

Abstract
Hydrochlorothiazide (HCTZ) is the most widely used thiazide diuretic for the treatment of hypertension either alone or in combination with other antihypertensives. HCTZ is mainly cleared by the kidney via tubular secretion, but the underlying molecular mechanisms are unclear. Using cells stably expressing major renal organic anion and cation transporters [human organic anion transporter 1 (hOAT1), human organic anion transporter 3 (hOAT3), human organic cation transporter 2 (hOCT2), human multidrug and toxin extrusion 1 (hMATE1), and human multidrug and toxin extrusion 2-K (hMATE2-K)], we found that HCTZ interacted with both organic cation and anion transporters. Uptake experiments further showed that HCTZ is transported by hOAT1, hOAT3, hOCT2, and hMATE2-K but not by hMATE1. Detailed kinetic analysis coupled with quantification of membrane transporter proteins by targeted proteomics revealed that HCTZ is an excellent substrate for hOAT1 and hOAT3. The apparent affinities (Km) for hOAT1 and hOAT3 were 112 ± 8 and 134 ± 13 μM, respectively, and the calculated turnover numbers (kcat) were 2.48 and 0.79 s−1, respectively. On the other hand, hOCT2 and hMATE2-K showed much lower affinity for HCTZ. The calculated transport efficiency (kcat/Km) at the single transporter level followed the rank order of hOAT1> hOAT3 > hOCT2 and hMATE2-K, suggesting a major role of organic anion transporters in tubular secretion of HCTZ. In vitro inhibition experiments further suggested that HCTZ is not a clinically relevant inhibitor for hOAT1 or hOAT3. However, strong in vivo inhibitors of hOAT1/3 may alter renal secretion of HCTZ. Together, our study elucidated the molecular mechanisms underlying renal handling of HCTZ and revealed potential pathways involved in the disposition and drug-drug interactions for this important antihypertensive drug in the kidney.
Keywords: hydrochlorothiazide, organic anion transporters, organic cation transporters, probenecid, renal tubular secretion, valsartan
INTRODUCTION
Hydrochlorothiazide (HCTZ), a drug on the World Health Organization (WHO) list of essential medicines, is the most commonly prescribed thiazide diuretic for the treatment of hypertension (16). In 2013, HCTZ was the 12th most commonly prescribed drug in the United States, with 50 million prescriptions (36). It belongs to the thiazide class of diuretics, which are the firstline treatment for patients with uncomplicated hypertension (9, 37). Thiazide diuretics lower blood pressure by blocking the electroneutral Na+/Cl− cotransporter located on the apical membrane of the early segment of the renal distal convoluted tubule, resulting in reduced Na+ reabsorption and increased water loss (42). Among the thiazide diuretics, HCTZ remains the cornerstone for treatment of hypertension, either alone or in combination with other classes of antihypertensive drugs, such as angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, β-blockers, and Ca2+ channel blockers (9, 42). While generally safe at low doses, HCTZ at high doses significantly increases the risk of hydroelectrolytic and metabolic adverse effects such as hypokalemia, hyperuricemia, and hyperglycemia (42).
HCTZ is a hydrophilic (log D7.4 of −0.57) small molecule drug (molecular wt: 297). Orally administrated HCTZ is rapidly absorbed in the gastrointestinal track with a bioavailability of ~60–80% (3). The drug is primarily eliminated by the kidney with >95% of the absorbed dose excreted unchanged in the urine (3, 42). The binding of HCTZ to plasma protein is ~58% (3, 42), resulting in a glomerular filtration clearance of ~50 ml/min. The reported renal clearance of HCTZ (~320 ml/min) is more than fivefold higher than its filtration clearance, suggesting a dominant role of tubular secretion in HCTZ renal excretion (2). Secretion is also important for delivering HCTZ to its site of action in the distal convoluted tubule (42). Despite the importance of renal secretion in HCTZ pharmacology, the mechanisms underlying HCTZ tubular secretion have not been clearly defined at the molecular level.
Tubular secretion of drug molecules is mediated by the sequential action of transporters located at the basolateral and apical membranes of proximal tubular cells (30). Two major secretion systems exist for organic anion and organic cation drugs (12, 48). In the human kidney, negatively charged drugs are first transported from blood into renal tubular cells by basolateral human organic anion transporters 1 and 3 (hOAT1 and hOAT3, respectively) (7, 51). Once inside the cells, these organic anion drugs are further effluxed into the lumen by apical membrane transporters such as human multidrug resistance-associated proteins 2 and 4 (hMRP2 and hMRP4, respectively) (23). In contrast, positively charged drugs are first transported into renal tubular cells by electrogenic basolateral human organic cation transporter 2 (hOCT2) and then effluxed into urine by apical proton/organic cation exchangers including human multidrug and toxin extrusion proteins 1 and 2-K (hMATE1 and hMATE2-K, respectively) (31). Although these transporters are generally charge selective, hOAT3 also transports certain positively charged drugs such as cimetidine (18), and hMATEs have been reported to transport some anionic and zwitterion drugs (29, 43, 50). These renal transporters are increasingly recognized as the target for clinically significant drug-drug interactions (DDIs) and are recommended by the United States Food and Drug Administration for assessing DDI potentials during drug development (44a).
Like most thiazide diuretics, HCTZ contains a central backbone of benzothiadiazine and two sulfonamide groups (42). At physiological pH, a significant portion of HCTZ is deprotonated, carrying a negative charge that is stabilized by resonance (46). HCTZ has been previously shown to be an in vitro substrate of hMRP4 (21). In Mrp4 knockout mice, HCTZ renal clearance is significantly reduced (21). These data suggest that apical efflux of HCTZ, the second step in its tubular secretion, is mediated by at least in part by hMRP4 (21). The molecular mechanisms of HCTZ uptake at the basolateral membranes have not been well characterized, although uptake of thiazide diuretics in general is assumed to be mediated by OATs since many of the these drugs exist as organic anions at physiologic pH (42). HCTZ has been previously shown to inhibit rat Oat1 as well as hOAT1 and hOAT3 with IC50 values ranging from ~70 to 1,000 µM (20, 45). In addition, a stop-flow peritubular capillary perfusion study (44) in the rat kidney suggested that HCTZ may also interact with the renal organic cation transport system. However, it is not known if HCTZ is a substrate of hOAT1, hOAT3, hOCT2, or hMATEs and which transporters are more important for human HCTZ renal clearance. In the present study, we investigated the molecular mechanisms involved in renal handling of HCTZ in humans. We first characterized the interaction of HCTZ with major renal organic anion and cation transporters. The detailed transport kinetics of HCTZ were analyzed in conjunction with transporter protein quantification with targeted LC-MS/MS proteomics. Finally, we evaluated the drug interaction potential of HCTZ at the site of hOAT1/3 using in vitro inhibition assays.
MATERIALS AND METHODS
Materials.
[14C]metformin (98 mCi/mmol) was purchased from Moravek Biochemicals (Brea, CA). [3H]estrone sulfate (ES; 50 Ci/mmol) and [3H]p-aminohippurate (PAH; 3 Ci/mmol) were purchased from American Radiolabeled Chemicals (St. Louis, MO). All other compounds were purchased from Sigma-Aldrich (St. Louis, MO) and were of analytic grade. Synthetic signature peptides (Supplemental Table S1; Supplemental Data are available online at https://doi.org/10.6084/m9.figshare.7871783.v1) for protein quantification were obtained from New England Peptides (Boston, MA), and the corresponding stable isotope-labeled (SIL) internal standards were obtained from ThermoFisher Scientific (Rockford, IL). Cell culture media and reagents were purchased from Invitrogen (Carlsbad, CA).
