

Abstract
Secondary Hyperparathyroidism (SHP) seen as a frequent complication in Chronic Kidney Disease (CKD) has many pathogenetic peculiarities that are still incompletely defined and understood. During the long course of chronic renal failure, SHP can also transform sometimes into the hypercalcemic state characterized by quasi-autonomous production of Parathyroid Hormone from the parathyroid glands: a disorder that is termed Tertiary Hyperparathyroidism. The clinical consequences of SHP in CKD are protean, encompassing bone and mineral abnormalities but as recently identified, also several metabolic and cardiovascular problems, the most important of which is vascular calcification. There have been several advances in the therapeutic armamentarium available for the treatment of SHP, though clear demonstration of a benefit regarding major clinical outcomes with any of the new agents is still lacking. This narrative review summarizes the current understanding about this disorder and highlights some of the recent research on the subject.
Keywords: CKD, FGF-23, parathyroid hyperplasia, renal failure, secondary hyperparathyroidism, tertiary hyperparathyroidism
INTRODUCTION
The parathyroid gland(s) play a pivotal role in bone mineral homeostasis through its secretion of parathyroid hormone (PTH). PTH increases calcium efflux from the bone, increases tubular reabsorption of calcium and phosphate excretion in the kidneys, and by stimulating the renal production of 1,25 dihydroxy vitamin D [(1,25 (OH)2D], increases gastrointestinal absorption of calcium.
It is important to distinguish between a primary disorder of the parathyroid glands in which there is dysregulated and excessive production of PTH (as in the case of primary hyperparathyroidism, PHPT), and situations in which the parathyroid glands respond secondarily to a stimulus such as malabsorption or renal failure and reacts by increasing PTH secretion. These latter forms of hyperparathyroidism are collectively known as secondary hyperparathyroidism (SHP) [Table 1].
Table 1.
Causes of secondary hyperparathyroidism
| Chronic Kidney Disease |
| Decreased Calcium Intake |
| Decreased Absorption of Calcium Vitamin D deficiency Bariatric Surgery Celiac disease Pancreatic diseases with fat malabsorption |
| Renal Calcium losses -Idiopathic hypercalciuria -Loop Diuretics |
| Secondary to Phosphate Replacement therapy in conditions such as X-linked Hypophosphatemia, Autosomal Dominant Hypophosphatemia, Tumour Induced Osteomalacia etc. |
Tertiary hyperparathyroidism refers to the hypercalcemic state in which, after longstanding SHP, the stimulated parathyroid glands assume a quasiautonomous role akin to that seen in PHPT. The differentiation of tertiary from PHPT is usually made possible since in the former; a clearly identifiable longstanding disorder such as malabsorption or renal failure is present predating the onset of hypercalcemia [Table 2].
Table 2.
Biochemical differentiation between primary, secondary and tertiary hyperparathyroidism
| Biochemical Parameter | Primary hyperparathyroidism | Secondary hyperparathyroidism | Tertiary hyperparathyroidism |
|---|---|---|---|
| Calcium | ↑ | ↓ | ↑ |
| phosphate | ↓ | ↑* | ↑ |
| iPTH | ↑ | ↑ | ↑ |
*N.B. SHP in patients with normal renal function (unlike as in those with CKD) is usually associated with low levels of phosphate given the inhibitory effect of PTH on Sodium-Phosphate co-transporters in the renal tubules
Though SHP and its eventual progression to tertiary hyperparathyroidism has many causes as outlined in Table 1, this review will focus on the pathogenic, clinical and therapeutic aspects of these conditions in the setting of chronic kidney disease (CKD).
The SHP associated with CKD is characterized by a complicated, multifaceted and as, yet incompletely understood pathophysiology. It is estimated that 30%-50% of stage 5 CKD patients have iPTH levels of >300 pg/ml.[1] As the kidneys fail, gross derangements in fluid and solute clearance occur. An initial adaptive response, it becomes maladaptive over time and leads to the clinical syndrome termed as Chronic Kidney Disease-Metabolic Bone disorder (CKD-MBD),[2] defined as a systemic disorder of mineral and bone metabolism, manifested by either one or a combination of the following:
Abnormalities of calcium, phosphorus, PTH or vitamin D metabolism.
Abnormalities in bone turnover, mineralization, volume, linear growth or strength
Vascular or other soft tissue calcification.
NORMAL PHYSIOLOGY
The secretion of PTH is closely regulated by extracellular ionized calcium through the calcium sensing receptor (CaSR) on parathyroid cells.[3] PTH synthesis and secretion are also influenced by 1,25 (OH)2D produced in renal proximal tubular cells by the conversion of 25(OH)D under the influence of the enzyme 1-alpha hydroxylase. 1,25 (OH)2D binds to the vitamin D receptor (VDR) in parathyroid tissue and inhibits PTH mRNA synthesis.[4] Inorganic Phosphate (Pi) may also act as an important regulator of PTH although the exact sensing mechanism through which it does this still remains to be elucidated. In addition, the relatively recently identified and characterized fibroblast growth factor 23 (FGF-23), an osteocyte and osteoblast derived phosphaturic hormone[5] has been shown to decrease PTH synthesis and secretion[6] by acting on the parathyroid glands through its receptor Klotho-FGF.[7,8]
PATHOPHYSIOLOGY
It was long considered that the failure of the kidney to excrete serum phosphate with resultant hyperphosphatemia is the key driver of SHP in CKD patients.[9] The formation of calciumphosphate salts with a reduction in serumionized calcium,[10] the inhibitory effect of Pi on the enzyme CYP27B1 (25(OH) D1alpha hydroxylase) that is involved in the conversion of 25hydroxy D to 1,25(OH)2 D in the proximal renal tubular cell[11] and the decreased viable renal mass.[12,13] in chronic renal insufficiency with resultant lesser 1alpha hydroxylase activity, all are believed to result in hypocalcemia and initiate the cascade of events that leads to dysregulation of parathyroid hormone in SHP. However, these postulations do not explain the clinical observation that serum 1,25(OH)2D begins to decline even in early kidney disease before overt hyperphosphatemia develops and that hyperparathyroidism develops early in chronic renal failure at a time when plasma calcium and phosphorous are within normal limits.
The identification and characterization of FGF-23 in the last decade has provided important clues towards understanding the early phases in the pathogenesis of SHP.[14,15] This 22.5 kDa protein, encoded by the FGF-23 gene located on chromosome 12 is secreted mainly by osteocytes and osteoblasts. Its synthesis and release is mainly stimulated by 1,25(OH)2D and also by Pi, PTH and calcium by as yet incompletely defined mechanisms though it is thought that PTH induces FGF-23 transcription through activation of the orphan receptor Nurr1 and through activation of PKA and Wnt signaling in bones thereby constituting a bone-parathyroid-endocrine loop.[16,17,18] FGF23 together with its obligate coreceptor, the membrane bound αKlotho function to induce phosphaturia through downregulation of sodiumphosphate cotransporters. The FGF-23 αKlotho complex also inhibits 1,25(OH)2D synthesis in the kidney by inhibiting 1alpha hydroxylase and, by stimulating 24hydroxylase, the catabolism of active vitamin sterols.[19] This leads to hypocalcemia and stimulation of the parathyroid gland [Figure 1]. A soluble and circulating form of αKlotho produced mainly by the kidney may also have additional autonomous (i.e., independent of FGF-23) phosphaturic and anti-calciuric effects.[20] A progressive renal reduction in production of both membrane-bound and circulating αklotho, increasing levels of FGF-23 secondary to its reduced renal clearance and due to Pi retention, and resistance to the phosphaturic effect of FGF-23 due to deficiency of αKlotho characterize CKD progression.
