

Abstract
Perampanel (PER) is a selective blocker of AMPA receptors showing efficacy in treating various epileptic disorders including brain tumor-related epilepsy and also potential in treating motor neuron disease. However, besides its inhibition of AMPA-induced currents, whether PER has any other direct ionic effects in different types of neurons remains largely unknown. We investigated the effects of PER and related compounds on ionic currents in different types of cells, including hippocampal mHippoE-14 neurons, motor neuron-like NSC-34 cells and U87 glioma cells. We found that PER differentially and effectively suppressed the amplitude of voltage-gated Na+ currents (INa) in mHippoE-14 cells. The IC50 values required to inhibit peak and late INa were 4.12 and 0.78 μM, respectively. PER attenuated tefluthrin-induced increases in both amplitude and deactivating time constant of INa. Importantly, PER also inhibited the amplitude of M-type K+ currents (IK(M)) with an IC50 value of 0.92 μM. The suppression of IK(M) was attenuated by the addition of flupirtine or ZnCl2 but not by L-quisqualic acid or sorafenib. Meanwhile, in cell-attached configuration, PER (3 μM) decreased the activity of M-type K+ channels with no change in single-channel conductance but shifting the activation curve along the voltage axis in a rightward direction. Supportively, PER suppressed IK(M) in NSC-34 cells and INa in U87 glioma cells. The inhibitory effects of PER on both INa and IK(M), independent of its antagonistic effect on AMPA receptors, may be responsible for its wide-spectrum of effects observed in neurological clinical practice.
Keywords: perampanel, M-type K+ current, Na+ current, hippocampal cell, motor neuron, glioma cell
1. Introduction
Perampanel (PER, Fycompa®) is the first approved anti-epileptic drug (AED) of the selective non-competitive antagonist of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-type receptor class [1,2,3]. This class of AMPA-type receptors is the major subtype of ionotropic glutamate receptors. PER has been demonstrated to reduce neuronal excitation primarily through the blockade of AMPA receptors [1,2,3]. As a once-daily oral AED, it has been approved globally for the adjunctive treatment of partial epilepsy and primary generalized tonic-clonic seizures [4,5,6], although the underlying mechanism of actions through which PER exerts its broad-spectrum antiepileptic effect has not been fully elucidated.
Several studies have shown that PER can ameliorate painful sensations such as spontaneous pain symptoms [7,8,9]. PER has also been demonstrated to potentially improve post-stroke functional and cognitive impairments through multiple pathways [10,11]. In addition, PER has also been reported to be beneficial for patients with Parkinson’s disease and motor neuron diseases [12,13,14,15] and also in treating glioma-associated epilepsy [16,17,18,19]. However, several reports have shown that some of the actions of PER may not be closely linked to its blocking effect on AMPA receptors [11,20].
Molecular studies of epileptogenesis have demonstrated that specific ion channels play important roles in both genetic and acquired forms of epilepsy, particularly voltage-gated Na+ (NaV) channels [21,22,23]. Nine isoforms (NaV1.1-1.9) are found in mammalian excitable tissues, including the central nervous system, peripheral nervous system, skeletal muscles and heart [24]. Moreover, inhibitors of the late component of voltage-gated Na+ currents (INa) such as ranolazine and eugenol have been reported to suppress seizure activity [25,26]. However, whether PER exerts any effects on the amplitude and gating of INa remains unclear.
Among the K+ channels, the KCNQ2, KCNQ3 and KCNQ5 genes are known to encode the core subunits of KV7.2, KV7.3 and KV7.5 channels, respectively. The enhanced activity of these voltage-gated (KV) channels can generate a population of K+ currents, namely M-type K+ currents (IK(M)), which display a slowly activating and deactivating property [27,28,29]. Targeting IK(M) has been reported to be an adjunctive management strategy for various neurological disorders including epilepsy [22,29]. The involvement of IK(M) has also been shown in motor neuron hyperexcitability in patient with motor neuron diseases [30]. Alternatively, the magnitude of delayed-rectifier K+ currents (e.g., Kv2.1- or KV1.1-encoded current) has been demonstrated to be linked to epileptogenesis [31,32]. However, whether PER exerts any actions on these types of ionic currents remains unexplored.
Therefore, in this study, we explored the possible modulatory effects of PER on different types of ionic currents (e.g., voltage-gated Na+ currents [INa], M-type K+ currents [IK(M)] and delayed-rectifier K+ currents [IK(DR)]) in hippocampal mHippoE-14 cells. We also investigated the effects of PER on IK(M) in motor neuron-like NSC-34 cells and on INa in U87 glioma cells. The findings showed that PER could differentially suppress the peak and late components of INa in both mHippoE-14 cells and U87 glioma cells and that it could suppress IK(M) in mHippoE-14 cells and NSC-34 cells. Therefore, in addition to its blocking effects on AMPA receptors, the actions revealed in this study may also contribute to its in vivo pharmacological actions.
2. Materials and Methods
2.1. Drugs and Solutions
Perampanel (PER, Fycompa®, 2-(2-oxo-1-phenyl-5-pyridin-2-yl-1,2-dihydropyridin-3-yl) benzonitrile, 3-(2-cyanophenyl)-5-(2-pyridyl)-1-phenyl-1,2-dihydropyridin-2-one, E2007, C23H15N3O) was obtained from Eisai Co., Ltd. (Tokyo, Japan). Diazoxide, 9-phenanthrol, tefluthrin, tetraethylammonium chloride (TEA) and tetrodotoxin (TTX) were purchased from Sigma-Aldrich (St. Louis, MO, USA). L-quisqualic acid (QA) and NBQX disodium salt (2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinozaline-7-sulfonamide disodium salt) were purchased from Tocris (Bristol, UK) and gefitinib and sorafenib were obtained from Selleck (Houston, TX, USA). Tissue culture media, fetal bovine serum, L-glutamine and trypsin were obtained from Invitrogen (Carlsbad, CA, USA) and all other chemicals including CsCl, CsOH and ZnCl2 were regular commercial chemicals and of reagent grade. Deionized water was made using a Milli-Q water purification system (Millipore, Bedford, MA, USA).
The bath solution (i.e., HEPES-buffered normal Tyrode’s solution) used in this study was composed of 136 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 0.53 mM MgCl2, 5.5 mM glucose and 5.5 mM HEPES-NaOH buffer, pH 7.4. To measure macroscopic K+ currents (e.g., IK(M) or IK(DR)) and to preclude contamination of Cl- currents, we filled patch pipettes with solution containing 130 mM K-aspartate, 20 mM KCl, 1 mM KH2PO4, 1 mM MgCl2, 3 mM Na2ATP, 0.1 mM Na2GTP, 0.1 mM EGTA and 5 mM HEPES-KOH buffer, pH 7.2. To record INa, we replaced K+ ions in the filling solution of the electrode with equimolar Cs+ ions and the pH was adjusted to 7.2 with CsOH. For measurements of whole-cell IK(M), we used a high K+-bathing solution consisting of 145 mM KCl, 0.53 mM MgCl2 and 5 mM HEPES-KOH buffer, pH 7.4, while the recording pipette was filled with a solution containing 145 mM KCl, 2 mM MgCl2 and 5 mM HEPES-KOH buffer, pH 7.2. To measure the activity of single KM channels, we used a pipette solution of NaCl 136.5 mM, KCl 5.4 mM, MgCl2 0.53 mM and HEPES-NaOH buffer 5 mM, pH 7.4. All solutions were prepared using deionized water from a Milli-Q water purification system (APS Water Services, Inc., Van Nuys, CA). The pipette solution and culture medium were filtered on the day of use with an Acrodisc® syringe filter with a 0.2 μm Supor® membrane (Pall Corp., Port Washington, NY, USA).