Cell lines and cell culture.
Flp-In human embryonic kidney (HEK)-293 cell lines stably expressing hOAT1, hOAT3, hOCT2, hMATE1, and hMATE2-K and pcDNA5 vector-transfected control HEK-293 cells were as previously established (13, 49). All expressed transporters showed high transport activity compared with control (49). Both transporter-expressing and control Flp-In HEK-293 cells were cultured in DMEM (high glucose) supplemented with 10% FBS, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 150 μg/ml hygromycin B. For better attachment of Flp-In HEK-293 cells, cell culture plastic surfaces were coated with 0.01% poly-d-lysine. Cells were maintained in a 37°C humidified incubator with 5% CO2.
Uptake and inhibition assays in HEK-293 cells.
Assays were performed as previously described (49) with some modifications. Control and transporter-expressing cells were seeded in poly-d-lycine-coated 96-well plates and allowed to grow for 2–3 days to reach 80–90% confluence. Uptake experiments were performed at 37°C in KRH buffer [containing (in mM) 5.6 glucose, 125 NaCl, 4.8 KCl, 1.2 KH2PO4, 1.2 CaCl2, 1.2 MgSO4, and 25 HEPES] with substrates. For uptake experiments in hMATE1- and hMATE2-K-expressing cells, KRH buffer was adjusted to pH 8.0 to generate an outwardly directed proton gradient because hMATEs function as proton/organic cation exchangers (33). For all other transporters, uptake experiments were performed at pH 7.4. After cells had been incubated with substrate for a specified time, uptake was terminated by washing cells three times with ice-cold KRH buffer. Cells were then solubilized with 100 μl of 1 M NaOH and neutralized by an equal volume of 1 M HCl. The radioactivity in the lysates was measured by a liquid scintillation counter (Tri-Carb B3110TR, Perkin-Elmer, Waltham, MA). For LC-MS/MS quantification of HCTZ uptake, cells were lysed with 100 μl of 80% acetonitrile and 20% H2O containing the internal standard (500 nM diclofenac) and then diluted 1:1 with HPLC water. For inhibition experiments, uptake was measured in both the absence and presence of inhibitors. Protein content in the cell lysates was measured by the BCA Protein Assay Kit (Pierce Chemical, Rockford, IL), and uptake in cells was normalized to their protein concentrations. Transporter-specific uptake was calculated by subtracting baseline uptake in control cells.
Quantification of HCTZ by LC-MS/MS.
HCTZ was quantified by LC-MS/MS using an AB Sciex 4000 QTRAP Mass Spectrometer (AB Sciex, Framingham, MA) coupled to an Acquity UPLC system (Waters, Milford, MA) and operated in electrospray ionization negative mode. Ten microliters of sample were injected into a Zorbax Eclipse Plus C18 column (1.8 µm, 2.1 × 50 mm, Agilent Technologies, Santa Clara, CA) running with 0.4 ml/min mobile phase. The mobile phase consisted of 0.1% (vol/vol) formic acid in water (A) and 0.1% (vol/vol) formic acid in acetonitrile (B). For gradient elution, the initial mobile phase conditions were 90% A and 10% B held for 0.1 min followed by a linear gradient from 10% B to 90% B over 0.01 min. The 90% mobile phase B was maintained for 1.39 min and then returned to 10% B within 0.01 min. This was followed by reequilibration for 1.49 min. Mass transitions (m/z) were 295.8 → 268.9 for HCTZ and 293.9 → 249.7 for diclofenac, the internal standard. The quantification limit of HCTZ was 5 ng/ml in cell lysates, and the method was linear up to 250 ng/ml. Instrument control and data processing were performed using Analyst software 1.6 (AB Sciex). Accuracy was within 15% for each batch (20% for the lower limit of quantification).
Membrane protein preparation and LC-MS/MS quantification of transporter protein.
Membrane proteins were prepared from transporter-transfected HEK-293 cells using the ProteoExtract Native Membrane Protein Extraction Kit (Calbiochem/EMD Millipore, San Diego, CA), and concentrations of membrane proteins were determined by the BCA Protein Assay Kit (Pierce Chemical). To quantify membrane transporter protein in each cell line, an LC-MS/MS proteomic approach (28, 35) was used. An Agilent 6460A Triple Quadrupole Mass Spectrometer coupled to the Agilent 1290 Infinity LC system (Agilent Technologies) operated in electrospray ionization-positive ionization mode was used for LC-MS/MS analysis of the peptides (Supplemental Table 1, available online at https://doi.org/10.6084/m9.figshare.7871783.v1). Approximately 1.5 µg or less of the trypsin digest (5 µl) were injected onto the column (Kinetex 2.6 µm, C18, 100 × 3 mm, Phenomenex, Torrance, CA) and eluted at 0.3 ml/min. The mobile phase gradient conditions were 97% A [0.1% (vol/vol) formic acid] and 3% B [acetonitrile containing 0.1% (vol/vol) formic acid] held for 2.5 min followed by three steps of linear gradient of mobile phase B concentrations of 3–8%, 8–21%, and 21–30%. The 30% mobile phase B was maintained for 2 min. This was followed by a washing step using 80% mobile phase B for 1.6 min and reequilibration for 3.3 min. The doubly/triply charged parent to singly charged product transitions for the analyte peptides and their respective SIL peptides were monitored using optimized LC-MS/MS parameters (Supplemental Table 1, available online at https://doi.org/10.6084/m9.figshare.7871783.v1). Data were processed by integrating the peak areas generated from the reconstructed ion chromatograms for the analyte peptides and the respective SIL internal standards using MassHunter software V (Agilent Technologies).
Data analysis.
All uptake and inhibition experiments were performed in triplicate, and experiments were repeated independently at least three times. Data are presented as means ± SD; n ≥ 3. Transport kinetics data were fitted by nonlinear regression using GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA). The Michaelis-Menten equation [V = Vmax × S/(Km + S), where V is the velocity of uptake, Vmax is the maximum velocity of uptake, S is the substrate concentration, and Km is the Michaelis-Menten constant] was fit to the data to obtain Km and Vmax. IC50 values were obtained by fitting the log (inhibitor concentration) versus normalized response equation to the data as follows: , where V is the rate of uptake in the presence of inhibitor, “bottom” is the residual noninhibitable baseline value, “top” is the rate of uptake in the absence of inhibitor, I is the inhibitor concentration, and nH is the Hill coefficient. Statistical analysis was performed using an unpaired Student’s t-test. P values of <0.05 were considered statistically significant.
RESULTS
Inhibitory effect of HCTZ on renal drug transporters.
To determine if HCTZ interacts with renal organic anion transporters hOAT1 and hOAT3 and organic cation transporters hOCT2, hMATE1, and hMATE2-K, the uptake of a probe substrate (PAH for hOAT1, ES for hOAT3, and metformin for hOCT2, hMATE1, and hMATE2-K) in the presence and absence of HCTZ (0.5 and 2 mM) was measured in both control and transporter-expressing cells. The uptake of probe substrate in the presence of HCTZ was expressed as a percentage of the specific uptake in the absence of HCTZ. As shown in Fig. 1, at a concentration of 0.5 mM, HCTZ only inhibited hOAT1 and hOAT3. At a concentration of 2 mM, all tested transporters were inhibited, although hOCT2, hMATE1, and hMATE2-K were inhibited to a lesser degree than hOAT1 and hOAT3.