Figure 1.
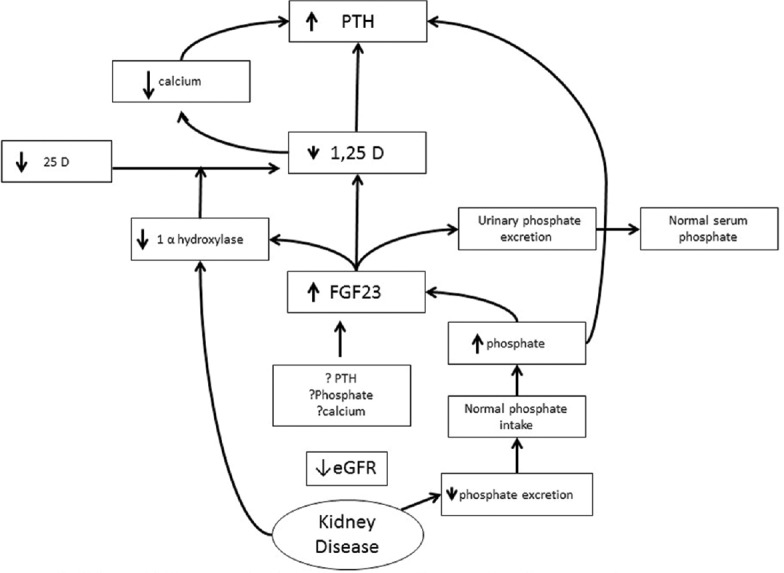
Schematic representation of the current understanding regarding the pathophysiology of SHP
As FGF-23 level increases, a “trade-off” occurs between maintaining normophosphatemia versus 1,25 hydroxy vitamin D deficiency, with the latter progressing relentlessly and causing SHP.[21] Phosphate level will start to rise only when the adaptive compensation by FGF-23 becomes inadequate. At this stage, hyperphosphatemia, continued decreased 1,25(OH)2D and hypocalcemia all contribute to increasing PTH mRNA levels and PTH synthesis. Initial diffuse parathyroid cell hyperplasia and ultimately nodular hyperplasia results. A separate CaSR independent mechanism for hypocalcemia to stimulate PTH secretion may also be through microRNA (miRNA) dysregulation within the parathyroid glands.[22]
Though conflicting data also exists,[23] FGF-23 may also act directly on the parathyroid gland to suppress PTH secretion through the Klotho–FGFR1 complex.[24] In patients with advanced SHP, the parathyroid expression of the Klotho–FGFR1 complex is downregulated.[25] This likely contributes to the resistance to the inhibitory effect of FGF-23 on PTH secretion in progressive and advanced SHP.
As parathyroid hyperplasia progresses, both CaSR and VDR on the parathyroid glands become downregulated and reduced expression of these has been observed in the most severe forms of SHP.[26,27,28,29,30] The size of the parathyroid glands progressively increases as SHP worsens and gland size is positively correlated with serum PTH levels [Figure 2]. The cellular etiology of tertiary hyperparathyroidism is unknown, but it is postulated to be due to a monoclonal expansion of parathyroid cells in which the set point of the CaSRs has been altered such that semi-autonomous secretion of PTH occurs despite high serum calcium levels. Monoclonal chief cell growth results in the formation of nodules. Nodular glands have less VDRs and CaSRs[26,27,28,29,30] compared to diffusely hyperplastic glands and this exacerbates parathyroid gland resistance to calcitriol and calcium.
Figure 2.
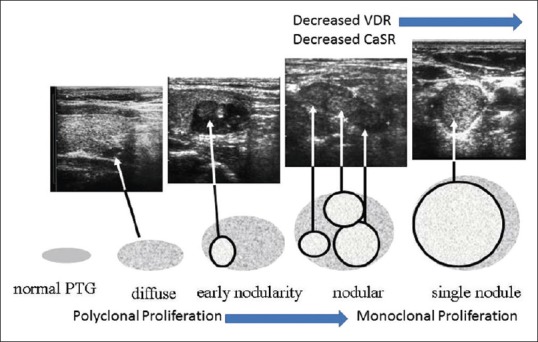
The stages in the evolution of secondary and tertiary hyperparathyroidism
CLINICAL FEATURES
Skeletal manifestations
PTH binds to the PTH/PTHrP receptor on osteoblasts and thus by indirectly stimulating osteoclastic activity leads to a high turnover bone disease. Fragility fractures have been reported to be 2-4 times more frequent in patients with SHP when compared with age- and gender-matched normal populations. This increased risk is associated with an increased risk of mortality[31] and an association between PTH levels and fracture risk has been observed, with intact PTH levels above 900 pg/ml shown to be independently associated with an increased risk of incident fractures in the DOPPS study.[32] It has to be remembered however that the bone fragility in CKD may have several causes other than SHP; such as metabolic acidosis, anemia, hypogonadism, inflammation, beta 2 microglobulin associated amyloidosis, vitamin D deficiency, bone formation inhibition secondary to Wnt inhibition in osteocytes, etc. to name a few.
Extra-skeletal manifestations
Elevated PTH levels may be associated with an increased sympathetic drive, and endothelial stress and SHP may play a causal role in the development of vascular calcifications, ischemic cardiovascular events and cardiac failure.[33] In contrast to the intimal calcification seen in aging individuals with normal renal function, what is seen in patients with CKD is calcification of the medial layer.
Elevated PTH levels have been found to be independently associated with anemia, which is a hallmark of CKD and severe SHP is associated with a resistance to erythropoietin therapy in CKD.[34] It should be borne in mind that, no clear causal relationship between SHP and these extra-skeletal manifestations has been established, neither has it been shown that correction of SHP can result in a complete remission of these clinical conditions.
