

Abstract
Background
Exposure to cigarette smoke (CS) is a major threat to human health worldwide. It is well established that smoking increases the risk of respiratory diseases, cardiovascular diseases and different forms of cancer, including lung, liver, and colon. CS-triggered inflammation is considered to play a central role in various pathologies by a mechanism that stimulates the release of pro-inflammatory cytokines. During this process, epigenetic alterations are known to play important roles in the specificity and duration of gene transcription.
Main text
Epigenetic alterations include three major modifications: DNA modifications via methylation; various posttranslational modifications of histones, namely, methylation, acetylation, phosphorylation, and ubiquitination; and non-coding RNA sequences. These modifications work in concert to regulate gene transcription in a heritable fashion. The enzymes that regulate these epigenetic modifications can be activated by smoking, which further mediates the expression of multiple inflammatory genes. In this review, we summarize the current knowledge on the epigenetic alterations triggered by CS and assess how such alterations may affect smoking-mediated inflammatory responses.
Conclusion
The recognition of the molecular mechanisms of the epigenetic changes in abnormal inflammation is expected to contribute to the understanding of the pathophysiology of CS-related diseases such that novel epigenetic therapies may be identified in the near future.
Keywords: Epigenetic, DNA methylation, Histone modification, miRNA, LncRNA, Inflammation
Background
Cigarette smoke (CS) presents a significant risk factor related to various diseases, such as cardiovascular diseases [1], chronic obstructive pulmonary disease (COPD) [2], and cancers [3]. Numerous chemical substances inside tobaccos have toxic effects, including polycyclic aromatic hydrocarbons (benzo[a]pyrene), N-nitrosamines, heavy metals (nickel, cadmium, chromium and arsenic), alkaloids (nicotine and its major metabolite, cotinine) and aromatic amines. The pathogenic mechanisms of CS include inflammation and immune changes, genetic alterations, oxidative damage, endothelial dysfunction, cell senescence, etc. [4]. However, tobacco use confers long-term risk of diseases, even decades after cessation, that are not well understood [5]. Protein-coding genes cannot account for all environmental diseases. Recently, epigenetic changes have been proposed as possible alternative causes of some diseases.
Epigenetic research is becoming a rapidly developing and exciting area in biology. Epigenetics is usually defined as heritable alterations in gene expression patterns that are not directly caused by modifications encoded in the nucleotide genome sequence but are caused by posttranslational modifications in DNA and histone proteins and by the regulation of non-coding RNAs (ncRNAs) [6]. The epigenetic mechanisms are shown in Fig. 1. DNA is methylated by the transfer of a methyl group from S-adenosyl-l-methionine to covalently bind to the cytosines in CpG dinucleotides. DNA hypermethylation always leads to transcriptional suppression and decreased gene expression, while DNA hypomethylation influences chromosome stability or enhanced aneuploidy [7]. Eukaryotic chromatin is made up of nucleosomes, which consist of ~ 147 bp of DNA wrapped around an octamer of two copies of four core histones (H2A, H2B, H3, and H4). Posttranslational modification of histones involves covalent modification of specific sites and residues that form specific three-dimensional arrangements of nucleosomes that control the accessibility to transcriptional factors to genes to induce gene expression. Histones are primarily modified at basic lysine and arginine residues, thereby marking active and inactive chromatin states [8]. Important histone modifications include acetylation, methylation, phosphorylation, and ubiquitination. NcRNAs, once thought to represent transcriptional noise, are now recognized as having important functions in cellular processes [9]. Indeed, only a small proportion of the human genome encodes protein (~ 2%); the majority of the genome is not translated into proteins and may instead code a range of ncRNA types (> 90% of the genome). These ncRNAs are classified into small ncRNAs, with transcript length of 200 or fewer nucleotides, and long ncRNAs (lncRNAs), with transcript lengths more of > 200 nucleotides [10]. Small RNAs include but are not limited to microRNAs (miRNAs), PIWI-interacting RNAs, small interfering RNAs, small nucleolar RNAs, and transfer RNAs. In recent years, growing evidence has suggested that ncRNAs, especially miRNAs and lncRNAs, play important roles in the pathogenesis of human diseases [11, 12].
Fig. 1.
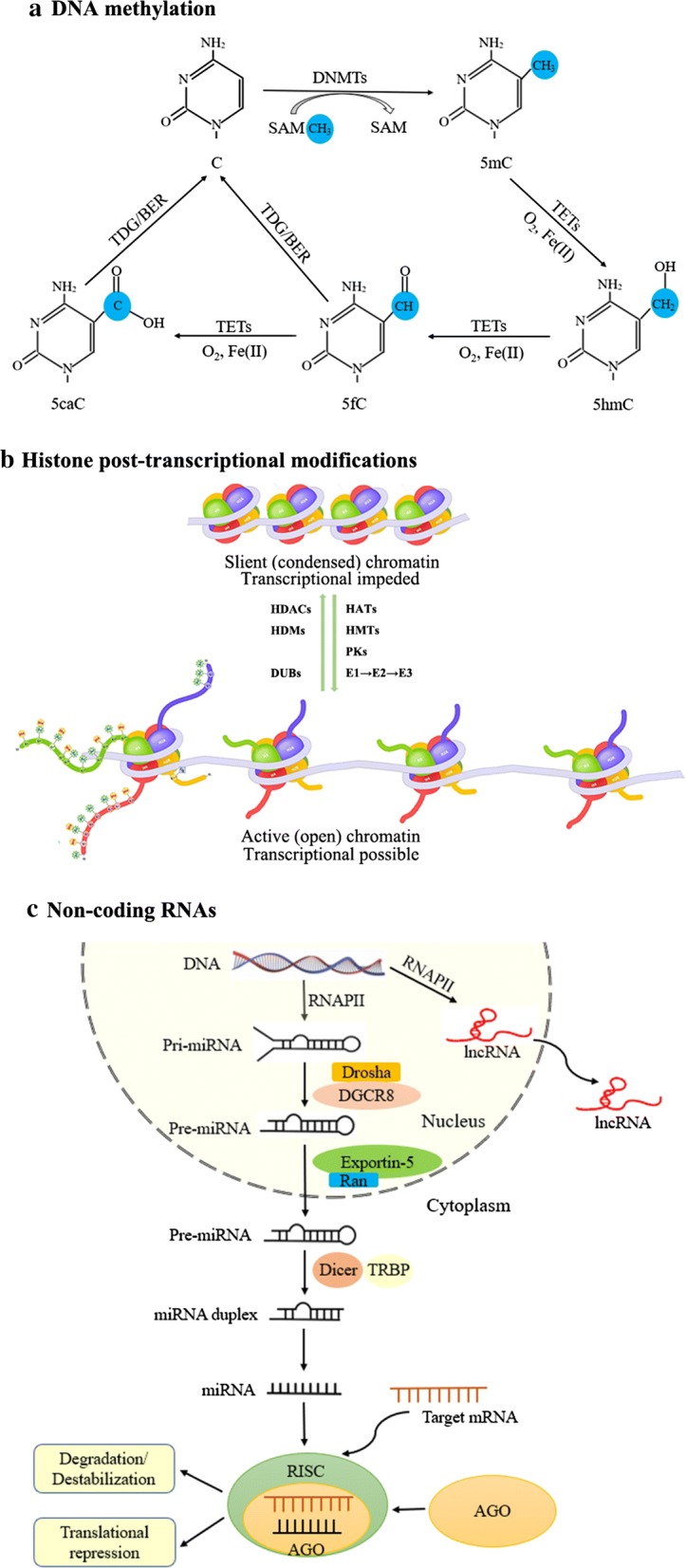
Epigenetic mechanisms. a DNA methylation. DNA methylation is catalyzed by DNA methyl transferases (DNMTs). DNA methylation involves the covalent transfer of a methyl group from S-adenosyl methionine (SAM) to 5′ position of cytosine residues in CG dinucleotides. DNA demethylation involves ten-eleven translocation (TET) proteins. TETs initiate DNA demethylation by oxidizing 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), which can be further oxidized to 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC). 5fC and 5caC can be recognized and excised by thymine DNA glycosylase (TDG), and the residual abasic site is repaired as unmodified C by base excision repair (BER) pathway to complete “active” demethylation. Furthermore, oxygen and Fe(II) are indispensable for the TET enzymes to perform the successive oxidation of 5mC, 5hmC and 5fC. b Histone posttranscriptional modifications. Histone modification is regulated by opposing enzymes. Histone acetylation is mediated by histone acetyltransferases (HATs) and deacetylation by histone deacetylases (HDACs). Methylation is mediated by histone methyltransferase (HMTs) and demethylation by histone demethylases (HDMs). Phosphorylation is mediated by phosphorylase kinases (PKs). Ubiquitination is carried out through three main enzymatic reactions performed stepwise by ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin ligase (E3) consecutively; deubiquitination is mediated by deubiquitinating enzymes (DUBs). c The biogenesis of miRNA and lncRNA. The miRNA is transcribed to create the primary miRNA (pri‐miRNA) by RNA polymerase II (RNAPII). Following cleaved by the Drosha and DRCG8, precursor miRNA (pre‐miRNA) is exported from the nucleus by exportin 5. Then, Dicer and TAR RNA‐binding protein (TRBP) will further process the molecule and form a double-stranded miRNA: miRNA duplex. One strand of the duplex, together with the argonaute (AGO) protein and the target messenger RNA, is incorporated in the RNA-induced silencing complex (RISC) and subsequently targets mRNAs for degradation or translational repression. The lncRNA is transcribed mostly by RNAPII and its biogenesis process is similar to miRNA
Epigenetic regulation of gene and protein expression is carried out by a set of highly conserved processes that contribute to normal development and adaptation to changes in cellular and organ homeostasis [13]. It is very important to note that the changes caused by epigenetic modifications of DNA and/or histones and changes in ncRNA expression represent dynamitic information that is markedly influenced by environmental stimuli, such as CS, air pollution and dietary changes. Epigenetic dysregulation that leads to inappropriate gene expression or silence has been shown to play a significant role in the CS-related diseases. Moreover, it can combine with other CS-related molecular mechanisms, such as oxidative stress, inflammation, apoptosis, and senescence. Understanding the basic mechanisms induced by CS and the relationships among them is invaluable to our knowledge of cellular differentiation and genome programming and the connection of these relationships with diseases. Herein, we provide a comprehensive overview of the current research on how epigenetic alterations are combined with other pathogenic mechanisms induced by CS during the disease processes.
The effect of cigarette smoke on epigenetics
Cigarette smoke and DNA methylation
Alterations in DNA methylation have recently been suggested to be one possible mechanism potentially mediating CS-induced diseases [14]. Several epigenome-wide association studies based on different sample sizes, races and areas have confirmed that the altered DNA methylation at multiple CpG sites is induced by CS [15–23] (Table 1) and can lead to changes in gene transcription and increased susceptibility to diseases. A recent meta-analysis of genome-wide DNA methylation assessed nearly 16,000 blood-derived DNA samples from participants in 16 cohorts and the results showed that 7201 annotated genes associated with 18,760 CpGs, representing almost one-third of all known human genes, were differentially methylated in the current smokers versus the number found in the never smokers [5]. DNA methylation altered by tobacco smoke is a reversible gene regulatory modification. After smoking cessation, the majority of the differentially methylated CpG sites observed in the analysis of the current versus never smokers returned to the level of the never smokers within 5 years of smoking cessation. However, some of the methylated genes did not return to the level of the never smokers even after 30 years of smoking cessation [5], suggesting that CS may lead to lasting damage to human health. In addition to the current smokers, human DNA methylation at specific CpG sites can also be influenced by maternal smoking. Howe et al. [24] analyzed the methylomes of sorted cord blood CD4+ cells derived from 20 Hispanic white newborns. Of the newborns, 10 were exposed to maternal tobacco smoke in pregnancy and 10 were not exposed. They found that 10,381 CpGs were differentially methylated by CS exposure. Five-hundred and fifty-seven differentially methylated regions were overrepresented in important regulatory regions, including those of enhancers [24]. Joubert et al. [25, 26] demonstrated that maternal smoking affects DNA methylation in newborn cord blood if the mother smoked past 18 weeks into her pregnancy, whereas significant effects on methylation were not observed for mothers who quit smoking before 18 gestational weeks. Given that smoking is an established risk factor for many chronic diseases, these loci could have important applications as objective biomarkers of both current and lifetime smoking exposure and for quantifying risks of smoking-related diseases. As DNA methylation is mainly catalyzed by DNA methyltransferases (DNMTs), researchers found that DNMT1 expression was significantly higher in the lung tissues of the smokers compared to the expression in the nonsmokers [27]. Furthermore, the inhibition of DNMT1 can restore the expression of genes that had been suppressed by a CS condensate through demethylation [28]. Thus, CS can induce gene hypermethylation by upregulating DNMT1, which in turn leads to the downregulation of target genes.
Table 1.
The changes of DNA methylation profiles affected by CS
| Sample size | Population | Tissue for DNA methylation analysis | Smoking status | Main result | References | Year |
|---|---|---|---|---|---|---|
| 596 | Chinese population | Whole blood | Current smoke | 318 CpG sites were differentially methylated due to cigarette smoking. | Zhu et al. [15] | 2016 |
| 745 | European women | Whole blood | Current smoke | 461 CpG sites were aberrant methylated due to cigarette smoking. Of them, 448 CpG sites were hypomethylated | Guida et al. [16] | 2015 |
| Former smoke | The methylation of 751 CpG sites were changed the recoded smoking status. Of them, 602 CpG sites reverted back to that of never smokers from up to 35 years after smoking cessation. 149 CpG sites remained differentially methylated > 35 years after smoking | |||||
| 123 | Arab population | Whole blood | Current smoke | Aberrant methylation was detected due to tobacco smoking. The exact number of CpG sites was not mentioned | Zaghlool et al. [17] | 2015 |
| 111 | African American women | Peripheral blood mononuclear cells | Current smoke | 910 loci were found to be differentially methylated in smokers | Dogan et al. [18] | 2014 |
| 192 | South Asian and European men | Whole blood | Current smoke | 29 CpG sites at 18 unique loci were differential methylated in smokers | Elliott et al. [19] | 2014 |
| 180 | Italian population | Peripheral white blood cells | Current smoke/former smoke | 17 and 19 loci in the breast cancer and colon cancer were differentially methylated between smokers, former smokers and never smokers. In former smokers, methylation levels at AHRR, 2q37 and 6p21 loci returned to the levels of nonsmokers with increasing time from cessation and those who had smoked more intensively had methylation levels that were closer to that of current smokers | Shenker et al. [20] | 2013 |
| 1793 | Augsburg population | Whole blood | Current smoke/FORMER smoke | 972 CpG sites were differentially methylated after smoking. Of which, 187 CpG sites were differentially methylated in current smokers. | Zeilinger et al. [21] | 2013 |
| 98 | African Americans | Lymphocyte cells | Current smoke | Aberrant methylation was detected due to tobacco smoking. The authors listed 30 most significant methylated CpG sites without mentioning the exact number of methylated CpG sites | Philibert et al. [22] | 2013 |
| 972 | African Americans | Peripheral leukocytes | Current smoke | 15 autosomal DNAm sites were significantly associated with current smoking using a Bonferroni corrected p value of 0.05. 89 DNAm sites associated with current smoking with FDR q value less than 0.05 | Sun et al. [23] | 2013 |
| 20 | Hispanic newborns | CD4+ cells from cord blood | In utero exposure to maternal tobacco smoke | 10,381 CpG sites were differentially methylated by tobacco smoking. Of them, 557 differentially methylated regions were overrepresented in important regulatory regions, including enhancers | Howe et al. [24] | 2019 |
| 1062 | Norwegian newborns | Cord blood | In utero exposure to maternal tobacco smoke | Differential DNA methylation of 26 CpG sites mapped to 10 genes were found in newborns born to smoking mothers compared to nonsmoking mothers | Joubert et al. [25] | 2012 |
| 1042 | Norwegian newborns | Cord blood | In utero exposure to maternal tobacco smoke | Maternal smoking affected DNA methylation of 26 CpG sites that mapped to 10 genes in newborn cord blood if the mother smokes past 18 weeks in pregnancy, whereas significant effects on methylation were not observed for mothers that quit before 18 gestational weeks | Joubert et al. [26] | 2014 |
Cigarette smoke and histone posttranscriptional modifications
Cigarette smoke and histone acetylation
Previous studies reported that CS caused histone hyperacetylation in the lungs of both human smokers [29, 30], mouse models exposed to CS [31], and human bronchial epithelial (HBE) cells treated with cigarette smoke extract (CSE) in vitro [32]. It was found that active smoking may promote acetylation of histone H4, while ex smokers showed increased histone H3 acetylation [29]. Histone acetylation is a dynamic process that depends on the balance of two opposing types of enzymes: histone acetyltransferases (HATs), which add an acetyl group, and histone deacetylases (HDACs), which remove acetyl groups from conserved lysine residues and nonhistone proteins [33]. Current studies have shown that CS induces increased histone acetylation mainly by decreasing the activity and expression of HDACs [29–32]. Moreover, there is a significant and negative relationship between HDAC activity and smoking exposure levels [34]. In the lung tissues of smokers with COPD, the expression of HDAC2, HDAC5, HDAC8 [29, 30], HDAC7 [35] and HDAC10 [36] was decreased significantly. An increased number of acetylated H3 and H4 proteins were shown to be associated with decreased expression of HDAC1 [37], HDAC2 and HDAC4 [38] in CS-treated rat lungs.
