

Abstract
[FeFe] hydrogenases are very active enzymes that catalyze the reversible conversion of molecular hydrogen into protons and electrons. Their active site, the H-cluster, contains a unique binuclear iron complex, [2Fe]H, with CN– and CO ligands as well as an aza-propane-dithiolate (ADT) moiety featuring a central amine functionality that mediates proton transfer during catalysis. We present a pulsed 13C-ENDOR investigation of the H-cluster in which the two methylene carbons of ADT are isotope labeled with 13C. We observed that the corresponding two 13C hyperfine interactions are of opposite sign and corroborated this finding using density functional theory calculations. The spin polarization in the ADT ligand is shown to be linked to the asymmetric coordination of the distal iron site with its terminal CN– and CO ligands. We propose that this asymmetry is relevant for the enzyme reactivity and is related to the (optimal) stabilization of the iron-hydride intermediate in the catalytic cycle.
The active site of [FeFe] hydrogenases, called the “H-cluster”, features a two-iron subcluster that is coordinated by CN– and CO ligands as well as a unique 2-aza-propane 1,3-dithiolate (ADT) bridging ligand.1 The [2Fe]H subcluster is linked to a generic [4Fe–4S] cubane cluster through a cysteine thiolate bridge (Figures 1 and 2). It is assumed that the amine moiety of the ADT bridge functions as a proton shuttle to and from the open coordination site at the iron site (Fed) distal to the [4Fe–4S]H subcluster. The catalytically active oxidized state (Hox) is characterized by a mixed valence redox configuration [Fe(II)Fe(I)] in the [2Fe]H subcluster. This state features a so-called “frustrated Lewis pair” (FLP),2 with the electrophilic Fed center adjacent to the Brønsted basic amine of the ADT.3 The FLP splits H2 heterolytically placing a proton on the amine and a hydride on Fed. This Fed–H species is immediately oxidized through the electron transport chain connected to the iron core of the H-cluster. This oxidation converts the hydridic Fed–H to an acidic Fed–H species. The subsequent stages in the catalytic cycle (Figure 1) feature several protonation, reduction, and proton coupled electron transfer (PCET) steps.4,5 This multistep sequence results in a very flat energy landscape, which explains the very high catalytic rates observed in [FeFe] hydrogenases (up to 10,000 s–1).6,7
Figure 1.
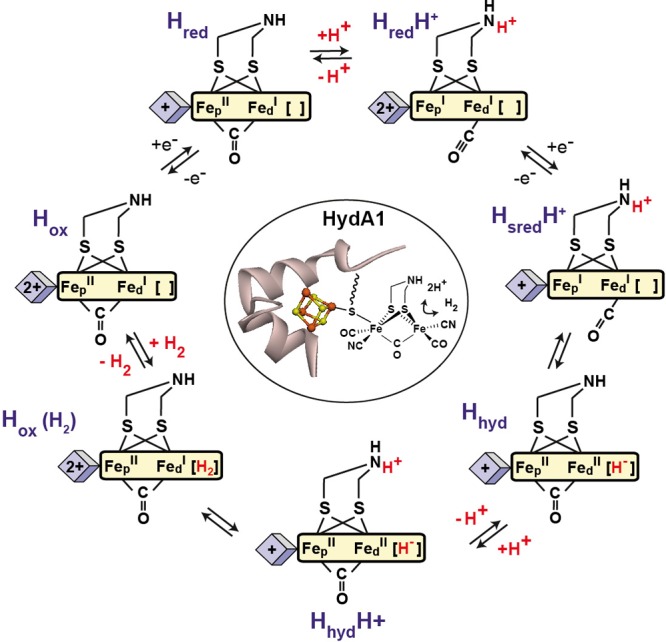
Structure of the H-cluster in [FeFe] hydrogenase from Chlamydomonas reinhardtii (CrHydA1) and the proposed catalytic cycle.
Figure 2.

Left: Schematic structure of the H-cluster in its Hox state, triply isotope labeled with 57Fe, as well as 13C and 2H in the aza-propane-dithiolate (ADT) ligand. Right: Q-band EPR spectrum (pseudo modulated FID detected) of our Hox preparation. Fitted g-values: (2.1008, 2.0398, 1.9966). A small contribution (<5%) from the Hox-CO state is evident from the feature at g = 2.006 and is marked by an asterisk. Full analysis of the EPR spectrum is presented in Figure S2.
