

Keywords: liver, oral digoxin, pyruvate kinase M2-HIF-1α axis, steatohepatitis, sterile inflammation
Abstract
The cardiac glycoside digoxin was identified as a potent suppressor of pyruvate kinase isoform 2-hypoxia-inducible factor-α (PKM2-HIF-1α) pathway activation in liver injury mouse models via intraperitoneal injection. We have assessed the therapeutic effects of digoxin to reduce nonalcoholic steatohepatitis (NASH) by the clinically relevant oral route in mice and analyzed the cellular basis for this effect with differential involvement of liver cell subsets. C57BL/6J male mice were placed on a high-fat diet (HFD) for 10 wk and started concurrently with the gavage of digoxin (2.5, 0.5, 0.125 mg/kg twice a week) for 5 wk. Digoxin significantly reduced HFD-induced hepatic damage, steatosis, and liver inflammation across a wide dosage range. The lowest dose of digoxin (0.125 mg/kg) showed significant protective effects against liver injury and sterile inflammation. Consistently, digoxin attenuated HIF-1α sustained NLRP3 inflammasome activation in macrophages. We have reported for the first time that PKM2 is upregulated in hepatocytes with hepatic steatosis, and digoxin directly improved hepatocyte mitochondrial dysfunction and steatosis. Mechanistically, digoxin directly bound to PKM2 and inhibited PKM2 targeting HIF-1α transactivation without affecting PKM2 enzyme activation. Thus, oral digoxin showed potential to therapeutically inhibit liver injury in NASH through the regulation of PKM2-HIF-1α pathway activation with involvement of multiple cell types. Because of the large clinical experience with oral digoxin, this may have significant clinical applicability in human NASH.
NEW & NOTEWORTHY This study is the first to assess the therapeutic efficacy of oral digoxin on nonalcoholic steatohepatitis (NASH) in a high-fat diet (HFD) mouse model and to determine the divergent of cell type-specific effects. Oral digoxin reduced liver damage, steatosis, and inflammation in HFD mice. Digoxin attenuated hypoxia-inducible factor (HIF)-1α axis-sustained inflammasome activity in macrophages and hepatic oxidative stress response in hepatocytes via the regulation of PKM2-HIF-1α axis pathway activation. Oral digoxin may have significant clinical applicability in human NASH.
INTRODUCTION
Overnutrition and obesity impair metabolic homeostasis, cause stress, and produce inflammatory process (6), playing a major role in the development of nonalcoholic fatty liver disease (NAFLD). NAFLD represents a wide range of pathologies in the liver, beginning with macrovesicular steatosis, which evolves and progresses to nonalcoholic steatohepatitis (NASH). NASH is characterized by the presence of hepatic steatosis and sustained sterile inflammation with hepatocyte injury (10, 19). Despite extensive research, there are no approved therapies for NAFLD/NASH. The emergence of sterile inflammation within the umbrella of metabolic syndrome is one of the most pressing medical challenges in NAFLD/NASH. The interconnection between innate immune cells and those cells resident in liver parenchyma have been highlighted. Macrophages represent a key cellular component of the liver and are essential for maintaining tissue homeostasis and ensuring rapid responses to hepatic injury through the modulation of immune cell function and inflammation (11). In contrast, hepatocytes, the major parenchymal cells in the liver, play pivotal roles in metabolism, detoxification, and orchestration of immune responses. Both cell types can modulate and interfere with critical processes implicated in metabolic disease and inflammatory pathways, initiating and sustaining liver damage (10). We took a two-step approach by first identifying core regulatory machinery, the activation of which is required for the development of sustained sterile inflammation in disease conditions. Second, we identified an approved drug, which can inhibit this pathway, allowing for the possibility of rapid transition to clinical use. The requirement of hypoxia-inducible factor-α (HIF-1α) pathway activation in macrophages for the development of sustained inflammatory response gives HIF-1α a key role in the core regulatory machinery, and enables cardiac glycosides to inhibit HIF-1α, fulfilling the second criteria of our approach (20, 21).
We have previously described a novel and potent therapeutic effect of digoxin by intraperitoneal injection in reducing the severity of steatosis, inflammation, and hepatocellular damage in livers undergoing both acute and chronic sterile inflammatory injury from multiple mouse models (21). Digoxin improves oxidative stress during liver injury by maintaining cellular redox homeostasis, as well as by suppressing HIF-1α pathway activation and downstream signature genes in the liver. We have further identified the key metabolic regulator pyruvate kinase isoform 2 (PKM2) as a digoxin-binding protein. PKM2 exists primarily as an enzymatically inactive monomer or in a dimer form. The dimer of PKM2 can translocate to the nucleus, where it interacts with HIF-1α and regulates the gene expression of numerous proglycolytic enzymes (15). Digoxin does not alter PKM2 kinase activity but suppresses PKM2-dependent HIF-1α transcriptional activity in immune cells (21). Further protein immunoprecipitation, combined with mass spectrometry, demonstrates that nuclear PKM2 directly interacts with multiple modified chromatin proteins, and digoxin reduces the binding of histones to PKM2, suggesting digoxin suppresses PKM2-promoted HIF-1α transactivation through chromatin modifications. These results identify PKM2-HIF1α pathway activation as a key in liver sterile inflammation and provide molecular basis for the observed effects of digoxin in antagonizing liver injury to the insults from a wide range of liver diseases (21). Despite the importance of PKM2-HIF1α pathway activation in the control of sustained sterile inflammation in immune cells, whether it also plays functional role in liver hepatocytes was unknown. Furthermore, in addition to immune cells, the contribution of hepatocytes to digoxin inhibition on liver injury was less understood. Because oral digoxin is routinely used in clinical practice, we determined the efficacy of the oral route of digoxin for the protection of liver damage, steatosis, and inflammation on a high-fat diet (HFD)-induced NAFLD/NASH model in mice, and further analyze the cellular basis for this effect with differential involvement of liver cell subsets.
MATERIALS AND METHODS
Animals and primary cell isolation.
C57BL/6J mice were from the National Cancer Institute. All experiments were performed in specific pathogen-free facilities and were performed in accordance with the regulations adopted by the National Institutes of Health (NIH) and approved by the Animal Care and Use Committee of the Yale University.
Parietal epithelial cells were isolated by peritoneal lavage 3 days after intraperitoneal injection of 4% thioglycollate solution (B255; Fluka). Cells were cultured in DMEM medium (10% FBS), penicillin-streptomycin, and l-glutamine. Mouse bone marrow-derived macrophages were isolated and differentiated for 7 days in complete RPMI 1640 medium supplemented with 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 μM 2-mercaptoethanol, 10% FBS, and 20 ng/ml macrophage colony-stimulating factor (M-CSF).
