

Abstract

We present a novel optical transient absorption and reflection microscope based on a diffraction-limited pump pulse in combination with a wide-field probe pulse, for the spatiotemporal investigation of ultrafast population transport in thin films. The microscope achieves a temporal resolution down to 12 fs and simultaneously provides sub-10 nm spatial accuracy. We demonstrate the capabilities of the microscope by revealing an ultrafast excited-state exciton population transport of up to 32 nm in a thin film of pentacene and by tracking the carrier motion in p-doped silicon. The use of few-cycle optical excitation pulses enables impulsive stimulated Raman microspectroscopy, which is used for in situ verification of the chemical identity in the 100–2000 cm–1 spectral window. Our methodology bridges the gap between optical microscopy and spectroscopy, allowing for the study of ultrafast transport properties down to the nanometer length scale.
The performance of all optoelectronics materials is critically dependent on efficient carrier transport. Our understanding of the associated transport characteristics commonly relies on the interpretation of indirect measurements such as microwave conductivity, photoluminescence quenching, terahertz photoconductivity, or time-of-flight techniques, which determine charge carrier mobilities or diffusion constants.1−5 While these methods successfully describe transport in spatially homogeneous materials, they lack access to nanoscopic morphologies which can affect the deduced transport parameters. In particular, next-generation semiconductors such as hybrid metal-halide perovskites,6 organic semiconductors,7 quantum dot films,8,9 and monolayer 2D semiconductors10 exhibit spatial heterogeneity on length scales as small as 10 nm, which need to be accessed to fully understand and optimize their charge carrier transport behavior in devices.
Furthermore, the photophysics of optoelectronic materials often involve sub-100 fs photodynamics, typically resolved using femtosecond transient absorption or reflection spectroscopy.7,11,12 These ultrafast processes are implicated to be of relevance in spatial charge carrier (or exciton) transport, but the associated transport parameters are indirectly inferred from the obtained kinetics, which are intrinsically averaged over all morphologies within the probe spot (50–100 μm).7,13,14 In contrast, spatially localized measurement techniques such as transient optical microscopy currently achieve a spatial accuracy of σ ≈ 10–50 nm but are limited to time resolutions of ∼100–300 fs, which are insufficient to access the earliest photodynamics.15−18
Here we overcome these limitations and explore the impact of ultrafast processes on the spatiotemporal carrier dynamics in optoelectronic materials by introducing a time-resolved transient absorption and reflection microscope capable of delivering sub-10 nm spatial accuracy with temporal resolutions down to 12 fs. In thin pentacene films, our technique resolves rapid spatial expansion occurring during the singlet fission process, which is completed within 200 fs after photoexcitation. In contrast, in p-doped silicon we observe charge carrier transport dynamics matching previously published reports even at the earliest time scales.19,20 In addition, our high temporal resolution allows us to chemically characterize the sample region in a universal fashion via impulsive stimulated Raman scattering.
Our experimental approach consists of an ultrafast pump–probe experiment coupled to a single-objective wide-field optical microscope (Figure 1a). Analogous to our previously reported implementation of wide-field femtosecond transient absorption microscopy,12 we generate temporally compressed broadband pulses spanning wavelengths from 520 to 640 nm (pump) and from 660 to 900 nm (probe). However, instead of using wide-field pump pulses we now focus the pump pulses tightly onto the sample by sending them through a high numerical aperture (NA = 1.1) oil-immersion objective, which results in a near-diffraction-limited Gaussian excitation profile (σ = 115 nm; Figure 1b and Supporting Information, section 1.1). After the pump pulses have photoexcited the sample and generated a localized carrier population, we spatially resolve the photoinduced transient response by imaging time-delayed wide-field probe pulses onto an emCCD.
Figure 1.

Setup and imaging channels employed in femtosecond transient absorption and reflection microscopy to monitor ultrafast charge carrier diffusion. (a) Schematic of the optical components. A pump–probe scheme is integrated into a wide-field optical microscope with a high numerical aperture (NA = 1.1) objective, and the transmitted or reflected probe is subsequently detected by an emCCD camera at a given probe wavelength. BS, beam splitter; LP, long-pass filter; TL, tube lens; BP, bandpass filter; M, mirror; WLC, white light continuum. (b) Spatial profile of the pump pulse obtained by monitoring the fluorescence intensity from a fluorescent bead scanned across the pump spot (black circles) and corresponding Gaussian fit (blue). (c) Temporal characterization of the pump (green) and probe (orange) pulses at the sample. Probe pulses are only compressed for transmission experiments. (d) Transmission geometry and (e) reflection geometry. CM, curved mirror; WL, wide-field lens; BS, beam splitter.
To achieve the highest temporal resolution, we employ a single-lens high-NA oil-immersion objective instead of the more conventional flat-field and chromatic aberration-corrected multicomponent objectives. This choice compromises the absolute imaging capability but only weakly affects a differential pump–probe experiment over a small field of view (see also Supporting Information, section 2), allowing us to compress our pump pulses to 9.8 fs with commercially available optical elements (Figure 1c, green line; Supporting Information, section 1.2). We remark that near-IR pulses (650–950 nm) can be temporally compressed to sub-10 fs for high-NA multicomponent microscope objectives using advanced pulse-shaping methods.21,22 Similar results for visible pulses (550–650 nm) are, however, lacking because of the significantly larger dispersion corrections required for this wavelength range.