In bronchial epithelial cells and alveolar macrophages exposed to CSE, the expression and activity of HDAC3 were reported to be decreased [39, 40]. In contrast to that of other HDACs, cytoplasmic HDAC6 expression is elevated in the lung tissues of chronic smokers with COPD, partly due to hypomethylation by HDAC6 [41]. Different results were found by Borgas et al. [42], who showed that HDAC6 protein levels were not changed in cultured lung endothelial cells exposed to CSE in vitro but significantly decreased in the lung tissues of mice exposed to CS for 3 weeks. Although there is a discrepancy regarding the impacts of CS on HDAC6 protein levels, HDAC6 may be activated both in vivo and in vitro through HDAC6 phosphorylation at Ser-22 [42]. Sirtuin (SIRT)1-7 belongs to the family of class III HDACs. It has been shown that the expression of SIRT1 was decreased when exposed to CS both in vivo and in vitro [43–45]. Furthermore, a statistically positive correlation was found between SIRT1 activity and two lung function parameters (FEV1 and FEV1/FVC) [43]. In the CS-induced hypertension mouse model, the expression of SIRT3 was reduced, as was the hyperacetylation of superoxide dismutase 2, and superoxide dismutase 2 activity was inhibited, which promoted mitochondrial oxidative stress [46]. Other SIRTs, such as SIRT4 [47], SIRT5 [48] and SIRT6 [49], were markedly downregulated when exposed to CS. In addition to its effect on HDACs, CS also alters the expression of HATs. In CS-treated bronchial epithelial cells, the expression of an HAT, cAMP-response element-binding protein (CBP/p300), was demonstrated to be increased and to induce a multitude of acetylating processes [50]. In short, decreased HDAC action and increased HAT action may account for CS-induced hyperacetylation.
Cigarette smoke and histone methylation
Previous researchers have reported that global levels of histone methylation were increased after CS exposure [32]. Both mono-and di-methylation of histone H3 residues (H3K27me2/3, H3K36me1/2, H3K56me2, and H3K79me1/2) and histone H4 residues (H4K20me1/2, H4R23me1, H4K31me2, H4R35me1/2, H4R36me1, H4R55me1 and H4K77me1) were increased in CS-exposed mouse lungs [32]. As the catalytic enzyme for histone methylation, histone methyltransferase (HMT) was also found to be regulated by CS. PRMT6 is a nuclear enzyme that specially catalyzes di-methylation of R2 in histone H3 (H3R2me2a). In previous studies, we demonstrated that CS may decrease the protein abundance of PRMT6 at the same time that H3R2me2a was decreased both in CS-exposed mouse lungs and in CSE-treated endothelial cells, which further influence cell apoptosis and inflammation [51, 52]. Enhancer of zeste homolog 2 (EZH2) is a specific HMT that catalyzes histone H3 lysine 27 tri-methylation (H3K27me3). It was shown that CSE can enhance the expression of EZH2 and increase the level of H3K27me3, which epigenetically controlled gene transcription [53]. To date, few studies have been concerned with changes in histone methylation caused by CS. Due to the regulating effects of histone methylation on gene transcription, understanding the role of histone methylation and their associated HMTs will provide insights into the clinical treatment of smoking-related diseases.
Histone phosphorylation
Phosphorylation of histone has been reported to be affected by various stimuli, such as arsenite, nickel, ultraviolet B rays and CS [54]. Treatment with CS markedly induced the phosphorylation of histone H3 at serine 10 and 28 residues [54], as well as histone H2AX [55], in a dose- and time-dependent manner. A number of kinases, such as ribosomal S6 kinase (RSK) and mitogen- and stress-activated kinase (MSK), were found to phosphorylate histone H3 depending on the specific stimulation or stress [56]. Sundar et al. [57] showed that CSE medicated the activation of MSK1, which was associated with the phosphorylation of histone H3S10 and further promotes the expression of pro-inflammatory genes. Knocking down MSK1 inhibited CSE-induced posttranslational modifications of histones and the transcription of pro-inflammatory genes [57]. MSKs are activated via a complex series of phosphorylation and autophosphorylation reactions by the extracellular signal-regulated kinase (ERK) and p38 mitogen-activated protein kinase (p38 MAPK) pathways [58]. A large number of studies have demonstrated that CS can activate the ERK and p38 pathways [59–61]. Another MAPK subfamily, c-jun N-terminal kinase (JNK), which mediates part of the stress response, was shown to induce phosphorylation of H3S10 [62]. The finding that CS activates the JNK pathway has been supported by numerous studies [61, 63, 64]. Taken together, these findings indicate that CS can induce histone phosphorylation alterations through the activation of various kinases.
Cigarette smoke and histone ubiquitination
Extensive studies have confirmed that CS exposure contributes to the development of skeletal muscle atrophy, which occurs when the level of protein breakdown exceeds protein synthesis [65]. Wasting of the muscle tissue is believed to the result of increased ubiquitin–proteasome system (UPS) activity, which can be induced by CS [66, 67]. Kim et al. [68] confirmed that CS induced protein degradation of Akt, which plays a critical role in cell survival and proliferation, by the UPS. Ubiquitination is carried out through three main enzymatic reactions performed stepwise by ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin ligase (E3) consecutively [69]. Muscle atrophy F-box (MAFbx/atrogin-1) and muscle ring finger-1 protein (MuRF1) have been identified as two muscle-specific E3 ligases. Both these two E3 ligases play central roles in muscle atrophy. Baptista et al. [70] found that leucine supplementation administered to rats with immobilized hindlimbs attenuated soleus muscle mass loss and minimized the MuRF1 and MAFbx/atrogin-1 gene expression. Moreover, mice deficient in either MAFbx or MuRF1 were protected from muscle atrophy, whereas overexpressed MAFbx in myotubes induced atrophy [71]. It has been shown that C2 myotubes exposed to CS upregulated MAFbx/atrogin-1 and MuRF1 by activating the p38 MAPK signaling pathway, which led to a reduction in myotube diameter and degradation of the main contractile proteins in a time- and dose-dependent manner [72]. Similar to its effect on the p38 MAPK pathway, CS exposure also resulted in nuclear factor kappa B (NF-κB) activation, which led to upregulated levels of MuRF1 and reduced myotube diameter of myotubes and time-dependent degradation of the main contractile protein, myosin heavy chain [73]. In addition to the E3 ligases, other ubiquitin-specific proteases (USPs), such as USP19, were also observed to be increased in CS-induced muscle atrophy [74]. These results suggest that CS can induce skeletal muscle atrophy and degradation via the activation of the UPS. Mitochondrial E3 ubiquitin protein ligase 1 (MUL1) is a novel Akt ubiquitin E3 ligase. CS can noticeably elevate MUL1 expression and Akt ubiquitination, while knocking down MUL1 suppressed CS-induced Akt ubiquitination/degradation and inhibited the reduction in cytoplasmic Akt and p-Akt [75]. USP17 has been shown to be an E3 deubiquitinase that inhibits proteasome-mediated degradation of transcription factors [76]. It has been shown that HDAC2 is excessively ubiquitinated and degraded in the proteasome, an action attributed to the low expression of USP17 in CS-exposed airway epithelial cells and macrophages, while overexpressed USP17 blocks the degradation of HDAC2 induced by CS [77].
Cigarette smoke and non-coding RNAs
Cigarette smoke and miRNAs
To date, more than 48,800 mature miRNAs have been reported in 271 species. Over 2650 human miRNAs are listed in the Sanger miRBase sequence database (ver. 22). Studies have revealed that miRNAs play key roles in regulating various physiological and pathological processes, such as cell growth, differentiation, proliferation, apoptosis, and organ development [78]. Approximately, 30% of known human protein-coding genes are predicted to be regulated by miRNAs [79]. Thus, an altered expression of miRNAs has been linked to various human diseases, and the profiles of tissue miRNAs suggest potential applications in diagnosis and prognosis. As an unstable single-stranded RNA, miRNAs can be easily influenced by environmental chemicals, and the effects of CS exposure on miRNA are among the most intensely studies areas of research. Recent studies have shown that exposure to CS leads to global alteration in miRNAs. Early in 2009, Izzotti et al. [80] first analyzed miRNA expression patterns in the lungs of rats exposed to environmental CS for 28 days. The researchers reported that, of the 484 miRNAs analyzed, 126 miRNAs (26.0%) were downregulated by at least twofold, and 24 miRNAs were downregulated more than threefold, and the difference between rats exposed to CS and sham group was significant. Only 7 miRNAs (1.4%) were upregulated. The same team also investigated the effects of CS exposure on miRNA expression in the lungs of mice [81]. Their findings were similar in that the miRNA alterations in the lungs of the mice were primarily downregulated. The dysregulation of miRNA expression was gender and age dependent. Moreover, a dose- and time-dependent effect of CS on miRNAs was also found by Izzotti and his colleagues [82, 83]. They evaluated the persistence of miRNA alterations after smoking cessation in mice that had been exposed to different doses of CS within 12 h after birth and continued daily for 4 weeks [82]. The two lowest doses (119 and 292 mg/m3 of total particulate matter) were not particularly effective, while the highest sublethal dose (438 mg/m3) produced extensive miRNA downregulation. Most of the altered miRNAs were restored 1 week after smoking cessation, although some of the mice recovered incompletely. However, when exposed to CS for 4 months, the miRNA changes detected in the mice persisted 3 months after smoking cessation [83]. These results suggest that high doses of and long-lasting exposure to CS are needed to induce irreversible miRNA alterations, which may be involved in CS-related diseases.
CS-induced dysregulation of miRNA has also been demonstrated in humans. Schembri et al. [84] examined whole-genome miRNA expression in bronchial airway epithelium from 10 current smokers and 10 never smokers. Of the 232 detected miRNAs, 28 miRNAs were found to be differentially expressed, with 82% being downregulated in current smokers. Downregulation in smokers compared to never smokers was also observed by Mascaux et al. [85]; they analyzed 60 biopsies of smokers and found that 69 miRNAs were differentially expressed in the smokers and the nonsmokers, the vast majority of which were downregulated. Similar results were detected in human peripheral blood mononuclear cells [86]. Twenty-five miRNAs in human mononuclear cell of peripheral blood were found to be differentially expressed before and after reduced smoke exposure, and most of them were upregulated after smoke reduction, suggesting a trend toward the recovery of miRNAs after smoking cessation. In addition, CS could also induce miRNA changes in human placenta [87] and human spermatozoa [88], which can further alter spermatozoal and/or embryonic gene expression. This finding may partly explain why CS-induced deleterious phenotypes can be transmitted to the progeny of smokers.
CS can also modify circulating miRNAs expression from patients. Takahashi et al. [89] determined the plasma miRNA profiles of 11 smokers and 7 nonsmokers and found that a larger number of miRNAs could be detected in smokers than could be detected in the nonsmokers, and two-thirds of the detected miRNAs (43 miRNAs) were significantly upregulated. Most of the changed miRNAs were previously reported to be potential biomarkers of diseases. Moreover, smoking cessation reversed the altered patterns of expression, which resembled those of the nonsmokers. Another study conducted by Shi et al. [90] also showed dysregulations in circulating miRNAs in smokers compared to the miRNAs in nonsmokers. They investigated the miRNA expression profiles of 28 smokers and 12 nonsmokers and found that 24 miRNAs were upregulated and 11 miRNAs were downregulated in smokers. Interestingly, the dysregulated miRNAs were related to the immune system and hormone regulation. Furthermore, most upregulated miRNAs were associated with hematologic cancers, while most miRNAs upregulated in the plasma of middle-aged smokers were associated with solid cancers. Thus, the CS-changed miRNAs would be potential biomarkers and targets for smoking-associated diseases.
As shown above, the miRNA changes induced by CS are reversible. The miRNA alterations can recover partially or completely when smoking is stopped early. In addition, a number of chemopreventive agents can also partially prevent CS-induced alterations of miRNAs. In a study conducted by Izzotti et al. [91], CS-exposed rats were treated with orally administered the chemopreventive agents N-acetylcysteine, oltipraz, indole-3-carbinol, 5,6-benzoflavone, and phenethyl isothiocyanate, as single agents or in combinations. The results showed that none of the above chemopreventive agents appreciably affected the baseline miRNA expression, but all of them attenuated CS-induced miRNA downregulation to a variable extent and in different patterns, indicating the potential safety of these agents and their potential efficacy in preventing miRNA downregulation. The effects of other chemopreventive agents on miRNAs, such as pioglitazone combined with bexarotene [92] and budesonide combined phenethyl isothiocyanate [93], were also observed in the lungs of mice in other studies.
Cigarette smoke and lncRNAs
LncRNA has generated considerable interest over the past decade. For a long time, lncRNAs were considered to be nonfunctional, but they are now believed to be involved in various biologic processes, such as oxidative stress, inflammation, cell growth and viability, apoptosis, migration and metabolism at both transcriptional and posttranscriptional levels [94, 95]. The abnormal expression of lncRNAs has been reported to affect the progression of many human diseases, including that of CS-related disorders [96]. Recently, a study of genome-wide lncRNA expression in the lung tissue of nonsmokers (n = 5) and smokers with (n = 5) or without (n = 5) COPD showed that, in smokers without COPD, 87 lncRNAs were significantly upregulated and 244 were downregulated compared to the lncRNAs expressed in the nonsmokers [97]. In the smokers with COPD, 120 lncRNAs were overexpressed and 43 were underexpressed compared with lncRNAs expressed in the smokers without COPD. These results suggest that the differentially expressed lncRNAs induced by CS in the lungs may participate in the pathogenesis and development of COPD and may provide new methods for the diagnosis and treatment of COPD. LncRNA expression profiles in the lung tissue of 5 CS-exposed mice and 5 control mice were also investigated by Zhang and his colleagues [98]. They found that 109 lncRNAs were differentially expressed after CS exposure, a finding that may provide insight useful for determining molecular mechanism of CS-associated oxidative stress and endoplasmic reticulum stress. In smoking-related lung squamous cell carcinoma, 21 lncRNAs were identified, 5 of which were upregulated and 16 of which were downregulated [99]. Further statistical analysis indicated that dysregulation of lncRNA AURKA, BIRC5, and LINC00094 indicated poor prognosis for patient with lung squamous cell carcinoma.
Several in vitro studies showed that CS is able to alter the expression of numerous lncRNAs in airway epithelial cells. Of these lncRNAs, the three most extensively studied with regard to CS are Hox transcript antisense intergenic RNA (HOTAIR), colon cancer-associated transcript-1 (CCAT1) and metastasis associated in lung adenocarcinoma transcript 1 (MALAT1) [100–103]. In CSE-treated HBE cells, the expression of HOTAIR and EZH2 was upregulated [100]. The inhibition of HOTAIR and EZH2 by siRNAs attenuated CSE-induced decreases in p21 levels, which led to cell cycle aberrations. In addition, in CSE-treated HBE cells, interleukin (IL)-6 activated phosphorylated signal transducer and activator of transcription 3 (STAT3) and increased the levels of HOTAIR, which has been found to be involved in the formation of cancer stem cells and malignant transformation [101]. Similarly, activated CCAT1 promotes an altered cell cycle transition during the malignant transformation of the HBE cells via epigenetic silencing of miR-218, suggesting a mechanism for lung cancer development [102]. MALAT1 increases were also induced by CS, which mediated the epithelial–mesenchymal transition via EZH2 and enhanced the CSE-induced epithelial–mesenchymal transition and malignant transformation of HBE cells [103]. Other examples that reveal changes in lncRNAs as a consequence of CS include smoke and cancer-associated lncRNA1 (SCAL1) [104] and lung cancer progression–association transcript 1 (LCPAT1) [105, 106], both of which were upregulated after exposure to CS treatment. Although many studies have investigated the role of lncRNAs in the development of CS-related diseases, the exact mechanism for their effect is not fully understood. The identification of lncRNAs in these processes could provide new clinical targets as well as diagnostic and prognostic tools for CS-related diseases.
Epigenetics and inflammation
DNA methylation and inflammation
CS causes multiple inflammatory diseases characterized by the influx of inflammatory cells and secretion of cytokines in susceptible individuals. In recent years, evidence has suggested that epigenetic modification mechanisms can possibly explain the link between CS exposure and inflammation. It has been shown that CS induced hypermethylation of the Runt-related transcription factor 3 (RUNX3), Janus kinase 3 (JAK3) and keratin 1 (KRT1) genes in people deficient for Alpha-1 antitrypsin, which is associated with C-reactive protein (CRP) levels [107]. Results from a meta-analysis of epigenome-wide association studies of serum CRP and DNA methylation revealed that the methylation at CpG sites that is associated with CRP differs by race or ethnicity [108]. Increased DNA methylation with the highest signal, at cg10636246 near Absent in melanoma 2 (AIM2) and involved in inflammasome response, was associated with lower expression of AIM2 and lower CRP levels. Hypomethylation at cg18181703 (suppressor of cytokine signaling 3, SOCS3), cg06126421 (tubulin beta, TUBB), and cg05575921 (aryl hydrocarbon receptor repressor, AHRR) was associated with higher CRP levels and increased risk of future CHD [108]. Moreover, AHRR can be hypomethylated by CS [109]. A 2-step epigenetic Mendelian randomization study conducted by Jhun et al. [110] in African Americans suggested that CS may increase serum IL-18 levels through a decrease in the DNA methylation levels of cg03636183 in the coagulation factor II (thrombin) receptor-like 3 gene. Systemic inflammation has been recognized as a pathogenetic hallmark of inflammatory bowel disease. Researchers found gut segment-specific differences in the DNA methylation profiles of primary intestinal epithelial cells from children with inflammatory bowel diseases versus those from controls, and some of the differentially methylated positions were independent of the mucosal inflammatory activity [111]. Similar results were also published by Somineni et al. [112]. In the blood of 164 pediatric patients with Crohn’s disease taken at diagnosis, they found 1189 CpG sites that were differentially methylated between the patients and controls. Moreover, of the 1189 CpG sites, 1155 (97%) exhibited directional consistency and 872 (73%) showed a statistically significant association with CRP.
Yu et al. [113] confirmed that elevated DNMT1 was correlated with increased methylation level of the peroxisome proliferator-activated receptor (PPAR)-γ promoter and decreased expression levels of PPAR-γ, a nuclear hormone receptor with anti-inflammatory effects, and increased pro-inflammatory cytokine production in peripheral blood monocytes. Moreover, the DNMT inhibitor 5-aza-2′-deoxycytidine (AZA) can promote alternative activation, suppress inflammation by macrophages [114], and inhibit the constitutive activation of pro-inflammatory NF-κB [115]. Tumor necrosis factor receptor-associated factor 6 (TRAF6) is a canonical transduction molecule that plays central roles in activating NF-κB and MAPK signaling pathways. Feng et al. [116] found that AZA pretreatment significantly enhanced the expression of IL-6 and IL-8 and activated the NF-κB and MAPK signaling pathways by reducing the methylation levels in the TRAF6 promoter in human dental pulp cells. In addition to regulating inflammatory factors, DNMT1 can also be activated by inflammatory factors, such as IL-6, transforming growth factor beta 1 [117] and IL-1β [118].