Fig. 1.
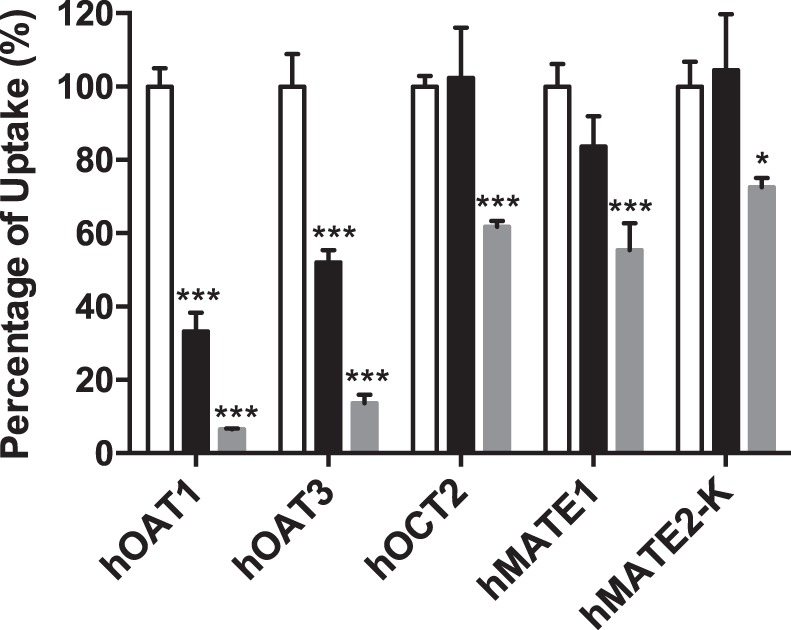
Inhibitory effect of hydrochlorothiazide (HCTZ) on the uptake of probe substrates by renal transporters. Uptake of 5.5 μM metformin by human organic cation transporter 2 (hOCT2) and human multidrug and toxin extrusion proteins 1 and 2-K (hMATE1 and hMATE2-K, respectively), 1 μM [3H]p-aminohippurate (PAH) by human organic anion transporter 1 (hOAT1), and 0.06 μM [3H]estrone sulfate (ES) by human organic anion transporter 3 (hOAT3) in the absence (white bar) and presence of 500 μM HCTZ (black bar) and 2 mM HCTZ (gray bar) was measured in both transporter-expressing and control human embryonic kidney-293 cells. Transporter-specific uptake was obtained by subtracting uptake in control cells from uptake in transporter-expressing cells. Uptake was measured after a 2-min incubation at 37°C. Data are presented as means ± SD. *P < 0.05 and ***P < 0.001, uptake in the presence of HCTZ was significantly lower than that in the absence of HCTZ.
Uptake of HCTZ by renal drug transporters.
To determine if HCTZ is a substrate of the five tested transporters, uptake of HCTZ (10 μM) was measured in both control and transporter-expressing cells. Compared with uptake in control cells, HCTZ uptake was significantly increased in cells expressing hOAT1, hOAT3, hOCT2, and hMATE2-K (Fig. 2). Cells expressing hOAT1 showed the highest uptake activity toward HCTZ. In contrast, no significant uptake of HCTZ was observed in cells expressing hMATE1.
Fig. 2.
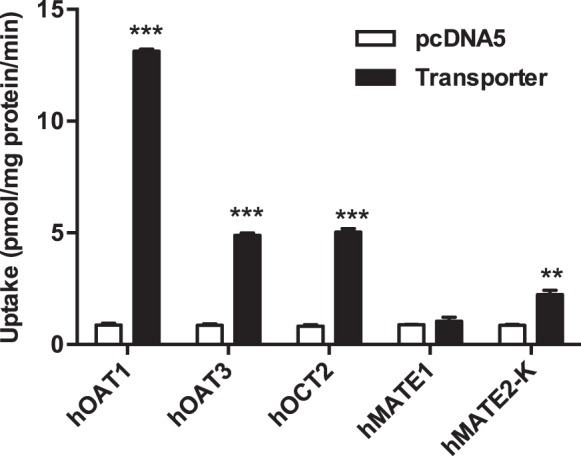
Uptake of hydrochlorothiazide (HCTZ) by renal transporters [human organic cation transporter 2 (hOCT2), human multidrug and toxin extrusion proteins 1 and 2-K (hMATE1 and hMATE2-K), and human organic anion transporters 1 and 3 (hOAT1 and hOAT3). Uptake of 10 μM HCTZ in transporter-expressing (black) and control (white) human embryonic kidney-293 cells was measured. Uptake was measured after a 5-min incubation at 37°C. Data are presented as means ± SD. **P < 0.01 and ***P < 0.001, uptake was significantly higher in transporter-expressing cells than in control cells.
HCTZ uptake kinetics by hOAT1 and hOAT3.
To determine the transport kinetics of HCTZ by renal organic anion transporters hOAT1 and hOAT3, time-dependent uptake of HCTZ (10 µM) was performed in KRH buffer (pH 7.4) at 37°C. Uptake in both hOAT1 and hOAT3 cells was linear up to 5–15 min (Fig. 3, A and B). Concentration-dependent uptake was thus performed over a 0- to 5-min incubation time, and specific uptake was obtained by subtracting uptake in control cells. hOAT1- and hOAT3-specific HCTZ uptake was saturable, and Km values derived from nonlinear regression fitting of the Michaelis-Menten equation to the data were 112 ± 8 and 134 ± 13 μM, respectively (Fig. 3, C and D, and Table 1). Eadie-Hofstee plots further revealed a pattern consistent with single enzyme kinetics for both transporters (Fig. 3, C and D, insets).
Fig. 3.

Hydrochlorothiazide (HCTZ) uptake kinetics by human organic anion transporters 1 and 3 (hOAT1 and hOAT3). Uptake of 10 μM HCTZ was measured in control and transporter-expressing human embryonic kidney (HEK)-293 cells at the specified time points (A and B). Concentration-dependent uptake of HCTZ was measured in both transporter-expressing and control HEK-293 cells after a 5-min incubation at 37°C (C and D). Transporter-specific uptake was obtained by subtracting uptake in control cells from uptake in transporter-expressing cells. The Michaelis-Menten equation was fit to the data using nonlinear regression. Results from a representative experiment are shown. Insets: Eadie-Hofstee transformation of the kinetics data. Each data point represents the means ± SD of replicate measures.
Table 1.
Kinetic parameters of hydrochlorothiazide uptake by hOAT1, hOAT3, hOCT2, and hMATE2-K
| Kinetic Parameter | Km, μM | Vmax, pmol·min−1·mg protein−1 |
|---|---|---|
| hOAT1 | 112 ± 8 | 134 ± 3 |
| hOAT3 | 134 ± 13 | 128 ± 4 |
| hOCT2 | 812 ± 85 | 420 ± 18 |
| hMATE2-K | 681 ± 101 | 78 ± 5 |
Results represent average parameter estimates (means ± SD) of 3 independent experiments. Km, Michaelis-Menten constant; Vmax, maximum velocity; hOAT1 and hOAT3, huamn organic anion transporter 1 and 3, respectively; hOCT2, human organic cation transporter 2; hMATE2-K, human multidrug and toxin extrusion protein 2-K.
HCTZ uptake kinetics by hOCT2 and hMATE2-K.
