, no expression of p-p65 (a marker of NF-κB activation) was observed (data not shown); no differences among any of the four groups were observed for expression of ETS1, HMGB1, p53, p16, H2AX, and p-p38 (data not shown). At this time point, expression of p21, p-ERK1/2, and p-JNK proteins was higher in both HO-2+/+ and HO-2−/− mice subjected to IR than sham IR, but there were no differences between HO-2+/+ and HO-2−/− groups (data not shown).
To determine whether the acute sensitivity of aged male HO-2−/− mice after IR was due, in part, to a failure in adaptive increments in nephroprotective mechanisms, we examined such changes in two other representative nephroprotectants, besides HO-1, in the kidney. As shown in Fig. 10, induction of SIRT1 and β-catenin proteins was markedly increased after the ischemic insult in aged male HO-2−/− mice compared with aged male HO-2+/+ mice. Thus, a failure or an attenuated increase in expression of these nephroprotectants does not underlie the increased sensitivity to IR in aged male HO-2−/− mice.
Fig. 10.

Renal expression of β-catenin and sirtuin (SIRT)1 in aged male mice with targeted disruption of the heme oxygenase (HO)-2 gene (HO-2−/− mice) and littermate control (HO-2+/+) mice 6 h after acute renal ischemia (IR). Renal expression of β-catenin and SIRT1 protein was determined by Western blot analysis. Values are means ± SE; n = 4 mice subjected to sham surgery and n = 7 HO-2+/+ and 8 HO-2−/− mice subjected to IR. *P < 0.05 vs. HO-2+/+ mice subjected to IR.
DISCUSSION
To the best of our knowledge, this study is the first to characterize the functional significance of HO-2 in experimental ischemic AKI. Our data demonstrate that when aged male mice are subjected to IR, the absence of HO-2 exacerbates ischemic AKI. However, the absence of HO-2 did not exacerbate ischemic injury in similarly stressed aged female mice or in aged female mice subjected to an even longer episode of IR. The deficiency of HO-2 also did not increase the sensitivity of young male or young female mice to IR. The exacerbating effect of HO-2 deficiency in aged male mice was evinced by concordant changes in two different filtration markers on 2 sequential days after IR, by renal histological findings and quantitative histological scoring on day 2 after IR, and by heightened upregulation of genes that are pathogenetically relevant to ischemic AKI also on day 2 after IR.
It is notable that the protective effect of HO-2 was evinced only in the setting of two other variables, namely, age and sex. The literature regarding susceptibility to clinical and experimental AKI supports the concept that older age is a risk factor for AKI for either sex (15, 18, 39, 53, 54, 64); our findings are consistent with these observations. For example, on days 1 and 2 after IR, mean levels of plasma creatinine (0.86 and 0.86 mg/dl) and BUN (73 and 94 mg/dl) in aged male HO-2+/+ mice were approximately twofold greater than plasma creatinine (0.38 and 0.28 mg/dl) and BUN (39 and 35 mg/dl) in young male HO-2+/+ mice. Similarly, on days 1 and 2 after IR, mean levels of plasma creatinine (0.45 and 0.30 mg/dl) and BUN (46 and 30 mg/dl) in aged female HO-2+/+ mice were ~50% greater than plasma creatinine (0.28 and 0.22 mg/dl) and BUN (21 and 19 mg/dl) in young female HO-2+/+ mice.
The basis for increased sensitivity to ischemic AKI conferred by age is unknown. One possible mechanism is that aging leads to the appearance of senescent cells in the kidney and other organs (54, 58). Senescent cells exhibit cell cycle arrest and resistance to apoptosis; notably, such cells characteristically display a senescence-associated secretory phenotype (SASP) that elaborates diverse proinflammatory cytokines, growth factors, vasoconstrictive agents, and proapoptotic species (54, 58). AKI itself elicits the appearance of a senescence phenotype in the injured kidney (24). It is thus possible that the preexisting SASP in the aged kidney renders the organ susceptible to AKI. Interestingly, in the experiments performed in aged male mice (Figs. 2–4), many of the genes induced by IR are regarded as SASP factors, and the deficiency of HO-2 exacerbated such induction. The functional significance of such upregulation of these genes in HO-2−/− mice after IR is at least twofold: such upregulation may impair the recovery from AKI induced by IR and/or may promote the transition to chronic kidney disease after IR-induced AKI.