2.2. Cell Preparations
Embryonic mouse hippocampal cell line (mHippoE-14, CLU198) was obtained from Cedarlane CELLutions Biosystems, Inc. (Burlington, ON, Canada) [29,33,34]. Cells were maintained at a density of 106/mL in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 2 mM L-glutamine. NSC-34 neuronal cells were kindly provided by Professor Dr. Yuh-Jyh Jong (Department of Pediatrics, Kaohsiung Medical University Hospital, Kaohsiung, Taiwan). They were grown in DMEM supplemented with 10% FBS. The glioblastoma multiforme cell line (U87) was obtained from American Type Culture Collection (ATCC, Manassas, VA) and the cells were grown in DMEM/F12 nutrient media (Invitrogen, Carlsbad, CA) supplemented with 10% FBS. The culture medium was changed every 2 to 3 days and the cells were passaged when they reached confluence. The experiments were usually performed 5 or 6 days after the cells had been cultured (60-80% confluence).
2.3. Electrophysiological Measurements
On the day of the measurements, mHippoE-14, NSC-34 or U78 cells were dissociated and an aliquot of cell suspension was immediately transferred to a custom-made recording chamber affixed to the stage of a DM-IL inverted microscope (Leica, Wetzlar, Germany). To visualize changes in cell size during the recordings, the microscope was coupled to a digital video system (DCR-TRV30; Sony, Japan) with a magnification of up to 1500×. The cells were bathed at room temperature (20–25 °C) in normal Tyrode’s solution, the composition of which is described above. The patch electrodes were fabricated with Kimax-51 capillaries with an outside diameter of 1.5 to 2.0 mm (#34500; Kimble, Vineland, NJ) using either a vertical PP-83 puller (Narishige, Tokyo, Japan) or a horizontal P-97 Flaming/Brown micropipette puller (Sutter, Novato, CA) and their tips were fire polished with an MF-83 microforge (Narishige). The electrodes used for the recordings had a tip resistance of 3-5 MΩ when filled with different internal solutions as described above. Ion currents were measured in whole-cell or cell-attached configurations of the standard patch-clamp technique with either an RK-400 (Bio-Logic, Claix, France) or an Axopatch-200B (Molecular Devices, Sunnyvale, CA, USA) amplifier [35]. All potentials were offset for liquid junction potentials which arose at the electrode tip when the composition of the pipette solution was different from that in the bath. Single KM-channel activity measured from the mHippoE-14 cells was analyzed using pCLAMP 10.7 software (Molecular Devices).
2.4. Data Recordings
Data comprising both potential and current traces were stored online in an Acer SPIN-5 touchscreen laptop computer (SP513-52N-55WE; Taipe, Taiwan) at 10 kHz equipped with a Digidata 1440A interface (Molecular Devices, Inc., Sunnyvale, CA), which was used for analog-to-digital/digital-to-analog conversion. During the recordings, the latter device was controlled by pCLAMP 10.7 software (Molecular Devices) run under Windows 10 (Redmond, WA, USA) and the signals were simultaneously monitored on an LCD monitor (MB169B+; ASUS, Taipei, Taiwan) through a USB type-C connection. Current signals were low-pass filtered at 2 kHz with a FL-4 four-pole Bessel filter (Dagan, Minneapolis, MN, USA) to minimize background noise. Through digital-to-analog conversion, various pCLAMP-generated voltage-clamp profiles with different waveforms were applied to establish the current-voltage (I-V) relationship of IK(M) or IK(DR). As high-frequency stimuli were needed, an Astro-med Grass S88X dual output pulse stimulator (Grass Technologies, West Warwick, RI, USA) was used.
2.5. Data Analysis
The digital signals were examined and analyzed offline using either pCLAMP 10.7 (Molecular Devices), 64-bit OriginPro 2016 (OriginLab, Northampton, MA, USA) or custom-made macros created in Microsoft Excel® 2013 which was run under Windows 10 (Redmond, WA, USA). The concentration-response data for the inhibition of either peak and late INa, IK(M) or IK(DR) in the mHippoE-14 cells were least-squares fitted to the Hill equation as follows:
| (1) |
where [PER] is the PER concentration, IC50 and nH are the concentrations required for a 50% inhibition and the Hill coefficient, respectively and Emax is the maximal suppression of both peak or late INa and IK(M) caused by the presence of PER.
2.6. Single-Channel Analysis
Single KM-channel currents recorded from mHippoE-14 neurons were analyzed using pCLAMP 10.7. Single-channel amplitudes measured with or without the addition of PER were determined by fitting Gaussian distributions to the amplitude histograms of the closed and open states. The single-channel conductance of KM channels was estimated using linear regression with averaged values of single-channel amplitudes measured at different levels of membrane potentials relative to the bath. To determine the voltage dependence of the inhibitory effect of PER on the activity of KM channels, the patch obtained with or without the addition of PER was held at different membrane potentials. The activation curves of KM-channel openings in the absence and presence of PER (3 μM) were appropriately fitted by the Boltzmann equation:
| (2) |
where Pmax is the maximal probability of KM-channel openings in the control (i.e., in the absence of PER) maintained at +70 mV relative to the bath, V1/2 is the voltage at which half-maximal activation of KM channels occurs, q is the apparent gating charge, F is Faraday’s constant, R is the universal gas constant, T is absolution temperature and F/RT = 0.04 mV−1.
2.7. Statistical Analysis
The data were analyzed and expressed as mean ± standard error of the mean (SEM). The paired or unpaired Student’s t-test or one-way analysis of variance (ANOVA) followed by post-hoc Fisher’s least-significance difference test for multiple-group comparisons were used for statistical evaluations of the differences among means. We used the non-parametric Kruskal-Wallis test, as the assumption of normality underlying ANOVA was violated. Statistical analyses were performed using IBM SPSS® version 20.0 (IBM Corp., Armonk, NY, USA). p < 0.05 was considered to be statistically significant.
3. Results
3.1. Inhibitory Effect of PER on Voltage-Gated Na+ Current (Ina) in Hippocampal Mhippoe-14 Cells
Although PER is a known antagonist of AMPA receptors, little is known regarding whether PER exerts any effects on ionic currents other than its antagonistic effect on AMPA-induced currents or Ca2+ rise. Therefore, in the initial set of experiments, cells were bathed in Ca2+-free Tyrode’s solution containing 10 mM tetraethylammonium chloride (TEA) and the recording pipette was loaded with a Cs+-containing solution. As shown in Figure 1, upon membrane depolarization from -80 to -10 mV, the whole-cell INa, which comprised the peak and late components of the current, was readily elicited in these cells. In particular, the addition of PER progressively and differentially suppressed peak and late INa in a concentration-dependent manner. For example, when mHippoE-14 cells were rapidly depolarized from −80 to −10 mV, the addition of PER (3 μM) decreased the peak amplitude of INa (i.e., at the beginning of the depolarizing pulse) from 469 ± 22 to 226 ± 26 pA (n = 13, p < 0.05); however, it also reduced the late component of INa (i.e., at the end of the depolarizing pulse) from 13.7 ± 2.7 to 4.2 ± 1.1 pA (n = 13, p < 0.05). After washout of the compound, the amplitudes of peak and late INa returned to 452 ± 21 and 12.3 ± 2.3 pA (n = 12, p < 0.05), respectively.
Figure 1.

Inhibitory effects of perampanel (PER) on the peak and late components of voltage-gated Na+ current (INa) in mouse hippocampal mHippoE-14 cells. In these experiments, we immersed the cells in Ca2+-free Tyrode’s solution containing 10 mM tetraethylammonium chloride (TEA) and the recording pipette was filled with a Cs+-containing solution. Once the whole-cell mode had been firmly established, the voltage protocol from −80 mV to different voltages with a duration of 80 msec was applied at a rate of 0.5 Hz. (A) Superimposed INa traces obtained in the controls (a) and presence (b) of 3 μM PER. The uppermost part shows the voltage protocol used. The lower part depicts an expanded record of late INa (INa,L) as indicated in the dashed box. (B) Concentration-response relationships for PER-induced suppression of INa measured at the beginning (□) and end (■) of the depolarizing pulses (mean ± SEM; n = 13 for each point). Smooth lines represent the least-squares fit to a modified Hill function as described in the Materials and Methods.