Mouse hepatocytes were isolated from 10- to 12- wk-old mice using collagenase perfusion, as previously described (2). Hepatocytes were maintained in collagen-sandwich culture to closely resemble in vivo conditions (2).
Human liver tissue slides consisting of normal and NASH cases were obtained from Yale tissue repository maintained by Yale Pathology Tissue Services (YPTS; Yale School of Medicine). The liver tissue slides were used for immunohistochemistry to detect PKM2 expression. Briefly, the paraffin sections were cleared, hydrated, and immunostained with rabbit anti-PKM2 antibody (1:500 dilution, D78A4, Cell Signaling Technologies) overnight and goat anti-rabbit HRP for 1 h followed by DAB reaction. Images were obtained using an Olympus BX 51 microscope (Olympus, Tokyo, Japan) under ×10 and ×40 objectives and processed using ImageJ software (ImageJ, NIH) to estimate PKM2 levels expressed as optical density values.
Human hepatocytes were obtained through the Liver Tissue Cell Distribution System, which was funded by NIH contract N01-DK-7–0004/HHSN267200700004C. The cells were maintained as previously reported (2).
Duolink proximity ligation assay.
Digoxin and PKM2 Duolink in situ red starter kit mouse/rabbit (DUO92101) was purchased from Sigma Aldrich. HeLa cells were pretreated with or without digoxin for 3 h followed by dimethyloxalylglycine (DMOG) treatment for 12 h. Cells were fixed in methanol, permeabilized with TBS with 0.25% Triton-X 100 and blocked with Duolink blocking solution. The cells were incubated with primary rabbit anti-PKM2 (D78A4) 1:500 dilution and primary mouse antidigoxin (DI-22; Sigma Aldrich) 1:250 dilution for overnight at 4°C. The cells were further incubated with Duolink Plus and Minus probes (anti-rabbit and anti-mouse antibodies tagged with complementary Oligo strands), ligated, and polymerized, as instructed by the manufacturer. Images were captured with Leica SP8 confocal laser-scanning microscope (Leica Microsystems) at the Center for Cellular and Molecular Imaging facility, Yale School of Medicine. The images were obtained as Z-stacks (25 stacks; each 400-nm thickness) with 512 × 512 pixel size, with excitation/emission settings for Duolink red signal (λAbs 594; λEmi624) and for DAPI (n = 6). The Z stacks were processed with Leica Application Suite X software to obtain superimposed multichannel three-dimensional images. The obtained images were analyzed using ImageJ software (NIH, Bethesda, MD) to calculate the number of proximity ligation assay (PLA) particles per 100 × field and the number of PLA particles in the nucleus.
Immunoprecipitation and Western blot analysis.
Whole cell lysates were obtained through RIPA lysis buffer, and isolation of nuclear extract was performed using a nuclear extraction kit. Total PKM2 was immunoprecipitated in nuclear extract from Raw264.7 cells using anti-PKM2 (D78A4) antibody followed by SDS-PAGE and Western blot analysis.
Quantitative real-time RT-PCR.
Total RNA was extracted using TRIzol (Invitrogen), and cDNA was generated with an oligo (dT) primer and the Superscript II system (Invitrogen) followed by analysis using LightCycler 480 system (Roche). Real-time PCR was performed using SYBR Green real-time PCR analysis (Roche). Expression of GAPDH was used to standardize the samples, and the results were expressed as a ratio relative to control. PCR primers used were as follows: Il6: forward tgcaagagacttccatccag and reverse tgaagtctcctctccggact; Tnfa: forward aaatggcctccctctcat and reverse cctccacttggtggtttg; Il1b: forward caactgtgaaatgccacct and reverse gaagcagcccttcatcttt; Ifng: forward acggcacagtcattgaaag and reverse catgtcaccatccttttgc; Il17: forward ctgcttttctaaatcgtgctg and reverse gtgtttgtgtgccttgtgata; Ccl2: forward caggtccctgtcatgctt and reverse gggatcatcttgctggtg; Slc2a1: forward: caatcgtaacgaggagaacc and reverse: ctttcatctcctgcaggtct; G6pc: forward caagggagaactcagcaagt and reverse gggcttcagagagtcaaaga; Pfkfb3: forward gactcgctacctcaactgga and reverse tgtactgcttcacagcctca; Srebpf1: forward gtgtactggcctttctgtgtc and reverse gtagcatcagagggagtgaga; Hif1a: forward ccaggccttgacaagcta and reverse cgcggagaaagagacaag; pgc1a: forward caggggcacatctgttct and reverse tcggatttcctggtcttg; Nox4: forward gacctggatttggatttctg and reverse acaggtttgttgctcctgat; Sirt1: forward gagacaacctcctgttga-ml and reverse gtgtgacgttctgtcatcgt; and b-actin: forward aaggactcctatgtgggtga and reverse atgtcgtcccagttggtaac.
HFD model of steatohepatitis and chronic liver injury.
Eight-week-old male C57BL/6J mice were placed on a high-fat diet (HFD; 45% fat, D12451 Research Diets) or chow. Animals were studied after 10–15 wk of feeding.
At the end of the protocols, the whole livers and serum were collected for histological, cytological, biochemical, and molecular analysis.
Preparation of the liver nonparenchymal cell suspension and flow cytometry (FACS).
Mouse liver was perfused with HBSS via the portal vein immediately before harvesting from animals. Tissues were cut into small pieces and passed through a 40-μm nylon cell strainer, and the cells were further centrifuged at 40 g for 3 min to pellet parenchymal cells. The single-cell suspensions were then isolated by Percoll gradient to collect the nonparenchymal cell fraction. Cells were incubated with antibodies against CD45-APC (clone 30-F11), mouse Ly-6G (Gr-1) Alexa Fluor 488 (clone RB6-8C5), and/or mouse CD11b-PE (clone M1/70) (all from BD Biosciences) in FACS buffer for 30 min. The cells were analyzed using flow cytometry. The dead cells were excluded by propidium iodide staining.
Statistical analyses.
Data are expressed as the means ± SD and were compared by one-way ANOVA and Student-Newman-Keuls test. Student’s t test was used for two-group analyses, except as mentioned specifically in the text. Differences in values were considered significant at P < 0.05.
RESULTS
Oral digoxin reduced the severity of liver damage and steatosis in HFD-induced NASH model in mice.