Our data also demonstrate that, for similarly aged mice, male sex increases the sensitivity to ischemic AKI. For example, on days 1 and 2 after IR, mean levels of plasma creatinine (0.86 and 0.86 mg/dl) and BUN (73 and 94 mg/dl) in aged male HO-2+/+ mice were considerably greater than plasma creatinine (0.45 and 0.30 mg/dl) and BUN (46 and 30 mg/dl) in similarly aged female HO-2+/+ mice. There is clear consensus that male sex is a risk factor for human AKI after general surgery and noncardiac surgery (18, 53). There are divergent findings regarding whether male sex confers such a risk after cardiothoracic surgery, and the Kidney Disease Improving Global Outcomes Clinical Practice Guidelines for AKI regard female, rather than male, sex as a risk for AKI. However, as recently and quite convincingly pointed out by Neugarten et al. (45), this lack of uniformity of opinion regarding the effect of sex on AKI after cardiothoracic surgery may reflect at least two issues: 1) there is heterogeneity in the criteria used to define AKI in such studies and 2) women generally have more severe cardiovascular disease and more impaired overall health than men by the time they undergo cardiac surgery. Moreover, in a recent systematic review and meta-analysis of studies that used uniform criteria for AKI (Acute Kidney Injury Network), Neugarten et al. (45) demonstrated that women exhibited a substantially lower risk for AKI than men after cardiothoracic surgery. Finally, female sex is associated with lower risk for AKI in hospitalized patients (43), for dialysis-requiring AKI (44), and for aminoglycoside-induced AKI (42).
In experimental AKI, female rodents are generally less sensitive than male rodents to AKI (27, 46, 47). Such relative resistance can be conferred by estrogens (22) and vitiated by testosterone in relatively large amounts (47), and the sensitivity of male rodents to AKI can be reduced by orchiectomy (46). This sexual dimorphism cannot be simply explained by the notion that testosterone is an exacerbating factor in males, whereas estrogen is an ameliorative factor in females. Specifically, as previously shown by Patil et al. (48) and Soljancic et al. (55), renal testosterone levels decrease with AKI, and testosterone administration in relatively low doses is protective, because it induces an anti-inflammatory renal response (IL-10) and suppresses a proinflammatory renal response (TNF-α).
Female sex, the HO system, and tissue response to injury have been linked in the current literature. For example, in the kidney, young female (compared with young male, aged male, and aged female) mice are resistant to cisplatin-induced nephrotoxicity, in part because of their profile of HO-1 induction and an accelerated renal recovery response in young female mice (10). Female rats, compared with male rats, exhibit a blunted pressor response and less cardiac ischemia in response to arginine vasopressin, a resistance mediated by HO activity (51). HO-1 and HO-2 protein expression, as well as HO activity, in the heart decrease with age and by ovariectomy, and HO inhibition worsens chemical-induced cardiac ischemia (52). Finally, in female mice, unlike in male mice, HO-2 deficiency does not exaggerate the increase in systemic blood pressure after renal artery stenosis (56).
In an attempt to uncover clues regarding the mechanism underlying this increased sensitivity of aged male mice to IR, we explored a variety of pathways at an earlier time point after IR (6 h). At 6 h, certain potentially nephropathic signaling pathways were induced by IR, but not differentially in HO-2+/+ and HO-2−/− mice, whereas others were not differentially induced by IR or HO-2 deficiency.
A notable finding was observed with p-STAT3, a proinflammatory signaling species implicated in the pathogenesis of ischemic AKI. In our present study, HO-2 deficiency markedly accentuated the upregulation of p-STAT3 in response to IR. While such differential upregulation of p-STAT3 may reflect differential severity of renal injury, we suggest that this exaggerated upregulation of p-STAT3 in aged male HO-2−/− mice contributed to the exacerbation of AKI observed in this setting for the following reasons. First, prior studies have shown that modalities that reduce p-STAT3 expression reduce AKI, whereas those that accentuate p-STAT3 expression exacerbate AKI (21, 33, 49, 63). Second, in our prior study (61) in which renal HO activity was reduced because of genetic deficiency of HO-1, p-STAT3 was markedly upregulated in the ischemic kidney and implicated in the attendant exacerbation of ischemic AKI caused by HO-1 deficiency. Third, certain STAT3 polymorphisms in humans associate with the occurrence of AKI after cardiopulmonary bypass (3). We recognize, however, that the behavior of p-STAT3 is context dependent in the injured kidney: p-STAT3 may induce expression of survivin in AKI (12) but exacerbate oxidant-induced cell death (5); p-STAT3 may mediate the salutary effects of IL-22 in AKI (32); and endothelial deletion of STAT3 worsens AKI (14), whereas unphosphorylated STAT3 mediates nicotine-induced renal inflammation (6). Finally, both STAT3 and p-STAT3 are potently profibrotic (6, 9); thus, the heightened upregulation of p-STAT3 in aged male HO-2−/− mice after IR may drive the AKI to CKD transition. Interestingly, HO-2 deficiency not only upregulated a potentially injurious species (p-STAT3) in aged male ischemic mice but also upregulated molecular species that are generally regarded as nephroprotectants, including HO-1 (35, 36), SIRT1 (15, 17, 19), and β-catenin (65); we speculate that the upregulation of these three cytoprotective species represents a countervailing response.