The relationships between PER concentration and the percentages of inhibition of the peak and late components of INa are illustrated in Figure 1B. The IC50 values required for the inhibitory effects of PER on peak and late INa were 4.12 and 0.78 μM, respectively. Therefore, PER exerted a significant action on the inhibition of peak and late INa in a concentration-dependent manner. More specifically, during cell exposure to PER, late INa in response to rapid depolarization was suppressed to a greater extent than peak INa.
3.2. The presence of Tefluthrin (Tef) and Tef Plus PER on Ina in Mhippoe-14 Cells
Tef, a type-I pyrethroid, has been demonstrated to activate INa [25,36,37]. We next explored whether in the continued presence of Tef, the subsequent addition of PER could suppress depolarization-induced INa. As shown in Figure 2A,B, consistent with previous observations [25,36], during cell exposure to 10 μM Tef, the peak amplitude of depolarization-elicited INa was progressively increased along with an increase in deactivating time constant (τdeact) measured at the level of −30 mV. Of note, the subsequent addition of PER (3 μM), still in the presence of 10 μM Tef, was effective both at suppressing Tef-stimulation of INa and at reversing its increase in τdeact. For example, the addition of Tef (10 μM) increased peak INa from 214 ± 11 to 276 ± 15 pA (n = 12, p < 0.05) and prolonged the slow component of τdeact from 8.7 ± 1.1 to 44.9 ± 6.5 msec (n = 12, p < 0.05). In the continued presence of Tef, the further addition of 3 μM PER significantly decreased peak INa and τdeact to 53 ± 7 pA and 11.2 ± 1.3 msec (n = 12, p < 0.05), respectively. Therefore, despite the presence of an INa stimulator (e.g., Tef), PER still remained effective at suppressing peak and late INa in mHippoE-14 cells.
Figure 2.

Effects of tefluthrin (Tef) and Tef plus PER on INa in mHippoE-14 cells. These experiments used the same protocol as described above. Tef (10 μM) was added to the bath and in the continued presence of Tef, PER was subsequently applied to the investigated cells at different concentrations. (A) Original INa traces obtained in the control (a) or in the presence of 10 μM Tef alone (b), 10 μM Tef plus 1 μM PER (c) and 10 μM Tef plus 3 μM PER (d). The upper part indicates the voltage-step protocol used; that is, the examined cells were depolarized from −80 to 0 mV for 30 msec, then repolarized to −30 mV. (B) and (C), respectively, depict the summary bar graphs indicating the peak amplitude and deactivation time constant (τdeact) of depolarization-induced INa obtained in the controls and during the exposure to Tef and Tef plus PER (mean ± SEM; n = 12 for each point). (1): controls (in the absence of Tef or PER); (2): 10 μM Tef alone; (3): 10 μM Tef plus 1 μM PER; (4): 10 μM Tef plus 3 μM PER. * Significantly different from the controls (i.e., bars (1)) (p < 0.05) and †significantly different from the 10 μM Tef alone group (i.e., bars (2)) (p < 0.05).
3.3. Effect of PER on M-type K+ Currents (IK(M)) in Mhippoe-14 Cells
To elucidate whether PER can perturb other types of ionic currents in neurons, we further studied its effects on IK(M) in response to long-lasting depolarizing pulses. To measure IK(M), cells were bathed in high-K+, Ca2+-free solution and the recording pipette was filled with K+-containing solution. Once the whole-cell mode had been established, voltage pulses from −50 to −10 mV with a duration of 1 sec were delivered to the cells and an IK(M) was readily evoked [29,38]. Of note, as the cells were exposed to increasing concentrations of PER, the IK(M) amplitude in response to membrane depolarization progressively decreased (Figure 3A,B). For example, the addition of PER (1 μM) decreased the current amplitude at the end of the depolarizing pulse from 141 ± 11 to 67 ± 9 pA (n = 12, p < 0.05). After washout of PER, the current amplitude returned to 137 ± 11 pA (n = 11, p < 0.05). Likewise, the presence of PER also decreased the τdeact value of IK(M) obtained upon return to −50 mV (Figure 3D). For example, the τdeact of IK(M) obtained under our experimental conditions was also significantly decreased from 89 ± 9 to 21 ± 5 msec (n = 12, p < 0.05) in the presence of PER (1 μM). As illustrated in Figure 3B, PER (0.03–10 μM) effectively suppressed the IK(M) amplitude in a concentration-dependent manner. From the Hill equation, the values of IC50 and Hill coefficient for PER-inhibited IK(M) were estimated to be 0.92 μM and 1.2, respectively. These results clearly showed that, in addition to the suppression of both peak and late components of INa, the presence of PER per se was effective at suppressing the amplitude of IK(M) in mHippoE-14 cells. However, NBQX disodium salt (10 µM), another antagonist of AMPA receptors, did not have any effect on IK(M) amplitude (143 ± 12 pA [in the control] versus 142 ± 13 pA [in the presence of 10 µM NBQX disodium salt], n = 8, p > 0.05). Of note and unexpectedly, as the cells were constantly exposed to PER, the subsequent addition of either flupirtine (10 μM) or ZnCl2 (10 μM) but not L-quisqualic acid (QA; 10 μM) or sorafenib (10 μM), significantly reversed the PER-mediated suppression of IK(M) (Figure 3C). Flupirtine and ZnCl2 have both been reported to activate IK(M) [29,39,40], QA is a known agonist of AMPA receptors [41] and sorafenib can suppress the activity of tyrosine kinases. Moreover, in mHippoE-14 cells which were pretreated with NBQX (10 µM) for 6 h, the inhibition by PER (1 µM) of IK(M) amplitude still remained efficacious, as evidenced by a significant reduction of the current from 146 ± 13 to 69 ± 11 pA (n = 11, p < 0.05). In keeping with these results, the presence of PER (1 µM) also decreased the τdeact value of the current. As cells were preincubated with NBQX, the activity of endogenous glutamate concentrations released from these cells could have been fully removed. Therefore, the PER-mediated suppression of IK(M) observed in these cells was not associated with either its antagonistic effect on AMPA receptors or changes in the activity of tyrosine kinases.
Figure 3.

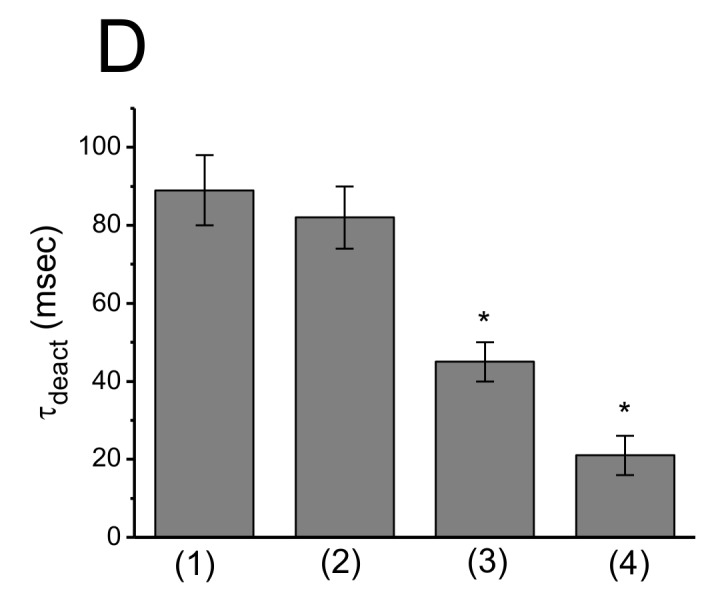
Concentration-dependent effect of PER on M-type K+ current (IK(M)) in mHippoE-14 cells. In these experiments, the cells were immersed in high-K+, Ca2+-free Tyrode’s solution and each pipette was filled with K+-containing solution. (A) Original IK(M) traces elicited by a long-lasting membrane depolarization from −50 to −10 mV (indicated in the upper part of (A)). a: controls; b: 0.1 μM PER; c: 0.3 μM PER; d: 1 μM PER. (B) Concentration-response relation for PER-induced inhibition of IK(M). Each point represents the mean ± SEM (n = 12–14). The smooth line indicates the best fit to the Hill equation. The IC50 value, the maximally inhibited percentage of the current and Hill coefficient were 0.92 μM, 100% and 1.2, respectively. (C) Summary bar graph showing the effects of PER, PER plus flupirtine, PER plus ZnCl2, PER plus L-quisqualic acid (QA) and PER plus sorafenib on IK(M) amplitude (mean ± SEM; n = 11 for each bar). Current amplitude was measured at the end of each depolarizing step. (1): control; (2): 1 μM PER; (3): 1 μM PER plus 10 μM flupirtine; (4): 1 μM PER plus 10 μM ZnCl2; (5) 1 μM PER plus 10 μM L-quisqualic acid (QA); (6) 1 μM PER plus 10 μM sorafenib. * Significantly different from the control (i.e., bar (1)) (p < 0.05) and Ϯsignificantly different from the 1 μM PER alone group (i.e., bar (2)) (p < 0.05). (D) Summary bar graph showing the effect of PER (0.1, 0.3 and 1 µM) on the τdeact value of IK(M) (mean ± SEM; n = 9 for each bar). * Significantly different from the control (p < 0.05). (1): control; (2) 0.1 µM PER; (3) 0.3 µM PER; (4) 1 µM PER.
