

Abstract
Hypomagnesemia is associated with reduced kidney function and life-threatening complications and sustains hypokalemia. The distal convoluted tubule (DCT) determines final urinary Mg2+ excretion and, via activity of the Na+-Cl− cotransporter (NCC), also plays a key role in K+ homeostasis by metering Na+ delivery to distal segments. Little is known about the mechanisms by which plasma Mg2+ concentration regulates NCC activity and how low-plasma Mg2+ concentration and K+ concentration interact to modulate NCC activity. To address this, we performed dietary manipulation studies in mice. Compared with normal diet, abundances of total NCC and phosphorylated NCC (pNCC) were lower after short-term (3 days) or long-term (14 days) dietary Mg2+ restriction. Altered NCC activation is unlikely to play a role, since we also observed lower total NCC abundance in mice lacking the two NCC-activating kinases, STE20/SPS-1-related proline/alanine-rich kinase and oxidative stress response kinase-1, after Mg2+ restriction. The E3 ubiquitin-protein ligase NEDD4-2 regulates NCC abundance during dietary NaCl loading or K+ restriction. Mg2+ restriction did not lower total NCC abundance in inducible nephron-specific neuronal precursor cell developmentally downregulated 4-2 (NEDD4-2) knockout mice. Total NCC and pNCC abundances were similar after short-term Mg2+ or combined Mg2+-K+ restriction but were dramatically lower compared with a low-K+ diet. Therefore, sustained NCC downregulation may serve a mechanism that enhances distal Na+ delivery during states of hypomagnesemia, maintaining hypokalemia. Similar results were obtained with long-term Mg2+-K+ restriction, but, surprisingly, NCC was not activated after long-term K+ restriction despite lower plasma K+ concentration, suggesting significant differences in distal tubule adaptation to acute or chronic K+ restriction.
Keywords: magnesium, Na+-Cl− cotransporter, potassium, transport
INTRODUCTION
Mg2+ is the second most abundant intracellular divalent cation and is essential for many cellular processes (16). It is required for activities of all ATP-dependent enzymes, many reactions involving kinases, regulation of mitochondrial function and ion channels, and neuromuscular excitability (15, 16, 28). As a result, hypomagnesemia frequently leads to neuromuscular hyperexcitability (ranging from tremors to convulsions) and neuropsychiatric disturbances (28). Hypomagnesemia is frequently associated with hypokalemia and/or hypocalcemia, which can result in cardiac arrhythmia and sudden death (5, 20). Chronic hypomagnesemia has adverse effects in many organs, including the kidney (48), and is associated with higher production of inflammatory and proatherogenic cytokines in endothelial cells that might lead to impaired renal function (37). In the setting of existing kidney disease, reduced plasma Mg2+ concentration may increase the risk of end-stage renal disease (68). A recent study (49) conducted in a cohort of healthy African-American and Caucasian adults showed that low dietary Mg2+ intake was associated with a rapid decline in kidney function.
Plasma Mg2+ concentration is strictly maintained by the interplay of intestinal absorption, renal transport, and bone exchange (67). About 80% of total plasma Mg2+ is filtered by the kidney, of which 90–95% is reabsorbed by various segments of the nephron (13). Most filtered Mg2+ undergoes passive paracellular reabsorption along the proximal tubule and thick ascending limb of the loop of Henle (6). Downstream of these segments is the distal convoluted tubule (DCT), which expresses the Na+-Cl− cotransporter (NCC), the target of thiazide diuretics. The DCT reabsorbs only 5–10% of the filtered Mg2+ by active transcellular transport via apical transient receptor potential M6/7 channels but plays a key role determining the final urinary excretion of Mg2+ (6, 13). This is demonstrated by the presentation of hypomagnesemia as a central feature of Gitelman syndrome caused by recessive mutations in slc12a3, which encodes NCC (59). Furthermore, pharmacological blockade of NCC by thiazides or genetic disruption of NCC in mice also causes hypomagnesemia (57).
The DCT also plays a central role in K+ homeostasis by acting as a sensor of plasma K+ concentration, which modulates NCC phosphorylation status (61, 62). When plasma K+ concentration decreases, K+ exits DCT cells through basolateral Kir4.1 channels; the resultant membrane depolarization increases Cl− efflux through ClC-Kb channels (62, 75, 76). Lower intracellular Cl− then disinhibits the Cl−-sensitive kinase with no lysine (K) kinase-4 (WNK4), leading to phosphorylation and activation of STE20/SPS-1-related proline/alanine-rich kinase (SPAK) and oxidative stress response kinase-1 (OSR1) (61, 62). SPAK/OSR1 directly phosphorylates the NH2-terminus of NCC, which activates NCC and decreases delivery of Na+ to distal segments. Because Na+ delivery generates the electrogenic drive for K+ secretion by distal segments, this results in reduced K+ secretion and maintenance of plasma K+ (3, 39, 60, 79). This dual role of the DCT in Mg2+ and K+ homeostasis is likely to be clinically important. Hypomagnesemia is one of the most frequently encountered forms of electrolyte dysregulation in critically ill patients; the prevalence is ∼10% of hospitalized patients and can be as high as 65% in intensive care patients (11, 55). Thiazide diuretics, which directly block NCC, cause both K+ and Mg2+ deficiencies (74). Importantly, Mg2+ deficiency exacerbates the effect of hypokalemia; hypokalemia with concomitant Mg2+ deficiency is often refractory to K+ supplement. Coadministration of Mg2+ is essential to rectify hypokalemia (29).
Many studies have shown that dietary interventions in addition to dietary K+ restriction have a robust influence on NCC activity (62). A K+-rich diet rapidly dephosphorylates and thereby deactivates NCC in a Cl−-and SPAK/OSR1-independent manner (47, 58). NCC abundance and phosphorylation are increased by low NaCl intake and decreased by high NaCl intake (12). Serum and glucocorticoid-inducible kinase 1 (SGK1) has been implicated in mediating low NaCl-mediated activation of NCC through the WNK-SPAK/OSR1 signaling pathway (65). A recent study (69) showed that mice displaying hyperactivation of NCC, induced by expression of constitutively active SPAK along the DCT, are normomagnesemic, suggesting an uncoupling of Na+ and Mg2+ reabsorption, at least in states of elevated NCC activity. In contrast, very little is known regarding how changes in plasma Mg2+ concentration modify NCC activity and DCT function.
One study (66) reported the effect of modifying plasma Mg2+ concentration on magnesiotropic genes, including slc12a3 in mice by chronically (14 days) providing Mg2+-deficient and high-Mg2+ diets. Whereas NCC mRNA levels did not differ, immunohistochemistry revealed lower abundance of total NCC after Mg2+ restriction compared with high-Mg2+ diet (66). However, this study focused only on total NCC and did not evaluate NCC phosphorylation, a key determinant of total NCC activity and stability (41, 54, 66). The mechanisms leading to lower NCC abundance and whether altered activity of the WNK-SPAK/OSR1 pathway plays a role have also not been determined. Finally, it is unknown whether the effects of dietary Mg2+ restriction on NCC occur rapidly, as occurs with changes in dietary K+, or whether there is an interplay between Mg2+ and K+ restriction. This is clinically relevant, since it has been proposed that increased Na+ delivery to K+-secreting segments, which may occur with lower NCC activity after Mg2+ restriction, is an important component of hypokalemia in the setting of hypomagnesemia (29). Therefore, in the present study, we investigated 1) the acute and chronic effects of dietary Mg2+ manipulation on total NCC and phosphorylated NCC (pNCC), 2) the contribution of the WNK-SPAK/OSR1 pathway, 3) whether the E3 ubiquitin-protein ligase neuronal precursor cell developmentally downregulated 4-2 (NEDD4-2) previously shown to play a role in NCC degradation (18, 53) mediates the effects of Mg2+ restriction, and 4) whether downregulation of NCC by dietary Mg2+ restriction overrides the strong stimulatory effect of K+ restriction on NCC.
MATERIALS AND METHODS
Animals.
C57BL/6J mice aged 3–5 mo maintained in a temperature-controlled and 12:12-h light-dark cycle room were used in this study. To investigate the effect of dietary Mg2+, mice were fed a normal Mg2+ [0.15% (wt/wt); NaCl ∼0.49%, K+ ~0.8%], Mg2+-deficient [0.0015–0.003% (wt/wt); NaCl ∼0.49%, K+ ∼0.8%], high-Mg2+ [0.48% (wt/wt); NaCl ∼0.49%, K+ ∼0.8%], Mg2+-K+-deficient (NaCl ∼0.49%), or K+-deficient diet (Mg2+ ∼0.15%, NaCl ∼0.49%) for 3 days (short-term study) or 14 days (long-term study). Normal and high-Mg2+ diets were prepared by adding MgO [to 0.15% (wt/wt) Mg2+] to a Mg2+-deficient diet (Teklad Custom Diet, TD.170002, Envigo). In short-term and long-term experiments to investigate the effect of dietary Mg2+ and K+ deficiencies, Mg2+-K+-deficient diet (Teklad Custom Diet, TD.170943, Envigo) was used. K+-deficient diet was prepared by adding MgO [to 0.15% (wt/wt) Mg2+]. To prepare normal or Mg2+-deficient diets, potassium citrate monohydrate [to 0.58% (wt/wt)], potassium sulfate [to 0.22% (wt/wt)], chromium potassium sulfate dodecahydrate [to 0.0001% (wt/wt)], and potassium iodate (1 N, 0.18 µl/100 g) were added to the Mg2+-K+-deficient diet, with the normal diet also having MgO added. All knockout (KO) mice were bred in house and were on a C57bl/6 background. The Pax8-rtTA/LC1 system was used to generate inducible renal epithelia-specific KO mice (64). SPAK and OSR1 double-KO (DKO) mice and OSR1fl/fl mice were bred with SPAK-KO mice until homozygous OSR1fl/fl/SPAK KO mice were obtained, which were then bred with Pax8-rtTA/LC1 mice. The resulting offspring were interbred to finally obtain OSR1fl/fl/SPAK KO/Pax8-rtTA/LC1 mice (21). Inducible nephron-specific NEDD4-2 KO mice were generated by breeding Pax8-rtTA/LC1 with NEDD4-2fl/fl mice (53). The resulting offspring were interbred to obtain NEDD4-2fl/fl/Pax8-rtTA/LC1 mice. For both SPAK/OSR1 DKO and NEDD4-2 KO mice, Cre-mediated recombination was induced by administration of doxycycline hyclate (5 mg/ml, 5% sucrose) in the drinking water for 2–3 wk. After doxycycline treatment, mice were returned to regular drinking water for ≥2 wk before experiments were performed. Littermate mice of the same age and genotype provided with 5% sucrose in drinking water were used as controls. Mice aged 2–5 mo were used, and both male and female mice were used as indicated in the figures.