The exacerbating effect of HO-2 deficiency on ischemic AKI occurred despite an exaggerated induction of HO-1 in the HO-2-deficient kidney 6 h after IR. This accentuated induction of HO-1 thus does not compensate for the diminution in HO activity caused by HO-2 deficiency. Prior studies have demonstrated that a mutant HO-2 protein lacking HO activity is cytoprotective in cellular models of oxidant stress through as yet undefined mechanisms (30) and that HO-2 and HO-1 exert distinct protective mechanisms in models of oxidant injury (31). Based on these findings, we suggest that the exacerbating effect of HO-2 deficiency, as observed in the present study, may reflect markedly reduced HO activity in the ischemic kidney as well as loss of the cytoprotective properties specifically emanating from the HO-2 protein itself.
Several properties of HO-2 recognized in other tissues may be germane to the exacerbating effect of HO-2 deficiency in aged male mice subjected to renal IR. First, HO-2 safeguards the viability of the endothelial cell in response to hypoxic and other types of stress (13, 20), and endothelial injury is a significant participant in the pathogenesis of renal IR injury. Second, HO-2 deficiency in tissues such as the cornea impairs their response to injury, leading to exaggerated inflammation, apoptosis, angiogenesis, and aberrant and delayed healing (7, 8). Finally, HO-2 is an oxygen sensor and is needed (at least in other tissues) to elicit homeostatic responses to hypoxic stress (1, 34). Hypoxia is a significant component in renal IR injury; thus, the absence of HO-2 may impair countervailing responses that contain the damaging effects of hypoxia in the ischemic kidney.
We suggest that the clinical implications of our present findings include the following considerations. There are recognized polymorphisms and variants in the HO-2 gene that associate with age-related macular degeneration (57), multiple sclerosis (4), Parkinson’s disease (59), and the ability to adapt to high altitude (62). It would be of interest to determine whether such polymorphisms and variants in the HO-2 gene may underlie the increased risk for AKI observed in aged male human patients. Additionally, as HO-2 expression declines with age in rodents (52), it is intriguing whether such decline also occurs with human aging and whether such decline is more marked in men, especially in the kidney. Finally, HO-2 is a constitutive gene that can be remarkably induced by glucocorticoids (36). A recent study (23) has demonstrated that glucocorticoid administration can reduce the risk for AKI after cardiovascular surgery. It would also be of interest whether these beneficial effects reflect the induction of HO-2 and whether such therapy may be considered, in particular, for aged male patients before and at the time of surgery or in other settings that predispose to AKI.
In summary, resident and readily available HO-2 protein and attendant HO activity in the unstressed kidney exert a protective role when the kidney is exposed to an acute ischemic insult, but additional risk factors, such as male sex and age, need to be present. Clinical AKI is rarely unifactorial, as it quite commonly requires the coexistence and confluence of several risk factors. Our present findings speak to this clinical axiom by demonstrating a specific instance wherein the beneficial effects of a putative protectant gene in renal ischemic injury become unveiled when the kidney is additionally stressed by age and male sex.
GRANTS
This work was supported by National Institutes of Health Grants DK-47060 (to K. A. Nath, J. P. Grande, and Z. S. Katusic), HL-136348 (to V. D. Garovic), DK-68055 (to G. Farrugia), and HL-114567 and HL-131065-03A11 (to G M. Vercellotti and J. D. Belcher).
DISCLOSURES
J. D. Belcher and G. M. Vercellotti are consultants with and receive research funding from Mitobridge-Astellas. J. D. Belcher and G. M. Vercellotti receive research funding from CSL Behring.
AUTHOR CONTRIBUTIONS
K.A.N. and A.J.C. conceived and designed research; A.J.C. and A.W.A. performed experiments; K.A.N., J.P.G., A.J.C., and A.W.A. analyzed data; K.A.N., V.D.G., J.P.G., A.J.C., A.W.A., G.F., Z.S.K., J.D.B., and G.M.V. interpreted results of experiments; K.A.N., A.J.C., and A.W.A. prepared figures; K.A.N. and A.J.C. drafted manuscript; K.A.N., V.D.G., J.P.G., A.J.C., A.W.A., G.F., Z.S.K., J.D.B., and G.M.V. edited and revised manuscript; K.A.N., V.D.G., J.P.G., A.J.C., A.W.A., G.F., Z.S.K., J.D.B., and G.M.V. approved final version of manuscript.