The electronic structure of the H-cluster is strongly affected by the spin exchange interactions between the two subclusters and within the [4Fe–4S]H subcluster as monitored by pulsed EPR and paramagnetic NMR studies.8−11 These effects certainly contribute to the facile, virtually barrier-free, electron transport over the H-cluster. The protonation of the bridging ADT ligand amine group appears to play a central role in modulating the electron transport between the two subclusters.4 Therefore, the electronic structure, as reflected by the magnetic spin parameters of the ADT ligand, may show features that can be related to the overall catalytic properties of the H-cluster. Indeed, paramagnetic NMR of Hox showed spin polarization of the methylene protons, i.e., positive and negative spin densities.11 In this communication, we investigate the 13C-labeled methylene carbons of the ADT ligand using electron nuclear double resonance (ENDOR) and electron nuclear nuclear triple resonance (TRIPLE) spectroscopy and show that spin-polarization also occurs for these nuclei. This phenomenon is explored using density functional theory (DFT) quantum chemical calculations and is found to be dependent on the asymmetry in the ligand coordination at Fed.
Isotope Labeling
For the 13C-ENDOR experiments described herein, we took advantage of a triply labeled (2 × 57Fe, 2 × 13C, 4 × 2H) synthetic precursor of [2Fe]H, initially prepared for ongoing nuclear resonance vibrational spectroscopy (NRVS) experiments. The new synthetic route for this compound is described in the Supporting Information (SI) section A and Scheme S1. The [FeFe] hydrogenase from Chlamydomonas reinhardtii (CrHydA1) was produced using artificial maturation12,13 of the apoenzyme expressed in E. coli with the triply labeled [2Fe]H precursor (see Figure 2). The enzyme was allowed to oxidize under an N2 atmosphere (so-called “auto-oxidation”) to produce the Hox state. This is characterized by a mixed valence Fe(II)Fe(I) binuclear subcluster showing a characteristic rhombic S = 1/2 EPR spectrum with g-parameters (2.1008, 2.0398, 1.9966) (see Figures 2 and S2).1,14 The EPR and FTIR spectra (Figure S1) are consistent with those obtained previously for the native Hox state.1
Mims-ENDOR
Orientation-selective 13C Mims ENDOR (see Experimental Section in the SI and Scheme 1) was recorded at Q-band (34 GHz) at field positions corresponding to the range from g = 2.0 to g = 2.1 (Figure 3). In order to facilitate spectral fitting, the ENDOR spectra were normalized to the same amplitude and symmetrized with respect to the Larmor frequency (indicated by the arrows in Figure 3).
Scheme 1. Pulse Sequence of the 13C Mims ENDOR Experiment.
Figure 3.

(a–i) Orientation-selective 13C Mims Q-band ENDOR (34 GHz) spectra of CrHydA1 in the Hox state recorded at 15 K. The indicated field positions are given in mT. π/2 pulses were 20 ns. The waiting time τ was set to 200 ns, while the 400W RF pulse had a length of 60 μs (see Scheme 1). The red traces represent spectral fits obtained using a home-written MATLAB script making use of first order perturbation theory to calculate the ENDOR transition frequencies (details in SI). For the raw (normalized) ENDOR traces (unsymmetrized) along with the number of scans, see Figure S3.
At the g = 2.0 and g = 2.1 positions (magnetic field values of 1219 and 1158.7 mT, respectively), only molecules with one of their g-axes (x or z, respectively) oriented along the magnetic field contribute to the ENDOR spectrum. These positions are called “pseudo single crystal positions”. In these spectra, clearly two doublets centered at the 13C nuclear Larmor frequency (12.85 MHz at 1200 mT) are observed corresponding to the nuclear spin transitions of the two 13C nuclei in the ADT ligand. The different splitting and line-width of the two doublets indicate that the two 13C nuclei have significantly different hyperfine interaction (HFI) tensors differing in magnitudes and orientations. ENDOR spectra recorded at intermediate field positions contain contributions from molecules with multiple orientations giving rise to an EPR transition at the effective g-value corresponding to the field position. These multiple orientations lead to a broadening of the ENDOR spectra showing “powder-type” features. The line-shapes at, e.g., 1165.8 and 1205.4 G are reminiscent of “near axial hyperfine anisotropy” for the 13C nuclei. Therefore, in order to reduce the number of free variables, spectral fitting was started assuming axial HFI tensors for both 13C nuclei. First order perturbation theory was employed to calculate the ENDOR frequencies. The (1 – cos(2πAτ)) dependence of the ENDOR amplitude was taken into account. This procedure provided a fair but nonoptimal fit to the experimental ENDOR spectra. Subsequently, the rhombicity parameter was relaxed, and individual scaling of the two ENDOR components was applied. The final fitted parameters are listed in Table 1 and Figure S4. Given the local mirror symmetry of the ADT ligand (Figure 2), it is surprising that the two 13C HFI-tensors have significantly different magnitudes (1.9 vs 1.2 MHz for the isotropic component).