3.4. Suppressive Effect of PER on The Activity of M-Type K+ (KM) Channels In Mhippoe-14 Cells
The PER-mediated inhibition of IK(M) described above may have arisen from its effects on the activity or gating of KM channels. To further determine the depressant action of PER on whole-cell IK(M), we further explored how PER influenced the activity of single KM-channel currents in these cells. In this set of single-channel current recordings, we bathed the cells in high-K+, Ca2+-free solution and the filling solution contained low-K+ solution, the composition of which is described above. As shown in Figure 4, when the potential was maintained at +30 mV relative to the bath, the activity of KM channels, which underwent open-closed transitions, was readily detected. As PER was applied to the bath, the probability of KM-channel opening was significantly decreased. For example, the addition of PER (3 μM) significantly reduced the channel activity from 0.058 ± 0.008 to 0.009 ± 0.001 (n = 11, p < 0.05). The further addition of flupirtine (10 μM), still in the presence of PER, was effective at increasing the KM-channel activity to 0.034 ± 0.004 (n=11, P<0.05). In addition, the mean open time of the channel in the presence of 3 μM PER was significantly decreased to 2.3 ± 0.3 msec (n = 11, p < 0.05) from a control time of 4.5 ± 0.8 msec (n=11).
Figure 4.


Effect of PER on the activity of M-type K+ (KM) channels recorded from mHippoE-14 cells. In these cell-attached single-channel recordings, the cells were bathed in high-K+, Ca2+-free solution, the recording pipette was filled with low-K+ (5.4 mM) solution and the potential was held at +30 mV relative to the bath. (A) Superimposed KM-channel traces in the absence (upper) and presence (lower) of 1 μM PER. Of note, the opening events caused the upper deflections of the trace in this on-cell patch. (B) Summary bar graph showing the effects of PER and PER plus flupirtine on the probabilities of KM channels that would be open (mean ± SEM; n = 11 for each bar). (1): controls; (2): 1 μM PER; (3): 3 μM PER; (4) 3 μM PER plus 10 μM flupirtine. * Significantly different from the controls (p < 0.05) and Ϯsignificantly different from the PER (3 μM) alone group (p < 0.05).
3.5. Effect of PER on I-V Relationships with KM Channels
We then investigated the effect of PER on the activity of KM channels at different levels of membrane potential. The single-channel amplitudes increased with greater depolarization and the presence of PER decreased the probability of KM-channel openings at different levels of membrane potential (Figure 5A). However, the single-channel conductance calculated from linear I-V relationships between the absence and presence of PER (3 μM) was only slightly perturbed (18.4 ± 0.9 pS [in the absence of PER] versus 18.3 ± 0.9 pS [in the presence of 3 μM PER], n = 11, p > 0.05). These results strongly indicated that despite its inhibitory effects on the probability of KM channel opening, PER exerted little or no effect on single-channel conductance of these channels recorded from mHippoE-14 cells.
Figure 5.

Effect of PER on current-voltage (I-V) relationships and voltage-dependent activation of KM channels in mHippoE-14 cells. In these single-channel recordings, the cells were bathed in high K+, Ca2+-free solution and the recording pipette was filled with low-K+ solution. (A) The I-V curves of KM channels in the absence (■) and presence (□) of 3 μM PER (mean ± SEM; n = 11 for each point). Each dashed line is pointed toward the level of the resting potential (i.e., −70 mV which is indicated by the arrowhead). Of note, the single-channel conductance (i.e. I-V relationship) of the channel in the controls was virtually overlaid with that during exposure to PER (3 μM). (B) The activation curve (i.e., relative channel open probability versus Δvoltage) of KM channels with or without the addition of 3 μM PER (mean ± SEM; n = 11 for each point). The smooth curve was least-squares fit to a Boltzmann function as described in the Materials and Methods. ■: controls; □: in the presence of 3 μM PER.
3.6. Rightward Shift of the Activation Curve of KM Channels Caused by PER
The voltage dependence of KM channels was further studied in the presence of PER. Figure 5B shows the activation curve of KM channels with or without the application of PER (3 μM). The plots of the relative open probability of KM channels as a function of the potential relative to the bath were least-squares fit with a Boltzmann function as described in the Materials and Methods. For the control, Pmax = 0.99 ± 0.01, V1/2 = 22 ± 4 mV, q = 6.5 ± 0.9 e (n = 11), whereas in the presence of 3 μM PER, Pmax = 0.48 ± 0.06, V1/2 = 33 ± 5 mV, q =6.4 ± 0.8 (n = 11). Therefore, the addition of PER caused an approximately 11-mV right shift along the voltage axis in the activation curve of KM channels; however, there was no significant modification of the gating charge (i.e., q) of the KM channels in its presence. These results clearly showed that the extent of the changes in KM-channel activity caused by PER was dependent on the level of membrane potential and that besides its reduction in the maximal probability of KM-channel opening, the addition of PER could modify the voltage dependence of KM channels.
3.7. Effect of PER on Delayed-Rectifier K+ Currents (IK(DR)) in Mhippoe-14 Cells
We also studied whether the presence of PER could modify delay-rectifier K+ currents (IK(DR)) in mHippoE-14 cells. This set of experiments was conducted with the cells bathed in Ca2+-free Tyrode’s solution containing 1 μM tetrodotoxin (TTX) and the recording pipette filled with K+-containing solution. The addition of PER at a concentration of 3 μM had little or no effect on IK(DR) elicited in response to membrane depolarization from −50 to +50 mV. As shown in Figure 6A,B, the IK(DR) examined at different depolarizing steps was suppressed by cell exposure to PER (10 μM). For example, on step depolarization from −50 to +50 mV, PER (10 μM) significantly decreased the amplitude of IK(DR) from 956 ± 68 to 689 ± 55 pA (n = 11, p < 0.05). However, the time course of IK(DR) inactivation in response to such long-lasting membrane depolarization was not modified in the presence of PER. Figure 6B illustrates the I-V relationships of IK(DR) measured at the end of each depolarizing pulse in the controls and during cell exposure to 10 μM PER.
Figure 6.

Effect of PER on delayed-rectifier K+ current (IK(DR)) in mHippoE-14 cells. The whole-cell current recordings were conducted in cells bathed in Ca2+-free Tyrode’s solution containing 1 μM tetrodotoxin (TTX). (A) Superimposed current traces in the absence (upper) and presence (lower) of 10 μM PER. The upper part indicates the voltage protocol applied. (B) Averaged I-V relationships of IK(DR) obtained in the controls (■) and during exposure to 10 μM PER (□) (mean ± SEM; n = 11 for each point). * Significantly different from the controls at the same level of membrane potential (p < 0.05). (C) Concentration-response curve for PER-induced inhibition of IK(DR) in mHippoE-14 cells. The examined cells were depolarized from −50 to +50 mV with a duration of 1 s and current amplitudes was measured at the end of depolarizing pulses during exposure to different PER concentrations was compared with the control value (mean ± SEM; n = 7–9 for each point). The continuous line represents the best fit to a Hill function. The values for IC50, maximally inhibited percentage of IK(DR) and the Hill coefficient were 25.3 µM, 100% and 1.2, respectively.