REFERENCES
- 1.Adachi T, Ishikawa K, Hida W, Matsumoto H, Masuda T, Date F, Ogawa K, Takeda K, Furuyama K, Zhang Y, Kitamuro T, Ogawa H, Maruyama Y, Shibahara S. Hypoxemia and blunted hypoxic ventilatory responses in mice lacking heme oxygenase-2. Biochem Biophys Res Commun 320: 514–522, 2004. doi: 10.1016/j.bbrc.2004.05.195. [DOI] [PubMed] [Google Scholar]
- 2.Agarwal A, Bolisetty S. Adaptive responses to tissue injury: role of heme oxygenase-1. Trans Am Clin Climatol Assoc 124: 111–122, 2013. [PMC free article] [PubMed] [Google Scholar]
- 3.Aghakhani Chegeni S, Rahimzadeh M, Montazerghaem H, Khayatian M, Dasturian F, Naderi N. Preliminary report on the association between STAT3 polymorphisms and susceptibility to acute kidney injury after cardiopulmonary bypass. Biochem Genet 56: 627–638, 2018. doi: 10.1007/s10528-018-9865-6. [DOI] [PubMed] [Google Scholar]
- 4.Agúndez JA, García-Martín E, Martínez C, Benito-León J, Millán-Pascual J, Díaz-Sánchez M, Calleja P, Pisa D, Turpín-Fenoll L, Alonso-Navarro H, Pastor P, Ortega-Cubero S, Ayuso-Peralta L, Torrecillas D, García-Albea E, Plaza-Nieto JF, Jiménez-Jiménez FJ. Heme oxygenase-1 and 2 common genetic variants and risk for multiple sclerosis. Sci Rep 6: 20830, 2016. doi: 10.1038/srep20830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arany I, Megyesi JK, Nelkin BD, Safirstein RL. STAT3 attenuates EGFR-mediated ERK activation and cell survival during oxidant stress in mouse proximal tubular cells. Kidney Int 70: 669–674, 2006. doi: 10.1038/sj.ki.5001604. [DOI] [PubMed] [Google Scholar]
- 6.Arany I, Reed DK, Grifoni SC, Chandrashekar K, Booz GW, Juncos LA. A novel U-STAT3-dependent mechanism mediates the deleterious effects of chronic nicotine exposure on renal injury. Am J Physiol Renal Physiol 302: F722–F729, 2012. doi: 10.1152/ajprenal.00338.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bellner L, Marrazzo G, van Rooijen N, Dunn MW, Abraham NG, Schwartzman ML. Heme oxygenase-2 deletion impairs macrophage function: implication in wound healing. FASEB J 29: 105–115, 2015. doi: 10.1096/fj.14-256503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bellner L, Patil KA, Castellano K, Halilovic A, Dunn MW, Schwartzman ML. Targeted suppression of HO-2 gene expression impairs the innate anti-inflammatory and repair responses of the cornea to injury. Mol Vis 17: 1144–1152, 2011. [PMC free article] [PubMed] [Google Scholar]
- 9.Bienaimé F, Muorah M, Yammine L, Burtin M, Nguyen C, Baron W, Garbay S, Viau A, Broueilh M, Blanc T, Peters D, Poli V, Anglicheau D, Friedlander G, Pontoglio M, Gallazzini M, Terzi F. Stat3 controls tubulointerstitial communication during CKD. J Am Soc Nephrol 27: 3690–3705, 2016. doi: 10.1681/ASN.2015091014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boddu R, Fan C, Rangarajan S, Sunil B, Bolisetty S, Curtis LM. Unique sex- and age-dependent effects in protective pathways in acute kidney injury. Am J Physiol Renal Physiol 313: F740–F755, 2017. doi: 10.1152/ajprenal.00049.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bolisetty S, Zarjou A, Agarwal A. Heme oxygenase 1 as a therapeutic target in acute kidney injury. Am J Kidney Dis 69: 531–545, 2017. doi: 10.1053/j.ajkd.2016.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen J, Chen JK, Conway EM, Harris RC. Survivin mediates renal proximal tubule recovery from AKI. J Am Soc Nephrol 24: 2023–2033, 2013. doi: 10.1681/ASN.2013010076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen RJ, Yuan HH, Zhang TY, Wang ZZ, Hu AK, Wu LL, Yang ZP, Mao YJ, Ji DJ, Zhu XR. Heme oxygenase-2 suppress TNF-α and IL6 expression via TLR4/MyD88-dependent signaling pathway in mouse cerebral vascular endothelial cells. Mol Neurobiol 50: 971–978, 2014. doi: 10.1007/s12035-014-8693-x. [DOI] [PubMed] [Google Scholar]