PCR genotyping.
Genomic DNA extracts were prepared from tail snips by heating overnight at 55°C in 300 μl digestion solution containing 5 mM EDTA, 200 mM NaCl, 100 mM Tris (pH 8.0), 0.2% SDS, and 0.4 mg/ml proteinase K followed by ethanol precipitation. The following primers were used: Pax8, forward 5′-CCATGTCTAGACTGGACAAGA-3′ and reverse 5′-CAGAAAGTCTTGCCATGACT-3′; Cre, forward 5′-TTTCCCGCAGAACCTGAACCTGAAGAT-3′ and reverse 5′-TCACCGGCATCAACGTTTTCTT-3′; SPAK, forward 5′-TTCTTAGCCACAGGGGGTGATG-3′ and reverse 5′-GAGTCATAGAAGAGCAGAATAGCAG-3′; OSR1flox, forward 5′-AGCTCAGGCTCCCTCCACGGAG-3′ and reverse 5′-AAGACACATTGATACTCTGTTT-3′; NEDD4-2, forward 5′-TGAGCTCATTGCTTCACTTCC-3′ and reverse 5′-TTCATGCTCGAAGCCTTAGC-3′, and NEDD4-2 flox, reverse 5′-TTTGTGAGGACAGCCTCTAGC-3′.
RNA extraction and quantitative PCR.
Snap-frozen kidneys were ground in a mortar and pestle under liquid nitrogen before RNA extraction with RNA universal mini kit (catalog no. 73404, Qiagen). Powdered kidney tissue was dissolved in Qiazol and further mechanically triturateed through a narrow syringe until the solution became homogenous before the manufacturer’s protocol for total RNA extraction was followed. RNA was then quantified on a spectrophotometer, and 1,000 ng of total RNA were converted to cDNA using the Maxima H Minus First Strand cDNA Synthesis Kit (catalog no. K1681, ThermoFischer Scientific) according to the manufacturer’s instructions. The resulting reaction was then diluted 1:10 so that 5 ng template cDNA was input into each quantitative PCR. Predesigned TaqMan probes were purchased from ABI for 18S rRNA and Slc12a3 (NCC). Duplicate quantitative PCRs were quantified on a QuantStudio 7 Flex (ABI). Expression levels for Slc12a3 were normalized to the levels of 18S rRNA and then normalized to the mean expression of Slc12a3 for mice on a control diet.
Antibodies.
Antibody sources, species, dilutions, and references are shown in Table 1.
Table 1.
Antibodies
| Antibody (Species-Antigen) | Use | Dilution | Source | Reference(s) |
|---|---|---|---|---|
| Rabbit-tNCC* | WB | 1:6,000 | Ellison Laboratory | 21, 22, 40, 42 |
| Rabbit-pNCC (T53)* | WB | 1:2,000 | Ellison Laboratory | 21, 22, 40, 42 |
| Rabbit-N-WNK4* | WB | 1:1,000 | Ellison Laboratory | 22, 42 |
| Rabbit-N-WNK4* | IF | 1:2,000 | Ellison Laboratory | 22, 42 |
| Rabbit-C-SPAK* | WB | 1:5,000 | McCormick Laboratory | 21, 40 |
| Rabbit-C-SPAK* | IF | 1:5,000 | McCormick Laboratory | 21, 40 |
| Rabbit-pSPAK (S383) | WB | 1:1,000 | Millipore (07-2273) | |
| Guinea pig-parvalbumin | IF | 1:2,000 | Swant, GP72 | 22 |
| Mouse-calbindin | IF | 1:2,000 | Swant, calbindin D-28, (300) | 22 |
| Rabbit-NEDD4L | WB | 1:1,000 | Cell Signaling Technology (no. 4013) | 53 |
| Rabbit-SGK1 | WB | 1:1,000 | Sigma-Aldrich | 26 |
tNCC, Na+-Cl− cotransporter (NCC); pNCC, phosphorylated NCC; WNK4, with-no-lysine (K) kinase-4; STE20/SPS-1-related proline/alanine-rich kinase; pSPAK, phosphorylated SPAK; NEDD4L, neuronal precursor cell developmentally downregulated 4L; SGK1, serum and glucocorticoid-induced kinase 1; WB, Western blot; IF, immunofluorescence.
NCC, pNCC, WNK4, SPAK, p-SPAK, and NEDD4L antibodies have been validated in knockout mice.
Kidney Western blot analysis.
Kidneys were snap frozen in liquid nitrogen after being harvested and stored at −80°C until homogenization using a Potter homogenizer in 1 ml cold homogenization buffer containing lysis buffer containing 300 mM sucrose, 50 mM Tris·HCl (pH 7.4), 1 mM EDTA, 1 mM EGTA, 1 mM NaVO4, 50 mM NaF, 1 mM ditiothreitol (Millipore, Sigma), 1 mM phenylmethane sulfonyl fluoride, 1 µg/ml aprotinin, and 4 µg/ml leupeptin. The homogenate was centrifuged at 6,000 g for 15 min at 4°C, and the supernatant was transferred to a new tube and stored at −80°C. Then, 20 μg protein was separated on a 4–12% bis-Tris acetate gel (Invitrogen) and transferred to a polyvinylidene fluoride membrane using the Trans-Blot Turbo Transfer System (Bio-Rad). The membrane was blocked with 5% nonfat milk in PBS-Tween followed by an incubation with primary antibody for either 1 h at room temperature or overnight at 4°C. Membranes were washed, incubated with horseradish peroxidase-goat anti-rabbit IgG (1:7,500, no. 65-6120, Invitrogen), washed again, and finally incubated with Western Lightning ECL (Perkin-Elmer). The ECL signal was detected with a Syngene Pxi4 imager. For donkey anti-rabbit IgG (1:15,000, LI-COR, IRDye 680RD) secondary antibody, an Odyssey CLx Near-Infrared Fluorescence Imaging System was used to scan the membrane, and densitometry was performed with ImageJ (http://rsbweb.nih.gov/ij/). Protein loading was adjusted by densitometric quantitation of total protein after Coomassie staining (see Supplemental Fig. S1 for an example; Supplemental Data is available online at http://dx.doi.org/10.6084/m9.figshare.8947001) (22, 43).
Blood analysis.
Blood was collected via cardiac puncture under isoflurane anesthesia and transferred into heparinized tubes; 80 μl were loaded into a Chem8+ cartridge for electrolyte measurement by an i-STAT analyzer (Abbot Point of Care). The remainder was centrifuged at 2,000 g for 5 min at room temperature, and plasma was removed and stored at −80°C. Plasma Mg2+ concentration was quantified by ratiometric assay (Pointe Science, Canton, MI). For some experiments, plasma K+ concentration was determined by flame photometry.
Immunofluorescence.
Animals were injected with an anesthesia cocktail [ketamine-xylazine-acepromazine (50:5:0.5 mg/kg)] and under deep anesthesia were perfusion fixed with 4% paraformaldehyde. After cryoprotection in 800 mosmol sucrose and freezing in OCT, 5-μm sections were cut. Sections were incubated overnight at room temperature with primary antibodies (Table 1). The secondary antibody used was Alexa Fluor 488 goat anti-guinea pig IgG (A11073, Invitrogen, 1:1,000), Cy3 goat anti-rabbit IgG (no. 81-6115, Zymed, 1:1,000), or FITC goat anti-mouse IgG (no. 81-6511, Zymed, 1:1,000). Secondary antibody was incubated for 1 h at room temperature. Images were captured with a Zeiss AXIO Imager M2 fluorescent microscope.
Statistics.
The null hypothesis was tested using two-tailed unpaired t-tests, one-way ANOVA, or two-way ANOVA for repeated measures using GraphPad Prism 7 as indicated in the figures. Post hoc analysis was performed using the Tukey multiple-comparison test. All data are plotted as means ± SE. P < 0.05 was considered significant.
Study approval.
Animal studies were performed in adherence to the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals (National Academies Press, 2011) and approved by the Oregon Health and Science University Institutional Animal Care and Use Committee (protocol IP00286) and New York Medical College Institutional Animal Care and Use Committee (protocol 60-8-1016H).
RESULTS
Long-term (14 days) dietary Mg2+ restriction downregulates NCC.