Tet-eleven translocation proteins (TETs) are dioxygenases that play important roles in decreasing the abundance of DNA methylation. Recently, TETs have gained attention for their roles in the inflammatory process. Upon Porphyromonas gingivalis (Pg.) lipopolysaccharide (LPS)/interferon (IFN)-γ and Escherichia coli (E. coli) LPS/IFN-γ stimulation, TET1 knockdown M1 macrophages exhibited a significant decrease in the production of pro-inflammatory markers such as IL-6 and tumor necrosis factor (TNF)-α because the NF-κB signaling pathway was suppressed [119]. Wang et al. [120] found that TET2 might be a positive regulator of LPS-induced inflammatory response in human dental pulp cells by epigenetically regulating the transcription of the signal transduction molecule MyD88 in the NF-κB signaling pathway. However, Zhang et al. [121] showed that TET2 recruited HDAC2 and negatively modulated the transcription of IL-6 via histone deacetylation in LPS-activated murine bone marrow-derived dendritic cells and peritoneal macrophages.
These findings indicate that DNA methylation regulates gene expression and may manipulate inflammatory genes to increase or decrease inflammation (Table 2). However, whether the alterations in the DNA methylation profiles are a cause or a consequence of inflammation is still debated. Somineni et al. [112] found that changes in the DNA methylation profiles of blood samples from inflammatory bowel disease patients (at diagnosis) were correlated with acute inflammation. However, with treatment, these changes closely resembled the patterns observed in patients without intestinal inflammation. These results indicate that inflammatory bowel disease-associated patterns of DNA methylation are a result of the inflammatory features of the disease and are less likely to contribute to the development or progression of the disease [112]. In addition, Wahl et al. [122] showed that obesity, an important risk of inflammatory disturbances, can influence DNA methylation. They used genetic association analyses to investigate the potential relationships between BMI (a key measure of adiposity) and DNA methylation in blood and found that the alterations in DNA methylation are predominantly the consequence of adiposity, rather than the cause of it. Therefore, it is possible that CS-induced inflammatory reactions may be a consequence rather than a cause of alterations in DNA methylation patterns. Future research into the identification of heritable epigenetic changes may assist in improving the current understanding of CS-related inflammatory diseases based on the expression of inflammatory markers and the interpretation of their methylation profiles. Understanding the changes in DNA methylation patterns in CS-induced inflammatory diseases may lead to the development of novel therapeutic/management strategies.
Table 2.
The role of CS-induced aberrant DNA methylation in inflammation
| Genes/CpG sites | Changes by CS | Functions on inflammation | Mechanism | Disease | References | Year |
|---|---|---|---|---|---|---|
| RUNX3 | Hypermethylation [107] | Pro-inflammation | Associated with the level of CRP | COPD | Siedlinski et al. [107] | 2012 |
| JAK3 | Hypermethylation [107] | Pro-inflammation | Associated with the level of CRP | COPD | Siedlinski et al. [107] | 2012 |
| KRT1 | Hypermethylation [107] | Pro-inflammation | Associated with the level of CRP | COPD | Siedlinski et al. [107] | 2012 |
| cg18181703 in SOCS3 | N/A | Anti-inflammation | Hypomethylation of the CpG site was associated with higher CRP levels | CHD | Ligthart et al. [108] | 2016 |
| cg06126421 in TUBB | N/A | Anti-inflammation | Hypomethylation of the CpG site was associated with higher CRP levels | CHD | Ligthart et al. [108] | 2016 |
| cg05575921 in AHRR | Hypomethylation [109] | Anti-inflammation | Hypomethylation of the CpG site was associated with higher CRP levels | CHD | Ligthart et al. [108] | 2016 |
| cg03636183 in F2RL3 | Hypomethylation [110] | Pro-inflammation | Increase IL-18 levels | Hypertension | Jhun et al. [110] | 2017 |
| DNMT1 | Upregulation [27] | Pro-inflammation | Increase methylation level of the PPAR-γ promoter and reduce expression of PPAR-γ | Atherosclerosis | Yu et al. [113] | 2016 |
| DNMT1 | Upregulation [27] | Pro-inflammation | Regulates IL-6 and TGFβ1; meanwhile, be activated by IL-6 and TGFβ | Benign prostatic hyperplasia | Xu et al. [117] | 2017 |
| DNMT1 | Upregulation [27] | Pro-inflammation | Be activated by IL-1β via MAPK and NF-κB pathways. | Benign meningiomas | Wang et al. [118] | 2016 |
| TET1 | N/A | Pro-inflammation | TET1 knockdown reduced IL-6 and TNF-α levels through downregulating NF-κB signaling pathway | Periodontal diseases | Huang et al. [119] | 2019 |
| TET2 | N/A | Pro-inflammation | Activate the NF-κB signaling pathway | Dental pulp inflammation | Wang et al. [120] | 2018 |
AML acute myeloid leukemia, ARHH aryl hydrocarbon receptor repressor, CHD coronary heart disease, COPD chronic obstructive pulmonary disease, CRP C-reactive protein, DNMT DNA methyltransferase, F2RL3 coagulation factor II (thrombin) receptor-like 3 gene, JAK3 Janus kinase 3, KRT1 keratin 1, MAPK mitogen‐activated protein kinase, NF-κB nuclear factor kappa B, PPAR peroxisome proliferator-activated receptor, RUNX3 runt-related transcription factor 3, SOCS3 suppressor of cytokine signaling 3, TET Tet-eleven translocation protein, TGFβ transforming growth factor beta 1, TNF-α tumor necrosis factor α, TUBB tubulin beta
Histone posttranscriptional modifications and inflammation
Histone acetylation and inflammation
The acetylation of both histones H3 and H4 has been associated with the promotion of the transcription of pro-inflammatory genes in inflammation. An increase in acetylated histone H4 was found in current smokers, while patients with COPD who had stopped smoking had increased histone H3 acetylation, suggesting that the persistent inflammation in the lungs of the COPD patients after smoking cessation may be regulated by histone H3 acetylation [29]. CS can alter the activity of both HATs and HDACs and thereby enhance histone acetylation, specifically by promoting the expression of NF-κB-dependent inflammatory genes [29]. In addition to histones, numerous nonhistone proteins can also be acetylated and deacetylated by HATs and HDACs, respectively [123]. Studies have shown that NF-κB can be acetylated at multiple lysine residues. The acetylation of the NF-κB p65 subunit at lysine 310 or lysine 221 increases the transcriptional activity or DNA-binding affinity of NF-κB, whereas acetylation at lysine 122 and lysine 123 facilitates the relocation of NF-κB from the nucleus [124]. Therefore, CS can promote inflammation by acetylating NF-κB by upregulating HATs and inhibiting HDACs.
Evidence is emerging that HATs/HDACs have important roles in the inflammatory response and that HDAC inhibitors may have clinical application in the treatment of inflammatory disease [125]. CBP/p300 is the most extensively studied HAT. Increased acetylation of histone (H3 and H4) and NF-κB by CBP/p300 is associated with CS-mediated pro-inflammatory cytokine release, which is responsible for the sustained pro-inflammatory response observed in COPD [126]. CS increased the expression of pro-inflammatory mediators IL-8, TNF-α and matrix metalloproteinase 9 through HDAC1 depression in rat lungs and in macrophages, while knocking down HDAC1 increases the expression of these genes [37]. In smokers with COPD, the levels and activities of HDAC2 are significantly reduced, leading to increased airway inflammation by promoting the expression of IL-17A [127] and/or the phosphorylation of Akt [128] and/or the activity of NF-κB [129]. Barnes et al. [130] concluded that decreased HDAC2 accounts for the amplified inflammation and resistance to the actions of corticosteroids. Furthermore, HDAC2-deficient mice are not responsive to budesonide, which inhibits LPS-induced lung inflammation [131]. Moreover, corticosteroids can suppress inflammation by recruiting HDAC2 at NF-κB-driven pro-inflammatory gene promoters, thereby inhibiting the transcription of these genes [132]. Enhanced HDAC activity by low-dose theophylline treatment can improve steroid responsiveness and reduce airway inflammation [133]. Thus, recruitment of HDAC2 to activated inflammatory genes is a major mechanism of inflammatory gene repression by corticosteroids. HDAC3 has been reported to be an important player in the recruitment of monocytes to sites of inflammation and in macrophage cytokine production. Acute smoke exposure has been shown to reduce HDAC3 activity in human alveolar macrophages, where it resulted in increased production of IL-8 and IL-1β [40]. HDAC4 functions as a corepressor of multiple immunologically related transcription factors, including c-Jun and IL-17A, and downregulates IL-6 expression [134]. Both Poralla et al. [135] and Zhao et al. [136] confirmed a pro-inflammatory capacity of HDAC5 in macrophages by activating NF-κB signaling pathway. Thus, HDAC5 overexpression promotes the production of inflammatory cytokine, and HDAC5 depletion inhibits the production of inflammatory cytokine. Studies on HDAC6 that is activated by CS [42] have shown a positive relationship between HDAC6 and inflammatory response. It has been shown that HDAC6 inhibition confers protective effects against LPS-induced inflammation, as is demonstrated by the reduced production of the pro-inflammatory cytokines TNF-α, IL-1β, and IL-6 and decreased leukocyte infiltration [137, 138]. Zhang et al. [139] found that You-Gui pills (You-Gui-Wan; YGW) reduced the production of inflammatory cytokines, such as IL-4, IL-5, and IL-13, by increasing the corresponding activity of HDAC7, HDAC710, HDAC11, and HDAC9–11, suggesting that HDAC7- and HDAC9–11-induced histone deacetylation of memory T lymphocytes has a protective role against lung inflammation and eosinophilic infiltration. HDAC8 has been shown to attenuate acetylation of Histone H3 and H4 in the IFN-β promoter, resulting in suppression of IFN-β transcription, which leads to the inhibition of innate IFN-β production [140]. Another study shows the opposite result: specific inhibition of HDAC8 reduces LPS-induced IL-1β, TNF-α, IL-6 secretion [141]. Evidence is emerging that inhibiting HDAC activity by HDAC inhibitors has clinical applications for anti-inflammatory treatment because of the suppressive effects of these inhibitors on pro-inflammatory molecules; that is, they can reduce the NF-κB-dependent production of pro-inflammatory cytokines, including IL-2, TNF-α IL-6, IL-1β, and IFN-γ, under various pathological conditions, which are triggered by respiratory syncytial virus, mitogenic anti-CD3, LPS, and phorbol myristate acetate [142]. Multiple HDACs are involved in inflammation. In some cases, more than two HDACs play opposite roles in regulating the inflammatory response, suggesting that pan-HDAC inhibitors may result in unwanted effects. Therefore, the detailed mechanism underlying the HDAC regulation of cytokine production and secretion need to be further investigated.
SIRTs, class III HDACs, have been identified as important regulators of the inflammatory process over the past few years. In TNF-α-stimulated human keratinocytes, the expression levels of SIRT1, SIRT2, SIRT3, SIRT4 and SIRT5 are downregulated, while those of SIRT6 and SIRT7 are upregulated, indicating a necessary function of SIRTs in inflammation [143]. Both SIRT1 and SIRT2 are reported to deacetylate the RelA/p65 subunit of NF-κβ at lysine 310, inhibiting NF-κβ signaling, while SIRT6 has been shown to interact with the p65/RelA that is bound to the NF-κβ promoter region to repress transcriptional activity, which results in reduced expression levels of inflammatory factors [144]. Further studies have shown that knocking down SIRT1, 2 or 6 both in vivo and in vitro results in increased expression of NF-κβ and simultaneous overexpression of inflammatory cytokines [145–147]. SIRT3 may serve as an important target that regulates inflammation. SIRT3 overexpression could ameliorate the activation of Nod-like receptor protein 3 (NLRP3) inflammasome, while SIRT3 knockdown accelerates the induction of NLRP3 expression [148]. It has been shown that SIRT3 can suppress inflammation by decreasing the expression levels of NF-kB, high mobility group box 1 (HMGB1), c-Jun, c-Fos, cyclooxygenase-2 (COX2), TNF-α, IL-1β and IL-6 [149]. SIRT4 is also found to negatively regulate CSE-induced NF-κB activation via inhibiting the degradation of inhibitory kappa B (IκB) kinase α [47]. The pro-inflammatory cytokines IL-1β, TNF-α, and IL-6, the downstream target genes of NF-κB, are inhibited by overexpression of SIRT4 [47]. Vakhrusheva et al. [150] showed that the absence of SIRT7 produces an increase in inflammation, illustrating that SIRT7 also functions to suppress inflammation. In contrast to other SIRTs, SIRT5 has a role in promoting inflammation. Qin et al. [151] showed that SIRT5 competes with SIRT2 to interact with NF-κB p65, in a deacetylase activity-independent manner to block the deacetylation of p65 by SIRT2, which leads to the increased acetylation of p65 and the activation of the NF-κB pathway and its downstream cytokines. In recent years, there have been an increasing number of studies with accumulating evidence confirming the importance of histone acetylation state in inflammation (Table 3). Targeting histone acetylation might be a new means of therapeutically targeting inflammatory processes.
Table 3.
Studies reporting aberrant HATs/HDACs induced by CS in inflammation
| HATs/HDACs | Changes by CS | Functions on inflammation | Mechanism | Disease | References | Year |
|---|---|---|---|---|---|---|
| CBP/p300 | Upregulation [50] | Pro-inflammation | Increase acetylation of histones (H3/H4) and NF-κB | COPD | Rajendrasozhan et al. [126] | 2009 |
| HDAC1 | Downregulation [37] | Anti-inflammation | Decrease level of acetylated H3K9 | COPD | Chen et al. [37] | 2015 |
| HDAC2 | Downregulation [29, 30, 38] | Anti-inflammation |
Inhibit IL-17A Suppress the phosphorylation of Akt Inhibit NF-κB |
COPD Asthma COPD |
Lai et al. [127] Xia et al. [128] To et al. [129] |
2018 2018 2017 |
| HDAC3 | Downregulation [39, 40] | Anti-inflammation | Repress synthesis of NF-κB -driven inflammatory cytokine | COPD | Winkler et al. [40] | 2012 |
| HDAC4 | Downregulation [38] | Anti-inflammation | Repress c-Jun and IL-17A | COPD | Lu et al. [134] | 2015 |
| HDAC5 | Downregulation [29, 30] | Pro-inflammation | Activate NF-κB | Chronic inflammatory diseases Mycoplasma pneumoniae pneumonia |
Poralla et al. [135] Zhao et al. [136] |
2015 2019 |
| HDAC6 |
Upregulation [41] Activation [42] |
Pro-inflammation |
Promote the increase of matrix metalloproteinase 9 expression and activate NF-κB by inducing IĸB phosphorylation Promote the phosphorylation of p38 MAPK |
Acute lung injury Brain inflammation |
Liu et al. [137] Song et al. [138] |
2019 2019 |
| HDAC7 | Downregulation [35] | Anti-inflammation | Increase histone deacetylation of memory T lymphocytes | Asthma | Zhang et al. [139] | 2015 |
| HDAC8 | Downregulation [29, 30] |
Anti-inflammation Pro-inflammation |
Promote the acetylation modification of IFNβ1 promoter, thus selectively increasing innate IFN-β production Increase the production of IL-1β, TNFα, and IL-6 |
Asthma Systemic juvenile idiopathic arthritis |
Meng et al. [140] Li et al. [141] |
2016 2015 |
| HDAC9 | N/A | Anti-inflammation | Increase histone deacetylation of memory T lymphocytes | Asthma | Zhang et al. [139] | 2015 |
| HDAC10 | Downregulation [36] | Anti-inflammation | Increase histone deacetylation of memory T lymphocytes | Asthma | Zhang et al. [139] | 2015 |
| HDAC11 | N/A | Anti-inflammation | Increase histone deacetylation of memory T lymphocytes | Asthma | Zhang et al. [139] | 2015 |
| SIRT1 | Downregulation [43] | Anti-inflammation | Deacetylate the RelA/p65 subunit of NF-κβ and attenuate NF-κB-mediated gene transcription | Chronic inflammatory diseases | Schug et al. [145] | 2010 |
| SIRT2 | N/A | Anti-inflammation | Deacetylate the RelA/p65 subunit of NF-κB at Lys310 and inhibit NF-κβ signaling | Inflammatory bowel disease | Lo et al. [146] | 2014 |
| SIRT3 | Downregulation [46] | Anti-inflammation |
Ameliorate NLRP3 inflammasome activation Decrease the expression levels of NF-κB, HMGB1, c-Jun, c-Fos, COX2, TNF-α, IL-1β and IL-6 |
Hyperlipidaemia Chronic kidney disease |
Liu et al. [148] Qiao et al. [149] |
2018 2018 |
| SIRT4 | Downregulation [47] | Anti-inflammation | Suppress NF-κB activating via inhibiting the degradation of IκBα | COPD | Chen et al. [47] | 2014 |
| SIRT5 | Downregulation [48] | Pro-inflammation | Compete with SIRT2 to interact with NF-κB p65 to block the deacetylation of p65 by SIRT2, leading to increased acetylation of p65 and the activation of NF-κB pathway | Sepsis | Qin et al. [151] | 2017 |
| SIRT6 | Downregulation [49] | Anti-inflammation | Interact with p65/RelA bound to the NF-κβ promoter region and repress transcriptional activity | Inflammatory vascular diseases | Lappas M. [147] | 2012 |
| SIRT7 | N/A | Anti-inflammation | Deacetylate p53, leading to inactivation of p53 | Heart diseases | Vakhrusheva et al. [150] | 2008 |
CBP cAMP-response element binding protein, COX2 cyclooxygenase-2, CS cigarette smoke, HAT histone acetyltransferases, HDACs histone deacetylases, HMGB1 high mobility group box 1, IFNβ1 interferon beta 1, IκBα inhibitory kappa B kinase α, IL interleukin, NLRP3 NOD-like receptor family, pyrin domain containing 3, SIRT sirtuin
Histone methylation and inflammation
HMTs have been proven to have significant regulatory effects on inflammatory responses. EZH2, the primary H3K27 tri-methyltransferase, was found to integrate the complex functions of TNF-α signaling to enhance the inflammatory response. EZH2 deficiency directly stimulates TRAF expression to enhance TNF-α-induced NF-κB signaling, thereby leading to uncontrolled inflammation [152]. Liang et al. [153] found that EZH2 can induce caspase-3 activation and production of pro-inflammatory molecules by activating p38. This effect can be reversed by inhibiting EZH2. Recently, PRMTs have been confirmed to be correlated with inflammatory responses. PRMT1, PRMT4, PRMT5 and PRMT6 are all reportedly involved in inflammatory responses in association with the pro-inflammatory factor NF-κB, although each of those has unique regulatory mechanisms [154]. In addition, elevated PRMT1, stimulated by IL-4, plays a crucial role in chronic antigen-induced pulmonary inflammation [155]. In LPS-induced macrophages, the dose of PRMT2 influences the lung airway hyper-responsiveness, the recruitment of neutrophils, and the expression of pro-inflammatory cytokines, such as IL-6 and TNF-α, through NF-κB [156]. PRMT5 was shown to bind and methylate HOXA9 in TNF-α-stimulated endothelial cells to regulate inflammatory processes of these cells [157]. The inhibition of PRMT5 by PRMT5 inhibitor or by siRNA-mediated knockdown prevents the phosphorylation of IκBβ and IκBα and inhibits the nucleus translocation of p65, leading to a reduction in IL-6 and IL-8 expression [158]. Our previous mouse model and in vitro studies revealed that PRMT6 exerts a negative effect on CS-induced pulmonary inflammation through H3R2me2a, while overexpression of PRMT6 inhibits CS-induced inflammation [51, 52]. Thus, HMTs play important roles in the process of inflammatory gene posttranslational modification. Given the importance of inflammation in a great number of diseases, HMTs appear to be a newly discovered potential therapeutic target to modulate the inflammatory response. However, current studies linking HMTs to inflammation are in a very nascent stage. Future studies are warranted to address the underlying molecular mechanism of histone methylation and HMTs in the regulation of inflammatory gene transcription.