CLINICAL EVALUATION
The diagnoses of secondary and tertiary hyperparathyroidism are purely biochemical and therefore, accurate measurement of PTH is essential. PTH is a hormone of 84 amino acids. Over the last few decades, three generations of PTH assays have been developed that measure different parts of the molecule [Figure 3]. The first radioimmunoassay for PTH was developed in 1963 by generating a single polyclonal antibody against epitopes in the carboxy terminal (C terminal) end of the PTH molecule.[35] The first-generation assays had poor specificity as the antibodies used mainly targeted the non-bioactive portion of the PTH molecule; the C-terminal which is retained in CKD. The second-generation assays that were developed to overcome this problem use two sets of antibodies: (a) capture antibodies against epitopes located within the C-terminal and (b) detection antibodies directed to amino acid sequences 12-20 within the amino terminal end.[36] This assay is currently the most widely used and is called intact PTH assay (iPTH) as it is assumed that it captures intact PTH 1-84. However, the detection antibodies have been found to cross-react with PTH 7-84 fragments that also tend to accumulate in patients with CKD.[37,38] It has also become apparent that high concentrations of 7-84 PTH and some other C-terminal PTH fragments may oppose the biochemical and bone-metabolic effects of 1-84 PTH; aggravating the potential undesirable clinical consequences of overestimating 1-84 PTH concentrations in renal failure patients,[39,40] that is, the physician might mistakenly assume the erroneously high PTH reading as the correct value and may institute further PTH lowering therapies with disastrous consequences. To overcome these shortcomings, third-generation PTH assays such as the whole PTH assay and the Bio-Intact PTH assay have been developed.[41] Though the capture antibody used in the third-generation assay is the same as that used in the second-generation assay, the detection antibody used is directed towards the first four amino acids. These assays do not thus recognize 7-84 PTH and are therefore considered more specific to 1-84 PTH than second-generation assays. Two automated third-generation PTH assays are now available.[42] However, there is little evidence to show that they provide any better clinical information than the second-generation assays with regard to the diagnosis of CKD-MBD[43] and therefore have not been adopted for use in current guidelines for management of SHP. In general, PTH levels measured with second-generation assays are higher than those obtained with third-generation ones. The ratio of whole (biointact) to intact PTH levels has been noted to be between 0.6-0.7 in dialysis patients,[44] though exceptions to this rule have been reported in patients with severe SHP with a new molecular form of PTH with an intact N-terminus[45,46] that can be detected by third-generation PTH assays but not by second-generation ones identified in these patients. Thus in these patients, PTH levels measured with third-generation assays are paradoxically higher than those with second-generation ones. Existing clinical data suggest that an over-production of N-PTH may be associated with rapid progression of SHP and that N-PTH has significant bio-activity.[47,48]
Figure 3.

Schematic representation of the three generations of PTH assays
KDIGO guidelines earlier recommended target iPTH levels of 2-9 times of the upper limit of normal for the given assay to noninvasively monitor bone status in dialysis patients. iPTH values above this target suggest high bone turnover bone disease with specificity of 86%, and values below the target values suggest low bone turnover with sensitivity of 66%. However, the latest update to the CKD-MBD guideline published by the KDIGO in 2017 suggest that in patients with CKD G3a–G5D, treatment for CKD-MBD should be based on serial assessments of phosphate, calcium, and PTH levels, considered together and not absolute values of any of these parameters.[49]
TREATMENT OPTIONS
A paradigm shift has occurred in the approach to the treatment of secondary and tertiary hyperparathyroidism with the understanding that the alterations in calcium and phosphate metabolism in CKD do not only cause renal osteodystrophy and bone abnormalities but also are linked to increased risk of cardiovascular disease and all-cause mortality potentially mediated through vascular calcification.[50]
The complex pathophysiology of secondary and tertiary hyperparathyroidism makes it necessary that their treatment should be multi-pronged. The three main targets are thus phosphate, 1,25 vitamin D and PTH.
Controlling Phosphate Levels
The management of hyperphosphatemia has formed the cornerstone of therapy for SHP for decades. Dietary phosphate restriction and treatment with oral phosphate binders can decrease PTH levels up to stages 3 and 4 CKD.[51] The reduction of protein intake (particularly protein of animal origin) has been the basis of dietary prescriptions in CKD. However, such a diet is difficult to maintain, and such dietary restrictions should be counter-balanced by the awareness that they may be associated with an increased risk of malnutrition in CKD patients. Nevertheless, all attempts should be made to have a diet that contains more vegetal than animal proteins and to avoid processed foods.
Most often, as CKD progresses and hyperphosphatemia ensues, dietary phosphate restriction alone is not helpful and phosphate binding medications are needed. An ever-increasing number of phosphate binders have been developed over the last few decades. They can be broadly grouped into calcium based and non–calcium-based agents. All these agents are more or less equally effective in the control of phosphate levels and SHP. Calcium-based phosphate binders such as calcium carbonate and calcium acetate are widely prescribed and very effectively lower Pi levels. However, their benefits must be weighed against possible adverse effects of hypercalcemia. KDOQI guidelines recommend that the total dose of elemental calcium provided through calcium-based phosphate binders should not exceed 1500 mg daily.[52]
One of the most commonly used non–calcium-based binders is Sevalamer; a phosphate binding resin. A meta-analysis of randomized controlled trials comparing calcium-based phosphate binders to non–calcium-based ones surmised that the use of calcium-based binders was associated with higher all-cause mortality compared to Sevalamer.[53] However, due to lack of placebo-controlled trials, the debate on whether calcium-based Pi binders are associated with higher risk for vascular calcifications and consequently for cardiovascular mortality continues.
Another non–calcium-based phosphate binder; Lanthanum carbonate has also been shown to control hyperphosphatemia. However, Lanthanum can be systemically absorbed and may accumulate in liver and bone. This limits its use as a first-line phosphate binder.
Enteral phosphate binders may lead to up-regulation of the intestinal sodium-phosphate NPT2b transporter thereby increasing active enteral phosphate absorption, off-setting some of their beneficial effects. Extended release Niacin, a NPT2b inhibitor has shown phosphate and FGF-23 lowering effects in adult CKD patients.[54] However, the recent CKD Optimal Management with Binders and Nicotinamide (COMBINE) study designed to assess whether the addition of NPT2b blockade to Lanthanum improves phosphate and FGF-23 levels showed disappointing results with no significant differences in any of the three active treatment groups (Nicotinamide plus Lanthanum Carbonate, Nicotinamide plus Placebo, or Lanthanum Carbonate plus Placebo) compared with the double placebo group though the findings could have been affected by the high non-compliance rates observed amongst the double-treatment group.[55] Tenapanor is a new agent that inhibits intestinal absorption of phosphate through the inhibition of intestinal sodium/hydrogen exchanger isoform 3 has recently been shown to effectively reduce phosphate in patients who are on maintenance dialysis.[56] It opens up a potential new therapeutic option in controlling of serum phosphate in patients with CKD.
Phosphate is also removed during dialysis. Hence it is vital that dialysis dose is adequate to optimize phosphate control. This can be achieved by adjusting dialysis time as well as blood flow settings during dialysis. It has been shown that patients who are on long/frequent dialysis have much better control of phosphate than their counterparts who are on conventional dialysis.[56]
Vitamin D analogues
Treatment with Vitamin D Receptor Activators (VDRAs) has long been a very important therapeutic strategy in the management of SHP. Calcitriol, the first synthetic VDRA to be developed decreases serum PTH levels[57,58] in CKD and also has been shown to reduce bone turnover and to thus ameliorate osteitis fibrosa in dialysis patients[59] though its effect on risk of fractures in CKD has not been adequately studied. The inhibitory effect on PTH synthesis is mediated through binding of calcitriol to its specific receptor (VDR) and subsequent regulation of gene transcription and inhibition of PTH mRNA synthesis. This is important to know because in advanced SHP, with nodular hyperplasia of the parathyroid glands, there is decreased expression of CaSR and VDRs in the parathyroid gland[29,30], and in such a situation, VDRAs are not as effective in suppressing PTH secretion.[60] The clinical utility of Calcitriol is limited by its potential to cause hypecalcemia. The newer selective VDRAs such as paricalcitol (19-nor-1,25-dihydroxyvitamin D2) and maxacalcitol (22-oxa-1,25-dihydroxyvitamin D3) may be preferable in this regard because they have more modest effects on serum calcium levels though it has to be noted that these agents can also cause hypercalcemia.