Mice were maintained on normal Mg2+, Mg2+-deficient, or high-Mg2+ diet for 14 days. As expected, plasma Mg2+ was significantly lower on the Mg2+-deficient diet compared with the normal diet and high-Mg2+ diet (Fig. 1A), but both the normal and high-Mg2+ diet groups had similar plasma Mg2+ concentration (Fig. 1A). In humans, maintenance of plasma Mg2+ concentration under a high Mg2+ load does not occur via increased renal excretion but by reduced gut absorption (30). To determine whether the high-Mg2+ diet increased urinary Mg2+ excretion to maintain plasma Mg2+ concentration in mice, we measured urinary Mg2+ excretion at baseline and after 1, 2, 3, or 14 days of normal or high-Mg2+ diet. We found no differences in urinary Mg2+ excretion between the normal diet and high-Mg2+ diet at any time point (Supplemental Fig. S2). Mg2+ inhibits renal outer medullary K+ (ROMK) channel-mediated K+ activity in vitro (38, 70, 80), so lower plasma Mg2+ concentration might be expected to lower plasma K+ concentration. However, plasma K+ concentration did not differ between the groups (Fig. 1A). Hypokalemia is not typically observed in isolated Mg2+ deficiency in experimental models (29, 73), for two possible reasons. First, chronic Mg2+ deficiency impairs Na+-K+-ATPase pump activity, which decreases K+ uptake by the muscle and kidney and thus maintains plasma K+ concentration (9, 29, 78). Moreover, increased K+ secretion through ROMK channels would cause hyperpolarization of the plasma membrane reducing K+ secretion through ROMK channels (24, 29). Plasma ionized Ca2+ concentration was lower in the Mg2+-deficient group than normal diet and high-Mg2+ diet groups (Fig. 1A), since chronic Mg2+ deficiency impairs the release of parathyroid hormone, leading to lower plasma ionized Ca2+ concentration (4, 77).
Fig. 1.

Long-term (14 days) dietary Mg2+ restriction downregulates total Na+-Cl− cotransporter (tNCC) abundance. A: plasma [K+] did not differ among the study groups (one-way ANOVA with Tukey’s multiple-comparison test). Plasma ionized [Ca2+] (i-[Ca2+]) was significantly lower on a Mg2+-deficient diet (D) compared with normal diet (N) and high-Mg2+ diet (H) (one-way ANOVA with Tukey’s multiple-comparison test). Plasma [Mg2+] was significantly lower on a Mg2+-deficient diet compared with normal diet and high-Mg2+ diet (one-way ANOVA with Tukey’s multiple-comparison test). B: Western blot analysis of whole kidney lysates showed that tNCC abundance was significantly lower on Mg2+-deficient diet than on normal diet and high-Mg2+ diet (one-way ANOVA with Tukey’s multiple-comparison test). C: phosphorylated (p)NCC abundance was also significantly lower on Mg2+-deficient diet compared with normal and high-Mg2+ diets (one-way ANOVA with Tukey’s multiple-comparison test). D: Western blot analysis of whole kidney lysates from female mice showed that tNCC abundance was significantly reduced on Mg2+-deficient diet than on normal diet (two-tailed unpaired t-test). For blot quantification, densitometric values were normalized using Coomassie-stained gels. Values are means ± SE; values in parentheses indicate n values. *P < 0.05; **P < 0.001.
Although plasma K+ concentration, a strong regulator of NCC, did not differ between groups (Fig. 1A), the abundance of total NCC in the kidney was dramatically lower in mice maintained on a Mg2+-deficient diet compared with those on normal or high-Mg2+ diets (Fig. 1B). The abundance of pNCC was also significantly lower in mice maintained on a Mg2+-deficient diet (Fig. 1C). However, the pNCC-to-total NCC ratio did not differ between groups, suggesting that the primary effect is on total NCC abundance. Although female mice display a greater abundance of total NCC and pNCC on a normal diet (52), dietary Mg2+ restriction also resulted in lower total NCC abundance in female mice (Fig. 1D), indicating no sex difference. Considering that no differences were observed in total NCC or pNCC between normal and high-Mg2+ diets, subsequent studies compared only the effects of normal and low-Mg2+ diets.
Dietary Mg2+ restriction does not affect the WNK4/SPAK pathway.
The stability of NCC at the plasma membrane and its overall abundance may depend on its phosphorylation status (54). To determine whether dietary Mg2+ deficiency altered activity of the canonical NCC-activating pathway, we first assessed the abundance of WNK4, the upstream kinase in the pathway. WNK4 abundance did not differ between normal and low-Mg2+ diets (Fig. 2A). SPAK and OSR1 lie downstream of WNK4 and directly phosphorylate the NH2-terminus of NCC (50). Abundances of pSPAK and total SPAK did not differ between normal and Mg2+-deficient diets (Fig. 2B). These data suggest that the WNK-SPAK/OSR1 pathway is neither activated nor inhibited by dietary Mg2+ restriction, but to further test this, we performed immunofluorescence to determine whether WNK4 and SPAK cellular localization changed. SPAK and WNK4 have been shown to localize to intracellular puncta (“WNK bodies”) in response to dietary K+ restriction (8, 22, 62). The composition and function of these puncta are not well defined, and it is unknown whether their formation plays a regulatory role in NCC and DCT function. Immunofluorescence showed that dietary Mg2+ restriction did not cause any change in the intracellular distribution of SPAK or WNK4 along DCT1, as shown by colocalization with parvalbumin (Fig. 2, C and D) or the DCT2/connecting segment (CNT), as shown by colocalization with calbindin (Supplemental Fig. S3).
Fig. 2.

Long-term (14 days) dietary Mg2+ restriction did not affect with-no-lysine (K) kinase-4 (WNK4) or STE20/SPS-1-related proline/alanine-rich kinase (SPAK). A: Western blot analysis of whole kidney lysates did not show any significant difference in the abundance of WNK4 between normal diet (N) and Mg2+-deficient (D) diet groups (two-tailed unpaired t-test). B: full-length SPAK (FL-SPAK) and phosphorylated SPAK (pSPAK) abundances also did not differ between normal and Mg2+-deficient diets (two-tailed unpaired t-test). C: immunofluorescence for SPAK (red) showed similar localization in normal and Mg2+-deficient diets. Colocalization with parvalbumin (PV; green) showed that SPAK was localized along the distal convoluted tubule (DCT). D: WNK4 (red) localization did not differ between normal diet and Mg2+-deficient diet. PV (green) was used to identify DCT in the same section. Note that on normal diet and low-Mg2+ diet, WNK4 signal is weak and diffuse along the DCT and DCT2/connecting segment (CNT), in contrast to what is observed after low-K+ diet (62) or genetic activation of the WNK4-SPAK pathway (22), so the images were not overlaid. Results are representative of 5 independent experiments. Scale bars in C and D = 40 μm. E: dietary Mg2+ restriction downregulated total Na+-Cl− cotransporter (tNCC) abundance in SPAK knockout (KO) mice. tNCC abundance was also significantly reduced in SPAK and oxidative stress-response kinase-1 (OSR1) double-KO (DKO) mice on Mg2+-deficient diet compared with normal diet (two-tailed unpaired t-test). For blot quantification, densitometric values were normalized using Coomassie-stained gels. Values are means ± SE; values in parentheses indicate n values. *P < 0.01; **P < 0.001.
Our analysis of SPAK and WNK4 abundance and distribution suggested that SPAK and WNK4 are not directly affected by dietary Mg2+ restriction and that altered activity of the pathway does not play a role in NCC downregulation. To further confirm this, we used SPAK KO and SPAK/OSR1 DKO mice. SPAK is the major NCC activator in vivo, since its disruption in mice leads to a dramatic reduction (∼90%) in pNCC and total NCC (27, 40, 82). SPAK KO mice were maintained on normal or Mg2+-deficient diets for 14 days. Dietary Mg2+ restriction further decreased total NCC abundance in SPAK KO mice (Fig. 2E). We have previously reported that OSR1 can compensate for a lack of SPAK to activate NCC in response to dietary K+ restriction (21). To exclude a compensatory role for OSR1, SPAK/OSR1 DKO mice were placed on a normal or Mg2+-deficient diet. After 14 days of dietary Mg2+ restriction, total NCC abundance was markedly lower in DKO mice (Fig. 2E). Seeing that total NCC abundance was lower after dietary Mg2+ restriction in the presence or absence of SPAK/OSR1, our data suggest that altered activity of the WNK-SPAK/OSR1 pathway is not necessary for the effect of low plasma Mg2+ concentration on NCC.
Short-term dietary Mg2+ restriction does not change plasma K+ concentration despite lower NCC abundance.
A previous study (66) examining the effect of dietary Mg2+ on NCC involved chronic manipulation (14 days). In contrast, studies examining the effects of manipulating K+ or NaCl have involved much shorter durations [typically 3–7 days for low K+ (21, 62, 71), 15 min to 4–7 days for high K+ (10, 12, 58, 65), or 7–10 days for low or high NaCl (12, 65)]. Furthermore, Ortega et al. (44) reported that short-term (3 days) dietary Mg2+ deficiency significantly increased plasma K+ concentration. Acute elevation of plasma K+ concentration after gavage of a KCl bolus caused rapid dephosphorylation of NCC (47, 58), which could affect total NCC abundance (54). Thus, one possibility is that in the short term, lower plasma Mg2+ concentration decreases total NCC abundance secondarily to an increase in plasma K+ concentration. Wild-type (WT) mice were placed on a normal or Mg2+-deficient diet for 3 days. In contrast to the previous report by Ortega et al. (44), plasma K+ concentration did not differ between normal and Mg2+-deficient groups (Fig. 3A), but total NCC and pNCC abundances were significantly lower in the Mg2+-deficient group (Fig. 3, B and C). No significant difference was observed in the abundance of total NCC in mice maintained on normal or high-Mg2+ diets for 3 days (data not shown). Because NCC abundance was lower after Mg2+ restriction, we measured 24-h urinary Na+ and K+ excretion. Short-term restriction did not alter body weight [26.6 ± 0.6 normal (n =6) vs. 27.4 ± 0.3 g low Mg2+ (n = 5), mean ± SE, P = 0.28], but food intake was lower (3.99 ± 0.16 vs. 3.36 ± 0.12 g, P = 0.01). However, normalized Na+ and K+ excretion did not differ (Na+: 3.29 ± 0.15 vs. 3.42 ± 0.11 μmol·g body wt−1·g food intake−1, P = 0.52; K+: 6.58 ± 0.32 vs. 6.36 ± 0.27 μmol·g body wt−1·g food intake−1, P = 0.63). With long-term Mg2+ restriction, there were no differences in body weight, food intake, or 24-h urinary Na+ or K+ excretion (Supplemental Fig. S4). These data are similar to those of Van Angelen et al. (66), who observed no difference in 24-h urinary Na+ and K+ excretion between mice on a Mg2+-deficient diet or high-Mg2+ diet after 14 days of restriction. With respect to functional effects on NCC, it should be noted that mice heterozygous for a Gitelman syndrome mutation (with 50% lower functional NCC) did not display changes in plasma or urinary electrolyte excretion at baseline (83).