The kinetics of HCTZ uptake by renal organic cation transporters hOCT2 and hMATE2-K were determined in a similar manner except that the uptake assay for hMATE2-K was performed in KRH buffer at pH 8.0 to provide an outwardly directed proton gradient (32, 33, 49). For hOCT2, uptake of HCTZ increased sharply in the first 2 min and then rapidly reached a plateau (Fig. 4A). Uptake in hMATE2-K cell increased progressively until 10 min and then reached a plateau (Fig. 4B). Concentration-dependent uptake was thus performed with 2-min incubation for both transporters. hOCT2- and hMATE2-K-mediated HCTZ uptake was saturable (Fig. 4, C and D), with the Km and Vmax values shown in Table 1. Compared with hOAT1 and hOAT3, hOCT2 and hMATE2-K showed a five- to sixfold lower affinity toward HCTZ. Time-dependent uptake of HCTZ was also performed in hMATE1-expressing cells. Consistent with the single time point experiment (Fig. 2), no significant uptake was observed with hMATE1 (Supplemental Fig. S1, available online at https://doi.org/10.6084/m9.figshare.7871783.v1).
Fig. 4.

Hydrochlorothiazide (HCTZ) uptake kinetics by human organic cation transporter 2 (hOCT2) and human multidrug and toxin extrusion protein 2-K (hMATE2-K). Uptake of 10 μM HCTZ was measured in control and transporter-expressing human embryonic kidney (HEK)-293 cells at the specified time points (A and B). Concentration-dependent uptake of HCTZ was measured in both transporter-expressing and control HEK-293 cells after a 2-min incubation at 37°C (C and D). Transporter-specific uptake was obtained by subtracting uptake in control cells from uptake in transporter-expressing cells. The Michaelis-Menten equation was fit to the data using nonlinear regression. Results from a representative experiment are shown. Insets: Eadie-Hofstee transformation of the kinetics data. Each data point represents the means ± SD of replicate measures.
Transporter quantification and turnover number determination.
To determine HCTZ transport efficiency at the single transporter level, we quantified transporter-specific protein content in membrane fractions prepared from transfected HEK-293 cell lines using targeted LC-MS/MS proteomics. As shown in Table 2, hOAT1 and hOAT3 had relatively lower expression levels, whereas hOCT2 and hMATE2-K had higher expression, in our Flp-In HEK-293 cell lines. The transporter turnover number (kcat), which represents the maximum number of substrate that a carrier can transport per second, was calculated by normalizing Vmax to moles of transporter present. As shown in Table 2, hOAT1 exhibited a kcat value three to seven times higher than hOAT3, hOCT2, and hMATE2-K. When the single transporter efficiency (kcat/Km) was compared, it followed the rank order of hOAT1 > hOAT3 > hOCT2 and hMATE2-K. These data suggest that, at the single transporter level, hOAT1 is the most efficient transporter for HCTZ, while hOCT2 and hMATE2-K are the least efficient. hOAT3 has an intermediate efficiency for HCTZ.
Table 2.
Turnover number and transport efficiency of hydrochlorothiazide uptake by hOAT1, hOAT3, hOCT2, and hMATE2-K
| Amount of Transporter, fmol/μg membrane | kcat, s−1 | kcat/Km, s−1·μM−1 | |
|---|---|---|---|
| hOAT1 | 1.3 ± 0.2 | 2.48 | 22.1 × 10−3 |
| hOAT3 | 3.2 ± 0.3 | 0.79 | 5.9 × 10−3 |
| hOCT2 | 41.1 ± 3.6* | 0.63 | 0.8 × 10−3 |
| hMATE2-K | 9.7 ± 1.3* | 0.34 | 0.5 × 10−3 |
Results represent average parameter estimates (means ± SD) of 3 independent experiments. Km, Michaelis-Menten constant; kcat, transporter turnover number; hOAT1 and hOAT3, huamn organic anion transporter 1 and 3, respectively; hOCT2, human organic cation transporter 2; hMATE2-K, human multidrug and toxin extrusion protein 2-K.
Amount of transporter protein for hOCT2 and hMATE2-K was determined in Yin et al. (49).
Inhibitory potential of HCTZ toward hOAT1 and hOAT3.
The relatively higher affinity of HCTZ toward hOAT1 and hOAT3 (Table 1) raised the possibility of inhibitory drug interactions with these transporters. To evaluate the potential for HCTZ to inhibit hOAT1 and hOAT3 in vivo, IC50 values for HCTZ inhibition of hOAT1- and hOAT3-mediated probe substrate transport were determined. The substrates used were PAH (1 μM) for hOAT1 and ES (0.06 μM) for hOAT3. As shown in Fig. 5, HCTZ exhibited similar potency toward hOAT1 and hOAT3, with IC50 values of 126 ± 13.7 and 213 ± 21.5 μM, respectively. These IC50 values were similar to the Km values of HCTZ shown in Table 1 but much higher than its circulating plasma concentrations (0.7 ~0.9 μM) after a typical 25-mg therapeutic dose (25, 46), suggesting that HCTZ is unlikely to significantly inhibit hOAT1 and hOAT3 activities in vivo.
Fig. 5.

Inhibition of human organic anion transporters 1 and 3 [hOAT1 (A) and hOAT3 (B)] by hydrochlorothiazide (HCTZ). Uptake of 1 μM [3H]p-aminohippurate (PAH) by hOAT1 and 0.06 μM [3H]estrone sulfate (ES) by hOAT3 in the absence and presence of HCTZ was measured in both transporter-expressing and control human embryonic kidney-293 cells. Transporter-specific uptake was obtained by subtracting uptake in control cells from uptake in transporter-expressing cells. The uptake was measured after a 2-min incubation at 37°C. Data are presented as means ± SD.
Inhibition of hOAT1- and hOAT3- mediated HCTZ transport by probenecid and valsartan.
Our data suggest that hOAT1 and hOAT3 play a major role in the renal secretion of HCTZ. Thus, inhibition of hOAT1 and hOAT3 by other drugs may alter the renal handling of HCTZ. We determined the inhibitory potency of two known hOAT inhibitors, probenecid and valsartan, toward hOAT1- and hOAT3-mediated HCTZ uptake in transfected HEK-293 cell lines. As shown in Fig. 6, probenecid and valsartan potently inhibited hOAT1- and hOAT3-mediated HCTZ transport, with IC50 values between 2.3 and 9.0 μM.
Fig. 6.

Inhibition of human organic anion transporters 1 and 3 (hOAT1 and hOAT3)-mediated hydrochlorothiazide (HCTZ) uptake by probenecid and valsartan. Uptake of 10 μM HCTZ by hOAT1 and hOAT3 in the presence of increasing concentration of probenecid (A and B) and valsartan (C and D) was measured in both transporter-expressing and control human embryonic kidney-293 cells. Transporter-specific uptake was obtained by subtracting uptake in control cells from uptake in transporter-expressing cells. Uptake was measured after a 5-min incubation at 37°C. Data are presented as means ± SD.