Although it has long been known that vitamin D deficiency is common in CKD patients, the common belief has been that the need for its correction is not as stringent in this clinical setting provided that active vitamin D is administered. However, the administration of native vitamin D may have other pleuripotent benefits and it has also been demonstrated that use of native vitamin D esters are effective in lowering PTH levels at least in the early stages of SHP.[61]
Calcimimetics
The introduction of calcimimetics, agents that allosterically activate the calcium sensing receptor, has significantly mitigated the need for high doses of activated vitamin D and the risk of hypercalcemia in SHP. These agents “mimic” calcium and increase the sensitivity of calcium sensing receptors on the parathyroid gland. Currently, the only oral calcimimetic approved by the FDA is Cinacalcet. Cinacalcet effectively reduces PTH levels and serum calcium levels in patients with SHP.[62] Notably, cinacalcet is effective even in patients with marked parathyroid hyperplasia[63] thus making it an acceptable alternative to parathyroidectomy for treatment of severe SHP. Cinacalcet may cause gastrointestinal adverse effects such as nausea and vomiting. The introduction of a new intravenous calcimimetic, Etelcalcetide offers a therapeutic alternative to oral cinacalcet.[64] Etelcalcetide has a longer half-life than cinacalcet and can be administered intravenously every other day at the end of dialysis treatment thus overcoming the problem of compliance with a daily oral regimen. Etelcalcetide has been shown to markedly decrease PTH levels in patients on hemodialysis with moderate to severe SHP and may be superior to Cinacalcet in this regard[65] though further studies are needed to assess clinical outcomes as well as long-term efficacy and safety of this agent.
The effects of Cinacalcet on bone turnover and bone histology in patients with CKD and evidence of high-turnover bone disease have been studied in the Bone Histomorphometry Assessment for dialysis patients with Secondary Hyperparathyroidism of End-Stage Renal Disease (BONAFIDE) study.[66] This study demonstrated that long-term treatment with cinacalcet lowers biochemical markers of high bone-turnover and improves bone histology in this setting. In the Evaluation of Cinacalcet Hydrochloride therapy to Lower Cardiovascular Events (EVOLVE) trial, a randomized controlled trial to assess the effects of cinacalcet on clinical outcomes, though no significant effect of cinacalcet in the primary intention to treat analysis was seen, a significant reduction in the risk of fracture in the cinacalcet group was found when differences in baseline characteristics, multiple fractures and/or events prompting discontinuation of study drug were taken into account.[67] The results of the EVOLVE trial also suggested a beneficial effect of Cinacalcet with regard to reduction in the risk of death or cardiovascular outcomes though it has to be noted that this again was not in the primary unadjusted intention to treat analysis but in the log-censoring analysis.[68]
Parathyroidectomy
Despite the availability of newer vitamin D analogues and calcimimetics, parathyroidectomy continues to be a necessity in certain patient groups. It is estimated that parathyroidectomy is required in about 15% of patients after 10 years and in 38% of patients after 20 years of ongoing dialysis therapy.[69] Successful surgical treatment results in a dramatic reduction in PTH levels and improvement of clinical symptoms such as bone pain and itching. Parathyroidectomy is also associated with better patient survival[70,71,72] and reduced risk of fractures[73] in patients with severe SHP.
A description of the surgical techniques for parathyroidectomy is beyond the scope of this article. The choice of surgical technique viz subtotal parathyroidectomy versus total parathyroidectomy with auto transplantation ultimately depends on operator experience and expertise and must be individualized to the patient. However, parathyroidectomy is not without its risk. The most commonly seen is the phenomenon of Hungry Bone Syndrome[74] characterized by severe hypocalcemia post parathyroidectomy. The abrupt withdrawal of very high and sustained levels of PTH following parathyroidectomy, turns off osteoclast activity and bone resorption in the remodeling space. However, osteoblast activity and new bone formation continues, which leads to the influx of calcium, phosphate and magnesium into bone resulting in their abrupt drops in the serum. This condition remains poorly defined and the prevalence of this condition has been reported to range from 8% to 87% following parathyroidectomy for SHP.[75] The other concern is the occurrence of adynamic bone disease and hypoparathyroidism post parathyroidectomy. Hypoparathyroidism typically is reported following surgery for PHPT and there is no study that reports the incidence or prevalence of this condition in patients who undergo parathyroidectomy for SHP. Low turnover bone disease, however, has been reported to occur post parathyroidectomy and has been associated with worsening of vascular calcification in hemodialysis patients.[76,77,78]
Chemical ablation of parathyroid gland
Percutaneous fine needle ethanol injection of parathyroid gland was first reported in 1985 in 12 patients with SHP.[79] In 2003, the Japanese Society of Parathyroid Intervention published its guideline for selective percutaneous ethanol injection therapy of the parathyroid glands in chronic dialysis patients in which they recommended that enlarged parathyroid glands with nodular hyperplasia could be “selectively” destroyed by ethanol injection, and other glands with diffuse hyperplasia could be then managed by medical therapy.[80]
Percutaneous injection using the Vitamin D analogue-Calcitriol instead of alcohol has also been described.[81] The rationale behind this approach is to introduce high level of vitamin D around the parathyroid gland without the systemic complications that could potentially be caused by its systemic administration.
These local approaches could be considered in patients who refuse or are not candidates for surgery although long-term control of SHP is unlikely to be obtained.