Fig. 3.

Short-term (3 days) dietary Mg2+ restriction downregulates total Na+-Cl− cotransporter (tNCC). A: plasma [K+] did not differ between the diets (two-tailed unpaired t-test). Plasma ionized [Ca2+] (i-[Ca2+]) was significantly lower on Mg2+-deficient diet (D) compared with normal diet (N) (two-tailed unpaired t-test), and plasma [Mg2+] was significantly lower on Mg2+-deficient diet than on normal diet (two-tailed unpaired t-test) B: Western blot analysis of whole kidney lysates showed that tNCC abundance was significantly lower on Mg2+-deficient diet than on normal diet (two-tailed unpaired t-test). C: abundance of phosphorylated (p)NCC was also significantly lower on Mg2+-deficient diet compared with normal diet (two-tailed unpaired t-test). For blot quantification, densitometric values were normalized using Coomassie-stained gels. Values are means ± SE; values in parentheses indicate n values. *P < 0.05; **P = 0.0001.
NEDD4-2 participates in NCC downregulation.
NEDD4-2 ubiquitinates NCC and the epithelial Na+ channel (ENaC), tagging them for proteasomal degradation (18, 53). The precise target of NEDD4-2 varies in response to different dietary manipulations, with NCC preferred during NaCl loading and ENaC preferred during dietary K+ restriction (1, 51). To determine whether NEDD4-2 plays a role in the dietary Mg2+ restriction-mediated downregulation of NCC, inducible nephron-specific NEDD4-2 KO mice were maintained on normal diet and Mg2+-deficient diet for 3 days. As previously reported, tNCC and pNCC abundances were higher in NEDD4-2-KO mice compared with control mice (53). However, in contrast to our observations in WT mice, dietary Mg2+ restriction did not lower the abundance of total NCC in NEDD4-2 KO mice (Fig. 4A). These data suggest that NEDD4-2 participates in the downregulation of NCC during Mg2+ deficiency. In this model, NEDD4-2 is knocked out in tubule segments but not in nontubule cells (53). Dietary Mg2+ restriction appeared to lead to higher abundance of nontubule NEDD4-2 in KO mice. Quantification of NEDD4-2 abundance and analysis by two-way ANOVA revealed no effect of dietary Mg2+ restriction on NEDD4-2 abundance in control mice [100 ± 9 normal (n = 3) vs. 105 ± 18% low Mg2+ (n = 3), mean ± SE, P = 0.98,]. NEDD4-2 abundance was significantly lower in KO mice versus control mice, but there were no differences in NEDD4-2 abundance between KO mice on a normal or low-Mg2+ diet (P = 0.47). However, comparison with control mice (with high NEDD4-2 abundance) likely masks an effect, so we directly compared KO mice on normal and low Mg2+ diets by an unpaired t-test and found significantly higher nontubule NEDD4-2 abundance after Mg2+ restriction [100 ± 13 normal (n = 3) vs. 401 ± 62% low Mg2+ (n = 3), mean ± SE, P = 0.008 ]. These data suggest that Mg2+ restriction exerts effects in nontubular cells in the kidney, which might also lead to the significantly higher abundance of total NCC in NEDD4-2 KO mice on a low-Mg2+ diet compared with normal diet (Fig. 4A).
Fig. 4.

Disruption of tubular E3 ubiquitin-protein ligase. Neuronal precursor cell developmentally downregulated 2 (NEDD4-2) prevents dietary Mg2+ restriction-mediated downregulation of Na+-Cl− cotransporter (NCC). A: control mice had lower abundance of total NCC (tNCC) and phosphorylated (p)NCC on Mg2+-deficient diet (D) compared with normal diet (N), whereas in inducible nephron-specific NEDD4-2 knockout (KO) mice, dietary Mg2+ restriction did not lead to lower abundance of NCC (two-way ANOVA with Tukey’s multiple-comparison test). B: abundance of serum and glucocorticoid-induced kinase 1 (SGK1) was comparable between normal diet and Mg2+-deficient diet after 3 days of dietary Mg2+ manipulation (two-tailed unpaired t-test). C: NCC mRNA did not differ among normal diet or short-term (3 days) and long-term (14 days) dietary Mg2+ restriction (one-way ANOVA with Tukey’s multiple-comparison test). For blot quantification, densitometric values were normalized using Coomassie-stained gels. Values are means ± SE; values in parentheses indicate n values. *P < 0.05; **P < 0.01; ***P < 0.0001.
Expression of serum and glucocorticoid-induced kinase 1 (SGK1) is strongly induced by aldosterone and phosphorylates and inhibits NEDD4-2 to increase ENaC abundance at the plasma membrane (33). There were no differences in SGK1 abundance after Mg2+ restriction (Fig. 4B), suggesting that the NEDD4-2 regulatory pathway differs between Na+ and Mg2+ restriction. It has been previously reported that NCC mRNA does not differ between mice fed low- or high-Mg2+ diets for 14 days (14, 66). Similarly, we found no significant differences in NCC mRNA between mice on low-Mg2+ or normal Mg2+ diets in the short term or long term (Fig. 4C), suggesting posttranscriptional regulation of NCC abundance.
Dietary Mg2+ restriction overrides the stimulatory effect of dietary K+ restriction on NCC.
Because dietary K+ restriction is a potent activator of NCC, we next determined whether Mg2+ deficiency affects K+ deficiency-mediated activation of NCC. WT mice were placed on K+-deficient, Mg2+-deficient, or Mg2+-K+-deficient diets for 3 days. Plasma Mg2+ concentration was markedly lower on Mg2+-deficient and Mg2+-K+-deficient diets compared with K+-deficient diet, and plasma K+ concentration was significantly decreased on K+-deficient and Mg2+-K+-deficient diets compared with Mg2+-deficient diet (Fig. 5A). There were no synergistic effects of dietary Mg2+-K+ restriction with regard to plasma Mg2+ or K+ concentrations. The abundances of total NCC and pNCC were lower on Mg2+-deficient and Mg2+-K+-deficient diets than on K+-deficient diet (Fig. 5, B and C), suggesting that dietary Mg2+ deficiency overrode the ability of dietary K+ restriction to induce NCC phosphorylation. To determine whether this effect persisted over the long term, WT mice were maintained on normal, Mg2+-K+-deficient, Mg2+-deficient, or K+-deficient diets for 14 days. Dietary Mg2+, K+, or Mg2+-K+ restriction resulted in lower plasma Mg2+ concentration and/or K+ concentration, with no synergistic effects (Fig. 6A). After 14 days of Mg2+ restriction, total NCC and pNCC abundances were significantly lower compared with control diet (Fig. 6B), consistent with Fig. 1B. Total NCC and pNCC abundances were also significantly lower after Mg2+-K+-deficient diet compared with control diet (Fig. 6B). As observed after short-term restriction (Fig. 5, B and C), abundances of total NCC and pNCC did not differ between mice placed on Mg2+-deficient or Mg2+-K+-deficient diets (Fig. 6B). Unexpectedly, given the well-established effect of short-term K+ restriction to strongly increase pNCC abundance, the abundances of total NCC and pNCC on the K+-deficient diet compared with the normal diet did not differ (Fig. 6B).
Fig. 5.

Short-term (3 days) dietary Mg2+ restriction overrides the effect of K+ deficiency on Na+-Cl− cotransporter (NCC). A: plasma [K+] was significantly lower on K+-deficient (D) diet and on Mg2+-K+ deficient diet compared with normal diet (N) and Mg2+-deficient diet (one-way ANOVA with Tukey’s multiple-comparison test). Plasma [Mg2+] was significantly lower on Mg2+-deficient and Mg2+-K+-deficient diet compared with normal diet and K+-deficient diet (one-way ANOVA with Tukey’s multiple-comparison test). B: Western blot analysis of whole kidney lysates showed that total NCC (tNCC) abundance was significantly lower on Mg2+-deficient diet and on Mg2+-K+-deficient diet than on normal diet and K+-deficient diet (one-way ANOVA with Tukey’s multiple-comparison test). C: phosphorylated (p)NCC abundance was significantly higher on K+-deficient diet than all other study groups (one-way ANOVA with Tukey’s multiple-comparison test). For blot quantification, densitometric values were normalized using Coomassie-stained gels. Values are means ± SE; values in parentheses indicate n values. *P < 0.01; **P < 0.001; ***P < 0.0001.
Fig. 6.