DISCUSSION
HCTZ is eliminated primarily by tubular secretion, but the molecular mechanisms underlying its renal handling have not been well characterized. In the present study, we systematically assessed the interaction of HCTZ with major renal organic anion and cation transporters and evaluated the drug interaction potential at the site of these transporters. Several novel findings were obtained from our study. First, our data demonstrated that HCTZ is not only a substrate of renal organic anion transporters hOAT1 and hOAT3 but is also transported by organic cation transporters hOCT2 and hMATE2-K. Second, by quantifying transporter protein levels in cultured cells, we determined the detailed intrinsic transport kinetics of HCTZ at the single transporter level. Our data revealed that compared with hOAT1/3, hOCT2 and hMATE2-K have lower affinity and lower transport efficiency for HCTZ. Finally, we showed that HCTZ is unlikely to be a clinically relevant inhibitor for renal hOATs or hOCTs. However, hOAT-mediated HCTZ secretion is likely to be affected by potent inhibitors of hOAT such as probenecid.
At high concentrations (e.g., 2 mM), HCTZ inhibited all tested renal transporters (Fig. 1). When tested as a substrate, we found that HCTZ is a substrate for hOAT1, hOAT3, hOCT2, and hMATE2-K but not hMATE1 (Fig. 2). While OATs and OCTs generally show distinct charge preference for anionic and cationic compounds, several studies have suggested that there is some overlap in substrate specificity (1, 41). For example, the antiviral drugs acyclovir and ganciclovir have been shown to be transported by both hOAT1 and hOCT1 (41). Cimetidine, a substrate and a classic inhibitor of OCTs, is a known substrate of hOAT3 (40). OATs and OCTs are both encoded by the SLC22 gene family and share similar 12 transmembrane domain structures (7, 27). The substrate overlap among these transporters may reflect some similarity of their substrate recognition sites likely due to a common ancestry. MATEs, which are encoded by SLC47, generally accept organic cations as substrates, but they are also capable of transporting certain zwitterion and anionic drugs (50). For example, the zwitterion drug cephalexin is a reported substrate of hOAT1/3 and hMATE1 (43, 47, 52). Here, we showed that HCTZ is a substrate for hOAT1, hOAT3, hOCT2, and hMATE2-K. Like other thiazide diuretics, HCTZ has a central backbone of benzothiadiazine and two sulfonamide groups (42). The amine center in the sulfonamide structure can lose one proton, resulting in a negatively charged conjugate that can be further stabilized by resonance (46). With a pKa of 7.9, ~30% of HCTZ exists in the negatively charged form, whereas the reminder is in the neutral form. Based on their charge preference and transport modes, hOAT1 and OAT3 likely transport the anionic form of HCTZ, whereas hOCT2 and hMATE2-K may prefer the uncharged form as substrate. More studies are needed to determine the specific HCTZ charge state that interacts with renal organic anion and cation transporters.
Our substrate data suggest that the first step of HCTZ renal secretion could be mediated by multiple uptake transporters (hOAT1, hOAT3, and hOCT2) at the basolateral membrane. In theory, the relative contribution of these transporters to HCTZ renal uptake can be assessed by the transport efficiency (kcat/Km) of each transporter and its total membrane expression in renal tubular epithelial cells. By performing detailed kinetic analysis coupled with absolute quantification of transporter protein levels, kcat/Km at the single transporter level was determined to follow the rank order of hOAT1> hOAT3 > hOCT2 (Table 2), suggesting that hOAT1 is the most efficient uptake transporter for HCTZ. mRNA transcripts of hOAT1, hOAT3, and hOCT2 are highly expressed in the human kidney (7, 27). Recently, the abundance of organic anion and cation transporters in the human kidney was quantified by targeted proteomics in cortex tissues isolated from 41 human kidneys (34). Mean protein expression for hOCT2, hOAT1, and hOAT3 was reported to be 7.4 ± 2.8, 5.3 ± 1.9, and 3.5 ± 1.6 pmol/mg total membrane protein, respectively. With the use of the assumption that all proteins are present at the basolateral membrane and combined with single transporter transport efficacy (Table 2), these data suggest that hOAT1 and hOAT3 are likely the major transporter responsible for HCTZ uptake into kidney tubular cells under normal physiological conditions. As HCTZ acts on Na+/Cl− cotransporter located on the luminal side of the renal distal tubule, these transporters may also be important for delivering the diuretic to its site of action. Interestingly and consistent with our kinetic data, a pharmacogenetics study in patients with hypertension treated with HCTZ suggested that an intergenic polymorphism (rs10792367) between the OAT1 and OAT3 genes was associated with antihypertensive responses to HCTZ (19).
In the kidney, basolateral hOAT1/3 and apical hMRP2/4 often work in tandem to secrete organic anions from blood into urine (51). Previously, hMRP4 has been identified as an efflux transporter for HCTZ (21). Renal clearance of HCTZ is significantly reduced in Mrp4 knockout mice, suggesting an important role of this transporter in HCTZ secretion (21). Our data showing that HCTZ is also transported by the apical organic efflux transporter hMATE2-K suggest that, in addition to MRP4, apical efflux of HCTZ may also be assisted by hMATE2-K (21). Based on these data, we proposed a molecular model for HCTZ secretion in renal proximal tubule cells consisting of at least two parallel pathways (Fig. 7). In this model, hOAT1/3 and hMRP4 form a major route while hOCT2 and hMATE2-K serve as a secondary pathway for HCTZ tubular secretion.
Fig. 7.

Proposed scheme of hydrochlorothiazide (HCTZ) transport in human renal proximal tubule cells. In this model, human organic anion transporters 1 and 3 (hOAT1 and hOAT3) and human multidrug resistance-associated protein 4 (hMRP4) form a major route (solid arrows), whereas human organic cation transporter 2 (hOCT2) and human multidrug and toxin extrusion protein 2-K (hMATE2-K) represent as a secondary pathway (dashed arrows) for HCTZ tubular secretion. DC, dicarboxylate.
It should be noted that there are several limitations of our experimental system in assessing transport kinetics for the human renal transporters. By definition, the turnover number (kcat) can be calculated by normalizing Vmax to the functional number of transporters present on the plasma membrane. Although the cell-based heterologous expression system is routinely used to determine transport kinetics, overexpression of a transporter protein may result in a significant fraction of the total protein residing in intracellular compartments. Since the ProteoExtract protocol is designed to maximize the yield of native membrane proteins rather than that restricted to the plasma membrane, our measured protein levels of hOAT1/3, hOCT2, and hMATE2-K may markedly overestimate functional transporter numbers on the cell surface. Consequently, calculated kcat values in our study may significantly underestimate true kcat values for these transporters. Furthermore, with respect to hMATE2-K, it has been shown that the transport kinetics for the MATE-mediated process is profoundly influenced by the availability of H+ in both cytosolic and extracellular compartments (11). In our study, the kinetic parameters for hMATE2-K were determined using uptake measurement at extracellular pH 8.0. While this approach has been typically used to characterize MATE-mediated transport kinetics, the use of a lower H+ concentration at pH 8.0 could significantly underestimate Km and Vmax at pH 7.4. Moreover, the intracellular pH of HEK-293 cells is ~7.3, whereas the luminal pH in renal proximal tubules under normal physiological conditions is ~6.8 (6). Consequently, Vmax and kcat for hMATE2-K-mediated HCTZ transport obtained from our experimental approach is likely to significantly underestimate the true values under physiological conditions. The combined impact likely results in a substantial underestimate of the efficiency of hMATE2-K-mediated luminal efflux of HCTZ in renal proximal tubule cells under normal physiological conditions.