Table 1. Experimental and DFT-Predicted 13C HFI Anisotropic and Isotropic (Fermi Contact) Spin Couplings (MHz) for the Two Methylene Carbons A1 and A2 in the ADT Ligand in the Hox Statea.
| A1 (MHz) |
A2 (MHz) |
|||||||
|---|---|---|---|---|---|---|---|---|
| X | Y | Z | iso | X | Y | Z | iso | |
| exptl | 1.00 | 1.30 | 3.30 | 1.87 | –1.49 | –1.75 | –0.45 | –1.23 |
| Anative | 1.20 | 1.46 | 3.08 | 1.91 | –1.34 | –1.09 | –0.26 | –0.90 |
| Enative | 0.46 | 0.80 | 2.19 | 1.15 | –1.51 | –1.23 | –0.30 | –1.02 |
| Ad-iso | –1.67 | –1.38 | –0.50 | –1.18 | 1.31 | 1.59 | 3.33 | 2.07 |
| Ap-iso | 1.23 | 1.50 | 3.20 | 1.97 | –1.71 | –1.41 | –0.57 | –1.23 |
| Apd-iso | –1.41 | –1.12 | –0.30 | –0.94 | 1.16 | 1.42 | 3.05 | 1.88 |
| Ad-rot | 0.84 | 1.06 | 2.74 | 1.55 | –2.31 | –2.09 | –0.38 | –1.60 |
| Ap-(CN)2 | –1.44 | –1.08 | 0.17 | –0.78 | –1.37 | –1.02 | 0.22 | –0.72 |
| Ad-(CN)2 | –0.46 | 0.07 | 0.40 | 0.01 | –0.13 | 0.38 | 0.78 | 0.34 |
The experimental (exptl) and representative DFT (Anative) values are in bold. Other values are from alternative isomeric DFT models shown in Figure S7.
Mims TRIPLE
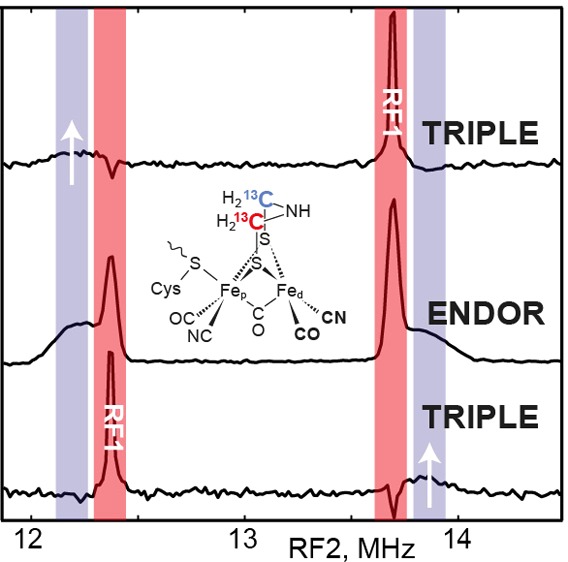
In order to determine the relative signs of the two fitted 13C HFIs, a triple resonance experiment was conducted at a field position corresponding to g = 2.02 (magnetic field position near 1218 mT, see Figure 3) where the two ENDOR doublet features are nicely separated and still relatively sharp (Figure 4 and Scheme 2). Adjusting the radiofrequency pumping pulse (RF1) to one of the ENDOR transitions inverts the spin state populations corresponding to this transition. This also affects (reduces) the population difference of the other nuclear spin transition in the same mS manifold resulting in a reduction of the ENDOR amplitude of the corresponding signal (see SI for more details).15,16 Pumping the high frequency line of the sharp 13C doublet leads to a reduction in amplitude of the low frequency line of the broad 13C doublet (upper spectra in Figure 4). Likewise, pumping the low frequency line of the sharp doublet causes a reduction in amplitude of the high frequency line of the broad doublet. We must, therefore, conclude that the signs of the two 13C hyperfine interactions are opposite (Tables 1 and S1). This implies that at one of the ADT carbon nuclei a negative (β) spin density is localized.
Figure 4.