The probe pulses in our setup are temporally compressed to 6.8 fs (Figure 1c, orange line) for the transmission channel, resulting in an effective time resolution of 11.9 fs (Figure 1d). In the reflection channel (Figure 1e), we use chirped probe pulses (∼700 fs), which in combination with bandpass-filtered detection results in a temporal resolution of ∼50 fs (see the Supporting Information, section 1.2 for details).23 While it is straightforward to compress the probe pulses for our microscope objective by adding an additional set of third-order compensated chirped mirrors into the beam path, as illustrated for the pump pulse, we opted to demonstrate that chirped probe pulses can be used equivalently if a simpler implementation with marginally lower temporal resolution (∼50 fs) is desired.
We incorporated two complementary probe channels to accommodate a broad range of samples irrespective of their optical properties. In the transmission channel, the probe is loosely focused onto the sample by a concave mirror (Figure 1d, Gaussian illumination area of σ ≈ 8 μm), whereas in the reflection channel, the probe is focused into the back-aperture of the high-NA objective with an additional wide-field lens (Figure 1e, Gaussian illumination area of σ ≈ 4 μm). Prior to detecting the probe, we insert narrow-bandpass filters into the detection path to gain access to wavelength-resolved spatiotemporal dynamics, while simultaneously removing optical artifacts associated with chromatic aberrations.
To demonstrate the capabilities of our microscope, we first imaged the spatiotemporal dynamics of a ∼120 nm thin film of the singlet fission material pentacene (Pc) in transmission. Following photoexcitation to S1, Pc undergoes singlet fission to form a correlated triplet–triplet (1TT) state on a sub-100 fs time scale, which can be followed by monitoring the growth of the photoinduced absorption band at 790 nm. We remark that this wavelength range is free from other overlapping spectral components, allowing direct insight into the spatiotemporal characteristics of the singlet fission dynamics (Supporting Information, section 3.2).24−27
A typical point-spread function recorded at this wavelength 1 ps after photoexcitation exhibits the expected negative differential transmission signal at its center, accompanied by periodic intensity changes as we move radially away from the center (Figure 2a). This point-spread function is intrinsic to wide-field microscopy, also known as a far-field Frauenhofer diffraction pattern, which arises from the optical interference pathways of the probe light simultaneously collected over all space by the detector. We note that the pulses employed here have an estimated spatial coherence length of ∼2 μm over which this interference effect can be observed. In our experiment, the tightly focused pump pulses generate a Gaussian-shaped aperture by changing the complex refractive index of the material, through which the probe pulses are subsequently diffracted, yielding the characteristic near-isotropic Airy disk point-spread function presented in Figure 2a. Closer inspection of our point-spread functions reveals a small degree of anisotropy in the intensity distribution of the first diffraction ring. We attribute this asymmetry to the reduced imaging capabilities provided by our microscope objective, causing spherical aberrations. This asymmetry does, however, not affect the central part of the point-spread function in our microscope, allowing us to confidently quantify time-resolved changes in its width (see Supporting Information, section 2 for a more detailed discussion).
Figure 2.

Femtosecond transient absorption microscopy of a thin pentacene film (∼120 nm) measured in transmission. (a) Differential transmission image at 1 ps after photoexcitation. Scale bar depicts 500 nm. (b) Transient kinetics at the central pixel of the point-spread function (gray line, left axis) and retrieved amplitude of a two-dimensional (2D) Gaussian fit (orange line, left axis, scaled for clarity). The corresponding retrieved 2D Gaussian standard deviation (green line, right axis) is also shown. (c) Mean-square displacement (MSD) curve relative to a time delay of 75 fs. The MSD is well-described by two linear diffusion regimes with the indicated slopes (see main text for corresponding diffusion coefficient). Early time delays (<75 fs) were ignored because of coherent artifact contributions (gray shaded region).
The transient photodynamics of pentacene at the central pixel of the point-spread function feature an initial coherent spike at zero-time delay, which evolves into a fast rise of the 1TT population with a τ = 76 ± 6 fs time constant (Figure 2b, gray line). After the initial rise, the differential signal amplitude remains largely constant, in agreement with previous transient absorption studies.24 To isolate the underlying spatial profiles, we described each transient absorption image with an isotropic two-dimensional Gaussian function and extracted the corresponding time-dependent amplitudes (Figure 2b, orange line) and standard deviations (Figure 2b, green line), in line with previous analysis methods.15,28 The retrieved amplitudes agree well with the central pixel amplitudes, but the standard deviations display an unexpected behavior characterized by an initial rise from ∼180 nm at 75 fs to ∼190 nm at 200 fs. This initial rise is followed by a significantly slower rise marginally above our signal-to-noise limit. We remark that this behavior is independent of fitting the point-spread function with an isotropic or anisotropic two-dimensional Gaussian function (Supporting Information, section 2).