HCTZ is frequently used in combination or coformulated as fixed dose combination pills (known as “polypills”) with other antihypertensive drugs to produce an additive effect in lowering blood pressure to treat patients with inadequately controlled hypertension by monotherapy (39). Some of the coadministered drugs may be inhibitors or substrates of renal organic anion or cation transporters, raising a concern of potential DDIs. For example, HCTZ is frequently used together with loop diuretics (e.g., furosemide) and β-blockers (e.g., atenolol) (39, 42). Furosemide and atenolol are also mainly eliminated by the kidney with involvement of renal organic anion or cation transporters (15, 49). The combination of HCTZ with valsartan, an angiotensin II receptor antagonist, has also been developed as fixed-dose tablets under the trade name Diovan HCT (10). Valsartan has been shown in vitro as a potent inhibitor of hOAT1 and hOAT3 (14, 24). Our data showed that both valsartan and probenecid potently inhibited hOAT1- and hOAT3-mediated HCTZ uptake in vitro with IC50 values in the low micromolar range (Fig. 6 and Table 3). However, valsartan is highly protein bound and has low plasma concentrations at clinically used doses (Table 3). With the assumption that only the unbound drug interacts with basolateral transporters, at a maximal plasma concentration after a typical dose of 320 mg (26), valsartan is predicted to only produce moderate inhibition of hOAT1 and hOAT3 (Table 3). Several clinical studies have examined the interaction between HCTZ and valsartan in healthy individuals, and two studies have reported no significant effect of valsartan on HCTZ pharmacokinetics (5, 22).
Table 3.
Prediction of percent inhibition of hOAT1- and hOAT3-mediated hydrochlorothiazide transport in vivo by probenecid and valsartan
| Inhibitor/Transporter | Imax, μM | fu, % | Imax,u, μM | IC50, μM | Percent Inhibition* |
|---|---|---|---|---|---|
| Probenecid† | 240 | 11 | 26.4 | ||
| hOAT1 | 7.1 ± 0.3 | 79 | |||
| hOAT3 | 5.8 ± 0.3 | 82 | |||
| Valsartan‡ | 24.6 | 5 | 1.2 | ||
| hOAT1 | 9.0 ± 0.4 | 12 | |||
| hOAT3 | 2.3 ± 0.2 | 34 | |||
Results for IC50 represent average parameter estimates (means ± SD) of 3 independent experiments. hOAT1 and hOAT3, human organic anion transporter 1 and 3; Imax, maximum plasma concentration; fu, unbound plasma concentration; Imax,u, maximum unbound plasma concentration (calculated by Imax × fu).
Percent inhibition was calculated as follows: [1 – 1/(1 + Imax,u/IC50)] × 100%.
Imax and fu of probenecid were obtained from Selen and Amidon (38); Imax was after a 1-g dose.
Compared with valsartan, probenecid is also highly protein bound but has much higher plasma concentrations at the clinically used dose. At a typical probenecid dose of 1 g (38), HCTZ uptake by hOAT1 and hOAT3 is predicted to be markedly inhibited by probenecid (Table 3). Probenecid is a well-established inhibitor of hOAT1/3 recommended by the United States Food and Drug Administration for the evaluation of DDI risk of hOAT1/3 substrates in vivo (8). To date, there is no report of a probenecid and HCTZ interaction in humans. However, in dogs, it has been shown that coadministration of probenecid reduced HCTZ renal clearance by >80% (4). At higher doses, HCTZ significantly increases the risk of hydroelectrolytic and metabolic adverse effects (42). Considering the importance of renal tubular secretion in HCTZ elimination, we should take caution when HCTZ is coadministered with strong in vivo hOAT1/3 inhibitors such as probenecid.
Finally, our inhibition experiments showed that HCTZ generally has low affinity toward hOAT1/3, hOCT2, and hMATEs (Figs. 1 and 5 and Table 1). With IC50 or Km values (>100–200 μM) much larger than the clinically encountered plasma concentrations (<1–10 μM), HCTZ is unlikely to significantly inhibit hOAT1/3, hOCT2, or hMATEs at clinically relevant doses. Therefore, concurrent use of HCTZ with a substrate drug of hOAT1/3 (e.g., furosemide) or hOCT2 (e.g., atenolol) may not affect the renal handling of these drugs.
In summary, we showed that HCTZ is an excellent substrate for renal organic anion transporters hOAT1 and hOAT3. It is also transported by renal organic cation transporters hOCT2 and hMATE2-K. Renal tubular secretion of HCTZ likely occurs mainly through the hOAT1/3 and hMRP4 pathway with hOCT2/hMATE2-K being a secondary pathway. Our data also suggest that the pharmacokinetics of HCTZ may be significantly affected by potent hOAT1/3 inhibitors. Together, our results elucidated the molecular mechanisms involved in renal handling of HCTZ and shed new light on the mechanisms involved in renal disposition and DDIs for HCTZ.
GRANTS
This work was supported in part by National Center for Advancing Translational Sciences Grants 1-UH2-TR-000504 and TL1-TR-000422 and the Drug Metabolism Transport and Pharmacogenetics Research Fund at the School of Pharmacy, University of Washington.
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
D. J. Wagner is an employee of AstraZeneca R&D. No conflicts of interest, financial or otherwise, are declared by the other authors.
AUTHOR CONTRIBUTIONS
J.Y., D.J.W., N.I., K.E.T., and J.W. conceived and designed research; J.Y., D.J.W., and B.P. performed experiments; J.Y., D.J.W., and B.P. analyzed data; J.Y., D.J.W., B.P., and J.W. interpreted results of experiments; J.Y. and D.J.W. prepared figures; J.Y., D.J.W., B.P., and J.W. drafted manuscript; J.Y., D.J.W., B.P., N.I., K.E.T., and J.W. edited and revised manuscript; J.Y., D.J.W., B.P., N.I., K.E.T., and J.W. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Kazuo Matsubara (Department of Clinical Pharmacology and Therapeutics, Kyoto University Hospital) for providing the hMATE2-K cDNA plasmid for this study.
Present address of D. J. Wagner: DMPK, 02451 Oncology R&D, Research and Early Development, AstraZeneca, Boston, MA 02451.