We have previously showed that C57BL/6J male mice on an ad libitum 45 kcal% fat diet (HFD) for 8–12 wk develop several features of NASH, including hepatic damage and steatosis, inflammatory cell infiltration, increased serum ALT level, and enhanced NAFLD activity scores. Mice were dosed with digoxin twice a week by intraperitoneal injection concurrently and after the HFD, resulting in a dramatic reduction in hepatic damage, steatosis, and inflammation, as determined by histological analysis, serum ALT, and leukocyte infiltration in the liver. To assess the therapeutic effects of digoxin by the clinical corresponding oral route, wild-type mice were placed on the same HFD for 10 wk and were started concurrently with oral digoxin administration (2.5, 0.5, 0.125 mg/kg twice a week) for 5 wk. The oral dosages were calculated from the 50–66% oral bioavailability from the previous studies (21). Digoxin (1 mg/kg ip daily) in mice results in the therapeutic serum levels achieved in humans (0.5–2 ng/ml) (29). INT-767 is a dual agonist on FXR/G protein-coupled receptor 5 (TGR5) and improves histological features of NASH (24). Oral INT-767 was, therefore, used as a positive control. Th wild-type mice were provided an ad libitum 45 kcal% fat diet for 15 wk, resulting in several features of NASH, including enhanced hepatic steatosis and inflammation (Fig. 1, A and B) and increased serum ALT level (Fig. 1C). Similar to the route of intraperitoneal injection, HFD-fed mice treated with a dose range of oral digoxin starting at week 10 after the HFD for a concurrent five more weeks showed dramatic reduction in hepatic damage and NAFLD activity scores, as determined by histological analysis and serum ALT in the liver (Fig. 1, A–C). These effects of digoxin were comparable with INT-767 (Fig. 1, A–C). Collectively, these data indicate that digoxin effectively protects chronic hepatic damage, steatosis, and inflammation in HFD-induced obese mice. Thus, we conclude that long-term treatment with oral digoxin reduces chronic liver damage, steatosis, and inflammation in experimental model of NASH.
Fig. 1.

Oral digoxin reduced hepatic damage and steatosis when given therapeutically. A–C: mice were fed with high-fat diet (HFD) for 10 wk, and then cocurrently started digoxin at multiple dosages (H, High: 2.5 mg/kg; M, middle: 0.5 mg/kg; L, low: 0.125 mg/kg), INT-767 (30 mg/kg) or vehicle twice a week by gavage feeding with continued HFD feeding for a total of 15 wk. A: digoxin reduces HFD-induced hepatic steatosis from histological analysis by hematoxylin-and-eosin (H&E)-stained sections. B: digoxin reduces HFD-induced hepatic steatosis and liver inflammation as scored quantitatively from H&E-stained sections. C: digoxin reduces HFD-induced hepatocellular damage, as measured by the serum ALT. All data in the figure show means ± SD from 8 to 10 mice in each group. *P < 0.05, **P < 0.01. Scale bar: 100 μm.
Oral digoxin reduced hepatic leukocyte infiltration in HFD-induced NASH model in mice.
The sterile inflammation reflected by inflammatory immune cell response is the key feature of NAFLD/NASH and contributes critically to the progress of steatohepatitis. We determined whether oral digoxin would protect the liver, as well as peripheral leukocyte infiltration. The wild-type mice were provided HFD for 15 wk, which resulted in increased neutrophil infiltration in liver and blood, but less in spleen (Fig. 2, A and B). Oral digoxin did not change neutrophil infiltration in liver, blood, and spleen under normal chow condition, but significantly reduced HFD-driven neutrophil infiltration in liver and blood (Fig. 2, A and B). The leukocyte infiltration of the monocyte population showed parallel reduction by oral digoxin in HFD-fed mice (Fig. 2C). Consistent with these changes, gene expression analyses revealed that proinflammatory markers such as Il1b, Il6, Tnf-a, Il17, and Ccl2 were significantly lower in liver tissue from digoxin-treated mice compared with vehicle controls (Fig. 2D). Furthermore, the gene expression levels relevant to the changes of hydrocarbon and glycolysis (SLC2a1, G6PC, Pfkfb3, Srebpf1, and Hif1a in Fig. 2F) were lower, and oxidation (Pgc1a, Nox4, and Sirt1 in Fig. 2E) were higher in digoxin-treated mouse liver. These data indicate that consistent with the reduction of hepatic damage and steatosis features, oral digoxin effectively reduces chronic inflammation and improves the abnormal metabolic states, even though the mice were maintained on the HFD.
Fig. 2.

Oral digoxin reduced high-fat diet (HFD)-induced liver inflammatory response. A and B: mice were fed with HFD for 10 wk, and then started digoxin at low dosage of 0.125 mg/kg or vehicle twice a week by gavage feeding with continued HFD feeding for a total of 15 wk. A: representative images of CD45.1+ Ly6G+ CD11b+ cells in nonparenchymal cell populations using flow cytometry from oral digoxin-treated HFD mice. B: digoxin reduced HFD-induced liver neutrophil infiltration as quantified by CD45.1+ Ly6G+ CD11b+ cells in nonparenchymal cell populations using flow cytometry (n = 3 repeats). *P < 0.05. C: digoxin reduced CD45.1+ Ly6G+ CD11b+ (neutrophil) vs. CD45.1+ Ly6C+ CD11b+ (monocyte) cells in nonparenchymal cell populations using flow cytometry from oral digoxin-treated HFD mice. D–F: changes of mRNA levels of indicated gene expression from oral digoxin-treated HFD mouse liver tissue.
Oral digoxin reduced body and liver weight in HFD-induced NASH model in mice.
We further measured whether oral digoxin affects the increase of total body weight and liver weight that occurs on HFD. Eight-week-old mice were provided an ad libitum 45 kcal% fat diet for 15 wk, resulting in significantly increased body weight, as well as liver weight (Fig. 3, A and B). HFD-fed mice treated with oral digoxin did not have the increase in total body weight and liver weight seen in the HFD control group (Fig. 3, A–C). During the assay, mice were provided HFD ad libitum, and food intake was comparable between the control and digoxin group (Fig. 3D).
Fig. 3.

Oral digoxin-reduced high-fat diet (HFD)-induced increase in body and liver weight. A–C: mice were fed with HFD for 10 wk, and then started digoxin at multiple dosages (H, High: 2.5 mg/kg; M, middle: 0.5 mg/kg; L, low: 0.125 mg/kg), INT-767 or vehicle twice a week by gavage feeding with continued HFD feeding for a total of 15 wk. A: digoxin reduces HFD-induced changes of body weight. B: digoxin reduces HFD-induced changes of liver weight. C: representative body morphological changes in digoxin-treated HFD mice. D: food intake changes in digoxin-treated HFD mice. All data throughout the figure are shown as the means ± SD from 8 to 10 mice in each group. *P < 0.05, **P < 0.01.