PERSISTENT HYPERPARATHYROIDISM AFTER KIDNEY TRANSPLANTATION
Persistent hyperparathyroidism even after renal transplantation is most likely to be present in patients with advanced SHP with nodular hyperplasia of the parathyroid glands before transplant. It is the most common cause of hypercalcemia[82] in renal transplant patients and may result in poor graft outcomes and progression of vascular calcification.[83] Surgical parathyroidectomy should be considered in kidney transplant patients with persistent hyperparathyroidism especially when it is associated with severe hypercalcemia. Cinacalcet appears to be a promising therapeutic option for patients with persistent hypercalcemia post transplantation at least as a bridging agent before parathyroidectomy.[84,85,86]
CONCLUSION
Our knowledge of the pathophysiology of secondary and tertiary hyperparathyroidism has vastly improved during the past few years. They may be caused by various conditions, however, that associated with CKD has been the one most studied and yet remains incompletely defined. The clinical consequences of these disorders of the parathyroid gland are not limited to the musculo-skeletal system but are multifold and systemic. It is however difficult to define clearly whether there is a causal relationship between the elevated levels of PTH seen in these disorders and the protean clinical manifestations or whether they simply are associations in a complex clinical setting. The number of available therapeutic options for the management of secondary and tertiary hyperparathyroidism has increased significantly and control of PTH, phosphate and calcium levels can be successfully achieved in most cases with these medications. However convincing benefits on major clinical outcomes such as prevention of fractures, cardiovascular events or survival have not been demonstrated so far and a significant percentage of patients still need parathyroidectomy – the approach to which should be undertaken on an individualized basis.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Hedgeman E, Lipworth L, Lowe K, Saran R, Do T, Fryzek J, et al. International burden of chronic kidney disease and secondary hyperparathyroidism: A systematic review of the literature and available data. Int J Nephrol. 2015;2015:184321. doi: 10.1155/2015/184321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Work Group. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD) Kidney Int Suppl. 2009:S1–130. doi: 10.1038/ki.2009.188. doi:10.1038/ki. 2009.188. [DOI] [PubMed] [Google Scholar]
- 3.Brown EM, Pollak M, Seidman CE, Seidman JG, Chou YH, Riccardi D, et al. Calcium-ion-sensing cell-surface receptors. N Engl J Med. 1995;333:234–40. doi: 10.1056/NEJM199507273330407. [DOI] [PubMed] [Google Scholar]
- 4.Dusso AS, Brown AJ, Slatopolsky E. Vitamin D. Am J Physiol Renal Physiol. 2005;289:F8–28. doi: 10.1152/ajprenal.00336.2004. [DOI] [PubMed] [Google Scholar]
- 5.Quarles LD. Endocrine functions of bone in mineral metabolism regulation. J Clin Invest. 2008;118:3820–8. doi: 10.1172/JCI36479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galitzer H, Ben-Dov I, Lavi-Moshayoff V, Naveh-Many T, Silver J. Fibroblast growth factor 23 acts on the parathyroid to decrease parathyroid hormone secretion. Curr Opin Nephrol Hypertens. 2008;17:363–7. doi: 10.1097/MNH.0b013e328303e172. [DOI] [PubMed] [Google Scholar]
- 7.Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–4. doi: 10.1038/nature05315. [DOI] [PubMed] [Google Scholar]
- 8.Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, et al. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem. 2006;281:6120–3. doi: 10.1074/jbc.C500457200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slatopolsky E, Caglar S, Pennell JP, Taggart DD, Canterbury JM, Reiss E, et al. On the pathogenesis of hyperparathyroidism in chronic experimental renal insufficiency in the dog. J Clin Invest. 1971;50:492–9. doi: 10.1172/JCI106517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown EM. Extracellular Ca2+sensing, regulation of parathyroid cell function, and role of Ca2+and other ions as extracellular ( first) messengers. Physiol Rev. 1991;71:371–411. doi: 10.1152/physrev.1991.71.2.371. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka Y, Deluca HF. The control of 25-hydroxyvitamin D metabolism by inorganic phosphorus. Arch Biochem Biophys. 1973;154:566–74. doi: 10.1016/0003-9861(73)90010-6. [DOI] [PubMed] [Google Scholar]
- 12.Llach F. Secondary hyperparathyroidism in renal failure: The trade-off hypothesis revisited. Am J Kidney Dis. 1995;25:663–79. doi: 10.1016/0272-6386(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 13.Slatopolsky E, Delmez JA. Pathogenesis of secondary hyperparathyroidism. Nephrol Dial Transplant. 1996;11(Suppl 3):130–5. doi: 10.1093/ndt/11.supp3.130. [DOI] [PubMed] [Google Scholar]
- 14.Elias RM, Dalboni MA, Coelho ACE, Moysés RMA. CKD-MBD: From the pathogenesis to the identification and development of potential novel therapeutic targets. Curr Osteoporos Rep. 2018;16:693–702. doi: 10.1007/s11914-018-0486-0. [DOI] [PubMed] [Google Scholar]
- 15.Gutiérrez OM. Fibroblast growth factor 23 and disordered vitamin D metabolism in chronic kidney disease: Updating the “trade-off” hypothesis. Clin J Am Soc Nephrol. 2010;5:1710–6. doi: 10.2215/CJN.02640310. [DOI] [PubMed] [Google Scholar]
- 16.Fukagawa M, Nii-Kono T, Kazama JJ. Role of fibroblast growth factor 23 in health and in chronic kidney disease. Curr Opin Nephrol Hypertens. 2005;14:325–9. doi: 10.1097/01.mnh.0000172717.49476.80. [DOI] [PubMed] [Google Scholar]
- 17.Lavi-Moshayoff V, Wasserman G, Meir T, Silver J, Naveh-Many T. PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: A bone parathyroid feedback loop. Am J Physiol Renal Physiol. 2010;299:F882–9. doi: 10.1152/ajprenal.00360.2010. [DOI] [PubMed] [Google Scholar]
- 18.Meir T, Durlacher K, Pan Z, Amir G, Richards WG, Silver J, et al. Parathyroid hormone activates the orphan nuclear receptor Nurr1 to induce FGF23 transcription. Kidney Int. 2014;86:1106–15. doi: 10.1038/ki.2014.215. [DOI] [PubMed] [Google Scholar]
- 19.Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19:429–35. doi: 10.1359/JBMR.0301264. [DOI] [PubMed] [Google Scholar]