REFERENCES
- 1.Ahn SY, Eraly SA, Tsigelny I, Nigam SK. Interaction of organic cations with organic anion transporters. J Biol Chem 284: 31422–31430, 2009. doi: 10.1074/jbc.M109.024489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beermann B, Groschinsky-Grind M. Pharmacokinetics of hydrochlorothiazide in man. Eur J Clin Pharmacol 12: 297–303, 1977. doi: 10.1007/BF00607430. [DOI] [PubMed] [Google Scholar]
- 3.Beermann B, Groschinsky-Grind M, Rosén A. Absorption, metabolism, and excretion of hydrochlorothiazide. Clin Pharmacol Ther 19: 531–537, 1976. doi: 10.1002/cpt1976195part1531. [DOI] [PubMed] [Google Scholar]
- 4.Beyer KH Jr, Baer JE. Enzymes, drugs and transport phenomena. Protoplasma 63: 112–120, 1967. doi: 10.1007/BF01248014. [DOI] [PubMed] [Google Scholar]
- 5.Bhad P, Ayalasomayajula S, Karan R, Leon S, Riviere GJ, Sunkara G, Jarugula V. Evaluation of pharmacokinetic interactions between amlodipine, valsartan, and hydrochlorothiazide in patients with hypertension. J Clin Pharmacol 51: 933–942, 2011. doi: 10.1177/0091270010376963. [DOI] [PubMed] [Google Scholar]
- 6.Brasen JC, Burford JL, McDonough AA, Holstein-Rathlou NH, Peti-Peterdi J. Local pH domains regulate NHE3-mediated Na+ reabsorption in the renal proximal tubule. Am J Physiol Renal Physiol 307: F1249–F1262, 2014. doi: 10.1152/ajprenal.00174.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burckhardt G. Drug transport by organic anion transporters (OATs). Pharmacol Ther 136: 106–130, 2012. doi: 10.1016/j.pharmthera.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 8.Center for Drug Evaluation and Research Clinical Drug Interaction Studies−Study Design, Data Analysis, and Clinical Implications. Silver Spring, MD: U.S. Food and Drug Administration, 2017. [Google Scholar]
- 9.Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL Jr, Jones DW, Materson BJ, Oparil S, Wright JT Jr, Roccella EJ; Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. National Heart, Lung, and Blood Institute; National High Blood Pressure Education Program Coordinating Committee . Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 42: 1206–1252, 2003. doi: 10.1161/01.HYP.0000107251.49515.c2. [DOI] [PubMed] [Google Scholar]
- 10.Chrysant SG. Fixed combination therapy of hypertension: focus on valsartan/hydrochlorothiazide combination (Diovan/HCT). Expert Rev Cardiovasc Ther 1: 335–343, 2003. doi: 10.1586/14779072.1.3.335. [DOI] [PubMed] [Google Scholar]
- 11.Dangprapai Y, Wright SH. Interaction of H+ with the extracellular and intracellular aspects of hMATE1. Am J Physiol Renal Physiol 301: F520–F528, 2011. doi: 10.1152/ajprenal.00075.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dresser MJ, Leabman MK, Giacomini KM. Transporters involved in the elimination of drugs in the kidney: organic anion transporters and organic cation transporters. J Pharm Sci 90: 397–421, 2001. doi: 10.1002/1520-6017(200104)90:4<397:AID-JPS1000>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 13.Duan H, Hu T, Foti RS, Pan Y, Swaan PW, Wang J. Potent and selective inhibition of plasma membrane monoamine transporter by HIV protease inhibitors. Drug Metab Dispos 43: 1773–1780, 2015. doi: 10.1124/dmd.115.064824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duan P, Li S, Ai N, Hu L, Welsh WJ, You G. Potent inhibitors of human organic anion transporters 1 and 3 from clinical drug libraries: discovery and molecular characterization. Mol Pharm 9: 3340–3346, 2012. doi: 10.1021/mp300365t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ebner T, Ishiguro N, Taub ME. The use of transporter probe drug cocktails for the assessment of transporter-based drug-drug interactions in a clinical setting-proposal of a four component transporter cocktail. J Pharm Sci 104: 3220–3228, 2015. doi: 10.1002/jps.24489. [DOI] [PubMed] [Google Scholar]
- 16.Feldman RD, Hussain Y, Kuyper LM, McAlister FA, Padwal RS, Tobe SW. Intraclass differences among antihypertensive drugs. Annu Rev Pharmacol Toxicol 55: 333–352, 2015. doi: 10.1146/annurev-pharmtox-010814-124446. [DOI] [PubMed] [Google Scholar]
- 17.Flesch G, Muller P, Lloyd P. Absolute bioavailability and pharmacokinetics of valsartan, an angiotensin II receptor antagonist, in man. Eur J Clin Pharmacol 52: 115–120, 1997. [DOI] [PubMed] [Google Scholar]
- 18.Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, Hoffmaster KA, Ishikawa T, Keppler D, Kim RB, Lee CA, Niemi M, Polli JW, Sugiyama Y, Swaan PW, Ware JA, Wright SH, Yee SW, Zamek-Gliszczynski MJ, Zhang L; International Transporter Consortium . Membrane transporters in drug development. Nat Rev Drug Discov 9: 215–236, 2010. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han YF, Fan XH, Wang XJ, Sun K, Xue H, Li WJ, Wang YB, Chen JZ, Zhen YS, Zhang WL, Zhou X, Hui R. Association of intergenic polymorphism of organic anion transporter 1 and 3 genes with hypertension and blood pressure response to hydrochlorothiazide. Am J Hypertens 24: 340–346, 2011. doi: 10.1038/ajh.2010.191. [DOI] [PubMed] [Google Scholar]
- 20.Hasannejad H, Takeda M, Taki K, Shin HJ, Babu E, Jutabha P, Khamdang S, Aleboyeh M, Onozato ML, Tojo A, Enomoto A, Anzai N, Narikawa S, Huang XL, Niwa T, Endou H. Interactions of human organic anion transporters with diuretics. J Pharmacol Exp Ther 308: 1021–1029, 2004. doi: 10.1124/jpet.103.059139. [DOI] [PubMed] [Google Scholar]
- 21.Hasegawa M, Kusuhara H, Adachi M, Schuetz JD, Takeuchi K, Sugiyama Y. Multidrug resistance-associated protein 4 is involved in the urinary excretion of hydrochlorothiazide and furosemide. J Am Soc Nephrol 18: 37–45, 2007. doi: 10.1681/ASN.2005090966. [DOI] [PubMed] [Google Scholar]
- 22.Hedaya MA, Helmy SA. Pharmacokinetic interactions of valsartan and hydrochlorothiazide: an open-label, randomized, 4-period crossover study in healthy Egyptian male volunteers. Clin Ther 35: 846–861, 2013. doi: 10.1016/j.clinthera.2013.04.014. [DOI] [PubMed] [Google Scholar]
- 23.Hillgren KM, Keppler D, Zur AA, Giacomini KM, Stieger B, Cass CE, Zhang L; International Transporter Consortium . Emerging transporters of clinical importance: an update from the International Transporter Consortium. Clin Pharmacol Ther 94: 52–63, 2013. doi: 10.1038/clpt.2013.74. [DOI] [PubMed] [Google Scholar]
- 24.Ingraham L, Li M, Renfro JL, Parker S, Vapurcuyan A, Hanna I, Pelis RM. A plasma concentration of α-ketoglutarate influences the kinetic interaction of ligands with organic anion transporter 1. Mol Pharmacol 86: 86–95, 2014. doi: 10.1124/mol.114.091777. [DOI] [PubMed] [Google Scholar]
- 25.Jeon H, Lim KS, Shin KH, Kim J, Yoon SH, Cho JY, Shin SG, Jang IJ, Yu KS. Assessment of the drug-drug interactions between fimasartan and hydrochlorothiazide in healthy volunteers. J Cardiovasc Pharmacol 59: 84–91, 2012. doi: 10.1097/FJC.0b013e318237389e. [DOI] [PubMed] [Google Scholar]
- 26.Jung JA, Noh YH, Jin S, Kim MJ, Kim YH, Jung JA, Lim HS, Bae KS. Pharmacokinetic interaction between pitavastatin and valsartan: a randomized, open-labeled crossover study in healthy male Korean volunteers. Clin Ther 34: 958–965, 2012. doi: 10.1016/j.clinthera.2012.01.026. [DOI] [PubMed] [Google Scholar]