Digoxin attenuated the succinate-mediated increase in oxidative stress.
The hepatic microenvironment is influenced by high levels of dietary fats and carbohydrates, and these high levels result in elevated oxidative stress and inflammation due, in part, to metabolic intermediators such as succinate (9, 27). Succinate is an important metabolite at the cross-roads of several metabolic pathways, and it is involved in the elimination of reactive oxygen species (ROS) and signal of inflammation. Succinate also acts as critical inflammatory signal that enhances interleukin-1β production through stabilizing HIF-1α in macrophages (26), and succinate concentrations are modestly elevated in NASH (9). The ability of digoxin to reduce HIF-1α activation predicts that digoxin will be able to reduce succinate-mediated increase in ROS production. To test whether digoxin reduced hepatic oxidative stress due to the relevant succinate-mediated effects, human monocyte/macrophage-like THP-1 cells were stimulated with dimethyl succinate, and the oxidative response was quantified by measuring total ROS production using CM H2DCFDA assays. Dimethyl succinate was used to trigger total ROS production, which was significantly inhibited by both indicated dosages of digoxin treatment (Fig. 4A). Consistently, we also detected an increased mitochondrial membrane potential (MMP) in THP-1 cells treated with dimethyl succinate, which digoxin reduced significantly (Fig. 4B). Collectively, these data support our previous finding that digoxin improves hepatic oxidative stress induced by a wide range of stimuli.
Fig. 4.

Digoxin improved succinate-induced increase in cellular reactive oxygen species (ROS). A: THP-1 cells were treated with digoxin at indicated concentrations or vehicle for 3 h and then with dimethyl succinate (5 mM) for an additional 3 h. Chloromethyl derivative of 2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) was added in the last 30 min and washed following kinetic analysis of intracellular ROS with fluorescent plate reader. B: THP-1 cells were treated with digoxin at indicated concentrations or vehicle for 3 h and then dimethyl succinate (5 mM) for an additional 3 h. Cells were then incubated with JC-1 for 30 min to monitor mitochondrial membrane potential. The kinetic analysis of mitochondrial membrane potential was performed using a fluorescent plate reader.
Digoxin attenuated HIF-1α-mediated sustained inflammasome activation in macrophages.
The amplitude of the sterile inflammation, however, varies widely between organs, and the liver is notable for developing an exceptionally strong sterile inflammatory response. This is seen in experimental models, including acetaminophen toxicity, alcoholic steatohepatitis, and metabolic syndrome-associated development of NASH (12, 17). Such high amplitude of sterile inflammation in the liver has major clinical consequences, as NASH is by far the most common liver disease in industrialized countries and is responsible for increasing amounts of liver damage and death (7). Inflammasome activation has recently emerged as a key feature in mediating sterile inflammatory response to a wide range of liver pathologies (12, 18). We have demonstrated that HIF-1α potentiates and sustains the amplitude of NLRP3 inflammasome activation and is vital for the transition from acute self-limiting to a sustained chronic inflammation (20). We have previously shown that pretreatment of mice with digoxin dramatically prevented LPS/D-GalN-induced NLRP3 inflammasome activation in vivo, which is consistent with reduced liver neutrophil infiltration and hepatocellular damage (21). We, therefore, determined whether digoxin has direct inhibitory effects on NLRP3 inflammasome activation in macrophage cultures. We have previously characterized that intracellular cAMP-HIF-1α pathway activation is critical for sustained inflammasome activation in response to oxidative stress stimulation (20). We further confirmed that dibutyryl cyclic AMP (db-cAMP), a cell-permeable analog of cAMP, prolonged pro-IL-1β gene expression compared with acute LPS response in macrophages over a time course (Fig. 5, A and B). Interestingly, digoxin significantly suppressed db-cAMP sustained pro-IL-1β gene expression at both mRNA and protein levels (Fig. 5, C, D, F, G). Consistent with previous findings, digoxin significantly suppressed LPS-induced HIF-1α protein expression levels (Fig. 5E). Under the typical NLRP3 inflammasome-active condition in vitro, digoxin inhibited IL-1β protein production, suggesting digoxin suppresses NLRP3 inflammasome activation through the inhibition of pro-IL-1β gene expression. Upon binding to inflammasome adaptor protein ASC, NLRP3 protein can translocate onto mitochondria, where it can cleave procaspase1 into mature caspase1. Confocal imaging showed that digoxin did not inhibit LPS/ATP-triggered NLRP3 translocation onto mitochondria (Fig. 5H). Collectively, these results indicate that digoxin effectively inhibits NLRP3 inflammasome activation through inhibition of HIF-1α-mediated pro-IL-1β gene transcription.
Fig. 5.

Digoxin suppressed HIF-1α sustained inflammasome activation. A–D: mouse parietal epithelial cells (PECs) were stimulated with LPS (100 ng/ml) for the indicated time course (A) or primed for 16 h and then treated with db-cAMP for the indicated time course (B), or pretreated with digoxin for 3 h following LPS for 16 h and further db-cAMP for 5 h (C and D). E: PECs were pretreated with digoxin (50 nm) following LPS for 12 h. F–H: mouse PECs were primed with LPS (100 ng/ml) for 16 h, and then pretreated with digoxin (50 nM). The cells were then challenged with ATP (5 mM) for 30 min (F and H), or the cells were pretreated with db-cAMP for 6 h before the challenge of LPS/ATP (G). The cells were harvested, and RNA was isolated after each treatment, and the gene expression of Il1b was quantified by real-time PCR using specific primers (A–C). Immunoblot analysis of the proIL-1β (D) and hypoxia-inducible factor (HIF)-1α (E) from cell lysate was performed by specific anti-IL-1β and anti HIF-1α antibodies. The cell supernatants were collected, and IL-1β was measured using ELISA kit (F and G). H: cells were stained with anti-NLRP3 and Tom-20 antibodies following confocal image analysis. Data are expressed as the means ± SD from three independent experiments. Immunoblots and confocal images shown are representative results from at least three independent experiments. *P < 0.05, **P < 0.01.
Digoxin improved oxidative stress response in primary hepatocyte cultures.