- 20.Kuro-O M. The Klotho proteins in health and disease. Nature reviews. Nephrology. 2019;15:27–44. doi: 10.1038/s41581-018-0078-3. [DOI] [PubMed] [Google Scholar]
- 21.Gutierrez O, Isakova T, Rhee E, Shah A, Holmes J, Collerone G, et al. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J Am Soc Nephrol. 2005;16:2205–15. doi: 10.1681/ASN.2005010052. [DOI] [PubMed] [Google Scholar]
- 22.Shilo V, Mor-Yosef Levi I, Abel R, Mihailović A, Wasserman G, Naveh-Many T, et al. Let-7 and MicroRNA-148 regulate parathyroid hormone levels in secondary hyperparathyroidism. J Am Soc Nephrol. 2017;28:2353–63. doi: 10.1681/ASN.2016050585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawakami K, Takeshita A, Furushima K, Miyajima M, Hatamura I, Kuro-O M, et al. Persistent fibroblast growth factor 23 signalling in the parathyroid glands for secondary hyperparathyroidism in mice with chronic kidney disease. Sci Rep. 2017;7:40534. doi: 10.1038/srep40534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krajisnik T, Björklund P, Marsell R, Ljunggren O, Akerström G, Jonsson KB, et al. Fibroblast growth factor-23 regulates parathyroid hormone and 1alpha-hydroxylase expression in cultured bovine parathyroid cells. J Endocrinol. 2007;195:125–31. doi: 10.1677/JOE-07-0267. [DOI] [PubMed] [Google Scholar]
- 25.Krajisnik T, Olauson H, Mirza MAI, Hellman P, Akerström G, Westin G, et al. Parathyroid Klotho and FGF-receptor 1 expression decline with renal function in hyperparathyroid patients with chronic kidney disease and kidney transplant recipients. Kidney Int. 2010;78:1024–32. doi: 10.1038/ki.2010.260. [DOI] [PubMed] [Google Scholar]
- 26.Tokumoto M, Tsuruya K, Fukuda K, Kanai H, Kuroki S, Hirakata H, et al. Reduced p21, p27 and vitamin D receptor in the nodular hyperplasia in patients with advanced secondary hyperparathyroidism. Kidney Int. 2002;62:1196–207. doi: 10.1111/j.1523-1755.2002.kid585.x. [DOI] [PubMed] [Google Scholar]
- 27.Yano S, Sugimoto T, Tsukamoto T, Chihara K, Kobayashi A, Kitazawa S, et al. Association of decreased calcium-sensing receptor expression with proliferation of parathyroid cells in secondary hyperparathyroidism. Kidney Int. 2000;58:1980–6. doi: 10.1111/j.1523-1755.2000.00370.x. [DOI] [PubMed] [Google Scholar]
- 28.Gogusev J, Duchambon P, Hory B, Giovannini M, Goureau Y, Sarfati E, et al. Depressed expression of calcium receptor in parathyroid gland tissue of patients with hyperparathyroidism. Kidney Int. 1997;51:328–36. doi: 10.1038/ki.1997.41. [DOI] [PubMed] [Google Scholar]
- 29.Kifor O, Moore FD, Wang P, Goldstein M, Vassilev P, Kifor I, et al. Reduced immunostaining for the extracellular Ca2+-sensing receptor in primary and uremic secondary hyperparathyroidism. J Clin Endocrinol Metab. 1996;81:1598–606. doi: 10.1210/jcem.81.4.8636374. [DOI] [PubMed] [Google Scholar]
- 30.Fukuda N, Tanaka H, Tominaga Y, Fukagawa M, Kurokawa K, Seino Y, et al. Decreased 1,25-dihydroxyvitamin D3 receptor density is associated with a more severe form of parathyroid hyperplasia in chronic uremic patients. J Clin Invest. 1993;92:1436–43. doi: 10.1172/JCI116720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tentori F, McCullough K, Kilpatrick RD, Bradbury BD, Robinson BM, Kerr PG, et al. High rates of death and hospitalization follow bone fracture among hemodialysis patients. Kidney Int. 2014;85:166–73. doi: 10.1038/ki.2013.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jadoul M, Albert JM, Akiba T, Akizawa T, Arab L, Bragg-Gresham JL, et al. Incidence and risk factors for hip or other bone fractures among hemodialysis patients in the Dialysis Outcomes and Practice Patterns Study. Kidney Int. 2006;70:1358–66. doi: 10.1038/sj.ki.5001754. [DOI] [PubMed] [Google Scholar]
- 33.Kestenbaum B, Katz R, de Boer I, Hoofnagle A, Sarnak MJ, Shlipak MG, et al. Vitamin D, parathyroid hormone, and cardiovascular events among older adults. J Am Coll Cardiol. 2011;58:1433–41. doi: 10.1016/j.jacc.2011.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanaka M, Komaba H, Fukagawa M. Emerging Association Between Parathyroid Hormone and Anemia in Hemodialysis Patients. Ther Apher Dial. 2018;22:242–5. doi: 10.1111/1744-9987.12685. [DOI] [PubMed] [Google Scholar]
- 35.Berson SA, Yalow RS, Aurbach GD, Potts JT. Immunoassay of bovine and human parathyroid hormone. Proc Natl Acad Sci U S A. 1963;49:613–7. doi: 10.1073/pnas.49.5.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nussbaum SR, Zahradnik RJ, Lavigne JR, Brennan GL, Nozawa-Ung K, Kim LY, et al. Highly sensitive two-site immunoradiometric assay of parathyrin, and its clinical utility in evaluating patients with hypercalcemia. Clin Chem. 1987;33:1364–7. [PubMed] [Google Scholar]
- 37.Brossard JH, Cloutier M, Roy L, Lepage R, Gascon-Barré M, D'Amour P, et al. Accumulation of a non-(1-84) molecular form of parathyroid hormone (PTH) detected by intact PTH assay in renal failure: Importance in the interpretation of PTH values. J Clin Endocrinol Metab. 1996;81:3923–9. doi: 10.1210/jcem.81.11.8923839. [DOI] [PubMed] [Google Scholar]
- 38.D'Amour P. Circulating PTH molecular forms: What we know and what we don't. Kidney Int Suppl. 2006;102:S29–33. doi: 10.1038/sj.ki.5001599. [DOI] [PubMed] [Google Scholar]
- 39.Nguyen-Yamamoto L, Rousseau L, Brossard JH, Lepage R, D'Amour P. Synthetic carboxyl-terminal fragments of parathyroid hormone (PTH) decrease ionized calcium concentration in rats by acting on a receptor different from the PTH/PTH-related peptide receptor. Endocrinology. 2001;142:1386–92. doi: 10.1210/endo.142.4.8093. [DOI] [PubMed] [Google Scholar]
- 40.Divieti P, John MR, Jüppner H, Bringhurst FR. Human PTH-(7-84) inhibits bone resorption in vitro via actions independent of the type 1 PTH/PTHrP receptor. Endocrinology. 2002;143:171–6. doi: 10.1210/endo.143.1.8575. [DOI] [PubMed] [Google Scholar]
- 41.Joly D, Drueke TB, Alberti C, Houillier P, Lawson-Body E, Martin KJ, et al. Variation in serum and plasma PTH levels in second-generation assays in hemodialysis patients: A cross-sectional study. Am J Kidney Dis. 2008;51:987–95. doi: 10.1053/j.ajkd.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 42.Cavalier E, Delanaye P, Lukas P, Carlisi A, Gadisseur R, Souberbielle JC, et al. Standardization of DiaSorin and Roche automated third generation PTH assays with an International Standard: Impact on clinical populations. Clin Chem Lab Med. 2014;52:1137–41. doi: 10.1515/cclm-2013-1027. [DOI] [PubMed] [Google Scholar]