Importantly,
the spatial resolution of our microscope remains diffraction-limited
at the probe wavelength (σ ≈ 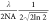 ≈ 152 nm at 790 nm), but
the spatial
accuracy, i.e. how well we can distinguish the standard deviations
of different transient point-spread functions, is dependent only on
the signal-to-noise ratio of the recorded images, as demonstrated
previously.15,28,29 Consequently, we can track changes to the profile of the imaged
point-spread function with high confidence. By combining the retrieved
standard deviation error from the two-dimensional Gaussian fit with
the temporal standard deviation error we retrieve a spatial accuracy
of ∼2.9 nm for this measurement. We refer the reader to section 2 of the Supporting Information for more
details (Figure S4). This spatial accuracy
is well below the 10 nm standard deviation expansion observed during
the first 200 fs for the pentacene films in Figure 2.
≈ 152 nm at 790 nm), but
the spatial
accuracy, i.e. how well we can distinguish the standard deviations
of different transient point-spread functions, is dependent only on
the signal-to-noise ratio of the recorded images, as demonstrated
previously.15,28,29 Consequently, we can track changes to the profile of the imaged
point-spread function with high confidence. By combining the retrieved
standard deviation error from the two-dimensional Gaussian fit with
the temporal standard deviation error we retrieve a spatial accuracy
of ∼2.9 nm for this measurement. We refer the reader to section 2 of the Supporting Information for more
details (Figure S4). This spatial accuracy
is well below the 10 nm standard deviation expansion observed during
the first 200 fs for the pentacene films in Figure 2.
We can roughly estimate the underlying two-dimensional diffusion dynamics from the mean-square displacement (MSD) curves according to the (linear) diffusion eq 1
| 1 |
where D is
the diffusion coefficient and t the time delay (Figure 2c).15,28,30 We note that we excluded early
time delays in our analysis (Figure 2b,c, gray shaded area), such that t0 = 75 fs, because the presence of coherent artifact contributions
indicates additional signal pathways, such as two-photon absorption
and cross-phase modulations, which can artificially reduce the retrieved
width.31,32 By analyzing the MSD of the previously identified
standard deviation regimes, we obtain diffusion coefficients with
values of Dfast = 136.5 ± 18.1 cm2 s–1 and Dslow = 3.2 ± 2.0 cm2 s–1. The initial
diffusion coefficient is furthermore independent of excitation density
in the linear absorption regime (Figure S9), eliminating carrier-recombination artifacts from our measurements.
Considering the diffusion coefficient and retrieved singlet fission
lifetime, we can estimate the effective carrier diffusion length for
this fully temporally resolved ultrafast regime as 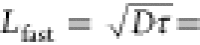 32.2 ± 2.1 nm.
32.2 ± 2.1 nm.
To generalize this result, we repeated the same experiment by detecting different probe wavelengths (Supporting Information, section 4). Within a probe range from 710 to 790 nm, coinciding with the photoinduced 1TT absorption band in Pc (Figure S7), we retrieve identical fast diffusion coefficients within error margins. These results firmly rule out refractive index artifacts33 or competing overlapping spectral features16 as responsible factors for this fast diffusion regime. The extraordinarily high diffusion coefficients (Dfast) and associated carrier lengths (Lfast) thus point to a previously unknown transport regime during the earliest times following photoexcitation in Pc.
While a detailed interpretation of the above results is beyond the scope of this work, the established photophysics of Pc allows us to provide a first qualitative explanation of our results.24,30,34,35 We speculate that the initially delocalized photoexcited S1 wave function could undergo ultrafast spatial broadening (Dfast) during the formation of a more delocalized 1TT wave function within a coherent transport regime. This rapid initial expansion is followed by a slower population expansion over several picoseconds, and the lower diffusion coefficient (Dslow) indicates an incoherent hopping mechanism which could be associated with the formation of an entangled but spatially separated 1T···T state.36 Further theoretical work is, however, critical to elucidate the origins of both the fast and slow regimes of the observed spatiotemporal behavior.
In addition to our ability to visualize exciton population transport in Pc during singlet fission, the high temporal resolution of our setup allows us to also monitor the chemical fingerprint of the excitation spot via impulsive stimulated Raman scattering for in situ verification of the chemical composition of the probed sample region.37−39 To highlight this capability, we conducted the same experiment on Pc at higher excitation densities, to achieve an increased signal-to-noise ratio, and resolved pronounced oscillatory modulations on top of the electronic background signal (Figure 3a, ∼25 fs period, see inset). To extract the associated impulsive Raman spectrum, we computed the Fourier power spectrum of the oscillatory modulations after removing the slowly varying electronic background signal (Figure 3b). The retrieved spectrum is in excellent agreement with the ground-state Raman spectrum of Pc thin films and is dominated by modes at 1193 (∼28 fs period) and 1371 cm–1 (25 fs period).40 The oscillatory components are also reproduced by applying a two-dimensional Gaussian fitting routine outlined above (Figure 3, orange line). We remark that the appearance of such coherent oscillations in the time domain corroborates the high temporal resolution deduced from our external pulse characterization (Supporting Information, section 1.2).