The pathogenesis of NASH is a complex process and implicates cell interactions between hepatocytes and immune cell populations in the liver. Despite the importance of digoxin on the inhibition of PKM2-HIF1α pathway activation in immune cells (21), its direct effect on hepatocytes was unclear. We have previously found that hepatocytes in the healthy liver had a very low level of PKM2; however, it was significantly elevated in humans and mice with steatosis (Fig. 6A). PKM2 protein level was also confirmed to be highly increased in hepatocytes isolated from HFD-fed mice (Fig. 6B). To further test whether digoxin has an inhibitory effect on steatosis in hepatocytes, we cultured both human and mouse primary hepatocytes and used palmitate as a NASH-relevant metabolic stress inducer. Palmitate induced remarkable lipid accumulation in hepatocytes from mice (Fig. 6C) and humans (Fig. 6D), and digoxin significantly reduced it (Fig. 6, C and D). The importance of ROS in liver injury has been shown by the fact that reduction of ROS by a range of manipulations protects the liver against oxidative stress (5, 14). Hepatocyte mitochondria are the major site for ROS generation in various forms of liver diseases, including NAFLD/NASH. We have previously demonstrated that digoxin has critical effects in improving abnormal intracellular redox status in vivo (21). We next determined whether digoxin has a direct effect on the inhibition of ROS production in hepatocyte cultures. Consistent with previous reports that both acute and chronic liver injury resulted in the production of high amounts of ROS from liver tissue, and mouse primary hepatocyte cultures, as evidenced by mitoSOX dye fluorescence. Hydrogen peroxide boosted mitoROS production, which was significantly inhibited by digoxin (Fig. 6, E and F). Collectively, these results indicate that digoxin has direct inhibitory effect on steatosis, as well as oxidative stress response in hepatocytes, in addition to its previous known effects in immune cells.
Fig. 6.

Human and mouse hepatocytes upregulate pyruvate kinase isoform 2 (PKM2) during steatosis, and digoxin reduced mitochondrial reactive oxygen species (ROS) and steatosis in primary hepatocyte cultures. A: immunohistochemistry of PKM2 (red arrow) liver samples from health donor and patients with moderate steatosis (A, left: representative images. A, right: quantifications. Values are expressed as means ± SD; n = 8. ***P < 0.001. B: PKM2 protein level in whole liver (L) and isolated hepatocytes (H) from mice fed a high-fat diet (HFD) and control mice fed regular chow for 12 wk (n = 3). C: mouse primary hepatocytes were pretreated with digoxin (10, 50 nM) for 3 h following palmitate for 24 h. D: human primary hepatocytes were pretreated with digoxin (10, 50 nM) for 3 h following palmitate for 24 h. Lipid staining by BODIPY 493/503; Nucleus by DAPI. C and D, left: representative images; C and D, right: quantifications. Values are expressed as means ± SD (n = 30 fields in C, and n = 20 fields in D). *P < 0.05, **P < 0.01. E and F: mouse primary hepatocytes were pretreated with digoxin (50 nM) or vehicle control for 3 h following MitoSOX and MitoTracker Green costaining for 30 min. The cells were washed and challenged with H2O2 for the indicated time course following imaged by confocal image system. E: representative results were shown from at least three independent experiments. F: density quantification of MitoSOX staining was shown in accounting five areas using ImageJ.
Digoxin inhibited PKM2-HIF-1α pathway activation in both macrophage and hepatocyte populations.
We have previously reported that digoxin reduces liver injury in NASH/ASH via a direct interaction with PKM2 in macrophages, leading to reduced cellular oxidative stress and liver inflammation (21). The small molecule TEPP-46 stabilizes the tetrameric form of PKM2 and reduces PKM2 nuclear translocation thus limiting its transactivation activity (1, 23). TEPP-46 treatment (50 mg/kg ip) before LPS significantly reduced liver hemorrhage, hepatocyte necrosis, and serum ALT level in D-GalN-sensitized mice (21). Digoxin did not block TEPP-46-induced tetramer formation (Fig. 7A), which is consistent with a previous finding that digoxin does not alter PKM2 enzymatic activity (21). In addition, digoxin also reduced the protein binding of PKM2 with HIF-1α in the nucleus by protein pull-down assay (Fig. 7B). The identified digoxin binding to PKM2 was further validated by Duolink in situ PLA assay, which relies on the very close proximity between interacting proteins (<40 nm). A strong PLA signal corresponding to digoxin-PKM2 interaction was observed in cells expressing the active form of PKM2 triggered by DMOG (Fig. 7, C and D), and the interaction can be visualized well in the nucleus (Fig. 7C). This is consistent with the reduction of PKM2 and histone interaction by digoxin as an important pathway in limiting liver inflammation and damage through an immune cell-mediated manner.
Fig. 7.

Digoxin inhibited pyruvate kinase isoform 2 (PKM2)-hypoxia-inducible factor (HIF)-1α axis targeting gene expression in both macrophages and hepatocytes. A and B: Raw264.7 cells were pretreated with digoxin or vehicle for 3 h following TEPP-46 (50 μM) for 6 h. A: cells were cross-linked using DSS, and the cell lysates were applied to Western blot using specific anti-PKM2 antibody. B: nuclear and cytosol fractions were isolated using commensal available kit following Western blot using anti-PKM2, HIF-1α, and Lamin A/C antibodies. C and D: HeLa cells were pretreated with or without digoxin for 3 h following dimethyloxalylglycine (DMOG) for 12 h. The cells were fixed and performed by proximity ligation assay (PLA) between digoxin and PKM2 following the manufacturer’s instructions provided with Duolink in situ red starter kit (DUO92101; Sigma). C: representative images. D: quantifications of PLA particles. Values are expressed as means ± SD; n = 10 fields, 3 repeats. ***P < 0.001. E: mouse primary hepatocytes were pretreated with digoxin or vehicle for 3 h following palmitate for 5 h. The NOX4 mRNA level was measured by quantitative RT-PCR (qRT-PCR) using specific primers. F: mouse primary hepatocytes were pretreated with digoxin or vehicle for 3 h following palmitate for 5 h. Reactive oxygen species (ROS) production was measured by the lucigenin chemiluminescence assay. The results were normalized for protein concentrations and showed increased ROS production. G: mouse primary hepatocytes were transfected with HIF-1α construct for 48 h following digoxin for 24 h. The NOX4 mRNA level was measured by qRT-PCR using specific primers. H: mouse primary hepatocytes were transfected with NOX4 luciferase promoter construct for 48 h following digoxin for 24 h. The luciferase activity was measured by dual-luciferase reporter assay. The relevant luciferase activity was based on Renilla control.