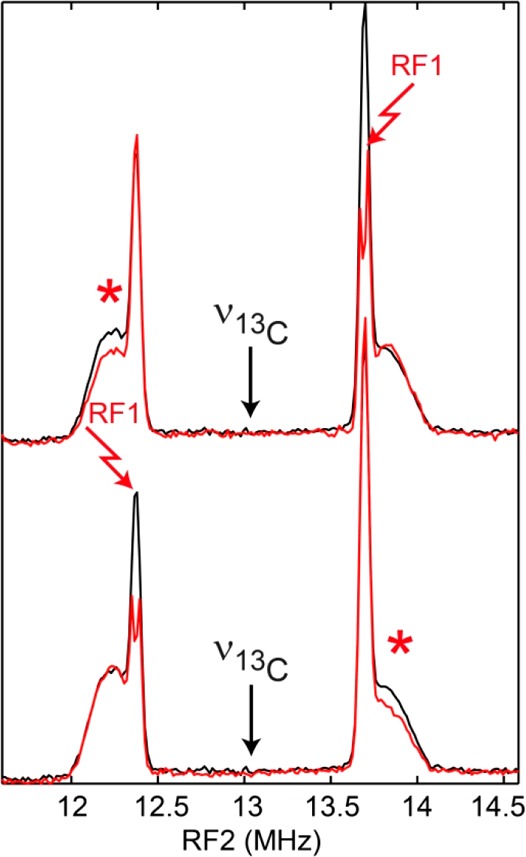
Mims TRIPLE resonance experiment (see pulse sequence in Scheme 2) at B = 1218 mT. The black traces represent the (unsymmetrized) 13C TRIPLE experiment with the pumping frequency RF1 off resonance at the 13C Larmor frequency, i.e., equivalent to the 13C ENDOR spectrum at B = 1218 mT (see Figure 3). The red traces represent the 13C Mims TRIPLE experiment with the RF1 pumping frequency tuned to one of the sharp ENDOR transitions. The “TRIPLE effect” is indicated by the red asterisks.
Scheme 2. Pulse Sequence of the 13C Mims Triple Resonance Experiment.
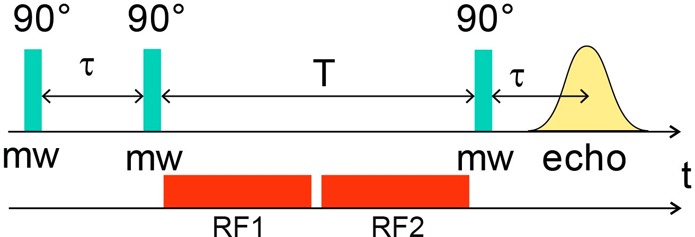
See Scheme S2 for extra details.
DFT Calculations
In order to obtain more insight into the underlying mechanism of this spin polarization phenomenon, we conducted DFT calculations using a [2Fe]H subcluster model in which the (oxidized) [4Fe–4S]H subcluster (S = 0) is modeled by a protonated thiol (ethyl-SH) group (Figure S5). The representative model is called “A” due to the “axial” conformation of the ADT–NH– moiety, with its proton pointing toward Fed. Using this modeling level, previously referred to as “S (small)”,17,18 the magnitude and sign of the experimental 13C-ADT HFIs for both carbon nuclei could be best reproduced. In Figure S6 the spin-density distribution of the (i) corresponding (see the DFT methods described in the SI) singly occupied molecular orbital (SOMO), (ii) formally doubly occupied MOs (Total-SOMO), as well as (iii) that of all MOs (Total) of the binuclear subsite are displayed with two contour values. A summary of these DFT results is presented in Figure 5.
Figure 5.