Figure 3.

Vibrational imaging in femtosecond transient absorption microscopy demonstrated on thin-film pentacene in transmission. (a) Detection of vibrational coherences superimposed on the electronic signal arising from impulsive stimulated Raman scattering. The oscillatory modulations are best visualized by either integrating the central image portion (8 × 8 pixels, gray line) or by carrying out a 2D Gaussian fit analysis (orange line). The signal is dominated by ∼25 fs periods (inset). (b) Fourier power spectrum after background removal of the electronic dynamics in panel a for the two analysis methods, displaying the dominant C–C and C=C stretching Raman modes of thin-film pentacene at 1193 and 1371 cm–1.
To highlight how our approach can be applied for optically thick or nontransparent materials, we investigated the ultrafast spatiotemporal properties of p-doped Silicon (Si) using the reflection channel of our microscope. Si is a prototypical, well-characterized material commonly used in solar cells, which is nontransparent to visible light and therefore cannot be measured in transmission. Here, we recorded transient reflection images probed above the optical bandgap (740 nm). A typical point-spread function at a time delay of 2.8 ps shows a negative, isotropic differential reflectivity signal in the form of an Airy disk, in agreement with previous reports (Figure 4a).33 The associated photodynamics exhibit an initial rise with a decay time constant of 207 ± 22 fs followed by a slower decay time of 11.29 ± 1.1 ps (Figure 4b, gray line).
Figure 4.

Femtosecond transient reflection microscopy of p-doped Silicon. (a) Differential reflectance image at 2.8 ps after photoexcitation. Scale bar depicts 500 nm. (b) Transient kinetics at the central pixel of the point-spread function (gray line, left axis) and retrieved amplitude of a two-dimensional (2D) Gaussian fit (orange line, left axis). The corresponding retrieved 2D Gaussian standard deviation (green line, right axis) is also shown. (c) Mean-square displacement (MSD) curve relative to a time delay of 90 fs. The MSD is well-described by a single linear diffusion regime with the indicated slope (see main text for corresponding diffusion coefficient). Early time delays (<90 fs) were ignored because of coherent artifact contributions (gray shaded region).
Analogous to our analysis on Pc, we described each transient reflection image with a two-dimensional isotropic Gaussian function to analyze the spatial profiles. The retrieved time-dependent amplitudes match the central pixel amplitudes (Figure 4b, orange line), as previously observed for Pc. Interestingly, despite the two temporal regimes identified in the signal amplitude, the corresponding standard deviation displays only a single rise from ∼190 nm at 100 fs to 205 nm at 3 ps (Figure 4b, green line), sufficient to be resolved with the spatial accuracy of ∼2.7 nm obtained in this experiment (see Supporting Information, section 2). The corresponding diffusion coefficient in our time window can be estimated from the MSD relative to t0 = 90 fs using eq 1 and yields D = 8.6 ± 0.5 cm2 s–1, in agreement with previously reported values (Figure 4c).19,33 Our results suggest that the diffusion characteristics in Si at the earliest times (∼100 fs to 3 ps) after photoexcitation are equivalent to the pico- or nanosecond time scales that have been reported with alternative measurement techniques.19,20,33
Apart from the improved temporal resolution, our transient absorption and reflection microscope differs significantly from previous implementations because of its wide-field imaging detection. Earlier studies15−18 almost exclusively rely on confocal microscopy,33 in which the transient image is reconstructed after scanning a tightly focused probe beam across the sample. The benefits and drawbacks of wide-field microscopy compared to confocal microscopy have been extensively reviewed in the literature and apply similarly for transient microscopes.41 While we have demonstrated an ultrafast wide-field microscope, because of its easier construction, our approach can readily improve the time resolution of a transient confocal microscope by simply replacing the relevant objective lenses with low-dispersion objectives and adding commercially available pulse compression optics (see Experimental Methods).
We emphasize that care has to be taken in the interpretation and comparison of the respective transient point-spread functions, which will appear as Airy disks for a wide-field microscope (e.g. Figure 2a) instead of a two-dimensional Gaussian function for a confocal microscope.15−18 Critically, while the underlying phase interference effects in our microscope may seem deleterious at first, they in principle allow the ultrafast tracking of three-dimensional carrier motion over the spatial coherence length of our probe pulses (∼2 μm) by carefully evaluating the corresponding point-spread functions (particularly in the reflection channel).33,42
Taken together, we have demonstrated ultrafast transient absorption and reflection microscopy which meets the time and length scale requirements of optoelectronic processes by achieving an effective temporal resolution down to 12 fs in transmission and ∼50 fs in reflection mode, with simultaneous sub-10 nm spatial accuracy. This spatiotemporal capability enabled us to uncover a previously unobserved ultrafast linear transport regime in Pc films indicative of a highly spatially delocalized 1TT wave function which corresponds to carrier transport lengths of up to 32 nm from the initial excitation spot within only 200 fs after photoexcitation. In contrast, p-doped Si did not show any ultrafast spatial expansion but instead followed its reported long time diffusive behavior. An inherent benefit of our approach is the ability to simultaneously monitor the chemical identity of the selected region via impulsive stimulated Raman scattering alongside the spatiotemporal dynamics. The combination of these functionalities provides comprehensive insight into charge carrier and exciton population transport in a wealth of materials with morphological and chemical specificity.4300 Consequently, our approach can be readily applied to heterojunctions and other inhomogeneous morphologies to elucidate interfacial population transport.