NOX4 is a target gene of PKM2, and NOX4 gene expression was induced by palmitate, which is the abundant fatty acid, and digoxin significantly inhibited the induction in primary hepatocyte cultures (Fig. 7E). In parallel, palmitate triggered ROS production, which was significantly inhibited by digoxin (Fig. 7F). Overexpression of HIF-1α in hepatocytes can mimic palmitate-induced NOX4 gene expression, and digoxin significantly inhibited this overexpression, suggesting the inhibitory effects of digoxin on NOX4 gene expression are in a HIF-1α-dependent manner (Fig. 7G). Digoxin significantly suppressed palmitate-induced NOX4 transcriptional promoter activity in primary hepatocyte cultures (Fig. 7H). Consistently, we confirmed that PKM2 protein was highly elevated in HFD-induced mouse liver tissue, as well as hepatocyte populations, indicating PKM2 may play a critical role in NASH development through the regulation of biological responses in multiple cell types. Collectively, our results indicate that digoxin reduces liver injury in NAFLD/NASH via a direct interaction with PKM2 and reducing PKM2-triggered transactivation in both immune and parenchymal cell populations, leading to reduced cellular oxidative stress and inflammation (Fig. 8).
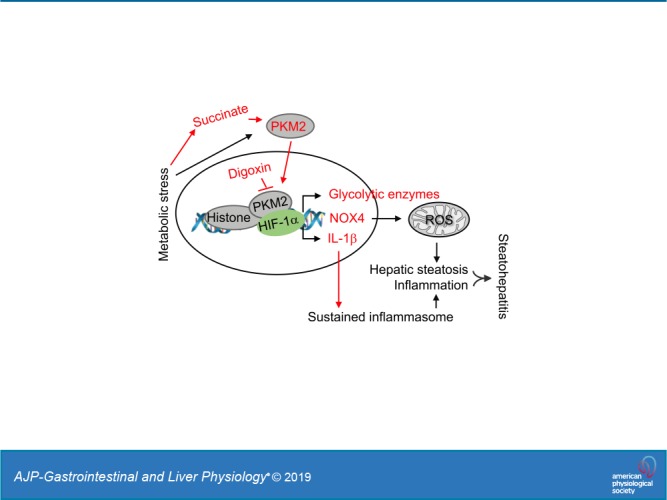
Fig. 8.

Working model of digoxin inhibited liver injury in nonalcoholic steatohepatitis (NASH). The cell metabolic stress generated from high-fat diet (HFD) intake in the mouse triggers pyruvate kinase M2 (PKM2) activation that can occur partially via metabolites like succinate. In addition to its pyruvate kinase function, PKM2 interacts with hypoxia-inducible factor-1α (HIF-1α) in the nucleus and functions as a transcriptional coactivator for HIF-1α, resulting in stimulation of HIF-1α-responsive genes. We have previously shown that chronic metabolic stress triggers cAMP pathway activation, which results in HIF-1α transactivation leading sustained IL-1β gene expression and inflammasome activation, and digoxin reduces this pathway-driven sustained inflammation. Thus, digoxin has the ability to improve nonalcoholic fatty liver disease (NAFLD)/nonalcoholic fatty liver disease (NASH) through the inhibition of PKM2-HIF1α pathway that targets both oxidative stress and proinflammatory gene expression. Labels and arrows highlighted in red indicate the new findings from our work.
DISCUSSION
Our results show that oral digoxin significantly reduced histological damage, NAFLD activity scores, neutrophil infiltration, serum ALT values, and body and liver weight in all dosage groups without a reduction in food intake from the NASH mouse model. Digoxin attenuated metabolite succinate-derived ROS generation, as well as the elimination of mitochondrial membrane potential (MMP). Digoxin attenuated HIF-1α-sustained inflammasome activation in macrophages and inhibited steatosis, as well as oxidative stress response in hepatocytes, indicating digoxin has an inhibitory effect on PKM2-HIF-1α axis activity in both immune and parenchymal cell populations in liver.
We have previously demonstrated that digoxin reduces oxidative stress during liver injury through maintaining cellular redox homeostasis and protects the liver from a wide variety of insults in multiple mouse models and does so by a route of intraperitoneal injection. In addition, our results show that the clinically relevant oral route of digoxin has therapeutically major comparable effects to improve hepatic damage, steatosis scores, and inflammatory immune cell infiltration in mouse NASH model.
The emergence of sterile inflammatory diseases within the umbrella of metabolic syndrome is one of the most pressing medical challenges. The liver is notably affected with the development of the related conditions NAFLD and NASH. The therapeutic development for NAFLD/NASH is limited by the relatively poor understanding of the initiating steps in sterile inflammation, as well as the dysregulation of a wide range of pathways, making it difficult to know which ones to target. The requirement of HIF-1α pathway activation in macrophages for the development of an inflammatory response made this candidate a core pathway, and the ability of cardiac glycosides, to inhibit HIF-1α fulfilled the second criteria. The fact that the mechanism by which digoxin inhibition on HIF-1α was unknown provided us a further opportunity to discover a new pathway regulating NAFLD. Inflammasome pathways are a key driver of sterile inflammation and important in multiple chronic diseases, but it is not known how the signaling is sustained after initiation. Inflammasome activation is dependent on stimuli such as LPS and ATP, which provide two distinct signals resulting in rapid production of IL-1β, with a lack of response to repeat stimulation. We have previously demonstrated that after receiving conventional signals 1 and 2, macrophages are dependent on adenosine through the A2A receptor-cAMP-PKA-HIF-1α pathway activation for initial and sustained inflammasome activity. Conceptually, this is significant, as it changes our interpretation of inflammasome activity to being caused by a lack of cytokine response by macrophages after LPS exposure from the cell being in an unresponsive state, to inflammasome activity being in a postactivation state, which is regulated by a different set of signals. Mechanistically and therapeutically, this is significant, as it requires a clearer understanding if tissue injury is due to the initial inflammasome activation or is due to maintenance of the activation state (20). Interestingly, we found that digoxin has direct effects on the inhibition of inflammasome activation. Digoxin has a small effect on typical inflammasome activity, while it strongly affects cAMP signaling-dependent sustained inflammasome activity and does so through the inhibition of HIF-1α pathway.
It has been reported that HIF-1α inhibition in intestine evokes impaired gut barrier homeostasis leading gut-liver axis dysregulation in alcoholic hepatitis (25). This may raise concerns about the risks of leaky gut caused by oral digoxin via HIF-1α inhibition. The role of intestine HIF-1α in hepatic lipid metabolism is the subject of conflicting reports, Xie et al. (28) showed that removal of HIF-2α from the intestinal epithelium improved hepatic steatosis and injury. In addition, they did not find a HIF-1α protein signal in lean and obese ileal biopsy tissue. The authors proposed that the response of NASH is either dominantly or exclusively dependent on the integrity of intestinal HIF-2α rather than HIF-1α. Thus, the protective role of oral digoxin on hepatic damage, liver steatosis, and inflammation in NASH was unlikely through the regulation of intestinal HIF-1α response. Furthermore, digoxin has been used in large numbers of people over many decades without any evidence of intestinal dysfunction.