The relationship between the PER concentration and the percentage inhibition of IK(DR) was determined and then constructed. As illustrated in Figure 6C, PER suppressed IK(DR) in a concentration-dependent manner with an IC50 value of 25.3 µM. Therefore, distinct from either INa or IK(M), the IK(DR) inherently in mHippoE-14 cells tended to be relatively resistant to suppression by PER.
3.8. Effect of PER on IK(M) in Motor Neuron-Like NSC-34 Cells
Earlier studies have demonstrated the effectiveness of PER in treating Parkinson’s disease and motor neuron diseases [1,12,13,14,15]. We thus wanted to explore whether the presence of PER could produce any effects on IK(M) in other types of central neurons (e.g., motor neuron-like NSC-34 cells). As shown in Figure 7A,B, under the same experimental conditions used for the mHippoE-14 cells, the depolarizing step from −50 to −10 mV elicited an IK(M) in NSC cells bathed in high-K+, Ca2+-free solution [38]. As the cells were exposed to different concentrations of PER, the amplitude of IK(M) in response to the long-lasting depolarizing step progressively decreased. In the continued presence of 1 μM PER, the subsequent addition of ZnCl2 (10 μM) was effective at reversing the PER-mediated suppression of IK(M); however, 9-phenanthrol, an activator of intermediate-conductance Ca2+-activated K+ (IKCa) channels [29], had a minimal effect (Figure 7B). These results indicated that the magnitude of PER-induced block of IK(M) in NSC-34 cells was very similar to that in the mHippoE-14 cells described above.
Figure 7.

Effect of PER on IK(M) in motor neuron-like NSC-34 cells. In this set of experiments, the cells were bathed in high-K+, Ca2+-free solution and the recording pipette was filled with K+-containing solution. (A) Superimposed IK(M) traces obtained in the controls (a) and during cell exposure to 0.3 µM PER (b) and 1 μM PER (c). The upper part indicates the step protocol applied. (B) Summary bar graph showing the effects of PER, PER plus 9-phenanthrol and PER plus ZnCl2 on IK(M) amplitude (mean ± SEM; n = 12 for each bar). IK(M) amplitude was measured at the end of each depolarizing step. (1): controls; (2): 0.3 μM PER; (3): 1 μM PER; (4): 1 μM PER plus 3 μM 9-phenanthrol; (5): 1 μM PER plus 10 μM ZnCl2. * Significantly different from the controls (p < 0.05) and †significantly different from the PER (1 μM) alone group (p < 0.05).
3.9. Effect of PER on INa in U87 Glioma Cells
It has recently been demonstrated that PER can effectively suppress the epileptic activity associated with glioma [16,17,18,19]. NaV channels are also functionally expressed in electrically non-excitable cells including glial cells [42]. Therefore, we conducted a final set of whole-cell experiments with U87 glioma cells to determine whether PER can perturb any effects on ion currents, particularly on INa. The cells were bathed in Ca2+-free Tyrode’s solution and the pipette was filled with Cs+-containing solution and the rapid depolarizing step from −80 to −10 mV evoked INa. As shown in Figure 8A and 8B, the peak and late components of INa in these glioma cells diminished during exposure to 1 and 3 μM PER. The NaV channels could be functionally expressed in U87 glioma cells and could create the expressed INa phenotype [42]. More importantly, similar to that described above in mHippoE-14 cells, INa recorded from glioma cells could still be suppressed by the presence of PER. The deactivating time course of the current measured at the level of −100 mV was also significantly shortened in its presence.
Figure 8.

Inhibitory effect of PER on INa in U87 glioma cells. The experimental protocol used was similar to that described above for mHippoE-14 cells. (A) Superimposed INa traces obtained in the controls (a) and during cell exposure to 3 μM PER (b). Inset indicates the voltage-clamp protocol used. (B) Summary bar graph showing the inhibitory effects of 1 or 3 μM PER on peak amplitude of INa in response to the rapid depolarizing step (mean ± SEM; n = 11 for each bar). * Significantly different from the controls (p < 0.05).
4. Discussion
The results of the present study demonstrate that in hippocampal mHippoE-14 cells, PER is capable of producing a depressant action on INa in a concentration- and time-dependent manner. PER preferentially suppressed the late over peak component of INa (IC50 value = 0.78 versus 4.12 μM), with a 5.3-fold selectivity for its suppression of late versus peak INa. Both ranolazine and eugenol, which are inhibitors of late INa, have previously been shown to suppress seizure activity [25,26]. Therefore, in addition to blockade of AMPA receptors, PER-induced block of INa may be another mechanism by which it depresses the excitability of neurons in vivo [22].
Several studies on healthy male volunteers who received 14 days of PER treatment reported blood PER levels ranging from 212 to 358 ng/mL (0.61–1.01 μM) with a 4-mg daily dose to 275 to 456 ng/mL (0.79–1.31 μM) with a 6-mg daily dose [43,44]. In addition, assuming 95% protein binding, free plasma PER concentrations ranged between 0.03 and 0.07 μM. However, at efficacious doses, PER concentrations at the receptors would be much lower than the IC50 value for the inhibition of neuronal transmission in hippocampal slices but still within the range of inhibitory effects on AMPA receptor-mediated responses [44]. Therefore, the concentration ranges used for the PER-mediated inhibition of peak and late INa or IK(M) appear to be clinically achievable.
Most IK(M) inherently in neurons is created by heteromultimers of KV7.2 and KV7.3. These channels have slow activation and deactivation kinetics, a relatively negative threshold for activation and little inactivation under physiological conditions [27,28,29]. Due to these distinctive features, KM-channel activity has a strong control over neuronal excitability. Thus, their negative threshold for activation allows a small number of KM channels to be open near the resting membrane potential, conferring strong control over the threshold for action potential firing. In turn, their slow gating kinetics and the absence of inactivation provide a mechanism for control over action potential firing (e.g., accommodation of firing rate). In the current study, the IC50 value for PER-mediated suppression of IK(M) was 0.92 μM, a value which is clinically achievable.
Moreover, the inhibition of INa could be indirectly altered by the PER-induced inhibition of IK(M), as the suppression of INa might be exacerbated by IK(M) inhibition. In this study, the presence of PER produced a rightward shift along the voltage axis in the activation curve of KM channels with no change in the gating charge of channel activation. As such, the PER molecule is capable of interacting with single KM channels in a voltage-dependent fashion and its inhibitory action would be dependent on the pre-existing level of resting potential, the PER concentration achieved or both. Therefore, it is conceivable that, at clinically relevant concentrations, PER influences the availability of ion channels responsible for maintaining the high electrical excitability of neurons.
In this study, in the continued presence of PER, the subsequent addition of either flupirtine or ZnCl2 reversed the PER-induced suppression of IK(M). However, neither 9-phenanthrol, an activator of IKCa channels [29], nor diazoxide, known to activate ATP-sensitive K+ channels, produced any effect. Consequently, the PER-induced decrease in IK(M) observed in this study seems unlikely to be predominantly linked to changes in the activity of IKCa or ATP-sensitive K+ channels, although these two types of ionic channels may be functionally active in central neurons [29,34,45].
Recent studies have linked the activity of tyrosine kinases to the deregulated trafficking of AMPA receptors in Huntington’s disease models [46]. However, in our study, as mHippoE-14 cells were continually exposed to PER, the subsequent addition of gefitinib or sorafenib, which are known to inhibit tyrosine kinases, did not exert any effects on the reduction of IK(M). Therefore, the PER-induced suppression of IK(M) observed in the mHippoE-14 cells appears to be independent of the activity of surface tyrosine kinases.
Epileptic seizures are known to be a frequent symptom in patients with glioma [47,48] and tumor-related seizures are strongly linked to the activity of NaV channels in glioma cells [47,49]. Of importance, certain antiepileptic drugs known to block NaV channels have recently been demonstrated to be efficacious in prolonging the survival of patients with malignant gliomas [48,50,51]. PER has previously been reported to have beneficial effects on glioma-associated epilepsy [16,17,18,19]. It is thus tempting to speculate that, despite its blockade of AMPA receptors, PER may be beneficial for tumor-related seizures, since it is capable of blocking the transient and persistent components of INa seen in glioma cells.