- 14.Dube S, Matam T, Yen J, Mang HE, Dagher PC, Hato T, Sutton TA. Endothelial STAT3 modulates protective mechanisms in a mouse ischemia-reperfusion model of acute kidney injury. J Immunol Res 2017: 4609502, 2017. doi: 10.1155/2017/4609502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fan H, Yang HC, You L, Wang YY, He WJ, Hao CM. The histone deacetylase, SIRT1, contributes to the resistance of young mice to ischemia/reperfusion-induced acute kidney injury. Kidney Int 83: 404–413, 2013. doi: 10.1038/ki.2012.394. [DOI] [PubMed] [Google Scholar]
- 16.Farrugia G, Lei S, Lin X, Miller SM, Nath KA, Ferris CD, Levitt M, Szurszewski JH. A major role for carbon monoxide as an endogenous hyperpolarizing factor in the gastrointestinal tract. Proc Natl Acad Sci USA 100: 8567–8570, 2003. doi: 10.1073/pnas.1431233100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao R, Chen J, Hu Y, Li Z, Wang S, Shetty S, Fu J. Sirt1 deletion leads to enhanced inflammation and aggravates endotoxin-induced acute kidney injury. PLoS One 9: e98909, 2014. doi: 10.1371/journal.pone.0098909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grams ME, Sang Y, Ballew SH, Gansevoort RT, Kimm H, Kovesdy CP, Naimark D, Oien C, Smith DH, Coresh J, Sarnak MJ, Stengel B, Tonelli M, Tonelli M, Hemmelgarn BR, James MT, Turin TC, Coresh J, Matsushita K, Grams M, Sang Y, Shlipak M, Sarnak MJ, Katz R, Wheeler DC, Emberson J, Landray MJ, Townend JN, Green J, Kirchner HL, Perkins R, Chang AR, Romundstad S, Aasarød K, Øien CM, Hallan S, Smith DH, Thorp ML, Johnson ES, Chodick G, Herzel E, Katz R, Shalev V, Gansevoort RT, Bakker SJL, Lambers Heerspink HJ, van der Harst P, Jee SH, Kimm H, Mok Y, Tangri N, Naimark D, Ärnlöv J, Larsson A, Lannfelt L, Kovesdy CP, Kalantar-Zadeh K, Coresh J, Grams M, Matsushita K, Gansevoort RT, de Jong PE, Iseki K, Levey AS, Sarnak MJ, Stengel B, Warnock D, Woodward M, Ballew SH, Coresh J, Grams M, Matsushita K, Sang Y, Woodward M; CKD Prognosis Consortium . A meta-analysis of the association of estimated GFR, albuminuria, age, race, and sex with acute kidney injury. Am J Kidney Dis 66: 591–601, 2015. doi: 10.1053/j.ajkd.2015.02.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hasegawa K, Wakino S, Yoshioka K, Tatematsu S, Hara Y, Minakuchi H, Sueyasu K, Washida N, Tokuyama H, Tzukerman M, Skorecki K, Hayashi K, Itoh H. Kidney-specific overexpression of Sirt1 protects against acute kidney injury by retaining peroxisome function. J Biol Chem 285: 13045–13056, 2010. doi: 10.1074/jbc.M109.067728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He JZ, Ho JJ, Gingerich S, Courtman DW, Marsden PA, Ward ME. Enhanced translation of heme oxygenase-2 preserves human endothelial cell viability during hypoxia. J Biol Chem 285: 9452–9461, 2010. doi: 10.1074/jbc.M109.077230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ibrahim SM, Al-Shorbagy MY, Abdallah DM, El-Abhar HS. Activation of α7 nicotinic acetylcholine receptor ameliorates zymosan-induced acute kidney injury in BALB/c mice. Sci Rep 8: 16814, 2018. doi: 10.1038/s41598-018-35254-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ikeda M, Swide T, Vayl A, Lahm T, Anderson S, Hutchens MP. Estrogen administered after cardiac arrest and cardiopulmonary resuscitation ameliorates acute kidney injury in a sex- and age-specific manner. Crit Care 19: 332, 2015. doi: 10.1186/s13054-015-1049-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacob KA, Leaf DE, Dieleman JM, van Dijk D, Nierich AP, Rosseel PM, van der Maaten JM, Hofland J, Diephuis JC, de Lange F, Boer C, Kluin J, Waikar SS. Intraoperative high-dose dexamethasone and severe AKI after cardiac surgery. J Am Soc Nephrol 