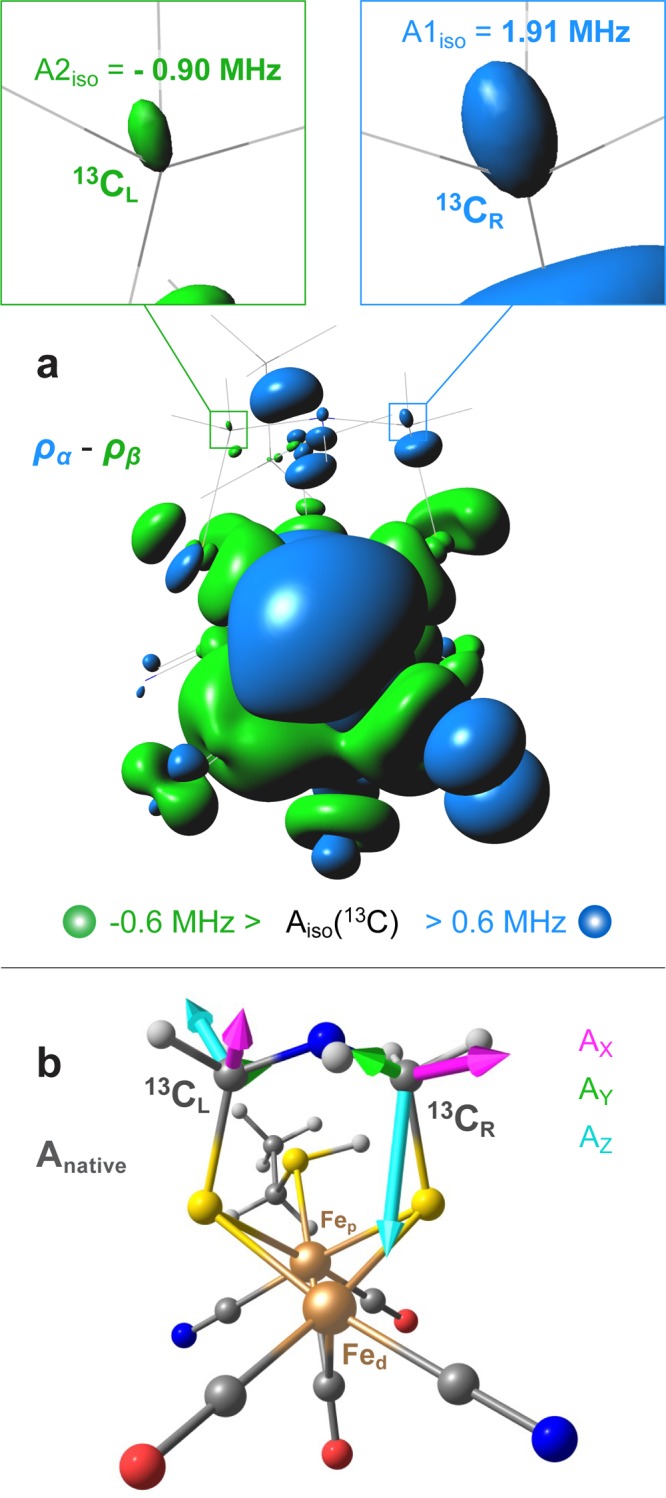
Top (a): Isosurfaces of total spin density at the 5 × 10–4 a.u. cutoff (corresponding to the Fermi contact HFI term Aiso(13C) = 0.6 MHz) for the representative HoxS = 1/2 DFT model Anative, showing positive (blue) density on the “right” 13C and negative (green) spin density on the “left” 13C nuclei. The model itself is shown in thin wire. Bottom (b): View of DFT model Anative along the Fep–Fed axis displaying the orientation of the calculated methylene 13C hyperfine tensors (Table S1). For extended information, see Figures S6 and S7.
Notably, the corresponding SOMO has 74% contribution from the Fed 3d shell. It turns out that the “left” methylene carbon, i.e., the carbon on the same side as the distal terminal CO ligand, has negative spin density due to spin polarization since the SOMO has only a vanishing contribution on this atom. The “right” carbon nucleus (on the same side as the distal CN– ligand) has positive spin density due to delocalization of the SOMO onto this center. No other MOs noticeably contribute to the spin densities at the two 13C nuclei. Although very small in their magnitude, the calculated atomic spin populations at the two ADT carbon nuclei (Table S3) are nevertheless consistent with the observed 13C HFI values. It is, therefore, tempting to assume that this asymmetric spin distribution is associated with the asymmetric ligand configuration at the distal iron center Fed. To verify this hypothesis, two isomeric DFT models were investigated in which the positions of the CN– and CO ligands were swapped at either Fep (“Ap-iso”) or Fed (“Ad-iso”), see Figure S7. Obviously, swapping CN– ligands at both Fe sites creates a pseudomirror image of the native [2Fe]H isomer (“Apd-iso”). A full mirror image can only be obtained if the protonation position of the ethyl-SH ligand is also swapped. Exchanging the CN– and CO ligand at the proximal iron Fep did not have an effect on the signs of the methylene 13C HFI. However, swapping the terminal CN– and CO ligands at the distal iron Fed changes the spin-polarization in the ADT ligand (Table 1). The behavior described is rationalized by the [Fe(II)pFe(I)d] electronic structure of the [2Fe]H subcluster in the Hox state with its S = 1/2 spin localization at the Fe(I)d distal iron site, while the Fe(II)p proximal site remains low-spin.19,20 Other investigated DFT models are “Enative”, having the −NH– proton in its alternative “equatorial” position pointing toward Fep,21 and “Ad-rot” with CN– at Fed rotated to the position trans to the bridging CO ligand;22 these two models represent [2Fe]H subcluster structural alternatives often discussed in the literature. While the two alternatives Enative and Ad-rot predict the same orientation of the 13C-ADT HFI tensors as seen in Anative, they produce an inferior match to the experimental HFI values (Figure S7, Tables 1 and S1). Interestingly, all the above-described ‘A’ (axial) isomeric models are found essentially equi-energetic within 1 kcal/mol, while the ‘E’ (equatorial) isomer is calculated +5 kcal/mol higher in its energy. Two additional intersite ligand swap isomers studied have both CN– ligands at either Fep (‘Ap-(CN)2’) or Fed (‘Ad-(CN)2’); these two structures are predicted to have significantly higher (+123/129 kcal/mol) relative energies. Notably, placing two negative CN– ligands at Fed in the Ad-(CN)2 isomer produces an inverted [Fe(I)pFe(II)d] oxidation pattern for the iron sites, having Fep as the spin center (Table S2).