Histone phosphorylation and inflammation
Previous studies indicated that the phosphorylation of histones is responsible for inflammatory processes [159]. COX2 is an important enzyme in activating the cellular inflammatory response. It has been shown that the phosphorylation of histone H3 at Ser10 and Ser28 plays a critical role in promoting COX2 expression [159]. Histone H3 phosphorylation at Ser10, which occurs at the promoters of the NF-κB-regulated cytokines, has been observed to increase the accessibility of NF-κB binding sides [160]. Chung et al. [161] find that the activation of IκBα is critical in the phospho-acetylation of histone H3 on the pro-inflammatory gene promoters in response to CS stimuli and the histone H3 phospho-acetylation is critical for the activation of NF-κB-directed gene expression. It has been shown that MSK1 can induce p65 phosphorylation at Ser276, which in turn induces IL-8/CXCL8 release in thrombin-stimulated human lung epithelial cells [162].
Chemokine fractalkine (CX3CL1) is essential in neuroinflammation. Galán-Ganga et al. [163] found that knocking down of MSK1 decreased the mRNA levels of IL-1β, TNF-α, which was triggered by stimulation with CX3CL1 through the inactivation of NF-κB signaling pathway. In addition, MSK1 was also shown to be an important downstream kinase involved in CS-induced NF-κB activation and the phospho-acetylation of histone H3 [57]. The NF-κB inhibitor dimethyl fumarate can inhibit NF-κB-dependent chemokine secretion by inhibiting histone H3 phosphorylation through MSK1 [160]. The IL-1 receptor antagonist (IL-1Ra) is homologous to IL-1 and is able to bind to, but not activate, the IL-1 receptor. It has been shown that MSK1 and MSK2 are required for the induced expression of both IL-1Ra mRNA and protein, while knocking out MSK in mice was found to result in a decreased IL-1Ra production [164]. These results indicate a promoting effect of MSK1 on inflammation. However, Zhong et al. [165] put forward a different opinion, suggesting that MSK1 is negatively correlated with astrocyte inflammation and can inhibit inflammatory cytokine production. They showed that MSK1 inhibition promoted the expression of TNF-α, IL-6, IL-10, and IL-1β. Thus, MSK1 might either promote or repress inflammation depending on the cell type or stimuli. RSK is another kind of kinase that catalyzes the phosphorylation of histone H3. Lin and his colleagues [166] found that the thrombin-induced release and activation of the inflammatory factor IL-8/CXCL8 can be inhibited by RSK1 siRNA, suggesting that RSK1 has an enhancing effect in the inflammatory process. Therefore, CS may induce inflammatory responses through the histone phosphorylation induced by activated kinases; these kinases may represent potential targets in therapy that controls the CS-mediated chronic inflammatory response.
Histone ubiquitination and inflammation
Over the last few years, evidence has accumulated showing that ubiquitination is a key process in regulating inflammation responses [167]. Studies in vivo and in vitro have revealed the involvement of the ubiquitin proteasome system in CS-induced inflammation. The muscle-specific E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1, which play important roles in the process of muscle-wasting degradation, were shown to be upregulated by CS [73]. This process is related to IκB-α degradation and NF-κB activation. NF-κB inhibition prevents CS-induced upregulation of MuRF1 and CS-induced reduction in the diameter and degradation of the MyHC-related C2 myotubes [2], suggesting that inflammation is responsible for CS-induced proteasomal degradation in skeletal muscle. Atrogin-1 is a ubiquitin protein ligase. Zeng et al. [168] found that atrogin-1 overexpression markedly inhibits the expression of pro-inflammatory-related genes (including IL-1β, IL-6, Ptgs2 and Serpinb2), while knocking down atrogin-1 by siRNA had the opposite effects. MUL1, an Akt ubiquitin E3 ligase that catalyze Akt ubiquitination/degradation, can be upregulated by CS [75]. Studies have shown that the knocking down MUL1 with a combination of siRNA and shRNA results in potentiated activation of both the IFN-β and NF-kB pathways [169]. HDAC2, which can suppress the inflammatory genes via deacetylation of histones and glucocorticoid receptors [130], was ubiquitinated and degraded in response to CS and oxidative stress [77]. It has been shown that, in CSE-exposed airway epithelial cells and macrophages, HDAC2 is excessively ubiquitinated and degraded in the proteasome due to the low expression of USP17, an important deubiquitinating enzyme [77]. Overexpressed USP17 blocks the degradation of HDAC2 induced by CSE, resulting in increased production of inflammatory factors. The NLRP3 inflammasome consists of a protein-nucleotide-binding domain and leucine-rich repeat pyrin-containing protein-3, an apoptosis-associated speck-like protein containing CARD and the pro-apoptotic protease caspase-1. Upon inflammasome activation, caspase-1 is cleaved and activated, and pro-inflammatory cytokines such as IL-1β and IL-18 are released [170]. Han et al. [171] reported that CSE can induce the degradation of NLRP3 protein via the ubiquitin proteasome system in human monocytes and macrophages, which in turn diminishes the formation of NLRP3 inflammasomes and prevents the subsequent release of IL-1β and IL-18. However, the levels of IL-1β and IL-18 were increased in the lungs, lavage fluid, and sputum of the COPD subjects and animals exposed to CS [172, 173]. It is possible that the response to CS could differ by cell type, model system, or kinetics [171]. Pulmonary cells such as lung epithelial cells or alveolar macrophages could have different responses to CSE in terms of cellular NLRP3 protein levels, while monocytes or monocyte-derived macrophages have decreased NLRP3 protein levels upon exposure to CS, leading to immunosuppression [171]. These results show that CS can elevate the level of ubiquitination through the upregulation of E3 ubiquitin ligases or the decrease of deubiquitinating enzymes, resulting in decreased inflammatory responses and immunosuppression. Thus, CS may suppress immune function by modifying the activity of the ubiquitin proteasome system, making smokers more susceptible to infection.
Non-coding RNAs and inflammation
MiRNAs and inflammation
In the last few years, there has been a growing interest in the role of miRNAs in the CS-related inflammatory process. MiRNAs have distinctive functions depending on their expression in different cell types, and they can directly or indirectly regulate the inflammatory response. Additionally, some proteins that are induced during inflammatory responses can regulate the processing of miRNAs [174]. The role of a few selected miRNAs changed by CS in inflammation is discussed below (Table 4). It is noteworthy that several miRNAs dysregulated due to CS, namely, including let-7, miR-16, miR-21, miR-24, miR-29b, miR-30, miR-132, miR-135b, miR-145, miR-146a, miR-149, miR-150, miR-181, miR-195, miR-200, miR-212, miR-218, miR-223, and miR-320 are found to be functionally related to inflammation. Among these, miR-132 is upregulated in the serum of smokers compared with its expression in the nonsmoker controls, and the miR-132 expression level was positively correlated with the levels of inflammatory cytokines (IL-1β and TNF-α). Knocking down miR-132 attenuates CS-induced inflammation in human monocyte-like cells by targeting the SOCS5, which acts to limit the duration of signaling responses [175]. MiR-195 was highly expressed in peripheral lung tissues from COPD patients compared to the levels found in never smokers. Knocking down miR-195 alleviates CS-induced lung injury and inflammatory cell infiltration, as well as production of TNF-α and IL-6, by regulating Akt signaling [176], indicating that miR-195 has a role in the promotion of inflammation. However, there are still disputes about the effects of miR-195 on inflammation. MiR-195 expression is decreased significantly in the serum samples of ulcerative colitis patients compared to the levels in normal subjects. In addition, the expression of miR-195 is further decreased in steroid-resistant ulcerative colitis patients compared with its expression in steroid-sensitive ulcerative colitis patients. Upregulating miR-195 represses Smad7 gene expression, which can aggravate inflammation through the inhibition of the TNF-β signal pathway [177]. These results indicate that the decrease in miR-195 might act in the steroid resistance of ulcerative colitis by activating inflammation through Smad7 signaling.
Table 4.
Implications of CS-related microRNAs on inflammation
| miRNAs | Changes by CS | Functions on inflammation | Mechanism | Disease | References | Year |
|---|---|---|---|---|---|---|
| Let-7c | Downregulation [80, 82] | Anti-inflammation |
Suppress NF-κB signaling Suppress STAT3 signaling |
Endometritis COPD |
Zhao et al. [181] Yu et al. [182] |
2019 2016 |
| miR-16 | Upregulation [196] | Pro-inflammation | Upregulated by NF-κB | Gastric cancer | Shin et al. [196] | |
| miR-21 | Upregulation [195, 196] | Pro-inflammation | Upregulated by NF-κB | Gastric cancer | Shin et al. [196] | |
| miR-24 | Downregulation [194] | Anti-inflammation | Suppress TNF-α, IL-1β and NF-κB | IPF | Ebrahimpour et al. [194] | 2019 |
| miR-29b | Downregulation [195] | Anti-inflammation | Reduce VEGF-A expression; meanwhile upregulated by NF-κB inhibitor | Breast cancer | Malik et al. [195] | 2018 |
| miR-30 | Downregulation [80, 82] | Anti-inflammation | Inhibit NF-κB | N/A | Izzotti et al. [80] | 2009 |
| miR-132 | Downregulation [175] | Anti-inflammation | Repress SOCS5 expression | COPD | Diao et al. [175] | 2018 |
| miR-135b | Upregulation [183] | Anti-inflammation | Upregulated by IL-1R1 and directly targets IL-1R1 in a negative regulatory feedback loop | N/A | Halappanavar et al. [183] | 2013 |
| miR-145 | Downregulation [187] | Anti-inflammation | Suppress Kruppel-like 5 and NF-κB | COPD | Dang et al. [187] | 2019 |
| miR-146a | Downregulation [80] | Anti-inflammation | Reduce CS-induced COX-2 production. | N/A | Zago et al. [180] | |
| miR-149 | Downregulation [188] | Anti-inflammation | Inhibit the TLR-4/NF-κB signaling pathway | COPD | Shen et al. [188] | 2017 |
| miR-150 | Downregulation [189] | Anti-inflammation | Inactivate NF-κB | COPD | Xue et al. [189] | 2018 |
| miR-181 | Downregulation [190] | Anti-inflammation | Inhibit CCN1 | COPD | Du et al. [190] | 2017 |
| miR-195 | Upregulation [176] |
Pro-inflammation Anti-inflammation |
Elevate Akt phosphorylation by suppressing PHLPP2 expression Inhibit NF-κB and JNK signaling by repressing Smad7 |
COPD Ulcerative colitis |
Gu et al. [176] Chen et al. [177] |
2018 2015 |
| miR-200 | Downregulation [197] | Anti-inflammation | Suppressed by NF-κB | Lung cancer | Zhao et al. [197] | 2013 |
| miR-212 | Downregulation [191] | Anti-inflammation | Promote the phosphorylation of Akt. | COPD | Jia et al. [191] | 2018 |
| miR-218 | Downregulation [192] | Anti-inflammation | Inactivate NF-κB | COPD | Conickx et al. [192] | 2017 |
| miR-223 |
Downregulation [80] Upregulation [186] |
Anti-inflammation Pro-inflammation |
Inhibit NF-κB by targeting IL-1 receptor-associated kinase 1 Downregulate NLRP3 Suppress HDAC2 |
Low back pain Inflammatory bowel disease COPD |
Wang et al. [184] Neudecker et al. [185] Leuenberger et al. [186] |
2018 2017 2016 |
| miR-320 | N/A | Anti-inflammation | Inhibit NF-κB | COPD | Faiz et al. [198] | 2019 |
CCN1 a member of CCN family, IPF idiopathic pulmonary fibrosis, PHLPP2 PH domain and leucine-rich repeat protein phosphatase 2, STAT3 signal transducer and activator of transcription 3, TLR toll-like receptor, VEGF vascular endothelial growth factor. JNK c-Jun N-terminal kinase
MiR-146a was originally identified as an inflammatory response miRNA that was known to target members of the NF-κB family [178]. The expression level of miR-146a was shown to be decreased after CS exposure [80]. RelB, an NF-κB protein, exhibited potent anti-inflammatory properties against CS by reducing the production of COX2 [179]. It has been shown that RelB inhibits CS-induced COX2 expression by upregulating miR-146a, which highlights the potential of the RelB-miR-146a axis as a novel regulatory pathway that can attenuate inflammation in response to CS [180]. Let-7c was previously reported to be downregulated in rat and mouse lungs upon CS exposure [80, 82]. It has been shown that let-7c can suppress LPS-induced inflammation by inhibiting NF-κB [181] and STAT3 [182]. Thus, let-7c may function as a protective effector by conferring protection against CS-induced inflammation. MiR-30 is downregulated remarkably, even at the lowest dose of CS exposure [80, 82]. This miRNA is involved in NF-κB activation, which represents a general defense mechanism against stress and toxic agents [80]. MiR-135b was found to be significantly upregulated in the lungs of CS-exposed mice and in lung epithelial cells following IL-1 receptor-1 stimulation [183]. MiR-135b can also bind to its own regulator (IL-1 receptor-1), as well as its downstream effector, Caspase-1, in an attempt to stop further expansion of the inflammatory process by other potent cytokines, such as IL-1β, implying that these molecules self-regulate in a negative feedback loop [183]. MiR-223 was shown to be downregulated in the lungs of rats exposed to CS [80]. It has been shown that the overexpression of miR-223 suppresses LPS-induced NF-κB signaling by targeting IL-1 receptor-associated kinase 1 [184]. Recent data have revealed the upregulation of miR-223 as a novel biomarker in subsets of patients with inflammatory bowel disease [185]. Mice genetically deficient in miR-223 displayed markedly exacerbated experimentally induced colitis, hyperactivated NLRP3, and IL-1β release, while mice with overexpressed miR-223 displayed the opposite effects [185], suggesting that miR-223 represents a novel target for reducing the inflammatory response. In contrast, Leuenberger et al. [186] found elevated expression levels of miR-223 in COPD patients and in CS-exposed mice. The increased expression of miR-223 repressed the expression and activity of HDAC2 in pulmonary cells, which, in turn, alter the expression profile of chemokines.
The levels of miR-145 [187], miR-149 [188], miR-150 [189], miR-181 [190], miR-212 [191] and miR-218 [192] were decreased significantly in vivo and in vitro during exposure to CS, and the expression of inflammatory genes was simultaneously increased. Overexpression of miR-145, miR-149, miR-150 or miR-218 suppressed the secretion of inflammatory cytokines that had been induced by CS through the inactivation of the NF-κB signaling pathway [187–189, 192]. miR-212 reduced inflammation by promoting the phosphorylation of Akt [191], while miR-181 decreased the inflammatory response, neutrophil infiltration, and inflammatory cytokine production by targeting CCN1 [190]. CCN1, also known as Cyr61, was found to be upregulated in lung epithelial cells by CSE via the induction of ROS and endoplasmic reticulum stress, which subsequently resulted in augmented IL-8 release through the activation of the Wnt pathway [193]. Nicotine reduced miR-24 expression in a dose-dependent manner and upregulated the expression of inflammatory cytokines such as TNF-α, IL-1β and NF-κB in primary human lung epithelial cells [194]. In addition, the induced expression of these inflammatory molecules by nicotine was mirrored by genetic ablation of miR-24, suggesting that nicotine controls these molecules, at least in part, through the inhibition of miR-24 [194].