26: 2947–2951, 2015. doi: 10.1681/ASN.2014080840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin H, Zhang Y, Ding Q, Wang SS, Rastogi P, Dai DF, Lu D, Purvis M, Cao C, Wang A, Liu D, Ren C, Elhadi S, Hu MC, Chai Y, Zepeda-Orozco D, Campisi J, Attanasio M. Epithelial innate immunity mediates tubular cell senescence after kidney injury. JCI Insight e125490: 4, 2019. doi: 10.1172/jci.insight.125490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Juncos JP, Grande JP, Croatt AJ, Hebbel RP, Vercellotti GM, Katusic ZS, Nath KA. Early and prominent alterations in hemodynamics, signaling, and gene expression following renal ischemia in sickle cell disease. Am J Physiol Renal Physiol 298: F892–F899, 2010. doi: 10.1152/ajprenal.00631.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Juncos JP, Grande JP, Murali N, Croatt AJ, Juncos LA, Hebbel RP, Katusic ZS, Nath KA. Anomalous renal effects of tin protoporphyrin in a murine model of sickle cell disease. Am J Pathol 169: 21–31, 2006. doi: 10.2353/ajpath.2006.051195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang KP, Lee JE, Lee AS, Jung YJ, Kim D, Lee S, Hwang HP, Kim W, Park SK. Effect of gender differences on the regulation of renal ischemia-reperfusion-induced inflammation in mice. Mol Med Rep 9: 2061–2068, 2014. doi: 10.3892/mmr.2014.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kang L, Grande JP, Farrugia G, Croatt AJ, Katusic ZS, Nath KA. Functioning of an arteriovenous fistula requires heme oxygenase-2. Am J Physiol Renal Physiol 305: F545–F552, 2013. doi: 10.1152/ajprenal.00234.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang L, Hillestad ML, Grande JP, Croatt AJ, Barry MA, Farrugia G, Katusic ZS, Nath KA. Induction and functional significance of the heme oxygenase system in pathological shear stress in vivo. Am J Physiol Heart Circ Physiol 308: H1402–H1413, 2015. doi: 10.1152/ajpheart.00882.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim YS, Doré S. Catalytically inactive heme oxygenase-2 mutant is cytoprotective. Free Radic Biol Med 39: 558–564, 2005. doi: 10.1016/j.freeradbiomed.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 31.Kim YS, Zhuang H, Koehler RC, Doré S. Distinct protective mechanisms of HO-1 and HO-2 against hydroperoxide-induced cytotoxicity. Free Radic Biol Med 38: 85–92, 2005. doi: 10.1016/j.freeradbiomed.2004.09.031. [DOI] [PubMed] [Google Scholar]
- 32.Kulkarni OP, Hartter I, Mulay SR, Hagemann J, Darisipudi MN, Kumar Vr S, Romoli S, Thomasova D, Ryu M, Kobold S, Anders HJ. Toll-like receptor 4-induced IL-22 accelerates kidney regeneration. J Am Soc Nephrol 25: 978–989, 2014. doi: 10.1681/ASN.2013050528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lv J, Wang X, Liu SY, Liang PF, Feng M, Zhang LL, Xu AP. Protective effect of Fenofibrate in renal ischemia reperfusion injury: Involved in suppressing kinase 2 (JAK2)/transcription 3 (STAT3)/p53 signaling activation. Pathol Biol (Paris) 63: 236–242, 2015. doi: 10.1016/j.patbio.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 34.Muñoz-Sánchez J, Chánez-Cárdenas ME. A review on hemeoxygenase-2: focus on cellular protection and oxygen response. Oxid Med Cell Longev 2014: 604981, 2014. doi: 10.1155/2014/604981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nath KA. Heme oxygenase-1 and acute kidney injury. Curr Opin Nephrol Hypertens 23: 17–24, 2014. doi: 10.1097/01.mnh.0000437613.88158.d3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nath KA. Heme oxygenase-1: a provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int 70: 432–443, 2006. doi: 10.1038/sj.ki.5001565. [DOI] [PubMed] [Google Scholar]