Our results show that oral digoxin significantly reduced body weight without the benefit of adequate food consumption. These data suggest that increased energy expenditure may be responsible for the efficacy of digoxin on the increased hepatic ectopic lipid accumulation in HFD mice. Decreased energy expenditure in rodents is associated with increased ectopic lipid content (3) and that increased energy expenditure resulting from increased mitochondrial activity (13) can protect mice from HFD-induced lipid content in the liver. The increased whole body energy expenditure could be responsible for the efficacy of digoxin on the inhibition of hepatic steatosis. This question should be addressed in the future by a comprehensive metabolic cage study and oxygen consumption assessment.
An imbalance in redox homeostasis, with the development of ROS is common to the development of a wide range of liver injury, including NASH. Cellular ROS can increase via a wide range of pathways, including mitochondrial damage, reduction in antioxidants, and subsequent to HIF-1α upregulation (4, 8, 22). ROS can cause hepatocyte dysfunction and apoptosis, infiltration of monocytes/macrophages into the liver, and activation of hepatic stellate cells. ROS are mainly generated from the electron transport chain in the mitochondria of hepatocytes. Thus, targeting excessive ROS accumulation is an effective method to attenuate oxidative stress-induced liver damage. We have previously shown the ability of digoxin to improve mitochondrial and cellular ROS, both in vivo and in vitro. The current results also show that digoxin has direct effects to improve oxidative stress in hepatocytes. This is consistent with the broad protective effects of digoxin in liver injury.
The ability of digoxin to bind to PKM2 is an unexpected finding and provides novel insights into PKM2 biology and the role of PKM2 in sterile inflammatory liver diseases. PKM2 is best known as the rate-limiting glycolytic enzyme that catalyzes the conversion of phosphoenopyruvate (PEP) to ADP to pyruvate and ATP (16). In addition to its pyruvate kinase function, PKM2 interacts with HIF-1α in the nucleus and functions as a transcriptional coactivator for HIF-1α, resulting in stimulation of HIF-1α-responsive genes (15). The current findings support the PKM2 protein interaction with HIF-1α via direct binding; digoxin significantly reduced the binding activity. Further, results showed that digoxin inhibits PKM2 targeting NOX4 gene transcription, even in the forced HIF-1α expression, suggesting that PKM2, but not HIF-1α, directly targets on NOX4, and digoxin inhibits it.
Collectively, these data identify that digoxin therapeutically improves hepatic damage, steatosis, and sterile inflammation from an experimental model of NAFLD/NASH by the clinically applicable oral route. We further demonstrate that digoxin can improve NAFLD/NASH through the regulation of PKM2-HIF1α pathway, which targets both oxidative stress and proinflammatory gene expression by multiple cell type-mediated responses.
GRANTS
Research reported in this publication was supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Number P30 DK-034989 (to X. Ouyang) and also by Grant R01 1R01AA025342 (to Y. Iwakiri).
DISCLOSURES
X. Ouyang is listed as inventor on a preliminary patent application by Yale University for the use of digoxin in hepatoprotection.
AUTHOR CONTRIBUTIONS
N.J.T., J. Li, Y.I., and X.O. conceived and designed research; P.Z., S.-N.H., M.N.Y., Y.C., M.S.M., and J. Liu performed experiments; P.Z., S.-N.H., S.A., M.N.Y., Y.Q., Y.C., and M.S.M. analyzed data; P.Z., S.-N.H.,M.N.Y., Y.C., M.S.M., and J. Liu interpreted the results of the experiment; P.Z., S.-N.H., M.N.Y., Y.C., M.S.M., and J. Liu prepared figures; P.Z., J. Li., X.O. drafted manuscript; S.A., Y.Q., and X.O. edited and revised manuscript; N.J.T., Y.I., and X.O. approved final version of manuscript.
REFERENCES
- 1.Anastasiou D, Yu Y, Israelsen WJ, Jiang JK, Boxer MB, Hong BS, Tempel W, Dimov S, Shen M, Jha A, Yang H, Mattaini KR, Metallo CM, Fiske BP, Courtney KD, Malstrom S, Khan TM, Kung C, Skoumbourdis AP, Veith H, Southall N, Walsh MJ, Brimacombe KR, Leister W, Lunt SY, Johnson ZR, Yen KE, Kunii K, Davidson SM, Christofk HR, Austin CP, Inglese J, Harris MH, Asara JM, Stephanopoulos G, Salituro FG, Jin S, Dang L, Auld DS, Park HW, Cantley LC, Thomas CJ, Vander Heiden MG. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol 8: 839–847, 2012. [Erratum in Nat Chem Biol 8: 1008, 2012.] doi: 10.1038/nchembio.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cai SY, Ouyang X, Chen Y, Soroka CJ, Wang J, Mennone A, Wang Y, Mehal WZ, Jain D, Boyer JL. Bile acids initiate cholestatic liver injury by triggering a hepatocyte-specific inflammatory response. JCI Insight 2: e90780, 2017. doi: 10.1172/jci.insight.90780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Camporez JP, Asrih M, Zhang D, Kahn M, Samuel VT, Jurczak MJ, Jornayvaz FR. Hepatic insulin resistance and increased hepatic glucose production in mice lacking Fgf21. J Endocrinol 226: 207–217, 2015. doi: 10.1530/JOE-15-0136. [DOI] [PubMed] [Google Scholar]
- 4.Cederbaum AI. Iron and CYP2E1-dependent oxidative stress and toxicity. Alcohol 30: 115–120, 2003. doi: 10.1016/S0741-8329(03)00104-6. [DOI] [PubMed] [Google Scholar]
- 5.Cederbaum AI, Lu Y, Wu D. Role of oxidative stress in alcohol-induced liver injury. Arch Toxicol 83: 519–548, 2009. doi: 10.1007/s00204-009-0432-0. [DOI] [PubMed] [Google Scholar]
- 6.Farrell GC, Haczeyni F, Chitturi S. Pathogenesis of NASH: How metabolic complications of overnutrition favour lipotoxicity and pro-inflammatory fatty liver disease. Adv Exp Med Biol 1061: 19–44, 2018. doi: 10.1007/978-981-10-8684-7_3. [DOI] [PubMed] [Google Scholar]