The findings of this study indicate that PER-induced blockade of IK(M) and INa may synergistically affect the functional activities of central neurons such as hippocampal and motor neurons. Therefore, caution should be taken with regards to its growing use as an antagonist of AMPA receptors [1,7,44,52]. In addition, to what extent the adverse effects (e.g., giddiness) caused by PER [4,53,54] can be relieved by ezogabine, levetiracetam or zinc remains to be determined, because these agents can activate IK(M) [40,55,56,57,58].
Interestingly, increasing evidence has shown that the gain of function in potassium channel variants is actually associated with epilepsy, including KCNQ2 and KCNQ3 [59]. The predominant hypothesis is that these variants dampen the excitability of interneurons [60] and KNCQ2/3 channels have been shown to be expressed in cortical interneurons [61]. An alternative explanation is that the left-shifted KCNQ2/3 channel gain of function hyperpolarizes the axon initial segment, thereby decreasing the steady-state inactivation of sodium channels [59]. Further clinical investigations are warranted to investigate the potential synergistic effects of PER on the modulation of IK(M) and INa, in addition to blockade of AMPA receptors on the wide-spectrum of epileptic and neurological disorders. Alternatively, most of established anti-epileptic drugs (e.g., phenytoin, carbamazepine, lamotrigine or others) are NaV channel blockers. However, whether they could perturb IK(M) or IK(DR) in central neurons is largely unknown and remains to be further investigated.
There are limitations in our study. Ideally, it would be interesting to evaluate the effect of PER on ionic currents, when the cells express AMPA receptors on their surface membranes and to investigate the simultaneous actions of all the ion channels of interest. However, under those experimental conditions, the overlapping by the PER binding to AMPA receptors, and/or by its simultaneous interactions with different types of ionic currents (e.g., [INa] and [IK(M)]) would potentially become too exaggerated to be accurately studied during the recordings.
Nonetheless, our studies indeed highlight the notion that, in addition to the antagonistic action on AMPA receptors, the PER molecules per se are capable of interacting directly with membrane ionic channels to modify the amplitude and gating of ionic currents. Since the modifications of INa and IK(M) significantly regulate the behaviors of electrically excitable cells, such actions on these currents are particularly of clinical, pharmacological or toxicological relevance.
Acknowledgments
The authors are grateful to Kaisen Lee for helpful assistance with the earlier work.
Abbreviations
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| DMEM | Dulbecco’s modified Eagle’s medium |
| FBS | fetal bovine serum |
| I-V | current versus voltage |
| IKCa channel | intermediate-conductance Ca2+-activated K+ channel |
| I K(DR) | delayed-rectifier K+ current |
| I K(M) | M-type K+ current |
| I Na | voltage-gated Na+ current |
| KM channel | M-type K+ channel |
| KV channel | voltage-gated K+ channel |
| NaV channel | voltage-gated Na+ channel |
| PER | perampanel |
| NBQX disodium salt | 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinozaline-7-sulfonamide disodium salt |
| QA | L-quisqualic acid |
| τdeact | deactivation time constant |
| TEA | tetraethylammonium chloride |
| Tef | tefluthrin |
| TTX | tetrodotoxin |
Author Contributions
M.-C.L., C.-W.H., and S.-N.W. performed experiments, assessed, and prepared the data. R.-C.T. assessed and interpreted data. C.-W.H. and S.-N.W. designed and coordinated the study, interpreted data and wrote the article. All authors revised the manuscript.
Funding
This study was in part supported by National Cheng Kung University (D106-35A13, D107-F2519 and NCKUH-10709001 to S. N. Wu), Tainan City, Taiwan. S. N. Wu received a Talent Award for Outstanding Researchers from the Ministry of Education, Taiwan. This work was also supported in part by grants from the Ministry of Science and Technology, Taiwan (107-2314-B-006-018-, 107-2320-B-006-019-, 108-2320-B-006-023- to C. W. Huang) and National Cheng Kung University Hospital (20180254, 20190160 to C. W. Huang).
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Hanada T., Hashizume Y., Tokuhara N., Takenaka O., Kohmura N., Ogasawara A., Hatakeyama S., Ohgoh M., Ueno M., Nishizawa Y. Perampanel: A novel, orally active, noncompetitive AMPA-receptor antagonist that reduces seizure activity in rodent models of epilepsy. Epilepsia. 2011;52:1331–1340. doi: 10.1111/j.1528-1167.2011.03109.x. [DOI] [PubMed] [Google Scholar]
- 2.Di Bonaventura C.D., Labate A., Maschio M., Meletti S., Russo E. AMPA receptors and perampanel behind selected epilepsies: current evidence and future perspectives. Exp. Opin. Pharmacother. 2017;18:1751–1764. doi: 10.1080/14656566.2017.1392509. [DOI] [PubMed] [Google Scholar]
- 3.Brigo F., Lattanzi S., Rohracher A., Russo E., Meletti S., Grillo E., Trinka E. Perampanel in the treatment of status epilepticus: A systematic review of the literature. Epilepsy Behav. 2018;86:179–186. doi: 10.1016/j.yebeh.2018.07.004. [DOI] [PubMed] [Google Scholar]
- 4.Potschka H., Trinka E. Perampanel: Does it have broad-spectrum potential? Epilepsia. 2018;60:22–36. doi: 10.1111/epi.14456. [DOI] [PubMed] [Google Scholar]
- 5.Lattanzi S., Striano P. The impact of perampanel and targeting AMPA transmission on anti-seizure drug discovery. Expert Opin. Drug. Discov. 2019;14:195–197. doi: 10.1080/17460441.2019.1566318. [DOI] [PubMed] [Google Scholar]
- 6.Tyrlikova I., Brazdil M., Rektor I., Tyrlik M. Perampanel as monotherapy and adjunctive therapy for focal onset seizures, focal to bilateral tonic-clonic seizures and as adjunctive therapy of generalized onset tonic-clonic seizures. Expert. Rev. Neurother. 2018;18:1–12. doi: 10.1080/14737175.2019.1555474. [DOI] [PubMed] [Google Scholar]
- 7.Murai N., Sekizawa T., Gotoh T., Watabiki T., Takahashi M., Kakimoto S., Takahashi Y., Iino M., Nagakura Y. Spontaneous and evoked pain-associated behaviors in a rat model of neuropathic pain respond differently to drugs with different mechanisms of action. Pharmacol. Biochem. Behav. 2016;141:10–17. doi: 10.1016/j.pbb.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 8.Khangura R.K., Bali A., Kaur G., Singh N., Jaggi A.S. Neuropathic pain attenuating effects of perampanel in an experimental model of chronic constriction injury in rats. Biomed. Pharmacother. 2017;94:557–563. doi: 10.1016/j.biopha.2017.07.137. [DOI] [PubMed] [Google Scholar]
- 9.Tringali G., Currò D., Navarra P. Perampanel inhibits calcitonin gene-related peptide release from rat brainstem in vitro. J. Headache Pain. 2018;19:107. doi: 10.1186/s10194-018-0940-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakajima M., Suda S., Sowa K., Sakamoto Y., Nito C., Nishiyama Y., Aoki J., Ueda M., Yokobori S., Yamada M., et al. AMPA receptor antagonist perampanel ameliorates post-stroke functional and cognitive impairments. Neuroscience. 2018;386:256–264. doi: 10.1016/j.neuroscience.2018.06.043. [DOI] [PubMed] [Google Scholar]