- 43.Lehmann G, Stein G, Hüller M, Schemer R, Ramakrishnan K, Goodman WG, et al. Specific measurement of PTH (1-84) in various forms of renal osteodystrophy (ROD) as assessed by bone histomorphometry. Kidney Int. 2005;68:1206–14. doi: 10.1111/j.1523-1755.2005.00513.x. [DOI] [PubMed] [Google Scholar]
- 44.Nakanishi S, Kazama JJ, Shigematsu T, Iwasaki Y, Cantor TL, Kurosawa T, et al. Comparison of intact PTH assay and whole PTH assay in long-term dialysis patients. Am J Kidney Dis. 2001;38(4 Suppl 1):S172–4. doi: 10.1053/ajkd.2001.27436. [DOI] [PubMed] [Google Scholar]
- 45.Arakawa T, D'Amour P, Rousseau L, Brossard JH, Sakai M, Kasumoto H, et al. Overproduction and secretion of a novel amino-terminal form of parathyroid hormone from a severe type of parathyroid hyperplasia in uremia. Clin J Am Soc Nephrol. 2006;1:525–31. doi: 10.2215/CJN.01391005. [DOI] [PubMed] [Google Scholar]
- 46.Tanaka M, Itoh K, Matsushita K, Fujii H, Fukagawa M. Normalization of reversed bio-intact-PTH (1-84)/intact-PTH ratio after parathyroidectomy in a patient with severe secondary hyperparathyroidism. Clin Nephrol. 2005;64:69–72. doi: 10.5414/cnp64069. [DOI] [PubMed] [Google Scholar]
- 47.Tanaka M, Komaba H, Itoh K, Matsushita K, Matshushita K, Hamada Y, et al. The whole-PTH/intact-PTH ratio is a useful predictor of severity of secondary hyperparathyroidism. NDT Plus. 2008;1(Suppl 3):iii59–62. doi: 10.1093/ndtplus/sfn089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Komaba H, Takeda Y, Shin J, Tanaka R, Kakuta T, Tominaga Y, et al. Reversed whole PTH/intact PTH ratio as an indicator of marked parathyroid enlargement: Five case studies and a literature review. NDT Plus. 2008;1(Suppl 3):iii54–58. doi: 10.1093/ndtplus/sfn088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Update Work Group. KDIGO 2017 clinical practice guideline update for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD) Kidney int Suppl. 2017;7:1–59. doi: 10.1016/j.kisu.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Palmer SC, Hayen A, Macaskill P, Pellegrini F, Craig JC, Elder GJ, et al. Serum levels of phosphorus, parathyroid hormone, and calcium and risks of death and cardiovascular disease in individuals with chronic kidney disease: A systematic review and meta-analysis. JAMA. 2011;305:1119–27. doi: 10.1001/jama.2011.308. [DOI] [PubMed] [Google Scholar]
- 51.Sprague SM, Abboud H, Qiu P, Dauphin M, Zhang P, Finn W, et al. Lanthanum carbonate reduces phosphorus burden in patients with CKD stages 3 and 4: A randomized trial. Clin J Am Soc Nephrol. 2009;4:178–85. doi: 10.2215/CJN.02830608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.National Kidney Foundation. K/DOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis. 2003;42(4 Suppl 3):S1–201. [PubMed] [Google Scholar]
- 53.Patel L, Bernard LM, Elder GJ. Sevelamer versus calcium-based binders for treatment of hyperphosphatemia in CKD: A meta-analysis of randomized controlled trials. Clin J Am Soc Nephrol. 2016;11:232–44. doi: 10.2215/CJN.06800615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rao M, Steffes M, Bostom A, Ix JH. Effect of niacin on FGF23 concentration in chronic kidney disease. Am J Nephrol. 2014;39:484–90. doi: 10.1159/000362424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ix JH, Isakova T, Larive B, Raphael KL, Raj DS, Cheung AK, et al. Effects of nicotinamide and lanthanum carbonate on serum phosphate and fibroblast growth factor-23 in CKD: The combine trial. J Am Soc Nephrol. 2019;30:1096–108. doi: 10.1681/ASN.2018101058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Palmer SC, Gardner S, Tonelli M, Mavridis D, Johnson DW, Craig JC, et al. Phosphate-binding agents in adults with CKD: A network meta-analysis of randomized trials. Am J Kidney Dis. 2016;68:691–702. doi: 10.1053/j.ajkd.2016.05.015. [DOI] [PubMed] [Google Scholar]
- 57.Palmer SC, McGregor DO, Craig JC, Elder G, Macaskill P, Strippoli GF, et al. Vitamin D compounds for people with chronic kidney disease requiring dialysis. Cochrane Database Syst Rev. 2009:CD005633. doi: 10.1002/14651858.CD005633.pub2. doi:10.1002/14651858.CD005633.pub2. [DOI] [PubMed] [Google Scholar]
- 58.Palmer SC, McGregor DO, Craig JC, Elder G, Macaskill P, Strippoli GF, et al. Vitamin D compounds for people with chronic kidney disease not requiring dialysis. Cochrane Database Syst Rev. 2009:CD008175. doi: 10.1002/14651858.CD008175. doi:10.1002/14651858.CD005633.pub2. [DOI] [PubMed] [Google Scholar]
- 59.Andress DL, Norris KC, Coburn JW, Slatopolsky EA, Sherrard DJ. Intravenous calcitriol in the treatment of refractory osteitis fibrosa of chronic renal failure. N Engl J Med. 1989;321:274–9. doi: 10.1056/NEJM198908033210502. [DOI] [PubMed] [Google Scholar]
- 60.Okuno S, Ishimura E, Kitatani K, Chou H, Nagasue K, Maekawa K, et al. Relationship between parathyroid gland size and responsiveness to maxacalcitol therapy in patients with secondary hyperparathyroidism. Nephrol Dial Transplant. 2003;18:2613–21. doi: 10.1093/ndt/gfg451. [DOI] [PubMed] [Google Scholar]
- 61.Miskulin DC, Majchrzak K, Tighiouart H, Muther RS, Kapoian T, Johnson DS, et al. Ergocalciferol supplementation in hemodialysis patients with vitamin D deficiency: A randomized clinical trial. J Am Soc Nephrol. 2016;27:1801–10. doi: 10.1681/ASN.2015040468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Block GA, Martin KJ, de Francisco ALM, Turner SA, Avram MM, Suranyi MG, et al. Cinacalcet for secondary hyperparathyroidism in patients receiving hemodialysis. N Engl J Med. 2004;350:1516–25. doi: 10.1056/NEJMoa031633. [DOI] [PubMed] [Google Scholar]
- 63.Komaba H, Nakanishi S, Fujimori A, Tanaka M, Shin J, Shibuya K, et al. Cinacalcet effectively reduces parathyroid hormone secretion and gland volume regardless of pretreatment gland size in patients with secondary hyperparathyroidism. Clin J Am Soc Nephrol. 2010;5:2305–14. doi: 10.2215/CJN.02110310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martin KJ, Bell G, Pickthorn K, Huang S, Vick A, Hodsman P, et al. Velcalcetide (AMG 416), a novel peptide agonist of the calcium-sensing receptor, reduces serum parathyroid hormone and FGF23 levels in healthy male subjects. Nephrol Dial Transplant. 2014;29:385–92. doi: 10.1093/ndt/gft417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Block GA, Bushinsky DA, Cheng S, Cunningham J, Dehmel B, Drueke TB, et al. Effect of etelcalcetide vs cinacalcet on serum parathyroid hormone in patients receiving hemodialysis with secondary hyperparathyroidism: A randomized clinical trial. JAMA. 2017;317:156–64. doi: 10.1001/jama.2016.19468. [DOI] [PubMed] [Google Scholar]