Benzo[a]pyrene, a constituent of CS, has been shown to promote inflammation pathways via TNF-α and NF-κB, leading to IL-6 upregulation, miRNA (let-7a, miR-21 and miR-29b) dysregulation and the activation of VEGF [195]. The downregulation of let-7a and miR-29b and the upregulation of miR-21 induced by benzo[a]pyrene in human mammary cells were associated with the altered expression of inflammation mediators. Moreover, pretreatment with an NF-κB inhibitor attenuated the benzo[a]pyrene-mediated dysregulation of the miRNAs. In addition, Shin et al. [196] found that the upregulation of miR-16 and miR-21 induced by nicotine stimulation can be restored by inhibiting NF-κB. Zhao et al. [197] showed that CS induced the activation of NF-κB and downregulated the expression of miR-200c. The inhibition of NF-κB activation also reversed the CS-induced decrease in miR-200c expression. These results suggest that let-7a, miR-16, miR-21, miR-29 and miR-200c are the downstream targets of NF-κB that are involved in NF-κB-mediated inflammation. CXCL8 is a key mediator in the neutrophil infiltration into airway tissue by acting as a neutrophil chemoattractant. It has been observed that miR-320d suppressed the CSE-induced release of CXCL8 via the inhibition of NF-κB activation, thus exhibiting its anti-inflammatory function [198]. Moreover, inhaled corticosteroids can induce increase in miR-320d. These studies open a window for certain miRNAs to serve as diagnostic and therapeutic agents against CS-induced inflammation.
LncRNAs and inflammation
In recent years, lncRNAs are emerging as biomarkers with diagnosis value in prognosis protocols or in the personalized treatment of inflammation-related alterations. Several reports have demonstrated the dysregulation of lncRNAs in the pathogenesis of various inflammatory diseases [199]. The abnormal expression of lncRNA in inflammatory diseases is related to the expression of many inflammatory genes and the activation or suppression of signaling pathways. However, until now, little has been known about lncRNAs in the context of CS-related inflammation. As mentioned above, the lncRNA expression profiles are changed when exposed to CS, and HOTAIR, MALAT1, and CCAT1 have been the most extensively studied of these lncRNAs. It has been reported that HOTAIR may be considered a promoter of inflammatory response [200]. In a sepsis mice model, LPS can induce the upregulation of HOTAIR in cardiomyocytes, in line with increased NF-κB activation and TNF-α production, which is reversed by HOTAIR silence [201]. Furthermore, HOTAIR-induced TNF-α production is suppressed by an NF-κB inhibitor, suggesting that HOTAIR can promote TNF-α production by activating the NF-κB signaling pathway [201]. Protein Kinase Resource (PKR) has been identified as a necessary protein kinase for the activation of one or several types of inflammasomes [202]. Ultraviolet B rays can promote the expression of HOTAIR in human keratinocytes, which in turn promotes the expression of PKR, TNF-α and IL-6 [203]. Suppressing HOTAIR decreases the expression of PKR as well as TNF-α and IL-6. A similar result was also found by Liu et al. [204]; that is, IL‐6, IL‐1β, COX2, and TNF-α protein levels are significantly depressed by silencing HOTAIR but are increased by upregulation of HOTAIR. Recently, lncRNA MALAT1 was also identified as a regulator of inflammatory cytokine production. MALAT1 can work as a novel inflammatory regulator that acts through the p38 MAPK/NF-κB pathway [205]. In the cecal ligation and puncture (CLP)-induced sepsis model, MALAT1 expression is significantly upregulated, which in turn aggravates cardiac dysfunction and inflammation by activating p38 MAPK/NF-κB, resulting in increased TNF-α, IL-1β, IL-6, IL-10, IL-17 and IFN-γ. In addition, these effects induced by CLP are reversed by knocking down MALAT1 [205]. A recent study also found that, under hyperglycemic conditions, MALAT1 expression is increased significantly in human umbilical vein endothelial cells, which is associated with a parallel increase in serum amyloid antigen 3, an inflammatory ligand and target of MALAT1 [206]. Knocking down MALAT1 downregulates serum amyloid antigen 3 activation, subsequently reducing the RNA and protein expressions of key inflammatory mediators (IL-6 and TNF-α) [206]. LncRNA MALAT1 was found to interact with NF-κB in the nucleus, thus inhibiting the DNA-binding activity of NF-κB and reducing the production of inflammatory cytokines. Knocking down MALAT1 can increase LPS-induced expression of TNF-α and IL-6 [207]. In addition to regulating the expression of inflammatory factors, both HOTAIR and MALAT1 can be upregulated by the pro-inflammatory cytokine IL-6 [101, 208]. Moreover, blocking IL-6 with an anti-IL-6 antibody significantly reduces MALAT1 expression [208]. These results suggest that a feedback regulation may involve IL-6, HOTAIR and MALAT1. CCAT1 is highly expressed in inflammatory bowel disease patients and in colorectal cancer cells. It has recently been shown that CCAT1 triggers TNF-α and IFN-γ expression by targeting miR-185 in human colorectal cancer cells. In addition, the expression of CCAT1 is significantly increased after TNF-α treatment, forming a positive regulatory feedback loop [209]. In summary, lncRNAs play roles in the regulation of gene transcription during the inflammatory response and are closely related to CS-induced inflammatory diseases (Table 5). Because of the complex pathogenesis of CS-induced inflammation and the impact of many factors, the function of many lncRNA is still not clear. Thus, substantial further researches are needed to illuminate the relationship between lncRNA and CS-induced inflammation. With the increased in-depth research on lncRNA function, it is expected to become a new target for the diagnosis of CS-related inflammatory diseases and a new therapeutic target.
Table 5.
CS‐regulated lncRNAs and their role in inflammation
| LncRNAs | Changes by CS | Functions on inflammation | Mechanism | Disease | References | Year |
|---|---|---|---|---|---|---|
| HOTAIR | Upregulation [100, 101] | Pro-inflammation |
Promote TNF-α production by activating NF-κB signaling Promote the expression of protein kinase resource, TNF-α and IL-6 Promote the expression of IL‐6, IL‐1β, COX2, and TNF-α Upregulated by pro-inflammatory cytokine IL-6 |
Sepsis Keratinocyte injury Atherosclerosis Lung cancer |
Wu et al. [201] Liu and Zhang [203] Liu et al. [204] Liu et al. [101] |
2016 2018 2019 2015 |
| MALAT1 | Upregulation [103] |
Pro-inflammation Anti-inflammation |
Activate p38 MAPK/NF-κB Increase serum amyloid antigen 3 Upregulated by pro-inflammatory cytokine IL-6 Inhibit the DNA binding activity of NF-κB |
Sepsis Diabetes Sepsis N/A |
Chen et al. [205] Puthanveetil et al. [206] Zhuang et al. [208] Zhao et al. [207] |
2018 2015 2017 2016 |
| CCAT1 | Upregulation [102] | Pro-inflammation | Trigger TNF-α, IFN-γ expression by targeting miR-185-3p, meanwhile upregulated by TNF-α | Inflammatory bowel disease | Ma et al. [209] | 2019 |
CCAT1 colon cancer-associated transcript-1, HOTAIR Hox transcript antisense intergenic RNA, MALAT1 metastasis associated in lung adenocarcinoma transcript 1
Conclusion
Genetic factors are unable to fully account for the risk and prognosis of CS-related diseases. A growing body of evidence supports the importance of epigenetic alterations in the initiation and development of CS-induced diseases. CS-mediated alterations in DNA methylation, histone modification, and ncRNAs can affect a variety of molecular and cellular processes, such as inflammation, cell cycle arrest, apoptosis, senescence, and autophagy. Based on our review, there is a wealth of evidence in support of the correlation between epigenetic changes and inflammatory responses. In particular, transcription regulation by NF-κB, a key pro-inflammatory molecule, appears to have a main function in CS-induced epigenetic changes in the mediation of inflammation. However, the role of epigenetic mechanisms in CS-mediated inflammation is very complex and not fully understood. Different changes of epigenetic alterations may play a pro-inflammatory or an anti-inflammatory role through the regulation of target gene expression, thereby participating in the occurrence and development of CS-medicated inflammatory diseases. The recognition of the alterations in DNA methylation, histone modifications, and ncRNAs provides a better understanding of the molecular basis for CS-medicated inflammation. With increasing levels of in-depth research on epigenetic functions, there is no doubt that, in the near future, epigenetic research will lead to the development of epigenomic tools that offer the potential for novel diagnostic and therapeutic agents for CS-medicated disorders.
Acknowledgements
Not applicable.
Abbreviations
- AZA
5-aza-2′-deoxycytidine
- CBP
c-AMP response element binding protein
- CCAT1
colon cancer-associated transcript-1
- COPD
chronic obstructive pulmonary disease
- COX2
cyclooxygenase-2
- COX II
cytochrome C oxidase subunit II
- CS
cigarette smoke
- DNMT
DNA methyl transferase
- ERK
extracellular signal-regulated kinase
- EZH2
enhancer of zeste homolog 2
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- HMT
histone methyltransferase
- HOTAIR
Hox transcript antisense intergenic RNA
- IκB
inhibitory kappa B
- IL-1Ra
interleukin-1 receptor antagonist
- LCPAT1
lung cancer progression–association transcript 1
- lncRNA
long non-coding RNA
- MAFbx
muscle atrophy F-box
- MALAT1
metastasis associated in lung adenocarcinoma transcript 1
- MAPK
mitogen-activated protein kinase
- MSK
mitogen- and stress-activated kinase
- MUL1
mitochondrial E3 ubiquitin protein ligase 1
- MuRF1
muscle ring finger-1 protein
- NF-κB
nuclear factor kappa B
- NLRP3
Nod-like receptor protein 3
- PPAR
peroxisome proliferator-activated receptor
- PRMT
protein arginine methyltransferase
- SCAL1
smoke and cancer-associated lncRNA1
- SIRT
sirtuin
- TETs
Tet-eleven translocation proteins
- UPS
ubiquitin–proteasome system
- USP
ubiquitin-specific protease
Authors’ contributions
DZ, XL, JL designed the drafting, acquired the materials and wrote the manuscript draft. RO and PC reviewed and edited the manuscript. All authors read and approved the final manuscript for publication.
Funding
This study was supported by grants from the Natural Science Foundation of Hunan Province (Grant No. 2018JJ3763) and the National Natural Science Foundation of China (Grant No. 81900042) and the National Key Clinical Specialty Construction Projects of China (Grant No. 2012-650).
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Dandan Zong, Email: zongdandan0402@csu.edu.cn.
Xiangming Liu, Email: liuxm_0410@outlook.com.
Jinhua Li, Email: jinhuali@csu.edu.cn.
Ruoyun Ouyang, Email: ouyangruoyun@csu.edu.cn.
Ping Chen, Email: pingchen0731@csu.edu.cn.
References
- 1.Lee J, Taneja V, Vassallo R. Cigarette smoking and inflammation: cellular and molecular mechanisms. J Dent Res. 2012;91:142–149. doi: 10.1177/0022034511421200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Teo KK, Ounpuu S, Hawken S, Pandey MR, Valentin V, Hunt D, et al. Tobacco use and risk of myocardial infarction in 52 countries in the INTERHEART study: a case-control study. Lancet. 2006;368:647–658. doi: 10.1016/S0140-6736(06)69249-0. [DOI] [PubMed] [Google Scholar]
- 3.Jindal SK, Aggarwal AN, Chaudhry K, Chhabra SK, D’Souza GA, Gupta D, et al. A multicentric study on epidemiology of chronic obstructive pulmonary disease and its relationship with tobacco smoking and environmental tobacco smoke exposure. Indian J Chest Dis Allied Sci. 2006;48:23–29. [PubMed] [Google Scholar]
- 4.Zhou Z, Chen P, Peng H. Are healthy smokers really healthy? Tob Induc Dis. 2016;14:35. doi: 10.1186/s12971-016-0101-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR, et al. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet. 2016;9:436–447. doi: 10.1161/CIRCGENETICS.116.001506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zong DD, Ouyang RY, Chen P. Epigenetic mechanisms in chronic obstructive pulmonary disease. Eur Rev Med Pharmacol Sci. 2015;19:844–856. [PubMed] [Google Scholar]
- 7.Pan Y, Liu G, Zhou F, Su B, Li Y. DNA methylation profiles in cancer diagnosis and therapeutics. Clin Exp Med. 2018;18:1–14. doi: 10.1007/s10238-017-0467-0. [DOI] [PubMed] [Google Scholar]
- 8.Kamrani A, Alipourfard I, Ahmadi-Khiavi H, Yousefi M, Rostamzadeh D, Izadi M, et al. The role of epigenetic changes in preeclampsia. Biofactors. 2019;45(5):712–724. doi: 10.1002/biof.1542. [DOI] [PubMed] [Google Scholar]
- 9.Xin S, Huang K, Zhu XG. Non-coding RNAs: regulators of glioma cell epithelial–mesenchymal transformation. Pathol Res Pract. 2019;215:152539. doi: 10.1016/j.prp.2019.152539. [DOI] [PubMed] [Google Scholar]
- 10.Awan HM, Shah A, Rashid F, Shan G. Primate-specific long non-coding RNAs and microRNAs. Genom Proteom Bioinform. 2017;15:187–195. doi: 10.1016/j.gpb.2017.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao C, Zhang Y, Popel AS. Mechanistic computational models of microRNA-mediated signaling networks in human diseases. Int J Mol Sci. 2019;20:421. doi: 10.3390/ijms20020421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kopp F. Molecular functions and biological roles of long non-coding RNAs in human physiology and disease. J Gene Med. 2019;21:e3104. doi: 10.1002/jgm.3104. [DOI] [PubMed] [Google Scholar]
- 13.Comer BS, Ba M, Singer CA, Gerthoffer WT. Epigenetic targets for novel therapies of lung diseases. Pharmacol Ther. 2015;147:91–110. doi: 10.1016/j.pharmthera.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao X, Jia M, Zhang Y, Breitling LP, Brenner H. DNA methylation changes of whole blood cells in response to active smoking exposure in adults: a systematic review of DNA methylation studies. Clin Epigenet. 2015;7:113. doi: 10.1186/s13148-015-0148-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu X, Li J, Deng S, Yu K, Liu X, Deng Q, et al. Genome-wide analysis of DNA methylation and cigarette smoking in a chinese population. Environ Health Perspect. 2016;124:966–973. doi: 10.1289/ehp.1509834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guida F, Sandanger TM, Castagne R, Campanella G, Polidoro S, Palli D, et al. Dynamics of smoking-induced genome-wide methylation changes with time since smoking cessation. Hum Mol Genet. 2015;24:2349–2359. doi: 10.1093/hmg/ddu751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zaghlool SB, Al-Shafai M, Al MW, Kumar P, Falchi M, Suhre K. Association of DNA methylation with age, gender, and smoking in an Arab population. Clin Epigenet. 2015;7:6. doi: 10.1186/s13148-014-0040-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dogan MV, Shields B, Cutrona C, Gao L, Gibbons FX, Simons R, et al. The effect of smoking on DNA methylation of peripheral blood mononuclear cells from African American women. BMC Genomics. 2014;15:151. doi: 10.1186/1471-2164-15-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elliott HR, Tillin T, McArdle WL, Ho K, Duggirala A, Frayling TM, et al. Differences in smoking associated DNA methylation patterns in South Asians and Europeans. Clin Epigenet. 2014;6:4. doi: 10.1186/1868-7083-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shenker NS, Ueland PM, Polidoro S, van Veldhoven K, Ricceri F, Brown R, et al. DNA methylation as a long-term biomarker of exposure to tobacco smoke. Epidemiology. 2013;24:712–716. doi: 10.1097/EDE.0b013e31829d5cb3. [DOI] [PubMed] [Google Scholar]
- 21.Zeilinger S, Kuhnel B, Klopp N, Baurecht H, Kleinschmidt A, Gieger C, et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS ONE. 2013;8:e63812. doi: 10.1371/journal.pone.0063812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Philibert RA, Beach SR, Lei MK, Brody GH. Changes in DNA methylation at the aryl hydrocarbon receptor repressor may be a new biomarker for smoking. Clin Epigenet. 2013;5:19. doi: 10.1186/1868-7083-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun YV, Smith AK, Conneely KN, Chang Q, Li W, Lazarus A, et al. Epigenomic association analysis identifies smoking-related DNA methylation sites in African Americans. Hum Genet. 2013;132:1027–1037. doi: 10.1007/s00439-013-1311-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Howe CG, Zhou M, Wang X, Pittman GS, Thompson IJ, Campbell MR, et al. Associations between maternal tobacco smoke exposure and the cord blood [formula: see text] DNA methylome. Environ Health Perspect. 2019;127:47009. doi: 10.1289/EHP3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joubert BR, Haberg SE, Nilsen RM, Wang X, Vollset SE, Murphy SK, et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect. 2012;120:1425–1431. doi: 10.1289/ehp.1205412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joubert BR, Haberg SE, Bell DA, Nilsen RM, Vollset SE, Midttun O, et al. Maternal smoking and DNA methylation in newborns: in utero effect or epigenetic inheritance? Cancer Epidemiol Biomark Prev. 2014;23:1007–1017. doi: 10.1158/1055-9965.EPI-13-1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwon YM, Park JH, Kim H, Shim YM, Kim J, Han J, et al. Different susceptibility of increased DNMT1 expression by exposure to tobacco smoke according to histology in primary non-small cell lung cancer. J Cancer Res Clin Oncol. 2007;133:219–226. doi: 10.1007/s00432-006-0160-2. [DOI] [PubMed] [Google Scholar]