Compared with normal diet, long-term (14 days) dietary K+ restriction did not increase phosphorylated Na+-Cl− cotransporter (pNCC) abundance. A: plasma [K+] was significantly lower on Mg2+-K+-deficient diet (D) and on K+-deficient diet compared with normal diet (N) and Mg2+-deficient diet (one-way ANOVA with Tukey’s multiple-comparison test). Plasma [Mg2+] was significantly lower on Mg2+-K+-deficient diet and on Mg2+-deficient diet compared with normal diet and K+-deficient diet (one-way ANOVA, Tukey’s multiple-comparison test). B: Western blot analysis of whole kidney lysates showed that long-term (14 days) dietary Mg2+ restriction downregulated total Na+-Cl− cotransporter (tNCC) abundance compared with normal diet and K+-deficient diet (one-way ANOVA with Tukey’s multiple-comparison test). Long-term (14 days) dietary Mg2+ restriction also lowered pNCC abundance compared with normal diet, whereas pNCC abundance was not higher on K+-deficient diet (one-way ANOVA with Tukey’s multiple-comparison test). For blot quantification, densitometric values were normalized using Coomassie-stained gels. Values are means ± SE; values in parentheses indicate n values. *P < 0.05; **P < 0.001; ***P < 0.0001.
DISCUSSION
The present study reveals that both long-term and short-term dietary Mg2+ deficiency downregulate total NCC abundance. Our observation of downregulation of total NCC protein abundance by dietary Mg2+ deficiency confirms previous findings (66) using Western blot analysis, rather than immunofluorescence, which is more difficult to accurately quantitate. In addition to using high- and low-Mg2+ diets, we also used a diet with a level of Mg2+ similar to that of regular chow and found that high Mg2+ did not affect NCC abundance. It was recently reported that mice expressing constitutively active SPAK, which display hyperactive NCC and mimic the disease familial hyperkalemic hypertension, are not hypermagnesemic (69). Therefore, NCC activity does not appear to be important for mediating long-term Mg2+ elimination during high Mg2+ intake. Our data also provide mechanistic insights. The effect of dietary Mg2+ restriction does not involve the WNK/SPAK/OSR1 pathway, the major NCC-activating pathway, but the E3 ubiquitin-protein ligase NEDD4-2 is required for Mg2+ deficiency-induced downregulation of NCC. We also found that NCC abundance and phosphorylation are dramatically lower after combined Mg2+-K+ restriction, despite the well-established effect of short-term K+ restriction to strongly activate NCC. Finally, we made the unexpected observation that total NCC and pNCC abundances are not higher than those in controls after long-term K+ restriction.
It is well established that Mg2+ restriction impairs the release of parathyroid hormone, leading to lower plasma ionized Ca2+ concentration, which might contribute to downregulation of NCC abundance. Using [3H]metolazone binding, Fanestil et al. (19) reported no effect of dietary Ca2+ restriction or loading on NCC density in the rat kidney. However, dietary Ca2+ manipulation had no effect on plasma ionized Ca2+ concentration. Supplementation of dietary Ca2+ to maintain plasma ionized Ca2+ concentration during Mg2+ restriction might help address this question.
Ortega et al. (44) reported that serum K+ concentration was significantly higher after short-term (3 days) dietary Mg2+ deficiency. Plasma K+ concentration potently regulates the abundance and phosphorylation status of NCC (21, 61, 62). High dietary K+ intake causes rapid dephosphorylation and, hence, inactivation of NCC (47, 58), which could contribute to degradation of NCC (54). However, in our hands, short-term dietary Mg2+ deficiency did not lead to a difference in plasma K+ concentration. This could be due to differences in technique (both studies used cardiac puncture) during the blood draw procedure, which may cause hemolysis. Ortega et al. (44) reported an average serum K+ concentration of ∼5 mmol/l at baseline, whereas in WT mice maintained on a normal diet, we typically observed a plasma K+ concentration of 3.5−4.5 mmol/l (21, 22). Moreover, our observation is consistent with the reports of other studies that in experimental models isolated Mg2+ deficiency does not cause any significant change in plasma K+ concentration (29, 73). Therefore, we conclude that Mg2+ deficiency does not lower total NCC abundance through an effect on plasma K+ concentration.
Unexpectedly, we did not observe increased abundances of total NCC and pNCC after 14 days of a K+-deficient diet. We repeated the experiment with a second batch of mice and obtained the same results (data not shown). Recent studies examining the effects of dietary K+ restriction on NCC have been performed over a period of 3−7 days (21, 62, 65, 71). A study from the early 1990s (17) has reported dramatic changes in tubule structure in rat kidney after long-term (18 days) dietary K+ restriction. Compared with controls, kidney weight was increased by 30% and glomerular filtration rate (GFR) decreased by 14% on a long-term K+-deficient diet. Striking hypertrophic changes were reported to occur in the outer medullary collecting duct (17). We recently reported that short-term (3 days) K+ restriction induces proliferation in early DCT and aquaporin 2-positive cells but does not determine the effects of sustained K+ restriction on proliferation or apoptosis in these segments (56). Thus, one possibility is that changes in NCC activity and DCT hypertrophy contribute to short/medium-term responses to K+ restriction, whereas remodeling of the K+-secreting CNT and cortical collecting duct becomes the predominant mechanism over the long term. Further studies will be required to further explore the precise changes in tubule structure at the molecular level after long-term K+ restriction and also whether Mg2+ restriction induces tubule remodeling and modulates K+ restriction-induced changes.
Short- or long-term Mg2+ restriction did not affect NCC mRNA levels, consistent with previous reports using only Mg2+-deficient and high-Mg2+ diets (14, 66). Therefore, we hypothesized that the underlying cause of downregulation of NCC after dietary Mg2+ restriction involves an effect on NCC protein degradation. The ubiquitin protein ligase NEDD4-2 is a well-known regulator of NCC abundance (18, 53). The primary regulatory target of NEDD4-2 action changes depending on dietary manipulation. NEDD4-2 primarily regulates NCC on dietary Na+ loading, whereas on dietary K+ restriction it primarily regulates ENaC (1, 51). We found that NCC abundance was not lower after Mg2+ restriction in NEDD4-2 KO mice, suggesting that NEDD4-2 may mediate the effect. NEDD4-2 can be activated by either directly binding Ca2+ via its Calb/C2-binding domain, which releases its autoinhibition, or after binding to substrate (31, 72). In the kidney, 100% of intracellular Mg2+ can be exchanged with the plasma within 3 to 4 h (36). Therefore, both short-term and long-term dietary Mg2+ restriction would be expected to significantly reduce intracellular Mg2+ content. Because intracellular Mg2+ concentration inversely regulates intracellular free ionized Ca2+ concentration (25, 84), dietary Mg2+ restriction likely elevates intracellular free ionized Ca2+ concentration. We speculate that along the DCT this might activate NEDD4-2, leading to NCC degradation. However, one caveat is that NEDD4-2 might prevent degradation of NCC in any condition. Mg2+ deficiency is also known to impair Na+-K+-ATPase pump activity (23, 34). Impaired Na+-K+-ATPase pump function diminishes NCC activity, and inactive NCC is more susceptible to degradation (34, 39, 54, 60, 81). Reduced K+ concentration also induces a K+-dependent decrease in Na+-K+-ATPase pump activity, which has been proffered to cause chronic dietary K+ restriction-mediated renal hypertrophy (7, 63). Further studies are required to determine the cellular mechanisms by which dietary Mg2+ restriction mediates NCC degradation.
There are limitations to our study. First, Mg2+ deficiency has effects on the vasculature that have been proposed to cause increased blood pressure (32), although it took 8 wk of Mg2+ restriction to cause a blood pressure increase. Downregulation of NCC could act as a compensatory mechanism to maintain blood pressure during the early phase of Mg2+ restriction. This could be tested by administration of the vasodilator hydralazine during Mg2+ restriction. Next, the recommended daily required Mg2+ intake for mice is inconsistent, but with a range of 500−2,600 mg/kg (43a). Whereas our normal diet contained 1,500 mg/kg and our high-Mg2+ diet contained 4,800 mg/kg, our low-Mg2+ diet provided only a maximum of 30 mg/kg, which is extremely low and nonphysiological. However, this low-Mg2+ diet resulted in plasma Mg2+ concentration of ∼0.4 mmol/l, which is in the range of values that have been observed with alcoholism, furosemide treatment, short bowel and other malabsorptive disorders, or rare diseases such as familial hypomagnesemia with hypercalciuria and nephrocalcinosis caused by claudin 16 or claudin 19 mutations. Nevertheless, future studies will require titration of dietary Mg2+ to minimize effects beyond NCC downregulation. Finally, we found that 14 days on a high-Mg2+ diet did not alter NCC abundance, but at the same time, this diet did not increase plasma Mg2+ concentration. Our findings are consistent with a previous report (66), and our data suggest that this may be due to lack of gut absorption, since urinary Mg2+ excretion did not differ compared with normal diet. However, a recent report by Liu et al. (35) showed that 6 wk of dietary Mg2+ loading increased plasma Mg2+ concentration by 61%. Together, these observations suggest that over time the mechanisms allowing the gut to limit Mg2+ absorption during a high-Mg2+ load may break down. Therefore, additional studies are required to determine whether hypermagnesemia induced by more chronic dietary Mg2+ loading alters NCC abundance.
In clinical cases of combined hypokalemia and hypomagnesemia, it is essential to coadminister Mg2+ to correct the hypokalemia (29). Huang and Kuo (29) proposed that increased Na+ delivery to the CNT and cortical collecting duct is a key stimulator of K+ wasting during hypomagnesemia. Downregulation of tNCC due to hypomagnesemia might be one mechanism leading to increased distal Na+ delivery, maintaining hypokalemia (45, 46, 60). However, isolated hypokalemia potently activates NCC (21, 62), which would be predicted to counter the effect of hypomagnesemia. Our data reveal that dietary Mg2+ can override the activating effect of dietary K+ restriction on NCC, which could sustain distal Na+ delivery and, hence, kaliuresis (Fig. 7). This provides a plausible explanation for the requirement to correct concomitant hypomagnesemia to normalize hypokalemia in the clinical setting.