Experimental Methods
Experimental Setup. Pulses were provided by a Yb:KGW amplifier (1030 nm, 200 fs, 200 kHz, 5 W, Pharos, Light Conversion) which seeded two white light continuum stages for probe and pump pulse generation. Probe pulses were produced in a 3 mm YAG crystal and spectrally filtered in a home-built 4F single-prism filter to adjust the bandwidth to 660–900 nm. Subsequently, the probe pulses were compressed to 6.8 fs with third-order corrected chirped mirrors (DCM9, Venton) and by optimizing the prism insertion in the spectral filter. Probe pulses were only compressed for the transmission channel (see further explanation in Supporting Information, section 1). Consequently, they experienced a temporal chirp of ∼700 fs in the spectral region from 660 to 900 nm in the reflection channel due to the additional dispersion caused by the wide-field and objective lens. Pump pulses were produced in a 3 mm sapphire crystal for increased bandwidth, spectrally adjusted by a 650 nm short-pass filter (FESH650, Thorlabs), and subsequently compressed to 9.8 fs by two sets of third-order compensated chirped mirrors (109811, Layertec) in combination with a wedge-prism pair, accounting for all optical components including the microscope objective. We additionally cleaned the spatial mode of the pump pulses by means of a 40 μm pinhole, which is imaged into the back-aperture of the objective to achieve near-diffraction-limited focusing at the sample. To set up the time-dependent pump–probe sequence, we modulated the pump pulses at 15 Hz via a mechanical chopper (Thorlabs) and temporally delayed the probe via a computer-controlled closed-looped piezo translation stage (P-625.1CL, PhysikInstrumente).
An emCCD (Rolera Thunder, QImaging) detected the transmitted or reflected probe pulses via a high numerical aperture oil-immersion objective (NA = 1.1, 100× magnification) in combination with an F = 500 mm tube lens (AC254-500-B, Thorlabs) resulting in 55.5 nm/pixel. To avoid pump light detection, we inserted a 650 nm long-pass filter (FELH650, Thorlabs) in front of the emCCD in addition to a narrow bandpass filter (FB790-10 for pentacene and FB740-10 for silicon, Thorlabs) as shown in Figure 1a.
An automatic focus control loop based on total internal reflection of a reference continuous wave laser (405 nm) was used to stabilize the focus position via an objective piezo (NP140, Newport) in pentacene.43 The same approach could not be applied to silicon. Instead, we measured eight different sample locations (experimental time 4 min/sample location) and averaged the results. During this time scale no significant focus drift was observed as verified by monitoring the autofocus performance during the measurements on pentacene.
The camera exposure time was set to 9 ms for pentacene and 12 ms for silicon at a camera frame rate of 30 Hz. For the experiments on pentacene shown in Figures 2 and 3, the photoexcited carrier densities were 2.90 × 1018 and 9.01 × 1018 cm–3,44 respectively, and 1.67 × 1018 cm–3 for silicon (Figure 4),20 as calculated from the peak energy of the pump pulse.33 Both experiments were conducted by adjusting the polarization of the pump and probe beams to magic angle.
Data Analysis. Normalized differential images were calculated from the raw images after subtraction of the dark counts and described for each time delay with an isotropic two-dimensional Gaussian function according to
where A encodes the signal amplitudes; x0 and y0 are the position, and σ is the standard deviation. Initial guesses for the position were taken from the center of mass position of each image; the amplitude was taken from the corresponding pixel amplitude, and we set σ = 100 nm and offset = 0.
We note that this approach does not describe the diffraction rings present in the Airy disk. This results in a systematic overestimation of the fitted signal amplitude, and we therefore scaled the reported amplitudes in Figures 2 and 4 to emphasize the correct reproduction of the transient kinetics. Importantly, our analysis focuses on evaluating time-dependent differences to the retrieved fit parameters. Especially for the standard deviation parameter, any error in describing the full point-spread function will translate into a small systematic deviation from the actual value, which will be inconsequential for assessing temporal changes.
Population kinetics were modeled with a sum of two exponentially decaying Gaussian functions, and stated error bars in the retrieved lifetimes correspond to the standard deviation error.
Acknowledgments
We acknowledge financial support from the EPSRC and the Winton Program for the Physics of Sustainability. This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (Grant Agreement No. 758826). C.S. acknowledges financial support by the Royal Commission of the Exhibition of 1851.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpclett.9b02437.
Further information regarding the temporal and spatial pulse characterization, illustration of the general fit performance and accuracy, sample preparation and characterization details, and discussion of point-spread function (PDF)
Author Contributions
§ C.S. and J.S. contributed equally to the work.