- 28.Yang W, Cui S, Ma J, Lu Q, Kong C, Liu T, et al. Cigarette smoking extract causes hypermethylation and inactivation of WWOX gene in T-24 human bladder cancer cells. Neoplasma. 2012;59:216–223. doi: 10.4149/neo_2012_028. [DOI] [PubMed] [Google Scholar]
- 29.Szulakowski P, Crowther AJ, Jimenez LA, Donaldson K, Mayer R, Leonard TB, et al. The effect of smoking on the transcriptional regulation of lung inflammation in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;174:41–50. doi: 10.1164/rccm.200505-725OC. [DOI] [PubMed] [Google Scholar]
- 30.Ito K, Ito M, Elliott WM, Cosio B, Caramori G, Kon OM, et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med. 2005;352:1967–1976. doi: 10.1056/NEJMoa041892. [DOI] [PubMed] [Google Scholar]
- 31.Marwick JA, Kirkham PA, Stevenson CS, Danahay H, Giddings J, Butler K, et al. Cigarette smoke alters chromatin remodeling and induces proinflammatory genes in rat lungs. Am J Respir Cell Mol Biol. 2004;31:633–642. doi: 10.1165/rcmb.2004-0006OC. [DOI] [PubMed] [Google Scholar]
- 32.Sundar IK, Nevid MZ, Friedman AE, Rahman I. Cigarette smoke induces distinct histone modifications in lung cells: implications for the pathogenesis of COPD and lung cancer. J Proteome Res. 2014;13:982–996. doi: 10.1021/pr400998n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Podobinska M, Szablowska-Gadomska I, Augustyniak J, Sandvig I, Sandvig A, Buzanska L. Epigenetic modulation of stem cells in neurodevelopment: the role of methylation and acetylation. Front Cell Neurosci. 2017;11:23. doi: 10.3389/fncel.2017.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Y, Huang P, Ai W, Li X, Guo W, Zhang J, et al. Histone deacetylase activity is decreased in peripheral blood monocytes in patients with COPD. J Inflamm (Lond) 2012;9:10. doi: 10.1186/1476-9255-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.To M, Yamamura S, Akashi K, Charron CE, Haruki K, Barnes PJ, et al. Defect of adaptation to hypoxia in patients with COPD due to reduction of histone deacetylase 7. Chest. 2012;141:1233–1242. doi: 10.1378/chest.11-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jeong I, Lim JH, Oh DK, Kim WJ, Oh YM. Gene expression profile of human lung in a relatively early stage of COPD with emphysema. Int J Chron Obstruct Pulmon Dis. 2018;13:2643–2655. doi: 10.2147/COPD.S166812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen X, Guan XJ, Peng XH, Cui ZL, Luan CY, Guo XJ. Acetylation of lysine 9 on histone H3 is associated with increased pro-inflammatory cytokine release in a cigarette smoke-induced rat model through HDAC1 depression. Inflamm Res. 2015;64:513–526. doi: 10.1007/s00011-015-0832-y. [DOI] [PubMed] [Google Scholar]
- 38.Sundar IK, Rahman I. Gene expression profiling of epigenetic chromatin modification enzymes and histone marks by cigarette smoke: implications for COPD and lung cancer. Am J Physiol Lung Cell Mol Physiol. 2016;311:L1245–L1258. doi: 10.1152/ajplung.00253.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferraro M, Di Vincenzo S, Dino P, Bucchieri S, Cipollina C, Gjomarkaj M, et al. Budesonide, aclidinium and formoterol in combination limit inflammaging processes in bronchial epithelial cells exposed to cigarette smoke. Exp Gerontol. 2019;118:78–87. doi: 10.1016/j.exger.2019.01.016. [DOI] [PubMed] [Google Scholar]
- 40.Winkler AR, Nocka KN, Williams CM. Smoke exposure of human macrophages reduces HDAC3 activity, resulting in enhanced inflammatory cytokine production. Pulm Pharmacol Ther. 2012;25:286–292. doi: 10.1016/j.pupt.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 41.Lam HC, Cloonan SM, Bhashyam AR, Haspel JA, Singh A, Sathirapongsasuti JF, et al. Histone deacetylase 6-mediated selective autophagy regulates COPD-associated cilia dysfunction. J Clin Invest. 2013;123:5212–5230. doi: 10.1172/JCI69636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Borgas D, Chambers E, Newton J, Ko J, Rivera S, Rounds S, et al. Cigarette smoke disrupted lung endothelial barrier integrity and increased susceptibility to acute lung injury via histone deacetylase 6. Am J Respir Cell Mol Biol. 2016;54:683–696. doi: 10.1165/rcmb.2015-0149OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Conti V, Corbi G, Manzo V, Malangone P, Vitale C, Maglio A, et al. SIRT1 activity in peripheral blood mononuclear cells correlates with altered lung function in patients with chronic obstructive pulmonary disease. Oxid Med Cell Longev. 2018;2018:9391261. doi: 10.1155/2018/9391261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ting WJ, Yang JJ, Kuo CH, Xiao ZJ, Lu XZ, Yeh YL, et al. Environmental tobacco smoke increases autophagic effects but decreases longevity associated with Sirt-1 protein expression in young C57BL mice hearts. Oncotarget. 2016;7:39017–39025. doi: 10.18632/oncotarget.9176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pace E, Di Vincenzo S, Ferraro M, Bruno A, Dino P, Bonsignore MR, et al. Carbocysteine counteracts the effects of cigarette smoke on cell growth and on the SIRT1/FoxO3 axis in bronchial epithelial cells. Exp Gerontol. 2016;81:119–128. doi: 10.1016/j.exger.2016.05.013. [DOI] [PubMed] [Google Scholar]
- 46.Dikalov S, Itani H, Richmond B, Vergeade A, Rahman S, Boutaud O, et al. Tobacco smoking induces cardiovascular mitochondrial oxidative stress, promotes endothelial dysfunction, and enhances hypertension. Am J Physiol Heart Circ Physiol. 2019;316:H639–H646. doi: 10.1152/ajpheart.00595.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen Y, Wang H, Luo G, Dai X. SIRT4 inhibits cigarette smoke extracts-induced mononuclear cell adhesion to human pulmonary microvascular endothelial cells via regulating NF-kappaB activity. Toxicol Lett. 2014;226:320–327. doi: 10.1016/j.toxlet.2014.02.022. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y, Zhu Y, Xing S, Ma P, Lin D. SIRT5 prevents cigarette smoke extract-induced apoptosis in lung epithelial cells via deacetylation of FOXO3. Cell Stress Chaperones. 2015;20:805–810. doi: 10.1007/s12192-015-0599-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takasaka N, Araya J, Hara H, Ito S, Kobayashi K, Kurita Y, et al. Autophagy induction by SIRT6 through attenuation of insulin-like growth factor signaling is involved in the regulation of human bronchial epithelial cell senescence. J Immunol. 2014;192:958–968. doi: 10.4049/jimmunol.1302341. [DOI] [PubMed] [Google Scholar]
- 50.Pace E, Di Vincenzo S, Ferraro M, Siena L, Chiappara G, Dino P, et al. Effects of carbocysteine and beclomethasone on histone acetylation/deacetylation processes in cigarette smoke exposed bronchial epithelial cells. J Cell Physiol. 2017;232:2851–2859. doi: 10.1002/jcp.25710. [DOI] [PubMed] [Google Scholar]
- 51.Kang N, Chen P, Chen Y, Zeng H, He X, Zhu Y. PRMT6 mediates CSE induced inflammation and apoptosis. Int Immunopharmacol. 2015;24:95–101. doi: 10.1016/j.intimp.2014.10.029. [DOI] [PubMed] [Google Scholar]
- 52.He X, Li T, Kang N, Zeng H, Ren S, Zong D, et al. The protective effect of PRMT6 overexpression on cigarette smoke extract-induced murine emphysema model. Int J Chron Obstruct Pulmon Dis. 2017;12:3245–3254. doi: 10.2147/COPD.S144881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang B, Liu Y, Luo F, Xu Y, Qin Y, Lu X, et al. Epigenetic silencing of microRNA-218 via EZH2-mediated H3K27 trimethylation is involved in malignant transformation of HBE cells induced by cigarette smoke extract. Arch Toxicol. 2016;90:449–461. doi: 10.1007/s00204-014-1435-z. [DOI] [PubMed] [Google Scholar]
- 54.Ibuki Y, Toyooka T, Zhao X, Yoshida I. Cigarette sidestream smoke induces histone H3 phosphorylation via JNK and PI3 K/Akt pathways, leading to the expression of proto-oncogenes. Carcinogenesis. 2014;35:1228–1237. doi: 10.1093/carcin/bgt492. [DOI] [PubMed] [Google Scholar]
- 55.Toyooka T, Ibuki Y. Cigarette sidestream smoke induces phosphorylated histone H2AX. Mutat Res. 2009;676:34–40. doi: 10.1016/j.mrgentox.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 56.He Z, Ma WY, Liu G, Zhang Y, Bode AM, Dong Z. Arsenite-induced phosphorylation of histone H3 at serine 10 is mediated by Akt1, extracellular signal-regulated kinase 2, and p90 ribosomal S6 kinase 2 but not mitogen- and stress-activated protein kinase 1. J Biol Chem. 2003;278:10588–10593. doi: 10.1074/jbc.M208581200. [DOI] [PubMed] [Google Scholar]
- 57.Sundar IK, Chung S, Hwang JW, Lapek JJ, Bulger M, Friedman AE, et al. Mitogen- and stress-activated kinase 1 (MSK1) regulates cigarette smoke-induced histone modifications on NF-kappaB-dependent genes. PLoS ONE. 2012;7:e31378. doi: 10.1371/journal.pone.0031378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reyskens KM, Arthur JS. Emerging roles of the mitogen and stress activated kinases MSK1 and MSK2. Front Cell Dev Biol. 2016;4:56. doi: 10.3389/fcell.2016.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang R, Jiang Y, Xia P, Luo G, Huang W, Hu Z, et al. Cigarette smoke extract promotes TIM4 expression in murine dendritic cells leading to th2 polarization through ERK-dependent pathways. Int Arch Allergy Immunol. 2019;178:219–228. doi: 10.1159/000494505. [DOI] [PubMed] [Google Scholar]
- 60.Zong D, Li J, Cai S, He S, Liu Q, Jiang J, et al. Notch1 regulates endothelial apoptosis via the ERK pathway in chronic obstructive pulmonary disease. Am J Physiol Cell Physiol. 2018;315:C330–C340. doi: 10.1152/ajpcell.00182.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liang Z, Wu R, Xie W, Zhu M, Xie C, Li X, et al. Curcumin reverses tobacco smoke induced epithelialmesenchymal transition by suppressing the MAPK pathway in the lungs of mice. Mol Med Rep. 2018;17:2019–2025. doi: 10.3892/mmr.2017.8028. [DOI] [PubMed] [Google Scholar]
- 62.Ke Q, Li Q, Ellen TP, Sun H, Costa M. Nickel compounds induce phosphorylation of histone H3 at serine 10 by activating JNK-MAPK pathway. Carcinogenesis. 2008;29:1276–1281. doi: 10.1093/carcin/bgn084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Du C, Lu J, Zhou L, Wu B, Zhou F, Gu L, et al. MAPK/FoxA2-mediated cigarette smoke-induced squamous metaplasia of bronchial epithelial cells. Int J Chron Obstruct Pulmon Dis. 2017;12:3341–3351. doi: 10.2147/COPD.S143279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liang Z, Wu R, Xie W, Xie C, Wu J, Geng S, et al. Effects of curcumin on tobacco smoke-induced hepatic MAPK pathway activation and epithelial–mesenchymal transition in vivo. Phytother Res. 2017;31:1230–1239. doi: 10.1002/ptr.5844. [DOI] [PubMed] [Google Scholar]
- 65.Degens H, Gayan-Ramirez G, van Hees HW. Smoking-induced skeletal muscle dysfunction: from evidence to mechanisms. Am J Respir Crit Care Med. 2015;191:620–625. doi: 10.1164/rccm.201410-1830PP. [DOI] [PubMed] [Google Scholar]
- 66.Caron MA, Morissette MC, Theriault ME, Nikota JK, Stampfli MR, Debigare R. Alterations in skeletal muscle cell homeostasis in a mouse model of cigarette smoke exposure. PLoS ONE. 2013;8:e66433. doi: 10.1371/journal.pone.0066433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Debigare R, Cote CH, Maltais F. Ubiquitination and proteolysis in limb and respiratory muscles of patients with chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2010;7:84–90. doi: 10.1513/pats.200906-051JS. [DOI] [PubMed] [Google Scholar]
- 68.Kim SY, Lee JH, Huh JW, Ro JY, Oh YM, Lee SD, et al. Cigarette smoke induces Akt protein degradation by the ubiquitin–proteasome system. J Biol Chem. 2011;286:31932–31943. doi: 10.1074/jbc.M111.267633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gallego LD, Ghodgaonkar SM, Polyansky AA, Schubert T, Zagrovic B, Zheng N, et al. Structural mechanism for the recognition and ubiquitination of a single nucleosome residue by Rad6-Bre1. Proc Natl Acad Sci USA. 2016;113:10553–10558. doi: 10.1073/pnas.1606863113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Baptista IL, Leal ML, Artioli GG, Aoki MS, Fiamoncini J, Turri AO, et al. Leucine attenuates skeletal muscle wasting via inhibition of ubiquitin ligases. Muscle Nerve. 2010;41:800–808. doi: 10.1002/mus.21578. [DOI] [PubMed] [Google Scholar]
- 71.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 72.Rom O, Kaisari S, Aizenbud D, Reznick A. Involvement of E3 ubiquitin ligases in cigarette smoke associated muscle catabolism. Free Radic Biol Med. 2014;75(Suppl 1):S5. doi: 10.1016/j.freeradbiomed.2014.10.835. [DOI] [PubMed] [Google Scholar]
- 73.Kaisari S, Rom O, Aizenbud D, Reznick AZ. Involvement of NF-kappaB and muscle specific E3 ubiquitin ligase MuRF1 in cigarette smoke-induced catabolism in C2 myotubes. Adv Exp Med Biol. 2013;788:7–17. doi: 10.1007/978-94-007-6627-3_2. [DOI] [PubMed] [Google Scholar]
- 74.Liu Q, Xu WG, Luo Y, Han FF, Yao XH, Yang TY, et al. Cigarette smoke-induced skeletal muscle atrophy is associated with up-regulation of USP-19 via p38 and ERK MAPKs. J Cell Biochem. 2011;112:2307–2316. doi: 10.1002/jcb.23151. [DOI] [PubMed] [Google Scholar]
- 75.Kim SY, Kim HJ, Park MK, Huh JW, Park HY, Ha SY, et al. Mitochondrial e3 ubiquitin protein ligase 1 mediates cigarette smoke-induced endothelial cell death and dysfunction. Am J Respir Cell Mol Biol. 2016;54:284–296. doi: 10.1165/rcmb.2014-0377OC. [DOI] [PubMed] [Google Scholar]
- 76.Han L, Yang J, Wang X, Wu Q, Yin S, Li Z, et al. The E3 deubiquitinase USP17 is a positive regulator of retinoic acid-related orphan nuclear receptor gammat (RORgammat) in Th17 cells. J Biol Chem. 2014;289:25546–25555. doi: 10.1074/jbc.M114.565291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Song H, Tao L, Chen C, Pan L, Hao J, Ni Y, et al. USP17-mediated deubiquitination and stabilization of HDAC2 in cigarette smoke extract-induced inflammation. Int J Clin Exp Pathol. 2015;8:10707–10715. [PMC free article] [PubMed] [Google Scholar]
- 78.Singh SK, Pal BM, Girschick HJ, Bhadra U. MicroRNAs—micro in size but macro in function. FEBS J. 2008;275:4929–4944. doi: 10.1111/j.1742-4658.2008.06624.x. [DOI] [PubMed] [Google Scholar]
- 79.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 80.Izzotti A, Calin GA, Arrigo P, Steele VE, Croce CM, De Flora S. Downregulation of microRNA expression in the lungs of rats exposed to cigarette smoke. FASEB J. 2009;23:806–812. doi: 10.1096/fj.08-121384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Izzotti A, Calin GA, Steele VE, Croce CM, De Flora S. Relationships of microRNA expression in mouse lung with age and exposure to cigarette smoke and light. FASEB J. 2009;23:3243–3250. doi: 10.1096/fj.09-135251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Izzotti A, Larghero P, Longobardi M, Cartiglia C, Camoirano A, Steele VE, et al. Dose-responsiveness and persistence of microRNA expression alterations induced by cigarette smoke in mouse lung. Mutat Res. 2011;717:9–16. doi: 10.1016/j.mrfmmm.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 83.Izzotti A, Larghero P, Balansky R, Pfeffer U, Steele VE, De Flora S. Interplay between histopathological alterations, cigarette smoke and chemopreventive agents in defining microRNA profiles in mouse lung. Mutat Res. 2011;717:17–24. doi: 10.1016/j.mrfmmm.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 84.Schembri F, Sridhar S, Perdomo C, Gustafson AM, Zhang X, Ergun A, et al. MicroRNAs as modulators of smoking-induced gene expression changes in human airway epithelium. Proc Natl Acad Sci USA. 2009;106:2319–2324. doi: 10.1073/pnas.0806383106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mascaux C, Laes JF, Anthoine G, Haller A, Ninane V, Burny A, et al. Evolution of microRNA expression during human bronchial squamous carcinogenesis. Eur Respir J. 2009;33:352–359. doi: 10.1183/09031936.00084108. [DOI] [PubMed] [Google Scholar]