- 37.Nath KA, Croatt AJ, Warner GM, Grande JP. Genetic deficiency of Smad3 protects against murine ischemic acute kidney injury. Am J Physiol Renal Physiol 301: F436–F442, 2011. doi: 10.1152/ajprenal.00162.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nath KA, Grande JP, Croatt AJ, Frank E, Caplice NM, Hebbel RP, Katusic ZS. Transgenic sickle mice are markedly sensitive to renal ischemia-reperfusion injury. Am J Pathol 166: 963–972, 2005. doi: 10.1016/S0002-9440(10)62318-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nath KA, Grande JP, Farrugia G, Croatt AJ, Belcher JD, Hebbel RP, Vercellotti GM, Katusic ZS. Age sensitizes the kidney to heme protein-induced acute kidney injury. Am J Physiol Renal Physiol 304: F317–F325, 2013. doi: 10.1152/ajprenal.00606.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nath KA, Haggard JJ, Croatt AJ, Grande JP, Poss KD, Alam J. The indispensability of heme oxygenase-1 in protecting against acute heme protein-induced toxicity in vivo. Am J Pathol 156: 1527–1535, 2000. doi: 10.1016/S0002-9440(10)65024-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nath KA, Hernandez MC, Croatt AJ, Katusic ZS, Juncos LA. Heme oxygenase activity as a determinant of the renal hemodynamic response to low-dose ANG II. Am J Physiol Regul Integr Comp Physiol 299: R1183–R1191, 2010. doi: 10.1152/ajpregu.00212.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neugarten J, Golestaneh L. The effect of gender on aminoglycoside-associated nephrotoxicity. Clin Nephrol 86: 183–189, 2016. doi: 10.5414/CN108927. [DOI] [PubMed] [Google Scholar]
- 43.Neugarten J, Golestaneh L. Female sex reduces the risk of hospital-associated acute kidney injury: a meta-analysis. BMC Nephrol 19: 314, 2018. doi: 10.1186/s12882-018-1122-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Neugarten J, Golestaneh L, Kolhe NV. Sex differences in acute kidney injury requiring dialysis. BMC Nephrol 19: 131, 2018. doi: 10.1186/s12882-018-0937-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neugarten J, Sandilya S, Singh B, Golestaneh L. Sex and the risk of AKI following cardio-thoracic surgery: a meta-analysis. Clin J Am Soc Nephrol 11: 2113–2122, 2016. doi: 10.2215/CJN.03340316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park KM, Cho HJ, Bonventre JV. Orchiectomy reduces susceptibility to renal ischemic injury: a role for heat shock proteins. Biochem Biophys Res Commun 328: 312–317, 2005. doi: 10.1016/j.bbrc.2004.12.177. [DOI] [PubMed] [Google Scholar]
- 47.Park KM, Kim JI, Ahn Y, Bonventre AJ, Bonventre JV. Testosterone is responsible for enhanced susceptibility of males to ischemic renal injury. J Biol Chem 279: 52282–52292, 2004. doi: 10.1074/jbc.M407629200. [DOI] [PubMed] [Google Scholar]
- 48.Patil CN, Wallace K, LaMarca BD, Moulana M, Lopez-Ruiz A, Soljancic A, Juncos LA, Grande JP, Reckelhoff JF. Low-dose testosterone protects against renal ischemia-reperfusion injury by increasing renal IL-10-to-TNF-α ratio and attenuating T-cell infiltration. Am J Physiol Renal Physiol 311: F395–F403, 2016. doi: 10.1152/ajprenal.00454.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peng J, Ren X, Lan T, Chen Y, Shao Z, Yang C. Renoprotective effects of ursolic acid on ischemia/reperfusion-induced acute kidney injury through oxidative stress, inflammation and the inhibition of STAT3 and NF-κB activities. Mol Med Rep 14: 3397–3402, 2016. doi: 10.3892/mmr.2016.5654. [DOI] [PubMed] [Google Scholar]
- 50.Pittock ST, Norby SM, Grande JP, Croatt AJ, Bren GD, Badley AD, Caplice NM, Griffin MD, Nath KA. MCP-1 is up-regulated in unstressed and stressed HO-1 knockout mice: pathophysiologic correlates. Kidney Int 68: 611–622, 2005. doi: 10.1111/j.1523-1755.2005.00439.x. [DOI] [PubMed] [Google Scholar]