Fig. 7.
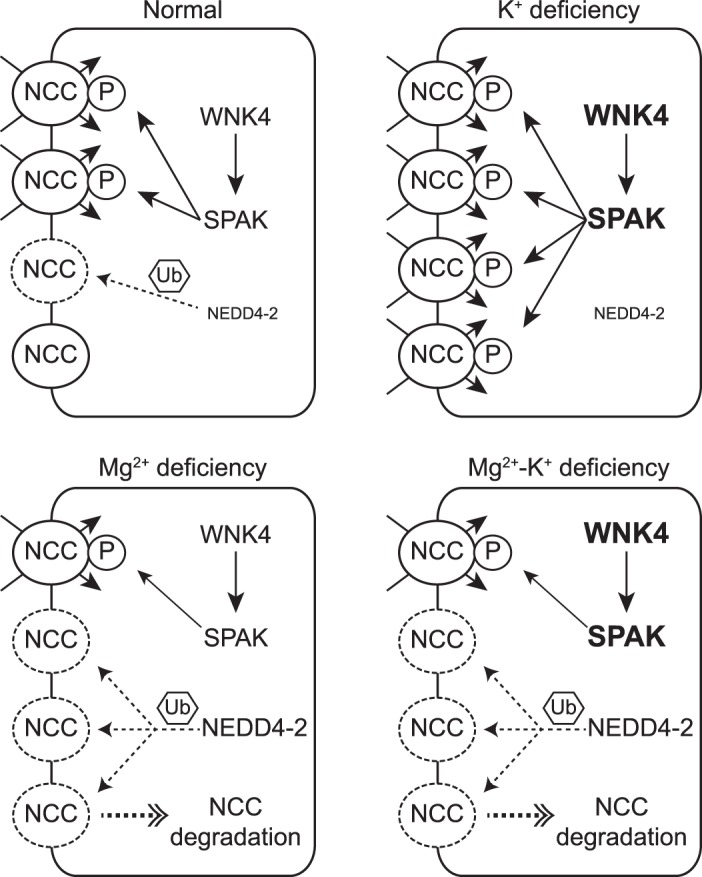
Model of the effect of dietary Mg2+ deficiency on total Na+-Cl− cotransporter (tNCC) in the setting of hypokalemia. With-no-lysine (K) kinase-4 (WNK4)-STE20/SPS-1-related proline/alanine-rich kinase (SPAK) is the major signaling pathway to activate NCC, whereas E3 ubiquitin-protein ligase neuronal precursor cell developmentally downregulated 4-2 (NEDD4-2) plays a key role in the catabolism of NCC. It is well established that dietary K+ deficiency activates NCC by phosphorylation through the WNK4-SPAK signaling pathway along the distal convoluted tubule. During K+ deficiency, NEDD4-2 spares NCC and mainly targets the epithelial Na+ channel. Dietary Mg2+ deficiency downregulates NCC, possibly through NEDD4-2. In the state of both Mg2+ and K+ deficiency, downregulation of NCC abundance, due to Mg2+ deficiency, prevents K+ deficiency-mediated NCC activation by phosphorylation through WNK4-SPAK. The effect of Mg2+ deficiency on NCC is independent of WNK4 and SPAK.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-098141 (to J. A. McCormick) and DK-54983 and DK-115366 (to D. Lin), American Heart Association Postdoctoral Fellowship 17POST33670206 (to M. Z. Ferdaus), and Swiss National Science Foundation Grant No. 310030_159735 and a grant from the Leducq Foundation (to O. Staub).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.Z.F., L.N.M., A.T., and J.A.M. conceived and designed research; M.Z.F., A.M., J.W.N., P.J.B., L.N.M., A.T., O.S., D.-H.L., and J.A.M. performed experiments; M.Z.F., A.M., J.W.N., P.J.B., L.N.M., A.T., O.S., D.-H.L., and J.A.M. analyzed data; M.Z.F., P.J.B., L.N.M., A.T., and J.A.M. interpreted results of experiments; M.Z.F., A.M., L.N.M., and D.-H.L. prepared figures; M.Z.F., L.N.M., and J.A.M. drafted manuscript; M.Z.F., J.W.N., P.J.B., L.N.M., O.S., D.-H.L., and J.A.M. edited and revised manuscript; M.Z.F., A.M., J.W.N., P.J.B., L.N.M., A.T., O.S., D.-H.L., and J.A.M. approved final version of manuscript.
REFERENCES
- 1.Al-Qusairi L, Basquin D, Roy A, Rajaram RD, Maillard MP, Subramanya AR, Staub O. Renal tubular ubiquitin-protein ligase NEDD4-2 is required for renal adaptation during long-term potassium depletion. J Am Soc Nephrol 28: 2431–2442, 2017. doi: 10.1681/ASN.2016070732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alessi DR, Zhang J, Khanna A, Hochdörfer T, Shang Y, Kahle KT. The WNK-SPAK/OSR1 pathway: master regulator of cation-chloride cotransporters. Sci Signal 7: re3, 2014. doi: 10.1126/scisignal.2005365. [DOI] [PubMed] [Google Scholar]
- 4.Anast CS, Mohs JM, Kaplan SL, Burns TW. Evidence for parathyroid failure in magnesium deficiency. Science 177: 606–608, 1972. doi: 10.1126/science.177.4049.606. [DOI] [PubMed] [Google Scholar]
- 5.Baker WL. Treating arrhythmias with adjunctive magnesium: identifying future research directions. Eur Heart J Cardiovasc Pharmacother 3: 108–117, 2017. doi: 10.1093/ehjcvp/pvw028. [DOI] [PubMed] [Google Scholar]
- 6.Blaine J, Chonchol M, Levi M. Renal control of calcium, phosphate, and magnesium homeostasis. Clin J Am Soc Nephrol 10: 1257–1272, 2015. doi: 10.2215/CJN.09750913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bowen JW, McDonough A. Pretranslational regulation of Na-K-ATPase in cultured canine kidney cells by low K+. Am J Physiol 252: C179–C189, 1987. doi: 10.1152/ajpcell.1987.252.2.C179. [DOI] [PubMed] [Google Scholar]
- 8.Boyd-Shiwarski CR, Shiwarski DJ, Roy A, Namboodiri HN, Nkashama LJ, Xie J, McClain KL, Marciszyn A, Kleyman TR, Tan RJ, Stolz DB, Puthenveedu MA, Huang CL, Subramanya AR. Potassium-regulated distal tubule WNK bodies are kidney-specific WNK1 dependent. Mol Biol Cell 29: 499–509, 2018. doi: 10.1091/mbc.E17-08-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carney SL, Wong NL, Dirks JH. Effect of magnesium deficiency and excess on renal tubular potassium transport in the rat. Clin Sci (Lond) 60: 549–554, 1981. doi: 10.1042/cs0600549. [DOI] [PubMed] [Google Scholar]
- 10.Castañeda-Bueno M, Cervantes-Perez LG, Rojas-Vega L, Arroyo-Garza I, Vázquez N, Moreno E, Gamba G. Modulation of NCC activity by low and high K+ intake: insights into the signaling pathways involved. Am J Physiol Renal Physiol 306: F1507–F1519, 2014. doi: 10.1152/ajprenal.00255.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chernow B, Bamberger S, Stoiko M, Vadnais M, Mills S, Hoellerich V, Warshaw AL. Hypomagnesemia in patients in postoperative intensive care. Chest 95: 391–397, 1989. doi: 10.1378/chest.95.2.391. [DOI] [PubMed] [Google Scholar]
- 12.Chiga M, Rai T, Yang SS, Ohta A, Takizawa T, Sasaki S, Uchida S. Dietary salt regulates the phosphorylation of OSR1/SPAK kinases and the sodium chloride cotransporter through aldosterone. Kidney Int 74: 1403–1409, 2008. doi: 10.1038/ki.2008.451. [DOI] [PubMed] [Google Scholar]
- 13.Dai LJ, Ritchie G, Kerstan D, Kang HS, Cole DE, Quamme GA. Magnesium transport in the renal distal convoluted tubule. Physiol Rev 81: 51–84, 2001. doi: 10.1152/physrev.2001.81.1.51. [DOI] [PubMed] [Google Scholar]
- 14.de Baaij JH, Groot Koerkamp MJ, Lavrijsen M, van Zeeland F, Meijer H, Holstege FC, Bindels RJ, Hoenderop JG. Elucidation of the distal convoluted tubule transcriptome identifies new candidate genes involved in renal Mg2+ handling. Am J Physiol Renal Physiol 305: F1563–F1573, 2013. doi: 10.1152/ajprenal.00322.2013. [DOI] [PubMed] [Google Scholar]
- 15.de Baaij JH, Hoenderop JG, Bindels RJ. Magnesium in man: implications for health and disease. Physiol Rev 95: 1–46, 2015. doi: 10.1152/physrev.00012.2014. [DOI] [PubMed] [Google Scholar]
- 16.Ebel H, Günther T. Magnesium metabolism: a review. J Clin Chem Clin Biochem 18: 257–270, 1980. [DOI] [PubMed] [Google Scholar]
- 17.Elger M, Bankir L, Kriz W. Morphometric analysis of kidney hypertrophy in rats after chronic potassium depletion. Am J Physiol 262: F656–F667, 1992. doi: 10.1152/ajprenal.1992.262.4.F656. [DOI] [PubMed] [Google Scholar]
- 18.Ellison DH. Ubiquitylation and the pathogenesis of hypertension. J Clin Invest 123: 546–548, 2013. doi: 10.1172/JCI66882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fanestil DD, Hyde RH, Blakely P, Vaughn DA. Dietary magnesium, not calcium, regulates renal thiazide receptor. J Am Soc Nephrol 10: 458–463, 1999. [DOI] [PubMed] [Google Scholar]
- 20.Fazekas T, Scherlag BJ, Vos M, Wellens HJ, Lazzara R. Magnesium and the heart: antiarrhythmic therapy with magnesium. Clin Cardiol 16: 768–774, 1993. doi: 10.1002/clc.4960161105. [DOI] [PubMed] [Google Scholar]
- 21.Ferdaus MZ, Barber KW, López-Cayuqueo KI, Terker AS, Argaiz ER, Gassaway BM, Chambrey R, Gamba G, Rinehart J, McCormick JA. SPAK and OSR1 play essential roles in potassium homeostasis through actions on the distal convoluted tubule. J Physiol 594: 4945–4966, 2016. doi: 10.1113/JP272311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferdaus MZ, Miller LN, Agbor LN, Saritas T, Singer JD, Sigmund CD, McCormick JA. Mutant Cullin 3 causes familial hyperkalemic hypertension via dominant effects. JCI Insight 2: e96700, 2017. doi: 10.1172/jci.insight.96700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fischer PW, Giroux A. Effects of dietary magnesium on sodium-potassium pump action in the heart of rats. J Nutr 117: 2091–2095, 1987. doi: 10.1093/jn/117.12.2091. [DOI] [PubMed] [Google Scholar]
- 24.Giebisch G. Renal potassium channels: function, regulation, and structure. Kidney Int 60: 436–445, 2001. doi: 10.1046/j.1523-1755.2001.060002436.x. [DOI] [PubMed] [Google Scholar]