The local CN–/CO ligand asymmetry at the distal iron site, which features the open coordination in the Hox state, seems to be the key to inducing the spin-polarization in the ADT ligand. A further result of this asymmetry is the spin polarization of the methylene protons as was observed in our paramagnetic NMR studies.11 Since in our [2Fe]H precursor the methylene protons have been labeled with 2H, we also performed 2H Mims ENDOR experiments at the three canonical field positions (gx, gy, gz) (see Figure S8). Signals of up to three 2H nuclei could be observed and show hyperfine couplings up to 0.6 MHz. The signals are, however, much weaker than those of the 13C nuclei, which, currently, precludes the application of a triple 2H ENDOR experiment.
To address the question of the relevance of the asymmetry in coordination of the distal iron atom for the catalytic cycle, we investigated how it influences the stabilization of the hydride-bound Hhyd-E state, which is formed from the 2e–-reduced (vs Hox), protonated HsredH+ state (see Figure 1). We calculated the energetics for this process for three isomers: the native configuration ‘A’ with one terminal CN– on each [2Fe]H iron, ‘p-(CN)2’ with both CN– ligands coordinated at the proximal iron, and ‘d-(CN)2’ with both CN– ligands coordinating the distal iron (see Figures S7 and S9). Clearly, the non-native isomeric configurations are of academic interest only since they do not occur in [FeFe] hydrogenase and cannot be synthesized. Moreover, the relative energies of the non-native isomers are around 130 kcal/mol higher than that of the native configuration (Figure S9). Since in our truncated model the [4Fe–4S]H subcluster is absent, HsredH+ is indistinguishable from HredH+. This also means that the models are diamagnetic, and no magnetic effects can be modeled. Nevertheless, it can be assumed that the thermodynamics of the Fed–H– hydride formation (i.e., the HsredH+-to-Hhyd-E intramolecular proton transfer from the ADT–NH2+– bridgehead to the distal iron) is well represented since the [4Fe–4S]H subcluster is unlikely to have a major impact on this process. It should be noted that the hydride formation can involve a transition from terminal CO in the H(s)redH+ state to bridging CO in Hhyd (see Figures 1 and S9). It is not clear if this ligand rearrangement also occurs to its full extent in the enzyme on the time scale of catalysis. For the ‘A’ native and p-(CN)2 isomers, the Hhyd-E hydride formation is predicted exothermic: ΔHnative = −3.4 kcal/mol, ΔHp-(CN)2 = −7.8 kcal/mol. For the d-(CN)2 isomer, the hydride formation is endothermic, ΔHd-(CN)2 = +16.8 kcal/mol. For the p-(CN)2 model, the distal iron Fed has a lack of charge density (in the absence of CN– coordination), providing extra stabilization of the hydride. However, model d-(CN)2 has excess charge density (with two CN– coordinating) at the distal iron, which prevents stabilization of the hydride at Fed. The charge density is more equally distributed over the two-iron core of [2Fe]H in the native configuration and seems to provide the perfect compromise to sufficiently stabilize the hydride state for fast catalysis yet not overstabilizing it, in order to avoid formation of a thermodynamic sink, which would strongly slow down the catalysis. We, therefore, conclude that the asymmetry of the ligand coordination at Fed ensures a flat energy landscape for the hydride formation and transfer during the reversible catalytic cycle of [FeFe] hydrogenase. This finding significantly advances the design strategies for artificial hydrogen conversion catalysts: Apart from the ligand geometry, also the charge balance at the catalytic iron center needs to be carefully controlled.