- 7.Fazel Y, Koenig AB, Sayiner M, Goodman ZD, Younossi ZM. Epidemiology and natural history of non-alcoholic fatty liver disease. Metabolism 65: 1017–1025, 2016. doi: 10.1016/j.metabol.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 8.Feldstein AE, Bailey SM. Emerging role of redox dysregulation in alcoholic and nonalcoholic fatty liver disease. Antioxid Redox Signal 15: 421–424, 2011. doi: 10.1089/ars.2011.3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kakimoto PA, Kowaltowski AJ. Effects of high fat diets on rodent liver bioenergetics and oxidative imbalance. Redox Biol 8: 216–225, 2016. doi: 10.1016/j.redox.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kazankov K, Jorgensen SMD, Thomsen KL, Moller HJ, Vilstrup H, George J, Schuppan D, Gronbaek H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol 16: 145–159, 2019. doi: 10.1038/s41575-018-0082-x. [DOI] [PubMed] [Google Scholar]
- 11.Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol 17: 306–321, 2017. doi: 10.1038/nri.2017.11. [DOI] [PubMed] [Google Scholar]
- 12.Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology 143: 1158–1172, 2012. doi: 10.1053/j.gastro.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 13.Lee HY, Choi CS, Birkenfeld AL, Alves TC, Jornayvaz FR, Jurczak MJ, Zhang D, Woo DK, Shadel GS, Ladiges W, Rabinovitch PS, Santos JH, Petersen KF, Samuel VT, Shulman GI. Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metab 12: 668–674, 2010. doi: 10.1016/j.cmet.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li S, Tan HY, Wang N, Zhang ZJ, Lao L, Wong CW, Feng Y. The role of oxidative stress and antioxidants in liver diseases. Int J Mol Sci 16: 26087–26124, 2015. doi: 10.3390/ijms161125942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, Cole RN, Pandey A, Semenza GL. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 145: 732–744, 2011. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo W, Semenza GL. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol Metab 23: 560–566, 2012. doi: 10.1016/j.tem.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marchesini G, Brizi M, Bianchi G, Tomassetti S, Bugianesi E, Lenzi M, McCullough AJ, Natale S, Forlani G, Melchionda N. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes 50: 1844–1850, 2001. doi: 10.2337/diabetes.50.8.1844. [DOI] [PubMed] [Google Scholar]
- 18.Mehal WZ. The inflammasome in liver injury and non-alcoholic fatty liver disease. Dig Dis 32: 507–515, 2014. doi: 10.1159/000360495. [DOI] [PubMed] [Google Scholar]
- 19.Musso G, Cassader M, Gambino R. Non-alcoholic steatohepatitis: emerging molecular targets and therapeutic strategies. Nat Rev Drug Discov 15: 249–274, 2016. doi: 10.1038/nrd.2015.3. [DOI] [PubMed] [Google Scholar]
- 20.Ouyang X, Ghani A, Malik A, Wilder T, Colegio OR, Flavell RA, Cronstein BN, Mehal WZ. Adenosine is required for sustained inflammasome activation via the A2A receptor and the HIF-1α pathway. Nat Commun 4: 2909, 2013. doi: 10.1038/ncomms3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ouyang X, Han SN, Zhang JY, Dioletis E, Nemeth BT, Pacher P, Feng D, Bataller R, Cabezas J, Stärkel P, Caballeria J, LePine Pongratz R, Cai SY, Schnabl B, Hoque R, Chen Y, Yang WH, Garcia-Martinez I, Wang FS, Gao B, Torok NJ, Kibbey RG, Mehal WZ. Digoxin suppresses pyruvate kinase M2-promoted HIF-1α transactivation in steatohepatitis. Cell Metab 27: 339–350, 2018. [Erratum in Cell Metab 27: 1156, 2018.] doi: 10.1016/j.cmet.2018.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paik YH, Kim J, Aoyama T, De Minicis S, Bataller R, Brenner DA. Role of NADPH oxidases in liver fibrosis. Antioxid Redox Signal 20: 2854–2872, 2014. doi: 10.1089/ars.2013.5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, van den Bosch MW, Quinn SR, Domingo-Fernandez R, Johnston DG, Jiang JK, Israelsen WJ, Keane J, Thomas C, Clish C, Vander Heiden M, Xavier RJ, O’Neill LA. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab 21: 65–80, 2015. doi: 10.1016/j.cmet.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roth JD, Feigh M, Veidal SS, Fensholdt LK, Rigbolt KT, Hansen HH, Chen LC, Petitjean M, Friley W, Vrang N, Jelsing J, Young M. INT-767 improves histopathological features in a diet-induced ob/ob mouse model of biopsy-confirmed non-alcoholic steatohepatitis. World J Gastroenterol 24: 195–210, 2018. doi: 10.3748/wjg.v24.i2.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shao T, Zhao C, Li F, Gu Z, Liu L, Zhang L, Wang Y, He L, Liu Y, Liu Q, Chen Y, Donde H, Wang R, Jala VR, Barve S, Chen SY, Zhang X, Chen Y, McClain CJ, Feng W. Intestinal HIF-1α deletion exacerbates alcoholic liver disease by inducing intestinal dysbiosis and barrier dysfunction. J Hepatol 69: 886–895, 2018. doi: 10.1016/j.jhep.2018.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, Zheng L, Gardet A, Tong Z, Jany SS, Corr SC, Haneklaus M, Caffrey BE, Pierce K, Walmsley S, Beasley FC, Cummins E, Nizet V, Whyte M, Taylor CT, Lin H, Masters SL, Gottlieb E, Kelly VP, Clish C, Auron PE, Xavier RJ, O’Neill LA. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496: 238–242, 2013. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tretter L, Patocs A, Chinopoulos C. Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis. Biochim Biophys Acta 1857: 1086–1101, 2016. doi: 10.1016/j.bbabio.2016.03.012. [DOI] [PubMed] [Google Scholar]
- 28.Xie C, Yagai T, Luo Y, Liang X, Chen T, Wang Q, Sun D, Zhao J, Ramakrishnan SK, Sun L, Jiang C, Xue X, Tian Y, Krausz KW, Patterson AD, Shah YM, Wu Y, Jiang C, Gonzalez FJ. Activation of intestinal hypoxia-inducible factor 2α during obesity contributes to hepatic steatosis. Nat Med 23: 1298–1308, 2017. doi: 10.1038/nm.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR, Rey S, Hammers H, Chang D, Pili R, Dang CV, Liu JO, Semenza GL. Digoxin and other cardiac glycosides inhibit HIF-1α synthesis and block tumor growth. Proc Natl Acad Sci USA 105: 19579–19586, 2008. doi: 10.1073/pnas.0809763105. [DOI] [PMC free article] [PubMed] [Google Scholar]