- 86.Su MW, Yu SL, Lin WC, Tsai CH, Chen PH, Lee YL. Smoking-related microRNAs and mRNAs in human peripheral blood mononuclear cells. Toxicol Appl Pharmacol. 2016;305:169–175. doi: 10.1016/j.taap.2016.06.020. [DOI] [PubMed] [Google Scholar]
- 87.Maccani MA, Avissar-Whiting M, Banister CE, McGonnigal B, Padbury JF, Marsit CJ. Maternal cigarette smoking during pregnancy is associated with downregulation of miR-16, miR-21, and miR-146a in the placenta. Epigenetics. 2010;5:583–589. doi: 10.4161/epi.5.7.12762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Marczylo EL, Amoako AA, Konje JC, Gant TW, Marczylo TH. Smoking induces differential miRNA expression in human spermatozoa: a potential transgenerational epigenetic concern? Epigenetics. 2012;7:432–439. doi: 10.4161/epi.19794. [DOI] [PubMed] [Google Scholar]
- 89.Takahashi K, Yokota S, Tatsumi N, Fukami T, Yokoi T, Nakajima M. Cigarette smoking substantially alters plasma microRNA profiles in healthy subjects. Toxicol Appl Pharmacol. 2013;272:154–160. doi: 10.1016/j.taap.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 90.Shi B, Gao H, Zhang T, Cui Q. Analysis of plasma microRNA expression profiles revealed different cancer susceptibility in healthy young adult smokers and middle-aged smokers. Oncotarget. 2016;7:21676–21685. doi: 10.18632/oncotarget.7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Izzotti A, Calin GA, Steele VE, Cartiglia C, Longobardi M, Croce CM, et al. Chemoprevention of cigarette smoke-induced alterations of MicroRNA expression in rat lungs. Cancer Prev Res (Phila). 2010;3:62–72. doi: 10.1158/1940-6207.CAPR-09-0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Izzotti A, Balansky R, D’Agostini F, Longobardi M, Cartiglia C, La Maestra S, et al. Relationships between pulmonary micro-RNA and proteome profiles, systemic cytogenetic damage and lung tumors in cigarette smoke-exposed mice treated with chemopreventive agents. Carcinogenesis. 2013;34:2322–2329. doi: 10.1093/carcin/bgt178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Izzotti A, Larghero P, Cartiglia C, Longobardi M, Pfeffer U, Steele VE, et al. Modulation of microRNA expression by budesonide, phenethyl isothiocyanate and cigarette smoke in mouse liver and lung. Carcinogenesis. 2010;31:894–901. doi: 10.1093/carcin/bgq037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–641. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 95.Zhang H, Chen Z, Wang X, Huang Z, He Z, Chen Y. Long non-coding RNA: a new player in cancer. J Hematol Oncol. 2013;6:37. doi: 10.1186/1756-8722-6-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Soares DAN, Cruz EMN, de Melo MB, Malagoli RR. Noncoding RNA profiles in tobacco- and alcohol-associated diseases. Genes (Basel) 2016;8:6. doi: 10.3390/genes8010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bi H, Zhou J, Wu D, Gao W, Li L, Yu L, et al. Microarray analysis of long non-coding RNAs in COPD lung tissue. Inflamm Res. 2015;64:119–126. doi: 10.1007/s00011-014-0790-9. [DOI] [PubMed] [Google Scholar]
- 98.Zhang H, Sun D, Li D, Zheng Z, Xu J, Liang X, et al. Long non-coding RNA expression patterns in lung tissues of chronic cigarette smoke induced COPD mouse model. Sci Rep. 2018;8:7609. doi: 10.1038/s41598-018-25702-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li S, Sun X, Miao S, Liu J, Jiao W. Differential protein-coding gene and long noncoding RNA expression in smoking-related lung squamous cell carcinoma. Thorac Cancer. 2017;8:672–681. doi: 10.1111/1759-7714.12510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu Y, Wang B, Liu X, Lu L, Luo F, Lu X, et al. Epigenetic silencing of p21 by long non-coding RNA HOTAIR is involved in the cell cycle disorder induced by cigarette smoke extract. Toxicol Lett. 2016;240:60–67. doi: 10.1016/j.toxlet.2015.10.016. [DOI] [PubMed] [Google Scholar]
- 101.Liu Y, Luo F, Xu Y, Wang B, Zhao Y, Xu W, et al. Epithelial–mesenchymal transition and cancer stem cells, mediated by a long non-coding RNA, HOTAIR, are involved in cell malignant transformation induced by cigarette smoke extract. Toxicol Appl Pharmacol. 2015;282:9–19. doi: 10.1016/j.taap.2014.10.022. [DOI] [PubMed] [Google Scholar]
- 102.Lu L, Xu H, Luo F, Liu X, Lu X, Yang Q, et al. Epigenetic silencing of miR-218 by the lncRNA CCAT1, acting via BMI1, promotes an altered cell cycle transition in the malignant transformation of HBE cells induced by cigarette smoke extract. Toxicol Appl Pharmacol. 2016;304:30–41. doi: 10.1016/j.taap.2016.05.012. [DOI] [PubMed] [Google Scholar]
- 103.Lu L, Luo F, Liu Y, Liu X, Shi L, Lu X, et al. Posttranscriptional silencing of the lncRNA MALAT1 by miR-217 inhibits the epithelial–mesenchymal transition via enhancer of zeste homolog 2 in the malignant transformation of HBE cells induced by cigarette smoke extract. Toxicol Appl Pharmacol. 2015;289:276–285. doi: 10.1016/j.taap.2015.09.016. [DOI] [PubMed] [Google Scholar]
- 104.Thai P, Statt S, Chen CH, Liang E, Campbell C, Wu R. Characterization of a novel long noncoding RNA, SCAL1, induced by cigarette smoke and elevated in lung cancer cell lines. Am J Respir Cell Mol Biol. 2013;49:204–211. doi: 10.1165/rcmb.2013-0159RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lin H, Zhang X, Feng N, Wang R, Zhang W, Deng X, et al. LncRNA LCPAT1 mediates smoking/particulate matter 2.5-induced cell autophagy and epithelial–mesenchymal transition in lung cancer cells via RCC2. Cell Physiol Biochem. 2018;47:1244–1258. doi: 10.1159/000490220. [DOI] [PubMed] [Google Scholar]
- 106.Gao S, Lin H, Yu W, Zhang F, Wang R, Yu H, et al. LncRNA LCPAT1 is involved in DNA damage induced by CSE. Biochem Biophys Res Commun. 2019;508:512–515. doi: 10.1016/j.bbrc.2018.11.171. [DOI] [PubMed] [Google Scholar]
- 107.Siedlinski M, Klanderman B, Sandhaus RA, Barker AF, Brantly ML, Eden E, et al. Association of cigarette smoking and CRP levels with DNA methylation in alpha-1 antitrypsin deficiency. Epigenetics. 2012;7:720–728. doi: 10.4161/epi.20319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ligthart S, Marzi C, Aslibekyan S, Mendelson MM, Conneely KN, Tanaka T, et al. DNA methylation signatures of chronic low-grade inflammation are associated with complex diseases. Genome Biol. 2016;17:255. doi: 10.1186/s13059-016-1119-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bergens MA, Pittman GS, Thompson I, Campbell MR, Wang X, Hoyo C, et al. Smoking-associated AHRR demethylation in cord blood DNA: impact of CD235a+ nucleated red blood cells. Clin Epigenet. 2019;11:87. doi: 10.1186/s13148-019-0686-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jhun MA, Smith JA, Ware EB, Kardia S, Mosley TJ, Turner ST, et al. Modeling the causal role of DNA methylation in the association between cigarette smoking and inflammation in African Americans: a 2-Step epigenetic mendelian randomization study. Am J Epidemiol. 2017;186:1149–1158. doi: 10.1093/aje/kwx181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Howell KJ, Kraiczy J, Nayak KM, Gasparetto M, Ross A, Lee C, et al. DNA methylation and transcription patterns in intestinal epithelial cells from pediatric patients with inflammatory bowel diseases differentiate disease subtypes and associate with outcome. Gastroenterology. 2018;154:585–598. doi: 10.1053/j.gastro.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Somineni HK, Venkateswaran S, Kilaru V, Marigorta UM, Mo A, Okou DT, et al. Blood-derived DNA methylation signatures of Crohn’s disease and severity of intestinal inflammation. Gastroenterology. 2019;156:2254–2265. doi: 10.1053/j.gastro.2019.01.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yu J, Qiu Y, Yang J, Bian S, Chen G, Deng M, et al. DNMT1-PPARgamma pathway in macrophages regulates chronic inflammation and atherosclerosis development in mice. Sci Rep. 2016;6:30053. doi: 10.1038/srep30053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang X, Cao Q, Yu L, Shi H, Xue B, Shi H. Epigenetic regulation of macrophage polarization and inflammation by DNA methylation in obesity. JCI Insight. 2016;1:e87748. doi: 10.1172/jci.insight.87748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fabre C, Grosjean J, Tailler M, Boehrer S, Ades L, Perfettini JL, et al. A novel effect of DNA methyltransferase and histone deacetylase inhibitors: NFkappaB inhibition in malignant myeloblasts. Cell Cycle. 2008;7:2139–2145. doi: 10.4161/cc.7.14.6268. [DOI] [PubMed] [Google Scholar]
- 116.Feng Z, Zhan M, Meng R, Wang X, Xu Q. 5-Aza-2′-deoxycytidine enhances lipopolysaccharide-induced inflammatory cytokine expression in human dental pulp cells by regulating TRAF6 methylation. Bioengineered. 2019;10:197–206. doi: 10.1080/21655979.2019.1621135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Xu H, Chen Y, Chen Q, Xu H, Wang Y, Yu J, et al. DNMT1 regulates IL-6- and TGF-beta1-induced epithelial mesenchymal transition in prostate epithelial cells. Eur J Histochem. 2017;61:2775. doi: 10.4081/ejh.2017.2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang B, Cui Z, Zhong Z, Sun Y, Yang GY, Sun Q, et al. The role and regulatory mechanism of IL-1beta on the methylation of the NF2 gene in benign meningiomas and leptomeninges. Mol Carcinog. 2016;55:2268–2277. doi: 10.1002/mc.22467. [DOI] [PubMed] [Google Scholar]
- 119.Huang Y, Tian C, Li Q, Xu Q. TET1 knockdown inhibits porphyromonas gingivalis LPS/IFN-gamma-induced M1 macrophage polarization through the NF-kappaB pathway in THP-1 cells. Int J Mol Sci. 2019;20:2023. doi: 10.3390/ijms20082023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang X, Feng Z, Li Q, Yi B, Xu Q. DNA methylcytosine dioxygenase ten-eleven translocation 2 enhances lipopolysaccharide-induced cytokine expression in human dental pulp cells by regulating MyD88 hydroxymethylation. Cell Tissue Res. 2018;373:477–485. doi: 10.1007/s00441-018-2826-x. [DOI] [PubMed] [Google Scholar]
- 121.Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. 2015;525:389–393. doi: 10.1038/nature15252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wahl S, Drong A, Lehne B, Loh M, Scott WR, Kunze S, et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature. 2017;541:81–86. doi: 10.1038/nature20784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chen HP, Zhao YT, Zhao TC. Histone deacetylases and mechanisms of regulation of gene expression. Crit Rev Oncog. 2015;20:35–47. doi: 10.1615/CritRevOncog.2015012997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang B, Lin L, Ai Q, Zeng T, Ge P, Zhang L. HAT inhibitor, garcinol, exacerbates lipopolysaccharide induced inflammation in vitro and in vivo. Mol Med Rep. 2016;13:5290–5296. doi: 10.3892/mmr.2016.5189. [DOI] [PubMed] [Google Scholar]
- 125.Royce SG, Karagiannis TC. Histone deacetylases and their inhibitors: new implications for asthma and chronic respiratory conditions. Curr Opin Allergy Clin Immunol. 2014;14:44–48. doi: 10.1097/ACI.0000000000000029. [DOI] [PubMed] [Google Scholar]
- 126.Rajendrasozhan S, Yao H, Rahman I. Current perspectives on role of chromatin modifications and deacetylases in lung inflammation in COPD. COPD. 2009;6:291–297. doi: 10.1080/15412550903049132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lai T, Tian B, Cao C, Hu Y, Zhou J, Wang Y, et al. HDAC2 suppresses IL17A-Mediated airway remodeling in human and experimental modeling of COPD. Chest. 2018;153:863–875. doi: 10.1016/j.chest.2017.10.031. [DOI] [PubMed] [Google Scholar]
- 128.Xia M, Xu H, Dai W, Zhu C, Wu L, Yan S, et al. The role of HDAC2 in cigarette smoke-induced airway inflammation in a murine model of asthma and the effect of intervention with roxithromycin. J Asthma. 2018;55:337–344. doi: 10.1080/02770903.2017.1337788. [DOI] [PubMed] [Google Scholar]
- 129.To M, Swallow EB, Akashi K, Haruki K, Natanek SA, Polkey MI, et al. Reduced HDAC2 in skeletal muscle of COPD patients. Respir Res. 2017;18:99. doi: 10.1186/s12931-017-0588-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Barnes PJ. Reduced histone deacetylase in COPD: clinical implications. Chest. 2006;129:151–155. doi: 10.1378/chest.129.1.151. [DOI] [PubMed] [Google Scholar]
- 131.Adenuga D, Caito S, Yao H, Sundar IK, Hwang JW, Chung S, et al. Nrf2 deficiency influences susceptibility to steroid resistance via HDAC2 reduction. Biochem Biophys Res Commun. 2010;403:452–456. doi: 10.1016/j.bbrc.2010.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ito K, Barnes PJ, Adcock IM. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol. 2000;20:6891–6903. doi: 10.1128/MCB.20.18.6891-6903.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cosio BG, Iglesias A, Rios A, Noguera A, Sala E, Ito K, et al. Low-dose theophylline enhances the anti-inflammatory effects of steroids during exacerbations of COPD. Thorax. 2009;64:424–429. doi: 10.1136/thx.2008.103432. [DOI] [PubMed] [Google Scholar]
- 134.Lu W, You R, Yuan X, Yang T, Samuel EL, Marcano DC, et al. The microRNA miR-22 inhibits the histone deacetylase HDAC4 to promote T(H)17 cell-dependent emphysema. Nat Immunol. 2015;16:1185–1194. doi: 10.1038/ni.3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Poralla L, Stroh T, Erben U, Sittig M, Liebig S, Siegmund B, et al. Histone deacetylase 5 regulates the inflammatory response of macrophages. J Cell Mol Med. 2015;19:2162–2171. doi: 10.1111/jcmm.12595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhao Y, Ma G, Yang X. HDAC5 promotes Mycoplasma pneumoniae-induced inflammation in macrophages through NF-kappaB activation. Life Sci. 2019;221:13–19. doi: 10.1016/j.lfs.2019.02.004. [DOI] [PubMed] [Google Scholar]
- 137.Liu L, Zhou X, Shetty S, Hou G, Wang Q, Fu J. HDAC6 inhibition blocks inflammatory signaling and caspase-1 activation in LPS-induced acute lung injury. Toxicol Appl Pharmacol. 2019;370:178–183. doi: 10.1016/j.taap.2019.03.017. [DOI] [PubMed] [Google Scholar]
- 138.Song Y, Qin L, Yang R, Yang F, Kenechukwu NA, Zhao X, et al. Inhibition of HDAC6 alleviating lipopolysaccharide-induced p38MAPK phosphorylation and neuroinflammation in mice. Pharm Biol. 2019;57:263–268. doi: 10.1080/13880209.2018.1563620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhang HP, Fu JJ, Fan T, Zhang WB, Wang ZL, Wang L, et al. Histone deacetylation of memory T lymphocytes by You-Gui-Wan alleviates allergen-induced eosinophilic airway inflammation in asthma. Chin Med. 2015;10:9. doi: 10.1186/s13020-015-0038-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Meng J, Liu X, Zhang P, Li D, Xu S, Zhou Q, et al. Rb selectively inhibits innate IFN-beta production by enhancing deacetylation of IFN-beta promoter through HDAC1 and HDAC8. J Autoimmun. 2016;73:42–53. doi: 10.1016/j.jaut.2016.05.012. [DOI] [PubMed] [Google Scholar]
- 141.Li S, Fossati G, Marchetti C, Modena D, Pozzi P, Reznikov LL, et al. Specific inhibition of histone deacetylase 8 reduces gene expression and production of proinflammatory cytokines in vitro and in vivo. J Biol Chem. 2015;290:2368–2378. doi: 10.1074/jbc.M114.618454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kumar P, Gogulamudi VR, Periasamy R, Raghavaraju G, Subramanian U, Pandey KN. Inhibition of HDAC enhances STAT acetylation, blocks NF-kappaB, and suppresses the renal inflammation and fibrosis in Npr1 haplotype male mice. Am J Physiol Renal Physiol. 2017;313:F781–F795. doi: 10.1152/ajprenal.00166.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Fan X, Yan K, Meng Q, Sun R, Yang X, Yuan D, et al. Abnormal expression of SIRTs in psoriasis: decreased expression of SIRT 1-5 and increased expression of SIRT 6 and 7. Int J Mol Med. 2019;44:157–171. doi: 10.3892/ijmm.2019.4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mendes KL, Lelis DF, Santos S. Nuclear sirtuins and inflammatory signaling pathways. Cytokine Growth Factor Rev. 2017;38:98–105. doi: 10.1016/j.cytogfr.2017.11.001. [DOI] [PubMed] [Google Scholar]
- 145.Schug TT, Xu Q, Gao H, Peres-da-Silva A, Draper DW, Fessler MB, et al. Myeloid deletion of SIRT1 induces inflammatory signaling in response to environmental stress. Mol Cell Biol. 2010;30:4712–4721. doi: 10.1128/MCB.00657-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lo SG, Menzies KJ, Mottis A, Piersigilli A, Perino A, Yamamoto H, et al. SIRT2 deficiency modulates macrophage polarization and susceptibility to experimental colitis. PLoS ONE. 2014;9:e103573. doi: 10.1371/journal.pone.0103573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lappas M. Anti-inflammatory properties of sirtuin 6 in human umbilical vein endothelial cells. Mediat Inflamm. 2012;2012:597514. doi: 10.1155/2012/597514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Liu P, Huang G, Wei T, Gao J, Huang C, Sun M, et al. Sirtuin 3-induced macrophage autophagy in regulating NLRP3 inflammasome activation. Biochim Biophys Acta Mol Basis Dis. 2018;1864:764–777. doi: 10.1016/j.bbadis.2017.12.027. [DOI] [PubMed] [Google Scholar]