The authors declare no competing financial interest.
Notes
The data underlying this work is available at https://doi.org/10.17863/CAM.44692.
Supplementary Material
References
- Herz L. M. Charge-Carrier Mobilities in Metal Halide Perovskites: Fundamental Mechanisms and Limits. ACS Energy Lett. 2017, 2, 1539–1548. 10.1021/acsenergylett.7b00276. [DOI] [Google Scholar]
- Savenije T. J.; Ferguson A. J.; Kopidakis N.; Rumbles G. Revealing the Dynamics of Charge Carriers in Polymer:Fullerene Blends Using Photoinduced Time-Resolved Microwave Conductivity. J. Phys. Chem. C 2013, 117, 24085–24103. 10.1021/jp406706u. [DOI] [Google Scholar]
- Ponseca C. S.; Savenije T. J.; Abdellah M.; Zheng K.; Yartsev A.; Pascher T.; Harlang T.; Chabera P.; Pullerits T.; Stepanov A.; et al. Organometal Halide Perovskite Solar Cell Materials Rationalized: Ultrafast Charge Generation, High and Microsecond-Long Balanced Mobilities, and Slow Recombination. J. Am. Chem. Soc. 2014, 136, 5189–5192. 10.1021/ja412583t. [DOI] [PubMed] [Google Scholar]
- Ward A. J.; Ruseckas A.; Samuel I. D. W. A Shift from Diffusion Assisted to Energy Transfer Controlled Fluorescence Quenching in Polymer–Fullerene Photovoltaic Blends. J. Phys. Chem. C 2012, 116, 23931–23937. 10.1021/jp307538y. [DOI] [Google Scholar]
- Tamai Y.; Ohkita H.; Benten H.; Ito S. Exciton Diffusion in Conjugated Polymers: From Fundamental Understanding to Improvement in Photovoltaic Conversion Efficiency. J. Phys. Chem. Lett. 2015, 6, 3417–3428. 10.1021/acs.jpclett.5b01147. [DOI] [PubMed] [Google Scholar]
- de Quilettes D. W.; Vorpahl S. M.; Stranks S. D.; Nagaoka H.; Eperon G. E.; Ziffer M. E.; Snaith H. J.; Ginger D. S. Impact of Microstructure on Local Carrier Lifetime in Perovskite Solar Cells. Science (Washington, DC, U. S.) 2015, 348, 683–686. 10.1126/science.aaa5333. [DOI] [PubMed] [Google Scholar]
- Gelinas S.; Rao A.; Kumar A.; Smith S. L.; Chin A. W.; Clark J.; van der Poll T. S.; Bazan G. C.; Friend R. H. Ultrafast Long-Range Charge Separation in Organic Semiconductor Photovoltaic Diodes. Science (Washington, DC, U. S.) 2014, 343, 512–516. 10.1126/science.1246249. [DOI] [PubMed] [Google Scholar]
- Liu M.; Chen Y.; Tan C.-S.; Quintero-Bermudez R.; Proppe A. H.; Munir R.; Tan H.; Voznyy O.; Scheffel B.; Walters G.; et al. Lattice Anchoring Stabilizes Solution-Processed Semiconductors. Nature 2019, 570, 96–101. 10.1038/s41586-019-1239-7. [DOI] [PubMed] [Google Scholar]
- Ning Z.; Gong X.; Comin R.; Walters G.; Fan F.; Voznyy O.; Yassitepe E.; Buin A.; Hoogland S.; Sargent E. H. Quantum-Dot-in-Perovskite Solids. Nature 2015, 523, 324–328. 10.1038/nature14563. [DOI] [PubMed] [Google Scholar]
- Yang J.; Wang Y.; Lagos M. J.; Manichev V.; Fullon R.; Song X.; Voiry D.; Chakraborty S.; Zhang W.; Batson P. E.; et al. Single Atomic Vacancy Catalysis. ACS Nano 2019, 13, 9958–9964. 10.1021/acsnano.9b05226. [DOI] [PubMed] [Google Scholar]
- Musser A. J.; Liebel M.; Schnedermann C.; Wende T.; Kehoe T. B.; Rao A.; Kukura P. Evidence for Conical Intersection Dynamics Mediating Ultrafast Singlet Exciton Fission. Nat. Phys. 2015, 11, 352–357. 10.1038/nphys3241. [DOI] [Google Scholar]
- Schnedermann C.; Lim J. M.; Wende T.; Duarte A. S.; Ni L.; Gu Q.; Sadhanala A.; Rao A.; Kukura P. Sub-10 fs Time-Resolved Vibronic Optical Microscopy. J. Phys. Chem. Lett. 2016, 7, 4854–4859. 10.1021/acs.jpclett.6b02387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X.-H.; Price M. B.; Finnegan J. R.; Boott C. E.; Richter J. M.; Rao A.; Menke S. M.; Friend R. H.; Whittell G. R.; Manners I. Long-Range Exciton Transport in Conjugated Polymer Nanofibers Prepared by Seeded Growth. Science (Washington, DC, U. S.) 2018, 360, 897–900. 10.1126/science.aar8104. [DOI] [PubMed] [Google Scholar]