- 27.Koepsell H, Lips K, Volk C. Polyspecific organic cation transporters: structure, function, physiological roles, and biopharmaceutical implications. Pharm Res 24: 1227–1251, 2007. doi: 10.1007/s11095-007-9254-z. [DOI] [PubMed] [Google Scholar]
- 28.Lee N, Hebert MF, Prasad B, Easterling TR, Kelly EJ, Unadkat JD, Wang J. Effect of gestational age on mRNA and protein expression of polyspecific organic cation transporters during pregnancy. Drug Metab Dispos 41: 2225–2232, 2013. doi: 10.1124/dmd.113.054072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsushima S, Maeda K, Inoue K, Ohta KY, Yuasa H, Kondo T, Nakayama H, Horita S, Kusuhara H, Sugiyama Y. The inhibition of human multidrug and toxin extrusion 1 is involved in the drug-drug interaction caused by cimetidine. Drug Metab Dispos 37: 555–559, 2009. doi: 10.1124/dmd.108.023911. [DOI] [PubMed] [Google Scholar]
- 30.Morrissey KM, Stocker SL, Wittwer MB, Xu L, Giacomini KM. Renal transporters in drug development. Annu Rev Pharmacol Toxicol 53: 503–529, 2013. doi: 10.1146/annurev-pharmtox-011112-140317. [DOI] [PubMed] [Google Scholar]
- 31.Motohashi H, Inui K. Organic cation transporter OCTs (SLC22) and MATEs (SLC47) in the human kidney. AAPS J 15: 581–588, 2013. doi: 10.1208/s12248-013-9465-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Müller F, König J, Hoier E, Mandery K, Fromm MF. Role of organic cation transporter OCT2 and multidrug and toxin extrusion proteins MATE1 and MATE2-K for transport and drug interactions of the antiviral lamivudine. Biochem Pharmacol 86: 808–815, 2013. doi: 10.1016/j.bcp.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 33.Otsuka M, Matsumoto T, Morimoto R, Arioka S, Omote H, Moriyama Y. A human transporter protein that mediates the final excretion step for toxic organic cations. Proc Natl Acad Sci USA 102: 17923–17928, 2005. doi: 10.1073/pnas.0506483102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prasad B, Johnson K, Billington S, Lee C, Chung GW, Brown CDA, Kelly EJ, Himmelfarb J, Unadkat JD. Abundance of drug transporters in the human kidney cortex as quantified by quantitative targeted proteomics. Drug Metab Dispos 44: 1920–1924, 2016. doi: 10.1124/dmd.116.072066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prasad B, Unadkat JD. Optimized approaches for quantification of drug transporters in tissues and cells by MRM proteomics. AAPS J 16: 634–648, 2014. doi: 10.1208/s12248-014-9602-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roush GC, Sica DA. Diuretics for Hypertension: A Review and Update. Am J Hypertens 29: 1130–1137, 2016. doi: 10.1093/ajh/hpw030. [DOI] [PubMed] [Google Scholar]
- 37.Salvetti A, Ghiadoni L. Thiazide diuretics in the treatment of hypertension: an update. J Am Soc Nephrol 17, Suppl 2: S25–S29, 2006. doi: 10.1681/ASN.2005121329. [DOI] [PubMed] [Google Scholar]
- 38.Selen A, Amidon GL, Welling PG. Pharmacokinetics of probenecid following oral doses to human volunteers. J Pharm Sci 71: 1238–1242, 1982. doi: 10.1002/jps.2600711114. [DOI] [PubMed] [Google Scholar]
- 39.Sica DA, Carter B, Cushman W, Hamm L. Thiazide and loop diuretics. J Clin Hypertens (Greenwich) 13: 639–643, 2011. doi: 10.1111/j.1751-7176.2011.00512.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tahara H, Kusuhara H, Endou H, Koepsell H, Imaoka T, Fuse E, Sugiyama Y. A species difference in the transport activities of H2 receptor antagonists by rat and human renal organic anion and cation transporters. J Pharmacol Exp Ther 315: 337–345, 2005. doi: 10.1124/jpet.105.088104. [DOI] [PubMed] [Google Scholar]
- 41.Takeda M, Khamdang S, Narikawa S, Kimura H, Kobayashi Y, Yamamoto T, Cha SH, Sekine T, Endou H. Human organic anion transporters and human organic cation transporters mediate renal antiviral transport. J Pharmacol Exp Ther 300: 918–924, 2002. doi: 10.1124/jpet.300.3.918. [DOI] [PubMed] [Google Scholar]
- 42.Tamargo J, Segura J, Ruilope LM. Diuretics in the treatment of hypertension. Part 1: thiazide and thiazide-like diuretics. Expert Opin Pharmacother 15: 527–547, 2014. doi: 10.1517/14656566.2014.879118. [DOI] [PubMed] [Google Scholar]
- 43.Tanihara Y, Masuda S, Sato T, Katsura T, Ogawa O, Inui K. Substrate specificity of MATE1 and MATE2-K, human multidrug and toxin extrusions/H+-organic cation antiporters. Biochem Pharmacol 74: 359–371, 2007. doi: 10.1016/j.bcp.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 44.Ullrich KJ, Fritzsch G, Rumrich G, David C. Polysubstrates: substances that interact with renal contraluminal PAH, sulfate, and NMeN transport: sulfamoyl-, sulfonylurea-, thiazide- and benzeneamino-carboxylate (nicotinate) compounds. J Pharmacol Exp Ther 269: 684–692, 1994. [PubMed] [Google Scholar]
- 44a.United States Food and Drug Administration In Vitro Metabolism- and Transporter-Mediated Drug-Drug Interaction Studies Guidance for Industry. Silver Spring, MD: U.S. Food and Drug Administration, 2017. [Google Scholar]
- 45.Uwai Y, Saito H, Hashimoto Y, Inui KI. Interaction and transport of thiazide diuretics, loop diuretics, and acetazolamide via rat renal organic anion transporter rOAT1. J Pharmacol Exp Ther 295: 261–265, 2000. [PubMed] [Google Scholar]
- 46.Vujić Z, Mulavdić N, Smajić M, Brborić J, Stankovic P. Simultaneous analysis of irbesartan and hydrochlorothiazide: an improved HPLC method with the aid of a chemometric protocol. Molecules 17: 3461–3474, 2012. doi: 10.3390/molecules17033461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Watanabe S, Tsuda M, Terada T, Katsura T, Inui K. Reduced renal clearance of a zwitterionic substrate cephalexin in MATE1-deficient mice. J Pharmacol Exp Ther 334: 651–656, 2010. doi: 10.1124/jpet.110.169433. [DOI] [PubMed] [Google Scholar]
- 48.Wright SH. Role of organic cation transporters in the renal handling of therapeutic agents and xenobiotics. Toxicol Appl Pharmacol 204: 309–319, 2005. doi: 10.1016/j.taap.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 49.Yin J, Duan H, Shirasaka Y, Prasad B, Wang J. Atenolol renal secretion is mediated by human organic cation transporter 2 and multidrug and toxin extrusion proteins. Drug Metab Dispos 43: 1872–1881, 2015. doi: 10.1124/dmd.115.066175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yonezawa A, Inui K. Importance of the multidrug and toxin extrusion MATE/SLC47A family to pharmacokinetics, pharmacodynamics/toxicodynamics and pharmacogenomics. Br J Pharmacol 164: 1817–1825, 2011. doi: 10.1111/j.1476-5381.2011.01394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.You G. The role of organic ion transporters in drug disposition: an update. Curr Drug Metab 5: 55–62, 2004. doi: 10.2174/1389200043489207. [DOI] [PubMed] [Google Scholar]
- 52.Zhang J, Wang C, Liu Q, Meng Q, Cang J, Sun H, Gao Y, Kaku T, Liu K. Pharmacokinetic interaction between JBP485 and cephalexin in rats. Drug Metab Dispos 38: 930–938, 2010. doi: 10.1124/dmd.110.032060. [DOI] [PubMed] [Google Scholar]