Acknowledgments
E.J.R, J.A.B., and W.L. would like to thank the Max Planck Society for continuous financial support. V.P. and M.K. acknowledge funding by the Deutsche Forschungsgemeinschaft under Germany’s Excellence Strategy–EXC 2008/1 (UniSysCat)–390540038. J.A.B. acknowledges funding from the DFG SPP 1927 “Iron–Sulfur for Life” project (Project No. BI 2198/1-1). The contributions of T.B.R. and C.P.R. were funded by the U.S. National Institutes of Health through GM61153. SPC was funded by NIH GM65440.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpclett.9b02354.
Author Contributions
⊥ E.J.R. and V.P. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Lubitz W.; Ogata H.; Rüdiger O.; Reijerse E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. 10.1021/cr4005814. [DOI] [PubMed] [Google Scholar]
- Stephan D. W.; Erker G. Frustrated Lewis Pair Chemistry: Development and Perspectives. Angew. Chem., Int. Ed. 2015, 54, 6400–6441. 10.1002/anie.201409800. [DOI] [PubMed] [Google Scholar]
- Birrell J. A.; Rüdiger O.; Reijerse E. J.; Lubitz W. Semisynthetic Hydrogenases Propel Biological Energy Research into a New Era. Joule 2017, 1, 61–76. 10.1016/j.joule.2017.07.009. [DOI] [Google Scholar]
- Sommer C.; Adamska-Venkatesh A.; Pawlak K.; Birrell J. A.; Rüdiger O.; Reijerse E. J.; Lubitz W. Proton Coupled Electronic Rearrangement within the H-Cluster as an Essential Step in the Catalytic Cycle of FeFe Hydrogenases. J. Am. Chem. Soc. 2017, 139, 1440–1443. 10.1021/jacs.6b12636. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Maciá P.; Pawlak K.; Rüdiger O.; Reijerse E. J.; Lubitz W.; Birrell J. A. Inter-cluster Redox Coupling Influences Protonation at the H-cluster in [FeFe] Hydrogenases. J. Am. Chem. Soc. 2017, 139, 15122–15134. 10.1021/jacs.7b08193. [DOI] [PubMed] [Google Scholar]
- Glick B. R.; Martin W. G.; Martin S. M. Purification and Properties of the Periplasmic Hydrogenase from Desulfovibrio desulfuricans. Can. J. Microbiol. 1980, 26, 1214–1223. 10.1139/m80-203. [DOI] [PubMed] [Google Scholar]
- Hatchikian E. C.; Forget N.; Fernández V. M.; Williams R.; Cammack R. Further Characterization of the [Fe]-Hydrogenase from Desulfovibrio desulfuricans ATCC 7757. Eur. J. Biochem. 1992, 209, 357–365. 10.1111/j.1432-1033.1992.tb17297.x. [DOI] [PubMed] [Google Scholar]
- Silakov A.; Reijerse E. J.; Albracht S. P. J.; Hatchikian E. C.; Lubitz W. The Electronic Structure of the H-cluster in the [FeFe]-hydrogenase from Desulfovibrio desulfuricans: A Q-band 57Fe ENDOR and HYSCORE Study. J. Am. Chem. Soc. 2007, 129, 11447–11458. 10.1021/ja072592s. [DOI] [PubMed] [Google Scholar]
- Silakov A.; Reijerse E. J.; Lubitz W. Unraveling the Electronic Properties of the Photoinduced States of the H-Cluster in the [FeFe] Hydrogenase from D. desulfuricans. Eur. J. Inorg. Chem. 2011, 2011, 1056–1066. 10.1002/ejic.201001080. [DOI] [Google Scholar]
- Silakov A.; Wenk B.; Reijerse E.; Albracht S. P. J.; Lubitz W. Spin Distribution of the H-cluster in the Hox-CO state of the [FeFe] Hydrogenase from Desulfovibrio desulfuricans: HYSCORE and ENDOR Study of 14N and 13C Nuclear Interactions. J. Biol. Inorg. Chem. 2009, 14, 301–313. 10.1007/s00775-008-0449-5. [DOI] [PubMed] [Google Scholar]