- 66.Behets GJ, Spasovski G, Sterling LR, Goodman WG, Spiegel DM, De Broe ME, et al. Bone histomorphometry before and after long-term treatment with cinacalcet in dialysis patients with secondary hyperparathyroidism. Kidney Int. 2015;87:846–56. doi: 10.1038/ki.2014.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moe SM, Abdalla S, Chertow GM, Parfrey PS, Block GA, Correa-Rotter R, et al. Effects of cinacalcet on fracture events in patients receiving hemodialysis: The evolve trial. J Am Soc Nephrol. 2015;26:1466–75. doi: 10.1681/ASN.2014040414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.EVOLVE Trial Investigators. Chertow GM, Block GA, Correa-Rotter R, Drüeke TB, Floege J, et al. Effect of cinacalcet on cardiovascular disease in patients undergoing dialysis. N Engl J Med. 2012;367:2482–94. doi: 10.1056/NEJMoa1205624. [DOI] [PubMed] [Google Scholar]
- 69.Lau WL, Obi Y, Kalantar-Zadeh K. Parathyroidectomy in the Management of Secondary Hyperparathyroidism. Clin J Am Soc Nephrol. 2018;13:952–61. doi: 10.2215/CJN.10390917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kestenbaum B, Andress DL, Schwartz SM, Gillen DL, Seliger SL, Jadav PR, et al. Survival following parathyroidectomy among United States dialysis patients. Kidney Int. 2004;66:2010–6. doi: 10.1111/j.1523-1755.2004.00972.x. [DOI] [PubMed] [Google Scholar]
- 71.Costa-Hong V, Jorgetti V, Gowdak LHW, Moyses RMA, Krieger EM, De Lima JJG, et al. Parathyroidectomy reduces cardiovascular events and mortality in renal hyperparathyroidism. Surgery. 2007;142:699–703. doi: 10.1016/j.surg.2007.06.015. [DOI] [PubMed] [Google Scholar]
- 72.Trombetti A, Stoermann C, Robert JH, Herrmann FR, Pennisi P, Martin PY, et al. Survival after parathyroidectomy in patients with end-stage renal disease and severe hyperparathyroidism. World J Surg. 2007;31:1014–21. doi: 10.1007/s00268-006-0693-1. [DOI] [PubMed] [Google Scholar]
- 73.Rudser KD, de Boer IH, Dooley A, Young B, Kestenbaum B. Fracture risk after parathyroidectomy among chronic hemodialysis patients. J Am Soc Nephrol. 2007;18:2401–7. doi: 10.1681/ASN.2007010022. [DOI] [PubMed] [Google Scholar]
- 74.Jain N, Reilly RF. Hungry bone syndrome. Curr Opin Nephrol Hypertens. 2017;26:250–5. doi: 10.1097/MNH.0000000000000327. [DOI] [PubMed] [Google Scholar]
- 75.Witteveen JE, van Thiel S, Romijn JA, Hamdy NAT. Hungry bone syndrome: Still a challenge in the post-operative management of primary hyperparathyroidism: A systematic review of the literature. Eur J Endocrinol. 2013;168:R45–53. doi: 10.1530/EJE-12-0528. [DOI] [PubMed] [Google Scholar]
- 76.Yajima A, Ogawa Y, Ikehara A, Tominaga T, Inou T, Otsubo O, et al. Development of low-turnover bone diseases after parathyroidectomy and autotransplantation. Int J Urol. 2001;8:S76–9. doi: 10.1046/j.1442-2042.2001.00340.x. [DOI] [PubMed] [Google Scholar]
- 77.Chan HWH, Chu KH, Fung SKS, Tang HL, Lee W, Cheuk A, et al. Prospective study on dialysis patients after total parathyroidectomy without autoimplant. Nephrology (Carlton) 2010;15:441–7. doi: 10.1111/j.1440-1797.2009.01257.x. [DOI] [PubMed] [Google Scholar]
- 78.Hernandes FR, Canziani MEF, Barreto FC, Santos RO, Moreira VDM, Rochitte CE, et al. The shift from high to low turnover bone disease after parathyroidectomy is associated with the progression of vascular calcification in hemodialysis patients: A 12-month follow-up study. PloS One. 2017;12:e0174811. doi: 10.1371/journal.pone.0174811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Giangrande A, Castiglioni A, Solbiati L, Allaria P. Ultrasound-guided percutaneous fine-needle ethanol injection into parathyroid glands in secondary hyperparathyroidism. Nephrol Dial Transplant. 1992;7:412–21. [PubMed] [Google Scholar]
- 80.Fukagawa M, Kitaoka M, Tominaga Y, Akizawa T, Kakuta T, Onoda N, et al. Guidelines for percutaneous ethanol injection therapy of the parathyroid glands in chronic dialysis patients. Nephrol Dial Transplant. 2003;18(Suppl 3):iii31–3. doi: 10.1093/ndt/gfg1008. [DOI] [PubMed] [Google Scholar]
- 81.Nakanishi S, Yano S, Nomura R, Tsukamoto T, Shimizu Y, Shin J, et al. Efficacy of direct injection of calcitriol into the parathyroid glands in uraemic patients with moderate to severe secondary hyperparathyroidism. Nephrol Dial Transplant. 2003;18(Suppl 3):iii47–9. doi: 10.1093/ndt/gfg1012. [DOI] [PubMed] [Google Scholar]
- 82.Gwinner W, Suppa S, Mengel M, Hoy L, Kreipe HH, Haller H, et al. Early calcification of renal allografts detected by protocol biopsies: Causes and clinical implications. Am J Transplant. 2005;5:1934–41. doi: 10.1111/j.1600-6143.2005.00938.x. [DOI] [PubMed] [Google Scholar]
- 83.Hernández D, Rufino M, Bartolomei S, González-Rinne A, Lorenzo V, Cobo M, et al. Clinical impact of preexisting vascular calcifications on mortality after renal transplantation. Kidney Int. 2005;67:2015–20. doi: 10.1111/j.1523-1755.2005.00303.x. [DOI] [PubMed] [Google Scholar]
- 84.Zavvos V, Fyssa L, Papasotiriou M, Papachristou E, Ntrinias T, Savvidaki E, et al. Long-term use of cinacalcet in kidney transplant recipients with hypercalcemic secondary hyperparathyroidism: A single-center prospective study. Exp Clin Transplant. 2018;16:287–93. doi: 10.6002/ect.2016.0342. [DOI] [PubMed] [Google Scholar]
- 85.Ważna-Jabłońska E, Gałązka Z, Durlik M. Treatment of persistent hypercalcemia and hyperparathyroidism with cinacalcet after successful kidney transplantation. Transplant Proc. 2016;48:1623–5. doi: 10.1016/j.transproceed.2016.01.044. [DOI] [PubMed] [Google Scholar]
- 86.Dulfer RR, Koh EY, van der Plas WY, Engelsman AF, van Dijkum EJMN, Pol RA, et al. Parathyroidectomy versus cinacalcet for tertiary hyperparathyroidism; a retrospective analysis. Langenbeck's Arch Surg. 2019;404:71–9. doi: 10.1007/s00423-019-01755-4. [DOI] [PMC free article] [PubMed] [Google Scholar]