- 11.Niu H.X., Wang J.Z., Wang D.L., Miao J.J., Li H., Liu Z.G., Yuan X., Liu W., Zhou J.R. The orally active noncompetitive AMPAR antagonist perampanel attenuates focal cerebral ischemia injury in rats. Cell Mol. Neurobiol. 2018;38:459–466. doi: 10.1007/s10571-017-0489-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duty S. Targeting glutamate receptors to tackle the pathogenesis, clinical symptoms and levodopa-induced dyskinesia associated with Parkinson’s disease. CNS Drugs. 2012;26:1017–1032. doi: 10.1007/s40263-012-0016-z. [DOI] [PubMed] [Google Scholar]
- 13.Akamatsu M., Yamashita T., Hirose N., Teramoto S., Kwak S. The AMPA receptor antagonist perampanel robustly rescues amyotrophic lateral sclerosis (ALS) pathology in sporadic ALS model mice. Sci. Rep. 2016;6:28649. doi: 10.1038/srep28649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sugiyama K., Aida T., Nomura M., Takayanagi R., Zeilhofer H.U., Tanaka K. Calpain-dependent degradation of nucleoporins contributes to motor neuron death in a mouse model of chronic excitotoxicity. J. Neurosci. 2017;37:8830–8844. doi: 10.1523/JNEUROSCI.0730-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lattanzi S., Grillo E., Brigo F., Silvestrini M. Efficacy and safety of perampanel in Parkinson’s disease. A systematic review with meta-analysis. J. Neurol. 2018;265:733–740. doi: 10.1007/s00415-017-8681-y. [DOI] [PubMed] [Google Scholar]
- 16.Vecht C., Royer-Perron L., Houillier C., Huberfeld G. Seizures and anticonvulsants in brain tumours: Frequency, mechanisms and anti-epileptic management. Curr. Pharm. Des. 2017;23:6464–6487. doi: 10.2174/1381612823666171027130003. [DOI] [PubMed] [Google Scholar]
- 17.Dunn-Pirio A.M., Woodring S., Lipp E., Herndon J.E., 2nd, Healy P., Weant M., Randazzo D., Desjardins A., Friedman H.S., Peters K.B. Adjunctive perampanel for glioma-associated epilepsy. Epilepsy Behav. Case Rep. 2018;10:114–117. doi: 10.1016/j.ebcr.2018.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Izumoto S., Miyauchi M., Tasaki T., Okuda T., Nakagawa N., Nakano N., Kato A., Fujita M. Seizures and tumor progression in glioma patients with uncontrollable epilepsy treated with perampanel. Anticancer Res. 2018;38:4361–4366. doi: 10.21873/anticanres.12737. [DOI] [PubMed] [Google Scholar]
- 19.Maschio M., Pauletto G., Zarabla A., Maialetti A., Ius T., Villani V., Fabi A., Koudriavtseva T., Giannarelli D. Perampanel in patients with brain tumour-related epilepsy in real-life clinical practice: a retrospective analysis. Int. J. Neurosci. 2018;129:593–597. doi: 10.1080/00207454.2018.1555160. [DOI] [PubMed] [Google Scholar]
- 20.Lv J.M., Guo X.M., Chen B., Lei Q., Pan Y.J., Yang Q. The noncompetitive AMPAR antagonist perampanel abrogates brain endothelial cell permeability in response to ischemia: involvement of claudin-5. Cell Mol. Neurobiol. 2016;36:745–753. doi: 10.1007/s10571-015-0257-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lerche H., Shah M., Beck H., Noebels J., Johnston D., Vincent A. Ion channels in genetic and acquired forms of epilepsy. J. Physiol. 2013;591:753–764. doi: 10.1113/jphysiol.2012.240606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Waszkielewicz A.M., Gunia A., Szkaradek N., Sloczyńska K., Krupińska S., Marona H. Ion channels as drug targets in central nervous system disorders. Curr. Med. Chem. 2013;20:1241–1285. doi: 10.2174/0929867311320100005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lai M.C., Lin K.M., Yeh P.S., Wu S.N., Huang C.W. The novel effect of immunomodulator-glatiramer acetate on epileptogenesis and epileptic seizures. Cell Physiol. Biochem. 2018;50:150–168. doi: 10.1159/000493965. [DOI] [PubMed] [Google Scholar]
- 24.Catterall W.A., Goldin A.L., Waxman S.G. International Union of Pharmacology, XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 2005;57:397–409. doi: 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- 25.Huang C.W., Chow J.C., Tsai J.J., Wu S.N. Characterizing the effects of Eugenol on neuronal ionic currents and hyperexcitability. Psychopharmacology. 2012;221:575–587. doi: 10.1007/s00213-011-2603-y. [DOI] [PubMed] [Google Scholar]
- 26.Anderson L.L., Thompson C.H., Hawkins N.A., Nath R.D., Petersohn A.A., Rajamani S., Bush W.S., Frankel W.N., Vanoye C.G., Kearney J.A., et al. Antiepileptic activity of preferential inhibitors of persistent sodium current. Epilepsia. 2014;55:1274–1283. doi: 10.1111/epi.12657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Selyanko A.A., Hadley J.K., Wood I.C., Abogadie F.C., Delmas P., Buckley N.J., London B., Brown D.A. Two types of K+ channel subunit, Erg1 and KCNQ2/3, contribute to the M-like current in a mammalian neuronal cell. J. Neurosci. 1999;19:7742–7756. doi: 10.1523/JNEUROSCI.19-18-07742.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shu Y., Yu Y., Yang J., McCormick D.A. Selective control of cortical axonal spikes by a slowing inactivating K+ current. Proc. Natl. Acad. Sci. 2007;104:11453–11458. doi: 10.1073/pnas.0702041104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen T.S., Lai M.C., Hung T.Y., Lin K.M., Huang C.W., Wu S.N. Pioglitazone, a PPAR-y activator, stimulates BKCa but suppresses IKM in hippocampal neurons. Front Pharmacol. 2018;9:977. doi: 10.3389/fphar.2018.00977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buskila Y., Kékesi O., Bellot-Saez A., Seah W., Berg T., Trpceski M., Yerbury J.J., Ooi L. Dynamic interplay between H-current and M-current controls motoneuron hyperexcitability in amyotrophic lateral sclerosis. Cell Death Dis. 2019;10:310. doi: 10.1038/s41419-019-1538-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simeone T.A., Simeone K.A., Samson K.K., Kim D.Y., Rho J.M. Loss of the Kv1.1 potassium channel promotes pathologic sharp waves and high frequency oscillations in in vitro hippocampal slices. Neurobiol. Dis. 2013;54:68–81. doi: 10.1016/j.nbd.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saitsu J.H., Akita T., Tohyama J., Goldberg-Stern H., Kobayashi Y., Cohen R., Kato M., Ohba C., Miyatake S., Tsurusaki Y., et al. De novo KCNB1 mutations in infantile epilepsy inhibit repetitive neuronal firing. Sci. Rep. 2015;5:15199. doi: 10.1038/srep15199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gingerich S., Kim G.L., Chalmers J.A., Koletar M.M., Wang X., Wang Y., Belsham D.D. Estrogen receptor α and G-protein coupled receptor 30 mediate the neuroprotective effects of 17β-estradiol in novel murine hippocampal cell models. Neuroscience. 2010;170:54–66. doi: 10.1016/j.neuroscience.2010.06.076. [DOI] [PubMed] [Google Scholar]
- 34.Huang C.W., Lin K.M., Hung T.Y., Chuang Y.C., Wu S.N. Multiple actions of rotenone, an inhibitor of mitochondrial respiratory chain, on ionic currents and miniature end-plate potential in mouse hippocampal (mHippoE-14) neurons. Cell Physiol. Biochem. 2018;47:330–343. doi: 10.1159/000489810. [DOI] [PubMed] [Google Scholar]
- 35.Wu S.N., Chern J.H., Shen S., Chen H.H., Hsu Y.T., Lee C.C., Chan M.H., Lai M.C., Shie F.S. Stimulatory actions of a novel thiourea derivative on large-conductance, calcium-activated potassium channels. J. Cell Physiol. 2017;232:3409–3421. doi: 10.1002/jcp.25788. [DOI] [PubMed] [Google Scholar]