- 25.Gimenez MS, Oliveros LB, Gomez NN. Nutritional deficiencies and phospholipid metabolism. Int J Mol Sci 12: 2408–2433, 2011. doi: 10.3390/ijms12042408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gleason CE, Frindt G, Cheng CJ, Ng M, Kidwai A, Rashmi P, Lang F, Baum M, Palmer LG, Pearce D. mTORC2 regulates renal tubule sodium uptake by promoting ENaC activity. J Clin Invest 125: 117–128, 2015. doi: 10.1172/JCI73935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grimm PR, Taneja TK, Liu J, Coleman R, Chen YY, Delpire E, Wade JB, Welling PA. SPAK isoforms and OSR1 regulate sodium-chloride co-transporters in a nephron-specific manner. J Biol Chem 287: 37673–37690, 2012. doi: 10.1074/jbc.M112.402800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gröber U, Schmidt J, Kisters K. Magnesium in prevention and therapy. Nutrients 7: 8199–8226, 2015. doi: 10.3390/nu7095388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang CL, Kuo E. Mechanism of hypokalemia in magnesium deficiency. J Am Soc Nephrol 18: 2649–2652, 2007. doi: 10.1681/ASN.2007070792. [DOI] [PubMed] [Google Scholar]
- 30.Jahnen-Dechent W, Ketteler M. Magnesium basics. Clin Kidney J 5, Suppl 1: i3–i14, 2012. doi: 10.1093/ndtplus/sfr163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lamothe SM, Zhang S. Chapter five−ubiquitination of ion channels and transporters. Prog Mol Biol Transl Sci 141: 161–223, 2016. doi: 10.1016/bs.pmbts.2016.02.005. [DOI] [PubMed] [Google Scholar]
- 32.Laurant P, Hayoz D, Brunner HR, Berthelot A. Effect of magnesium deficiency on blood pressure and mechanical properties of rat carotid artery. Hypertension 33: 1105–1110, 1999. doi: 10.1161/01.HYP.33.5.1105. [DOI] [PubMed] [Google Scholar]
- 33.Lee IH, Campbell CR, Cook DI, Dinudom A. Regulation of epithelial Na+ channels by aldosterone: role of Sgk1. Clin Exp Pharmacol Physiol 35: 235–241, 2008. doi: 10.1111/j.1440-1681.2007.04844.x. [DOI] [PubMed] [Google Scholar]
- 34.Li H, Sun S, Chen J, Xu G, Wang H, Qian Q. Genetics of magnesium disorders. Kidney Dis (Basel) 3: 85–97, 2017. doi: 10.1159/000477730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu M, Jeong EM, Liu H, Xie A, So EY, Shi G, Jeong GE, Zhou A, Dudley SC Jr. Magnesium supplementation improves diabetic mitochondrial and cardiac diastolic function. JCI Insight 4: e123182, 2019. doi: 10.1172/jci.insight.123182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maguire ME, Cowan JA. Magnesium chemistry and biochemistry. Biometals 15: 203–210, 2002. doi: 10.1023/A:1016058229972. [DOI] [PubMed] [Google Scholar]
- 37.Massy ZA, Nistor I, Apetrii M, Brandenburg VM, Bover J, Evenepoel P, Goldsmith D, Mazzaferro S, Urena-Torres P, Vervloet MG, Cozzolino M, Covic A, Era-Edta OB. Magnesium-based interventions for normal kidney function and chronic kidney disease. Magnes Res 29: 126–140, 2016. doi: 10.1684/mrh.2016.0412. [DOI] [PubMed] [Google Scholar]
- 38.Matsuda H, Saigusa A, Irisawa H. Ohmic conductance through the inwardly rectifying K channel and blocking by internal Mg2+. Nature 325: 156–159, 1987. doi: 10.1038/325156a0. [DOI] [PubMed] [Google Scholar]
- 39.McCormick JA, Ellison DH. Distal convoluted tubule. Compr Physiol 5: 45–98, 2015. doi: 10.1002/cphy.c140002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCormick JA, Mutig K, Nelson JH, Saritas T, Hoorn EJ, Yang CL, Rogers S, Curry J, Delpire E, Bachmann S, Ellison DH. A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab 14: 352–364, 2011. doi: 10.1016/j.cmet.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCormick JA, Nelson JH, Yang CL, Curry JN, Ellison DH. Overexpression of the sodium chloride cotransporter is not sufficient to cause familial hyperkalemic hypertension. Hypertension 58: 888–894, 2011. doi: 10.1161/HYPERTENSIONAHA.110.167809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCormick JA, Yang CL, Zhang C, Davidge B, Blankenstein KI, Terker AS, Yarbrough B, Meermeier NP, Park HJ, McCully B, West M, Borschewski A, Himmerkus N, Bleich M, Bachmann S, Mutig K, Argaiz ER, Gamba G, Singer JD, Ellison DH. Hyperkalemic hypertension-associated cullin 3 promotes WNK signaling by degrading KLHL3. J Clin Invest 124: 4723–4736, 2014. doi: 10.1172/JCI76126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McDonough AA, Veiras LC, Minas JN, Ralph DL. Considerations when quantitating protein abundance by immunoblot. Am J Physiol Cell Physiol 308: C426–C433, 2015. doi: 10.1152/ajpcell.00400.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43a.National Research Council Nutrient Requirements of Laboratory Animals: Fourth Revised Edition, 1995. Washington, DC: National Academies Press, 1995. [PubMed] [Google Scholar]
- 44.Ortega B, MacWilliams JR, Dey JM, Courtright VB. Hyperphosphatemia, hypocalcemia and increased serum potassium concentration as distinctive features of early hypomagnesemia in magnesium-deprived mice. Magnes Res 28: 126–135, 2015. doi: 10.1684/mrh.2015.0394. [DOI] [PubMed] [Google Scholar]
- 45.Palmer LG, Schnermann J. Integrated control of Na transport along the nephron. Clin J Am Soc Nephrol 10: 676–687, 2015. doi: 10.2215/CJN.12391213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pearce D, Soundararajan R, Trimpert C, Kashlan OB, Deen PM, Kohan DE. Collecting duct principal cell transport processes and their regulation. Clin J Am Soc Nephrol 10: 135–146, 2015. doi: 10.2215/CJN.05760513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Penton D, Czogalla J, Wengi A, Himmerkus N, Loffing-Cueni D, Carrel M, Rajaram RD, Staub O, Bleich M, Schweda F, Loffing J. Extracellular K+ rapidly controls NaCl cotransporter phosphorylation in the native distal convoluted tubule by Cl−-dependent and independent mechanisms. J Physiol 594: 6319–6331, 2016. doi: 10.1113/JP272504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pham PC, Pham PA, Pham SV, Pham PT, Pham PM, Pham PT. Hypomagnesemia: a clinical perspective. Int J Nephrol Renovasc Dis 7: 219–230, 2014. doi: 10.2147/IJNRD.S42054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rebholz CM, Tin A, Liu Y, Kuczmarski MF, Evans MK, Zonderman AB, Crews DC. Dietary magnesium and kidney function decline: the healthy aging in neighborhoods of diversity across the life span study. Am J Nephrol 44: 381–387, 2016. doi: 10.1159/000450861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Richardson C, Rafiqi FH, Karlsson HK, Moleleki N, Vandewalle A, Campbell DG, Morrice NA, Alessi DR. Activation of the thiazide-sensitive Na+-Cl- cotransporter by the WNK-regulated kinases SPAK and OSR1. J Cell Sci 121: 675–684, 2008. doi: 10.1242/jcs.025312. [DOI] [PubMed] [Google Scholar]
- 51.Rizzo F, Staub O. NEDD4-2 and salt-sensitive hypertension. Curr Opin Nephrol Hypertens 24: 111–116, 2015. doi: 10.1097/MNH.0000000000000097. [DOI] [PubMed] [Google Scholar]
- 52.Rojas-Vega L, Reyes-Castro LA, Ramírez V, Bautista-Pérez R, Rafael C, Castañeda-Bueno M, Meade P, de Los Heros P, Arroyo-Garza I, Bernard V, Binart N, Bobadilla NA, Hadchouel J, Zambrano E, Gamba G. Ovarian hormones and prolactin increase renal NaCl cotransporter phosphorylation. Am J Physiol Renal Physiol 308: F799–F808, 2015. doi: 10.1152/ajprenal.00447.2014. [DOI] [PubMed] [Google Scholar]
- 53.Ronzaud C, Loffing-Cueni D, Hausel P, Debonneville A, Malsure SR, Fowler-Jaeger N, Boase NA, Perrier R, Maillard M, Yang B, Stokes JB, Koesters R, Kumar S, Hummler E, Loffing J, Staub O. Renal tubular NEDD4-2 deficiency causes NCC-mediated salt-dependent hypertension. J Clin Invest 123: 657–665, 2013. doi: 10.1172/JCI61110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rosenbaek LL, Kortenoeven ML, Aroankins TS, Fenton RA. Phosphorylation decreases ubiquitylation of the thiazide-sensitive cotransporter NCC and subsequent clathrin-mediated endocytosis. J Biol Chem 289: 13347–13361, 2014. doi: 10.1074/jbc.M113.543710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ryzen E. Magnesium homeostasis in critically ill patients. Magnesium 8: 201–212, 1989. [PubMed] [Google Scholar]