- Menke S. M.; Holmes R. J. Exciton Diffusion in Organic Photovoltaic Cells. Energy Environ. Sci. 2014, 7, 499–512. 10.1039/C3EE42444H. [DOI] [Google Scholar]
- Block A.; Liebel M.; Yu R.; Spector M.; Sivan Y.; García de Abajo F. J.; van Hulst N. F. Tracking Ultrafast Hot-Electron Diffusion in Space and Time by Ultrafast Thermomodulation Microscopy. Sci. Adv. 2019, 5, eaav8965. 10.1126/sciadv.aav8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z.; Wan Y.; Yang M.; Snaider J.; Zhu K.; Huang L. Long-Range Hot-Carrier Transport in Hybrid Perovskites Visualized by Ultrafast Microscopy. Science (Washington, DC, U. S.) 2017, 356, 59–62. 10.1126/science.aam7744. [DOI] [PubMed] [Google Scholar]
- Hartland G. V. Ultrafast Studies of Single Semiconductor and Metal Nanostructures through Transient Absorption Microscopy. Chem. Sci. 2010, 1, 303–309. 10.1039/c0sc00243g. [DOI] [Google Scholar]
- Piland G.; Grumstrup E. M.. High-Repetition Rate Broadband Pump–Probe Microscopy. J. Phys. Chem. A 2019. 10.1021/acs.jpca.9b03858. [DOI] [PubMed] [Google Scholar]
- Jacoboni C.; Canali C.; Ottaviani G.; Alberigi Quaranta A. A Review of Some Charge Transport Properties of Silicon. Solid-State Electron. 1977, 20, 77–89. 10.1016/0038-1101(77)90054-5. [DOI] [Google Scholar]
- Green M. A. Self-Consistent Optical Parameters of Intrinsic Silicon at 300K Including Temperature Coefficients. Sol. Energy Mater. Sol. Cells 2008, 92, 1305–1310. 10.1016/j.solmat.2008.06.009. [DOI] [Google Scholar]
- Pawłowska M.; Goetz S.; Dreher C.; Wurdack M.; Krauss E.; Razinskas G.; Geisler P.; Hecht B.; Brixner T. Shaping and Spatiotemporal Characterization of Sub-10-Fs Pulses Focused by a High-NA Objective. Opt. Express 2014, 22, 31496–31510. 10.1364/OE.22.031496. [DOI] [PubMed] [Google Scholar]
- Goetz S.; Li D.; Kolb V.; Pflaum J.; Brixner T. Coherent Two-Dimensional Fluorescence Micro-Spectroscopy. Opt. Express 2018, 26, 3915–3925. 10.1364/OE.26.003915. [DOI] [PubMed] [Google Scholar]
- Kovalenko S. A.; Dobryakov A. L.; Ruthmann J.; Ernsting N. P. Femtosecond Spectroscopy of Condensed Phases with Chirped Supercontinuum Probing. Phys. Rev. A: At., Mol., Opt. Phys. 1999, 59, 2369–2384. 10.1103/PhysRevA.59.2369. [DOI] [Google Scholar]
- Wilson M. W. B.; Rao A.; Clark J.; Kumar R. S. S.; Brida D.; Cerullo G.; Friend R. H. Ultrafast Dynamics of Exciton Fission in Polycrystalline Pentacene. J. Am. Chem. Soc. 2011, 133, 11830–11833. 10.1021/ja201688h. [DOI] [PubMed] [Google Scholar]
- Musser A. J.; Clark J. Triplet-Pair States in Organic Semiconductors. Annu. Rev. Phys. Chem. 2019, 70, 323–351. 10.1146/annurev-physchem-042018-052435. [DOI] [PubMed] [Google Scholar]
- Wilson M. W. B.; Rao A.; Ehrler B.; Friend R. H. Singlet Exciton Fission in Polycrystalline Pentacene: From Photophysics toward Devices. Acc. Chem. Res. 2013, 46, 1330–1338. 10.1021/ar300345h. [DOI] [PubMed] [Google Scholar]
- Rao A.; Wilson M. W. B.; Albert-Seifried S.; Di Pietro R.; Friend R. H. Photophysics of Pentacene Thin Films: The Role of Exciton Fission and Heating Effects. Phys. Rev. B: Condens. Matter Mater. Phys. 2011, 84, 195411. 10.1103/PhysRevB.84.195411. [DOI] [Google Scholar]
- Zhu T.; Snaider J. M.; Yuan L.; Huang L. Ultrafast Dynamic Microscopy of Carrier and Exciton Transport. Annu. Rev. Phys. Chem. 2019, 70, 219–244. 10.1146/annurev-physchem-042018-052605. [DOI] [PubMed] [Google Scholar]
- Akselrod G. M.; Deotare P. B.; Thompson N. J.; Lee J.; Tisdale W. A.; Baldo M. A.; Menon V. M.; Bulović V. Visualization of Exciton Transport in Ordered and Disordered Molecular Solids. Nat. Commun. 2014, 5, 3646. 10.1038/ncomms4646. [DOI] [PubMed] [Google Scholar]