- 36.Wu S.N., Wu Y.H., Chen B.S., Lo Y.C., Liu Y.C. Underlying mechanism of actions of tefluthrin, a pyrethroid insecticide, on voltage-gated ion currents and on action currents in pituitary tumor (GH3) cells and GnRH-secreting (GT1-7) neurons. Toxicology. 2009;258:70–77. doi: 10.1016/j.tox.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 37.So E.C., Wu S.N., Lo Y.C., Su K. Differential regulation of tefluthrin and telmisartan on the gating charges of INa activation and inactivation as well as on resurgent and persistent INa in a pituitary cell line (GH3) Toxicol. Lett. 2018;285:104–112. doi: 10.1016/j.toxlet.2018.01.002. [DOI] [PubMed] [Google Scholar]
- 38.Liu Y.Y., Hiao H.T., Wang J.C., Liu Y.C., Wu S.N. Parecoxib, a selective blocker of cyclooxygenase-2, directly inhibits neuronal delayed-rectifier K+ current, M-type K+ current and Na+ current. Eur. J. Pharmacol. 2019;844:95–101. doi: 10.1016/j.ejphar.2018.12.005. [DOI] [PubMed] [Google Scholar]
- 39.Wu S.N., Hsu M.C., Liao Y.K., Wu F.T., Jong Y.J., Lo Y.C. Evidence for inhibitory effects of flupirtine, a centrally acting analgesic, on delayed rectifier K+ currents in motor neuron-like cells. Evid. Based Complement. Alternat. Med. 2012;2012:148403. doi: 10.1155/2012/148403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao H., Boillat A., Huang D., Liang C., Peers C., Gamper N. Intracellular zinc activates KCNQ channels by reducing their dependence on phosphatidylinositol 4,5-bisphosphate. Proc. Natl. Acad. Sci. 2017;114:E6410–E6419. doi: 10.1073/pnas.1620598114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas L.S., Jane D.E., Harris J.R., Croucher M.J. Metabotropic glutamate autoreceptors of the mGlu(5) subtype positively modulate neuronal glutamate release in the rat forebrain in vitro. Neuropharmacology. 2000;39:1554–1566. doi: 10.1016/S0028-3908(99)00223-3. [DOI] [PubMed] [Google Scholar]
- 42.Schrey M., Codina C., Kraft R., Beetz C., Kalff R., Wolfl S., Patt S. Molecular characterization of voltage-gated sodium channels in human gliomas. Neuroreport. 2002;13:2493–2498. doi: 10.1097/00001756-200212200-00023. [DOI] [PubMed] [Google Scholar]
- 43.Patsalos P.N., Gougoulaki M., Sander J.W. Perampanel serum concentrations in adults with epilepsy: effect of dose, age, sex, and concomitant anti-epileptic drugs. Ther. Drug. Monit. 2016;38:358–364. doi: 10.1097/FTD.0000000000000274. [DOI] [PubMed] [Google Scholar]
- 44.Rogawski M.A., Hanada T. Preclinical pharmacology of perampanel, a selective non-competitive AMPA receptor antagonist. Acta. Neurol. Scan. Suppl. 2013;127:19–24. doi: 10.1111/ane.12100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang C.W., Huang C.C., Wu S.N. The opening effect of pregabalin on ATP-sensitive potassium channels in differentiated hippocampal neuron-derived H19-7 cells. Epilepsia. 2006;47:720–726. doi: 10.1111/j.1528-1167.2006.00498.x. [DOI] [PubMed] [Google Scholar]
- 46.Zhang H., Zhang C., Vincent J., Zala D., Benstaali C., Sainlos M., Grillo-Bosch D., Daburon S., Coussen F., Cho Y., et al. Modulation of AMPA receptor surface diffusion restores hippocampal plasticity and memory in Huntington’s disease models. Nat. Commun. 2018;9:4272. doi: 10.1038/s41467-018-06675-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sontheimer H., Black J.A., Waxman S.G. Voltage-gated Na+ channels in glia: Properties and possible functions. Trends Neurosci. 1996;19:325–331. doi: 10.1016/0166-2236(96)10039-4. [DOI] [PubMed] [Google Scholar]
- 48.Guthrie G., Eljamel S. Impact of particular antiepileptic drugs on the survival of patients with glioblastoma multiforme. J. Neurosurg. 2013;118:859–865. doi: 10.3171/2012.10.JNS12169. [DOI] [PubMed] [Google Scholar]
- 49.Joshi A.D., Parsons D.W., Velculescu V.E., Riggins G.J. Sodium ion channel mutations in glioblastoma patients correlate with shorter survival. Mol. Cancer. 2011;10:17. doi: 10.1186/1476-4598-10-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsai H.C., Wei K.C., Tsai C.N., Huang Y.C., Chen P.Y., Chen S.W., Lu Y.J., Lee S.T. Effect of valproic acid on the outcome of glioblastoma multiforme. Br. J. Neurosurg. 2012;26:347–354. doi: 10.3109/02688697.2011.638996. [DOI] [PubMed] [Google Scholar]
- 51.Weller M., Stupp R., Wick W. Epilepsy meets cancer: When, why, and what to do about it? Lancet Oncol. 2012;13:E375–E382. doi: 10.1016/S1470-2045(12)70266-8. [DOI] [PubMed] [Google Scholar]
- 52.Chen T., Dai S.H., Jiang Z.Q., Luo P., Jiang X.F., Fei Z., Gui S.B., Qi Y.L. The AMPAR antagonist perampanel attenuates traumatic brain injury through anti-oxidative and anti-inflammatory activity. Cell Mol. Neurobiol. 2017;37:43–52. doi: 10.1007/s10571-016-0341-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Juhl S., Rubboli G. Add-on perampanel and aggressive behavior in severe drug-resistant focal epilepsies. Funct. Neurol. 2017;32:215–220. [PMC free article] [PubMed] [Google Scholar]
- 54.Youn S.E., Kim S.H., Ko A., Lee S.H., Lee Y.M., Kang H.C., Lee J.S., Kim H.D. Adverse events during perampanel adjunctive therapy in intractable epilepsy. J. Clin. Neurol. 2018;14:296–302. doi: 10.3988/jcn.2018.14.3.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang C.W., Tsai J.J., Huang C.C., Wu S.N. Experimental and simulation studies on the mechanisms of levetiracetam-mediated inhibition of delayed-rectifier potassium current (KV3.1): contribution of the firing of action potentials. J. Physiol. Pharmacol. 2009;60:37–47. [PubMed] [Google Scholar]
- 56.Faulkner M.A., Burke R.A. Safety profile of two novel antiepileptic agents approved for the treatment of refractory partial seizures: Ezogabine (retigabine) and perampanel. Exp. Opin. Drug. Saf. 2013;12:847–855. doi: 10.1517/14740338.2013.823399. [DOI] [PubMed] [Google Scholar]
- 57.Hansen C.C., Ljung H., Brodtkorb E., Reimers A. Mechanisms underlying aggressive behavior induced by antiepileptic drugs: focus on topiramate, levetiracetam, and perampanel. Behav. Neurol. 2018;2018:2064027. doi: 10.1155/2018/2064027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kanemura H., Sano F., Aihara M. Usefulness of perampanel with concomitant levetiracetam for patients with drug-resistant epilepsy. Eur. J. Paediatr. Neurol. 2018;23:197–203. doi: 10.1016/j.ejpn.2018.10.004. [DOI] [PubMed] [Google Scholar]
- 59.Niday Z., Tzingounis A. Potassium channel gain of function in epilepsy: An unresolved paradox. Neuroscientist. 2018;24:368–380. doi: 10.1177/1073858418763752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miceli F., Soldovieri M.V., Ambrosino P., De Maria M., Migliore M., Migliore R., Taglialatela M. Early-onset epileptic encephalopathy caused by gain-of function mutations in the voltage sensor of Kv7.2 and Kv7.3 potassium channel subunits. J. Neurosci. 2015;35:3782–3793. doi: 10.1523/JNEUROSCI.4423-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cooper E.C., Harrington E., Jan Y.N., Jan L.Y. M channel KCNQ2 subunits are localized to key sites for control of neuronal network oscillations and synchronization in mouse brain. J. Neurosci. 2001;21:9529–9540. doi: 10.1523/JNEUROSCI.21-24-09529.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]