- 56.Saritas T, Puelles VG, Su XT, McCormick JA, Welling PA, Ellison DH. Optical clearing in the kidney reveals potassium-mediated tubule remodeling. Cell Reports 25: 2668–2675.e3, 2018. doi: 10.1016/j.celrep.2018.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schultheis PJ, Lorenz JN, Meneton P, Nieman ML, Riddle TM, Flagella M, Duffy JJ, Doetschman T, Miller ML, Shull GE. Phenotype resembling Gitelman’s syndrome in mice lacking the apical Na+-Cl- cotransporter of the distal convoluted tubule. J Biol Chem 273: 29150–29155, 1998. doi: 10.1074/jbc.273.44.29150. [DOI] [PubMed] [Google Scholar]
- 58.Shoda W, Nomura N, Ando F, Mori Y, Mori T, Sohara E, Rai T, Uchida S. Calcineurin inhibitors block sodium-chloride cotransporter dephosphorylation in response to high potassium intake. Kidney Int 91: 402–411, 2017. doi: 10.1016/j.kint.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 59.Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, Vaara I, Iwata F, Cushner HM, Koolen M, Gainza FJ, Gitleman HJ, Lifton RP. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet 12: 24–30, 1996. doi: 10.1038/ng0196-24. [DOI] [PubMed] [Google Scholar]
- 60.Subramanya AR, Ellison DH. Distal convoluted tubule. Clin J Am Soc Nephrol 9: 2147–2163, 2014. doi: 10.2215/CJN.05920613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Terker AS, Zhang C, Erspamer KJ, Gamba G, Yang CL, Ellison DH. Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int 89: 127–134, 2016. doi: 10.1038/ki.2015.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Terker AS, Zhang C, McCormick JA, Lazelle RA, Zhang C, Meermeier NP, Siler DA, Park HJ, Fu Y, Cohen DM, Weinstein AM, Wang WH, Yang CL, Ellison DH. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab 21: 39–50, 2015. doi: 10.1016/j.cmet.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Toback FG, Walsh-Reitz MM, Mendley SR, Kartha S. Kidney epithelial cells release growth factors in response to extracellular signals. Pediatr Nephrol 4: 363–371, 1990. doi: 10.1007/BF00862521. [DOI] [PubMed] [Google Scholar]
- 64.Traykova-Brauch M, Schönig K, Greiner O, Miloud T, Jauch A, Bode M, Felsher DW, Glick AB, Kwiatkowski DJ, Bujard H, Horst J, von Knebel Doeberitz M, Niggli FK, Kriz W, Gröne HJ, Koesters R. An efficient and versatile system for acute and chronic modulation of renal tubular function in transgenic mice. Nat Med 14: 979–984, 2008. doi: 10.1038/nm.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vallon V, Schroth J, Lang F, Kuhl D, Uchida S. Expression and phosphorylation of the Na+-Cl- cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am J Physiol Renal Physiol 297: F704–F712, 2009. doi: 10.1152/ajprenal.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van Angelen AA, San-Cristobal P, Pulskens WP, Hoenderop JG, Bindels RJ. The impact of dietary magnesium restriction on magnesiotropic and calciotropic genes. Nephrol Dial Transplant 28: 2983–2993, 2013. doi: 10.1093/ndt/gft358. [DOI] [PubMed] [Google Scholar]
- 67.van der Wijst J, Bindels RJ, Hoenderop JG. Mg2+ homeostasis: the balancing act of TRPM6. Curr Opin Nephrol Hypertens 23: 361–369, 2014. doi: 10.1097/01.mnh.0000447023.59346.ab. [DOI] [PubMed] [Google Scholar]
- 68.Van Laecke S, Nagler EV, Verbeke F, Van Biesen W, Vanholder R. Hypomagnesemia and the risk of death and GFR decline in chronic kidney disease. Am J Med 126: 825–831, 2013. doi: 10.1016/j.amjmed.2013.02.036. [DOI] [PubMed] [Google Scholar]
- 69.van Megen WH, Grimm PR, Welling PA, van der Wijst J. Renal sodium and magnesium reabsorption are not coupled in a mouse model of Gordon syndrome. Physiol Rep 6: e13728, 2018. doi: 10.14814/phy2.13728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vandenberg CA. Inward rectification of a potassium channel in cardiac ventricular cells depends on internal magnesium ions. Proc Natl Acad Sci USA 84: 2560–2564, 1987. doi: 10.1073/pnas.84.8.2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wade JB, Liu J, Coleman R, Grimm PR, Delpire E, Welling PA. SPAK-mediated NCC regulation in response to low-K+ diet. Am J Physiol Renal Physiol 308: F923–F931, 2015. doi: 10.1152/ajprenal.00388.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang J, Peng Q, Lin Q, Childress C, Carey D, Yang W. Calcium activates Nedd4 E3 ubiquitin ligases by releasing the C2 domain-mediated auto-inhibition. J Biol Chem 285: 12279–12288, 2010. doi: 10.1074/jbc.M109.086405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Warnock DG. Renal genetic disorders related to K+ and Mg2+. Annu Rev Physiol 64: 845–876, 2002. doi: 10.1146/annurev.physiol.64.081501.160000. [DOI] [PubMed] [Google Scholar]
- 74.Weiner ID, Wingo CS. Hypokalemia−consequences, causes, and correction. J Am Soc Nephrol 8: 1179–1188, 1997. [DOI] [PubMed] [Google Scholar]
- 75.Weinstein AM. A mathematical model of rat distal convoluted tubule. I. Cotransporter function in early DCT. Am J Physiol Renal Physiol 289: F699–F720, 2005. doi: 10.1152/ajprenal.00043.2005. [DOI] [PubMed] [Google Scholar]
- 76.Weinstein AM. A mathematical model of rat distal convoluted tubule. II. Potassium secretion along the connecting segment. Am J Physiol Renal Physiol 289: F721–F741, 2005. doi: 10.1152/ajprenal.00044.2005. [DOI] [PubMed] [Google Scholar]
- 77.Wolf MT. Inherited and acquired disorders of magnesium homeostasis. Curr Opin Pediatr 29: 187–198, 2017. doi: 10.1097/MOP.0000000000000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wong NL, Sutton RA, Mavichak V, Quamme GA, Dirks JH. Enhanced distal absorption of potassium by magnesium-deficient rats. Clin Sci (Lond) 69: 625–630, 1985. doi: 10.1042/cs0690625. [DOI] [PubMed] [Google Scholar]
- 79.Wu A, Wolley M, Stowasser M. The interplay of renal potassium and sodium handling in blood pressure regulation: critical role of the WNK-SPAK-NCC pathway. J Hum Hypertens 33: 508–523, 2019. doi: 10.1038/s41371-019-0170-6. [DOI] [PubMed] [Google Scholar]
- 80.Yang L, Frindt G, Palmer LG. Magnesium modulates ROMK channel-mediated potassium secretion. J Am Soc Nephrol 21: 2109–2116, 2010. doi: 10.1681/ASN.2010060617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yang SS, Fang YW, Tseng MH, Chu PY, Yu IS, Wu HC, Lin SW, Chau T, Uchida S, Sasaki S, Lin YF, Sytwu HK, Lin SH. Phosphorylation regulates NCC stability and transporter activity in vivo. J Am Soc Nephrol 24: 1587–1597, 2013. doi: 10.1681/ASN.2012070742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang SS, Lo YF, Wu CC, Lin SW, Yeh CJ, Chu P, Sytwu HK, Uchida S, Sasaki S, Lin SH. SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J Am Soc Nephrol 21: 1868–1877, 2010. doi: 10.1681/ASN.2009121295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang SS, Lo YF, Yu IS, Lin SW, Chang TH, Hsu YJ, Chao TK, Sytwu HK, Uchida S, Sasaki S, Lin SH. Generation and analysis of the thiazide-sensitive Na+-Cl− cotransporter (Ncc/Slc12a3) Ser707X knockin mouse as a model of Gitelman syndrome. Hum Mutat 31: 1304–1315, 2010. doi: 10.1002/humu.21364. [DOI] [PubMed] [Google Scholar]
- 84.Zhang A, Cheng TP, Altura BM. Magnesium regulates intracellular free ionized calcium concentration and cell geometry in vascular smooth muscle cells. Biochim Biophys Acta 1134: 25–29, 1992. doi: 10.1016/0167-4889(92)90024-6. [DOI] [PubMed] [Google Scholar]