- 51.Pósa A, Kupai K, Ménesi R, Szalai Z, Szabó R, Pintér Z, Pálfi G, Gyöngyösi M, Berkó A, Pávó I, Varga C. Sexual dimorphism of cardiovascular ischemia susceptibility is mediated by heme oxygenase. Oxid Med Cell Longev 2013: 521563, 2013. doi: 10.1155/2013/521563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pósa A, Szabó R, Csonka A, Veszelka M, Berkó AM, Baráth Z, Ménesi R, Pávó I, Gyöngyösi M, László F, Kupai K, Varga C. Endogenous estrogen-mediated heme oxygenase regulation in experimental menopause. Oxid Med Cell Longev 2015: 429713, 2015. doi: 10.1155/2015/429713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rewa O, Bagshaw SM. Acute kidney injury-epidemiology, outcomes and economics. Nat Rev Nephrol 10: 193–207, 2014. doi: 10.1038/nrneph.2013.282. [DOI] [PubMed] [Google Scholar]
- 54.Schmitt R, Melk A. Molecular mechanisms of renal aging. Kidney Int 92: 569–579, 2017. doi: 10.1016/j.kint.2017.02.036. [DOI] [PubMed] [Google Scholar]
- 55.Soljancic A, Ruiz AL, Chandrashekar K, Maranon R, Liu R, Reckelhoff JF, Juncos LA. Protective role of testosterone in ischemia-reperfusion-induced acute kidney injury. Am J Physiol Regul Integr Comp Physiol 304: R951–R958, 2013. doi: 10.1152/ajpregu.00360.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stout JM, Gousset MU, Drummond HA, Gray W III, Pruett BE, Stec DE. Sex-specific effects of heme oxygenase-2 deficiency on renovascular hypertension. J Am Soc Hypertens 7: 328–335, 2013. doi: 10.1016/j.jash.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Synowiec E, Szaflik J, Chmielewska M, Wozniak K, Sklodowska A, Waszczyk M, Dorecka M, Blasiak J, Szaflik JP. An association between polymorphism of the heme oxygenase-1 and -2 genes and age-related macular degeneration. Mol Biol Rep 39: 2081–2087, 2012. doi: 10.1007/s11033-011-0955-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest 123: 966–972, 2013. doi: 10.1172/JCI64098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tian S, Yang X, Zhao Q, Zheng J, Huang H, Chen Y, An R, Xu Y. Association between a heme oxygenase-2 genetic variant and risk of Parkinson’s disease in Han Chinese. Neurosci Lett 642: 119–122, 2017. doi: 10.1016/j.neulet.2017.02.008. [DOI] [PubMed] [Google Scholar]
- 60.Tracz MJ, Alam J, Nath KA. Physiology and pathophysiology of heme: implications for kidney disease. J Am Soc Nephrol 18: 414–420, 2007. doi: 10.1681/ASN.2006080894. [DOI] [PubMed] [Google Scholar]
- 61.Tracz MJ, Juncos JP, Croatt AJ, Ackerman AW, Grande JP, Knutson KL, Kane GC, Terzic A, Griffin MD, Nath KA. Deficiency of heme oxygenase-1 impairs renal hemodynamics and exaggerates systemic inflammatory responses to renal ischemia. Kidney Int 72: 1073–1080, 2007. doi: 10.1038/sj.ki.5002471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang D, Peng Y, Ouzhuluobu, Bianbazhuoma, Cui C, Bianba, Wang L, Xiang K, He Y, Zhang H, Zhang X, Liu J, Shi H, Pan Y, Duojizhuoma, Dejiquzong, Cirenyangji, Baimakangzhuo, Gonggalanzi, Liu S, Gengdeng, Wu T, Chen H, Qi X, Su B. HMOX2 functions as a modifier gene for high-altitude adaptation in Tibetans. Hum Mutat 37: 216–223, 2016. doi: 10.1002/humu.22935. [DOI] [PubMed] [Google Scholar]
- 63.Yang N, Luo M, Li R, Huang Y, Zhang R, Wu Q, Wang F, Li Y, Yu X. Blockage of JAK/STAT signalling attenuates renal ischaemia-reperfusion injury in rat. Nephrol Dial Transplant 23: 91–100, 2008. doi: 10.1093/ndt/gfm509. [DOI] [PubMed] [Google Scholar]
- 64.Zager RA, Alpers CE. Effects of aging on expression of ischemic acute renal failure in rats. Lab Invest 61: 290–294, 1989. [PubMed] [Google Scholar]
- 65.Zhou D, Li Y, Lin L, Zhou L, Igarashi P, Liu Y. Tubule-specific ablation of endogenous β-catenin aggravates acute kidney injury in mice. Kidney Int 82: 537–547, 2012. doi: 10.1038/ki.2012.173. [DOI] [PMC free article] [PubMed] [Google Scholar]