- Rumpel S.; Ravera E.; Sommer C.; Reijerse E.; Fares C.; Luchinat C.; Lubitz W. 1H NMR Spectroscopy of [FeFe] Hydrogenase: Insight into the Electronic Structure of the Active Site. J. Am. Chem. Soc. 2018, 140, 131–134. 10.1021/jacs.7b11196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berggren G.; Adamska A.; Lambertz C.; Simmons T. R.; Esselborn J.; Atta M.; Gambarelli S.; Mouesca J. M.; Reijerse E. J.; Lubitz W.; Happe T.; Artero V.; Fontecave M. Biomimetic Assembly and Activation of (FeFe)-Hydrogenases. Nature 2013, 499, 66–69. 10.1038/nature12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esselborn J.; Lambertz C.; Adamska-Venkatesh A.; Simmons T.; Berggren G.; Noth J.; Siebel J. F.; Hemschemeier A.; Artero V.; Reijerse E. J.; Fontecave M.; Lubitz W.; Happe T. Spontaneous Activation of [FeFe]-Hydrogenases by an Inorganic [2Fe] Active Site Mimic. Nat. Chem. Biol. 2013, 9, 607–609. 10.1038/nchembio.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubitz W.; Reijerse E.; van Gastel M. [NiFe] and [FeFe] Hydrogenases Studied by Advanced Magnetic Resonance Techniques. Chem. Rev. 2007, 107, 4331–4365. 10.1021/cr050186q. [DOI] [PubMed] [Google Scholar]
- Schweiger A.; Jeschke G.. Principles of Pulse Electron Paramagnetic Resonance; Oxford University Press: Oxford, 2001. [Google Scholar]
- Harmer J. R. Hyperfine Spectroscopy - ENDOR. Emagres 2016, 5, 1493–1514. 10.1002/9780470034590.emrstm1515. [DOI] [Google Scholar]
- Pham C. C.; Mulder D. W.; Pelmenschikov V.; King P. W.; Ratzloff M. W.; Wang H.; Mishra N.; Alp E. E.; Zhao J.; Hu M. Y.; Tamasaku K.; Yoda Y.; Cramer S. P. Terminal Hydride Species in [FeFe]-Hydrogenases Are Vibrationally Coupled to the Active Site Environment. Angew. Chem., Int. Ed. 2018, 57, 10605–10609. 10.1002/anie.201805144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijerse E. J.; Pham C. C.; Pelmenschikov V.; Gilbert-Wilson R.; Adamska-Venkatesh A.; Siebel J. F.; Gee L. B.; Yoda Y.; Tamasaku K.; Lubitz W.; Rauchfuss T. B.; Cramer S. P. Direct Observation of an Iron-Bound Terminal Hydride in [FeFe]-Hydrogenase by Nuclear Resonance Vibrational Spectroscopy. J. Am. Chem. Soc. 2017, 139, 4306–4309. 10.1021/jacs.7b00686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedler A. T.; Brunold T. C. Computational Studies of the H-Cluster of Fe-Only Hydrogenases: Geometric, Electronic, and Magnetic Properties and Their Dependence on the [Fe4S4 ] Cubane. Inorg. Chem. 2005, 44, 9322–9334. 10.1021/ic050946f. [DOI] [PubMed] [Google Scholar]
- Greco C.; Silakov A.; Bruschi M.; Ryde U.; De Gioia L.; Lubitz W. Magnetic Properties of [FeFe]-Hydrogenases: A Theoretical Investigation Based on Extended QM and QM/MM Models of the H-Cluster and Its Surroundings. Eur. J. Inorg. Chem. 2011, 2011, 1043–1049. 10.1002/ejic.201001058. [DOI] [Google Scholar]
- Pelmenschikov V.; Birrell J. A.; Pham C. C.; Mishra N.; Wang H.; Sommer C.; Reijerse E.; Richers C. P.; Tamasaku K.; Yoda Y.; Rauchfuss T. B.; Lubitz W.; Cramer S. P. Reaction Coordinate Leading to H2 Production in [FeFe]-Hydrogenase Identified by Nuclear Resonance Vibrational Spectroscopy and Density Functional Theory. J. Am. Chem. Soc. 2017, 139, 16894–16902. 10.1021/jacs.7b09751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton M. L.; Bhuvanesh N.; Reibenspies J. H.; Darensbourg M. Y. Synthetic Support of De Novo Design: Sterically Bulky [FeFe]-Hydrogenase Models. Angew. Chem., Int. Ed. 2008, 47, 9492–9495. 10.1002/anie.200803939. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.