- 149.Qiao Y, Xu L, Tao X, Yin L, Qi Y, Xu Y, et al. Protective effects of dioscin against fructose-induced renal damage via adjusting Sirt3-mediated oxidative stress, fibrosis, lipid metabolism and inflammation. Toxicol Lett. 2018;284:37–45. doi: 10.1016/j.toxlet.2017.11.031. [DOI] [PubMed] [Google Scholar]
- 150.Vakhrusheva O, Smolka C, Gajawada P, Kostin S, Boettger T, Kubin T, et al. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ Res. 2008;102:703–710. doi: 10.1161/CIRCRESAHA.107.164558. [DOI] [PubMed] [Google Scholar]
- 151.Qin K, Han C, Zhang H, Li T, Li N, Cao X. NAD(+) dependent deacetylase Sirtuin 5 rescues the innate inflammatory response of endotoxin tolerant macrophages by promoting acetylation of p65. J Autoimmun. 2017;81:120–129. doi: 10.1016/j.jaut.2017.04.006. [DOI] [PubMed] [Google Scholar]
- 152.Liu Y, Peng J, Sun T, Li N, Zhang L, Ren J, et al. Epithelial EZH2 serves as an epigenetic determinant in experimental colitis by inhibiting TNFalpha-mediated inflammation and apoptosis. Proc Natl Acad Sci USA. 2017;114:E3796–E3805. doi: 10.1073/pnas.1700909114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Liang H, Huang Q, Liao MJ, Xu F, Zhang T, He J, et al. EZH2 plays a crucial role in ischemia/reperfusion-induced acute kidney injury by regulating p38 signaling. Inflamm Res. 2019;68:325–336. doi: 10.1007/s00011-019-01221-3. [DOI] [PubMed] [Google Scholar]
- 154.Kim JH, Yoo BC, Yang WS, Kim E, Hong S, Cho JY. The role of protein arginine methyltransferases in inflammatory responses. Mediat Inflamm. 2016;2016:4028353. doi: 10.1155/2016/4028353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sun Q, Liu L, Roth M, Tian J, He Q, Zhong B, et al. PRMT1 upregulated by epithelial proinflammatory cytokines participates in COX2 expression in fibroblasts and chronic antigen-induced pulmonary inflammation. J Immunol. 2015;195:298–306. doi: 10.4049/jimmunol.1402465. [DOI] [PubMed] [Google Scholar]
- 156.Dalloneau E, Pereira PL, Brault V, Nabel EG, Herault Y. Prmt2 regulates the lipopolysaccharide-induced responses in lungs and macrophages. J Immunol. 2011;187:4826–4834. doi: 10.4049/jimmunol.1101087. [DOI] [PubMed] [Google Scholar]
- 157.Bandyopadhyay S, Harris DP, Adams GN, Lause GE, McHugh A, Tillmaand EG, et al. HOXA9 methylation by PRMT5 is essential for endothelial cell expression of leukocyte adhesion molecules. Mol Cell Biol. 2012;32:1202–1213. doi: 10.1128/MCB.05977-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chen D, Zeng S, Huang M, Xu H, Liang L, Yang X. Role of protein arginine methyltransferase 5 in inflammation and migration of fibroblast-like synoviocytes in rheumatoid arthritis. J Cell Mol Med. 2017;21:781–790. doi: 10.1111/jcmm.13020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Keum YS, Kim HG, Bode AM, Surh YJ, Dong Z. UVB-induced COX-2 expression requires histone H3 phosphorylation at Ser10 and Ser28. Oncogene. 2013;32:444–452. doi: 10.1038/onc.2012.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Seidel P, Roth M, Ge Q, Merfort I, S’Ng CT, Ammit AJ. IkappaBalpha glutathionylation and reduced histone H3 phosphorylation inhibit eotaxin and RANTES. Eur Respir J. 2011;38:1444–1452. doi: 10.1183/09031936.00129610. [DOI] [PubMed] [Google Scholar]
- 161.Chung S, Sundar IK, Hwang JW, Yull FE, Blackwell TS, Kinnula VL, et al. NF-kappaB inducing kinase, NIK mediates cigarette smoke/TNFalpha-induced histone acetylation and inflammation through differential activation of IKKs. PLoS ONE. 2011;6:e23488. doi: 10.1371/journal.pone.0023488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lin CH, Shih CH, Chen BC. Thrombin-induced IL-8/CXCL8 release is mediated by CK2, MSK1, and NF-kappaB pathways in human lung epithelial cells. Eur J Pharmacol. 2015;767:135–143. doi: 10.1016/j.ejphar.2015.10.018. [DOI] [PubMed] [Google Scholar]
- 163.Galan-Ganga M, Garcia-Yague AJ, Lastres-Becker I. Role of MSK1 in the induction of NF-kappaB by the chemokine CX3CL1 in microglial cells. Cell Mol Neurobiol. 2019;39:331–340. doi: 10.1007/s10571-019-00664-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Darragh J, Ananieva O, Courtney A, Elcombe S, Arthur JS. MSK1 regulates the transcription of IL-1ra in response to TLR activation in macrophages. Biochem J. 2010;425:595–602. doi: 10.1042/BJ20091062. [DOI] [PubMed] [Google Scholar]
- 165.Zhong ZX, Feng SS, Chen SZ, Chen ZM, Chen XW. Inhibition of MSK1 promotes inflammation and apoptosis and inhibits functional recovery after spinal cord injury. J Mol Neurosci. 2019;68:191–203. doi: 10.1007/s12031-019-01298-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lin CH, Nai PL, Bien MY, Yu CC, Chen BC. Thrombin-induced CCAAT/enhancer-binding protein beta activation and IL-8/CXCL8 expression via MEKK1, ERK, and p90 ribosomal S6 kinase 1 in lung epithelial cells. J Immunol. 2014;192:338–348. doi: 10.4049/jimmunol.1203323. [DOI] [PubMed] [Google Scholar]
- 167.Wu Y, Kang J, Zhang L, Liang Z, Tang X, Yan Y, et al. Ubiquitination regulation of inflammatory responses through NF-kappaB pathway. Am J Transl Res. 2018;10:881–891. [PMC free article] [PubMed] [Google Scholar]
- 168.Zeng Y, Li J, Wang HX, Guo SB, Yang H, Zeng XJ, et al. Transcriptional effects of E3 ligase atrogin-1/MAFbx on apoptosis, hypertrophy and inflammation in neonatal rat cardiomyocytes. PLoS ONE. 2013;8:e53831. doi: 10.1371/journal.pone.0053831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Jenkins K, Khoo JJ, Sadler A, Piganis R, Wang D, Borg NA, et al. Mitochondrially localised MUL1 is a novel modulator of antiviral signaling. Immunol Cell Biol. 2013;91:321–330. doi: 10.1038/icb.2013.7. [DOI] [PubMed] [Google Scholar]
- 170.Ma X, Zheng X, Pan L, Zhang X. NLRP3 inflammasome activation in liver cirrhotic patients. Biochem Biophys Res Commun. 2018;505:40–44. doi: 10.1016/j.bbrc.2018.09.055. [DOI] [PubMed] [Google Scholar]
- 171.Han S, Jerome JA, Gregory AD, Mallampalli RK. Cigarette smoke destabilizes NLRP3 protein by promoting its ubiquitination. Respir Res. 2017;18:2. doi: 10.1186/s12931-016-0485-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Bafadhel M, McKenna S, Terry S, Mistry V, Reid C, Haldar P, et al. Acute exacerbations of chronic obstructive pulmonary disease: identification of biologic clusters and their biomarkers. Am J Respir Crit Care Med. 2011;184:662–671. doi: 10.1164/rccm.201104-0597OC. [DOI] [PubMed] [Google Scholar]
- 173.Imaoka H, Hoshino T, Takei S, Kinoshita T, Okamoto M, Kawayama T, et al. Interleukin-18 production and pulmonary function in COPD. Eur Respir J. 2008;31:287–297. doi: 10.1183/09031936.00019207. [DOI] [PubMed] [Google Scholar]
- 174.Contreras J, Rao DS. MicroRNAs in inflammation and immune responses. Leukemia. 2012;26:404–413. doi: 10.1038/leu.2011.356. [DOI] [PubMed] [Google Scholar]
- 175.Diao X, Zhou J, Wang S, Ma X. Upregulation of miR-132 contributes to the pathophysiology of COPD via targeting SOCS5. Exp Mol Pathol. 2018;105:285–292. doi: 10.1016/j.yexmp.2018.10.002. [DOI] [PubMed] [Google Scholar]
- 176.Gu W, Yuan Y, Yang H, Wu H, Wang L, Tang Z, et al. Role of miR-195 in cigarette smoke-induced chronic obstructive pulmonary disease. Int Immunopharmacol. 2018;55:49–54. doi: 10.1016/j.intimp.2017.11.030. [DOI] [PubMed] [Google Scholar]
- 177.Chen G, Cao S, Liu F, Liu Y. MiR-195 plays a role in steroid resistance of ulcerative colitis by targeting Smad7. Biochem J. 2015;471:357–367. doi: 10.1042/BJ20150095. [DOI] [PubMed] [Google Scholar]
- 178.O’Connell RM, Rao DS, Baltimore D. MicroRNA regulation of inflammatory responses. Annu Rev Immunol. 2012;30:295–312. doi: 10.1146/annurev-immunol-020711-075013. [DOI] [PubMed] [Google Scholar]
- 179.McMillan DH, Baglole CJ, Thatcher TH, Maggirwar S, Sime PJ, Phipps RP. Lung-targeted overexpression of the NF-kappaB member RelB inhibits cigarette smoke-induced inflammation. Am J Pathol. 2011;179:125–133. doi: 10.1016/j.ajpath.2011.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zago M, Rico DSA, Hecht E, Rousseau S, Hamid Q, Eidelman DH, et al. The NF-kappaB family member RelB regulates microRNA miR-146a to suppress cigarette smoke-induced COX-2 protein expression in lung fibroblasts. Toxicol Lett. 2014;226:107–116. doi: 10.1016/j.toxlet.2014.01.020. [DOI] [PubMed] [Google Scholar]
- 181.Zhao G, Zhang T, Wu H, Jiang K, Qiu C, Deng G. MicroRNA let-7c improves LPS-Induced outcomes of endometritis by suppressing NF-kappaB signaling. Inflammation. 2019;42:650–657. doi: 10.1007/s10753-018-0922-4. [DOI] [PubMed] [Google Scholar]
- 182.Yu JH, Long L, Luo ZX, Li LM, You JR. Anti-inflammatory role of microRNA let-7c in LPS treated alveolar macrophages by targeting STAT3. Asian Pac J Trop Med. 2016;9:72–75. doi: 10.1016/j.apjtm.2015.12.015. [DOI] [PubMed] [Google Scholar]
- 183.Halappanavar S, Nikota J, Wu D, Williams A, Yauk CL, Stampfli M. IL-1 receptor regulates microRNA-135b expression in a negative feedback mechanism during cigarette smoke-induced inflammation. J Immunol. 2013;190:3679–3686. doi: 10.4049/jimmunol.1202456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Wang H, Hao P, Zhang H, Xu C, Zhao J. MicroRNA-223 inhibits lipopolysaccharide-induced inflammatory response by directly targeting Irak1 in the nucleus pulposus cells of intervertebral disc. IUBMB Life. 2018;70:479–490. doi: 10.1002/iub.1747. [DOI] [PubMed] [Google Scholar]
- 185.Neudecker V, Haneklaus M, Jensen O, Khailova L, Masterson JC, Tye H, et al. Myeloid-derived miR-223 regulates intestinal inflammation via repression of the NLRP3 inflammasome. J Exp Med. 2017;214:1737–1752. doi: 10.1084/jem.20160462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Leuenberger C, Schuoler C, Bye H, Mignan C, Rechsteiner T, Hillinger S, et al. MicroRNA-223 controls the expression of histone deacetylase 2: a novel axis in COPD. J Mol Med (Berl). 2016;94:725–734. doi: 10.1007/s00109-016-1388-1. [DOI] [PubMed] [Google Scholar]
- 187.Dang X, Yang L, Guo J, Hu H, Li F, Liu Y, et al. MiR-145-5p is associated with smoke-related chronic obstructive pulmonary disease via targeting KLF5. Chem Biol Interact. 2019;300:82–90. doi: 10.1016/j.cbi.2019.01.011. [DOI] [PubMed] [Google Scholar]
- 188.Shen W, Liu J, Zhao G, Fan M, Song G, Zhang Y, et al. Repression of Toll-like receptor-4 by microRNA-149-3p is associated with smoking-related COPD. Int J Chron Obstr Pulm Dis. 2017;12:705–715. doi: 10.2147/COPD.S128031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Xue H, Li MX. MicroRNA-150 protects against cigarette smoke-induced lung inflammation and airway epithelial cell apoptosis through repressing p53: microRNA-150 in CS-induced lung inflammation. Hum Exp Toxicol. 2018;37:920–928. doi: 10.1177/0960327117741749. [DOI] [PubMed] [Google Scholar]
- 190.Du Y, Ding Y, Chen X, Mei Z, Ding H, Wu Y, et al. MicroRNA-181c inhibits cigarette smoke-induced chronic obstructive pulmonary disease by regulating CCN1 expression. Respir Res. 2017;18:155. doi: 10.1186/s12931-017-0639-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Jia Q, Chang J, Hong Q, Zhang JJ, Zhou H, Chen FH. MiR-212-5p exerts a protective effect in chronic obstructive pulmonary disease. Discov Med. 2018;26:173–183. [PubMed] [Google Scholar]
- 192.Conickx G, Mestdagh P, Avila CF, Verhamme FM, Maes T, Vanaudenaerde BM, et al. MicroRNA profiling reveals a role for microRNA-218-5p in the pathogenesis of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2017;195:43–56. doi: 10.1164/rccm.201506-1182OC. [DOI] [PubMed] [Google Scholar]
- 193.Moon HG, Zheng Y, An CH, Kim YK, Jin Y. CCN1 secretion induced by cigarette smoking extracts augments IL-8 release from bronchial epithelial cells. PLoS ONE. 2013;8:e68199. doi: 10.1371/journal.pone.0068199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ebrahimpour A, Shrestha S, Bonnen MD, Eissa NT, Raghu G, Ghebre YT. Nicotine modulates growth factors and MicroRNA to promote inflammatory and fibrotic processes. J Pharmacol Exp Ther. 2019;368:169–178. doi: 10.1124/jpet.118.252650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Malik DE, David RM, Gooderham NJ. Mechanistic evidence that benzo[a]pyrene promotes an inflammatory microenvironment that drives the metastatic potential of human mammary cells. Arch Toxicol. 2018;92:3223–3239. doi: 10.1007/s00204-018-2291-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Shin VY, Jin H, Ng EK, Cheng AS, Chong WW, Wong CY, et al. NF-kappaB targets miR-16 and miR-21 in gastric cancer: involvement of prostaglandin E receptors. Carcinogenesis. 2011;32:240–245. doi: 10.1093/carcin/bgq240. [DOI] [PubMed] [Google Scholar]
- 197.Zhao Y, Xu Y, Li Y, Xu W, Luo F, Wang B, et al. NF-kappaB-mediated inflammation leading to EMT via miR-200c is involved in cell transformation induced by cigarette smoke extract. Toxicol Sci. 2013;135:265–276. doi: 10.1093/toxsci/kft150. [DOI] [PubMed] [Google Scholar]
- 198.Faiz A, Steiling K, Roffel MP, Postma DS, Spira A, Lenburg ME, et al. Effect of long-term corticosteroid treatment on microRNA and gene-expression profiles in COPD. Eur Respir J. 2019;53:1801202. doi: 10.1183/13993003.01202-2018. [DOI] [PubMed] [Google Scholar]
- 199.Mathy NW, Chen XM. Long non-coding RNAs (lncRNAs) and their transcriptional control of inflammatory responses. J Biol Chem. 2017;292:12375–12382. doi: 10.1074/jbc.R116.760884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Carrion K, Dyo J, Patel V, Sasik R, Mohamed SA, Hardiman G, et al. The long non-coding HOTAIR is modulated by cyclic stretch and WNT/beta-CATENIN in human aortic valve cells and is a novel repressor of calcification genes. PLoS ONE. 2014;9:e96577. doi: 10.1371/journal.pone.0096577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Wu H, Liu J, Li W, Liu G, Li Z. LncRNA-HOTAIR promotes TNF-alpha production in cardiomyocytes of LPS-induced sepsis mice by activating NF-kappaB pathway. Biochem Biophys Res Commun. 2016;471:240–246. doi: 10.1016/j.bbrc.2016.01.117. [DOI] [PubMed] [Google Scholar]
- 202.Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundback P, et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature. 2012;488:670–674. doi: 10.1038/nature11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Liu G, Zhang W. Long non-coding RNA HOTAIR promotes UVB-induced apoptosis and inflammatory injury by up-regulation of PKR in keratinocytes. Braz J Med Biol Res. 2018;51:e6896. doi: 10.1590/1414-431X20186896. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 204.Liu J, Huang GQ, Ke ZP. Silence of long intergenic noncoding RNA HOTAIR ameliorates oxidative stress and inflammation response in ox-LDL-treated human macrophages by upregulating miR-330-5p. J Cell Physiol. 2019;234:5134–5142. doi: 10.1002/jcp.27317. [DOI] [PubMed] [Google Scholar]
- 205.Chen H, Wang X, Yan X, Cheng X, He X, Zheng W. LncRNA MALAT1 regulates sepsis-induced cardiac inflammation and dysfunction via interaction with miR-125b and p38 MAPK/NFkappaB. Int Immunopharmacol. 2018;55:69–76. doi: 10.1016/j.intimp.2017.11.038. [DOI] [PubMed] [Google Scholar]
- 206.Puthanveetil P, Chen S, Feng B, Gautam A, Chakrabarti S. Long non-coding RNA MALAT1 regulates hyperglycaemia induced inflammatory process in the endothelial cells. J Cell Mol Med. 2015;19:1418–1425. doi: 10.1111/jcmm.12576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Zhao G, Su Z, Song D, Mao Y, Mao X. The long noncoding RNA MALAT1 regulates the lipopolysaccharide-induced inflammatory response through its interaction with NF-kappaB. FEBS Lett. 2016;590:2884–2895. doi: 10.1002/1873-3468.12315. [DOI] [PubMed] [Google Scholar]
- 208.Zhuang YT, Xu DY, Wang GY, Sun JL, Huang Y, Wang SZ. IL-6 induced lncRNA MALAT1 enhances TNF-alpha expression in LPS-induced septic cardiomyocytes via activation of SAA3. Eur Rev Med Pharmacol Sci. 2017;21:302–309. [PubMed] [Google Scholar]
- 209.Ma D, Cao Y, Wang Z, He J, Chen H, Xiong H, et al. CCAT1 lncRNA promotes inflammatory bowel disease malignancy by destroying intestinal barrier via downregulating miR-185-3p. Inflamm Bowel Dis. 2019;25:862–874. doi: 10.1093/ibd/izy381. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.