- Wan Y.; Guo Z.; Zhu T.; Yan S.; Johnson J.; Huang L. Cooperative Singlet and Triplet Exciton Transport in Tetracene Crystals Visualized by Ultrafast Microscopy. Nat. Chem. 2015, 7, 785–792. 10.1038/nchem.2348. [DOI] [PubMed] [Google Scholar]
- Fischer M. C.; Wilson J. W.; Robles F. E.; Warren W. S. Invited Review Article: Pump-Probe Microscopy. Rev. Sci. Instrum. 2016, 87, 031101 10.1063/1.4943211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydova D.; de la Cadena A.; Akimov D.; Dietzek B. Transient Absorption Microscopy: Advances in Chemical Imaging of Photoinduced Dynamics. Laser Photon. Rev. 2016, 10, 62–81. 10.1002/lpor.201500181. [DOI] [Google Scholar]
- Delor M.; Weaver H. L.; Yu Q.; Ginsberg N. S. Imaging Material Functionality through 3D Nanoscale Tracking of Energy Flow. Nat. Mater. 2019, 10.1038/s41563-019-0498-x. [DOI] [PubMed] [Google Scholar]
- Folie B. D.; Haber J. B.; Refaely-Abramson S.; Neaton J. B.; Ginsberg N. S. Long-Lived Correlated Triplet Pairs in a π-Stacked Crystalline Pentacene Derivative. J. Am. Chem. Soc. 2018, 140, 2326–2335. 10.1021/jacs.7b12662. [DOI] [PubMed] [Google Scholar]
- Refaely-Abramson S.; da Jornada F. H.; Louie S. G.; Neaton J. B. Origins of Singlet Fission in Solid Pentacene from an Ab Initio Green’s Function Approach. Phys. Rev. Lett. 2017, 119, 267401. 10.1103/PhysRevLett.119.267401. [DOI] [PubMed] [Google Scholar]
- Scholes G. D. Correlated Pair States Formed by Singlet Fission and Exciton–Exciton Annihilation. J. Phys. Chem. A 2015, 119, 12699–12705. 10.1021/acs.jpca.5b09725. [DOI] [PubMed] [Google Scholar]
- Ruhman S.; Joly A. G.; Nelson K. A. Coherent Molecular Vibrational Motion Observed in the Time Domain through Impulsive Stimulated Raman Scattering. IEEE J. Quantum Electron. 1988, 24, 460–469. 10.1109/3.146. [DOI] [Google Scholar]
- Liebel M.; Schnedermann C.; Wende T.; Kukura P. Principles and Applications of Broadband Impulsive Vibrational Spectroscopy. J. Phys. Chem. A 2015, 119, 9506–9517. 10.1021/acs.jpca.5b05948. [DOI] [PubMed] [Google Scholar]
- Kuramochi H.; Takeuchi S.; Tahara T. Femtosecond Time-Resolved Impulsive Stimulated Raman Spectroscopy Using Sub-7-Fs Pulses: Apparatus and Applications. Rev. Sci. Instrum. 2016, 87, 043107 10.1063/1.4945259. [DOI] [PubMed] [Google Scholar]
- Hart S. M.; Silva W. R.; Frontiera R. R. Femtosecond Stimulated Raman Evidence for Charge-Transfer Character in Pentacene Singlet Fission. Chem. Sci. 2018, 9, 1242–1250. 10.1039/C7SC03496B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handbook of Biological Confocal Microscopy; Pawley J. B., Ed.; Springer US: Boston, MA, 2006. [Google Scholar]
- Taylor R. W.; Mahmoodabadi R. G.; Rauschenberger V.; Giessl A.; Schambony A.; Sandoghdar V. Interferometric Scattering Microscopy Reveals Microsecond Nanoscopic Protein Motion on a Live Cell Membrane. Nat. Photonics 2019, 13, 480–487. 10.1038/s41566-019-0414-6. [DOI] [Google Scholar]
- Sung J.; Schnedermann C.; Ni L.; Sadhanala A.; Chen R. Y. S.; Cho C.; Priest L.; Lim J. M.; Kim H.-K.; Monserrat B.;. et al. Long-Range Wavelike Propagation of Carriers in Methylammonium Lead Iodide Perovskites Thin Films. Nat. Phys., 2019, accepted. [Google Scholar]
- Ortega Arroyo J.; Cole D.; Kukura P. Interferometric Scattering Microscopy and Its Combination with Single-Molecule Fluorescence Imaging. Nat. Protoc. 2016, 11, 617–633. 10.1038/nprot.2016.022. [DOI] [PubMed] [Google Scholar]
- Nishikawa Y.; Nakata Y.; Ikehara S.; Fujii A.; Ozaki M. Evaluation of Ambipolar Carrier Mobility in Alkyl-Substituted Phthalocyanine Thin Film. J. Photonics Energy 2018, 8, 032214 10.1117/1.JPE.8.032214. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.