

Abstract
As the first biologics produced by recombinant deoxyribonucleic acid (DNA) technology were approved in the late 1980s and consequently the exclusive marketing rights of most of these biological medicinal products have expired or will expire very shortly, it is quite evident that biosimilars are being developed and marketed in developed as well as developing countries in line with these expiries. Hence, there is an explosion of published papers and scientific programs on biological medicinal products and biosimilar insulins in the last decade or so. Each of these papers or scientific programs generated more questions than providing clinically useful answers. The specific aim of the medical literature or scientific programs were blurred due to lot of attention (created by the innovators) directed towards confusing terminologies, past mishaps with biosimilars (in the era with the absence of regulatory guidelines for biosimilars) diverting our attention from the matters relevant to clinicians and patients. One of the principle reason behind this phenomenon has been our poor understanding of the manufacturing process, regulatory pathways, and study endpoints involved in developing a biosimilar in the present era. This drawback resulted in a nonsystematic approach in analyzing the biosimilars and apparently resulting in confusion. This review attempts at demystifying certain facets of frequently encountered information on biosimilars and acquire a personal understanding on the same, rather than depending on conflicting versions floated at different continuing medical educations (CMEs) and Diabetes Congresses.
Keywords: Biologics, biosimilar, european medicines agency, food and drug administration, insulin
INTRODUCTION
Due to recent advancement in the field of biotechnology and research, many serious disorders viz. diabetes, cancer, rheumatoid arthritis and so on are now treatable with a novel category of drugs called biologicals. However, there is still confusion in the mind of many clinicians with regards to biologicals. Biologicals are often referred to as biopharmaceuticals or therapeutic biologicals or biologics. These are produced by living entities, such as organisms, cells, or tissues through sophisticated techniques (such as recombinant DNA technology) or are naturally sourced. Biologics are highly complex, unstable, and high molecular weight substances [Figure 1]. Examples: Blood products, vaccines, recombinant proteins, monoclonal antibodies (mAbs) etc.[1,2]
Figure 1.
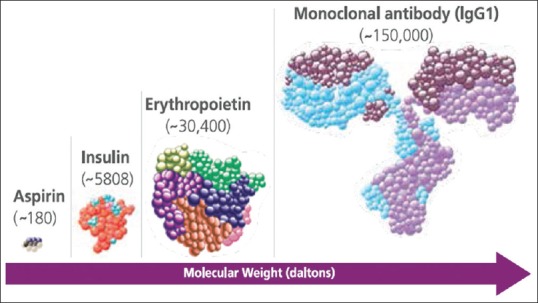
Biologics like insulin, erythropoietin, mAbs are high molecular weight substances that are far more complex than conventional chemical compounds (e.g., Aspirin). As the complexity of the molecule increases, the complexity of its manufacturing process increases[4]
Overview of biosimilars
Biosimilars are a successor to a biologic medicine that has lost exclusivity or patent protection. These are not simple generics due to complexity in size, structure and manufacturing process. Biosimilars are approved via stringent regulatory pathway demonstrating comparability with an innovator.[4] Similar biological medicinal products, similar biologics, follow-on biologics, subsequent entry biologics, similar biotherapeutic product are the various synonyms of biosimilars coined by the regulatory authorities [Table 1].
Table 1.
Definition of Biosimilars as per different regulatory agencies
| Agency | Definition |
|---|---|
| US-FDA | Highly similar to US-licensed reference biological product not withstanding minor differences in clinically inactive components, and for which there are no clinically meaningful differences in terms of safety, purity, and potency.[4] |
| EMA | Similarity to reference medicinal product in terms of quality characteristics, biological activity, safety and efficacy based on a comprehensive comparability exercise needs to be established.[4] |
| PMDA (Japan) | Biotechnological drug product developed by a different company to be comparable to an approved biotechnology-derived product of innovator. Comparability with respect to quality, safety and efficacy, or other relevant data should be established.[5] |
| CDSCO (India) | “Similar” in terms of safety, efficacy and quality to a reference biologic, which has been granted marketing authorization in India by DCGI or is approved in ICH countries (i.e., EU, Japan, US, Canada etc.).[6] |
| WHO | A biotherapeutic product that is similar in terms of quality, safety, and efficacy to an already licensed reference biotherapeutic product[7] |
Differentiation of biosimilars with generics
Biosimilars cannot be considered “generic” equivalents of innovator products [Table 2]. Generic medicines are copies of “original brand-name products” which contains the same active pharmaceutical ingredient in the same purity and are the same as those brand name drugs with regards to dosage form, strength, route of administration, quality, performance features, and intended use. It implies that the “original brand-name products” and the generics are bioequivalent and are absorbed in the “systemic circulation at a similar rate and extent, with bioavailability in the range of 80%–125%”. On the other hand, biosimilars are highly like the reference product they were compared to but have allowable differences because they are manufactured from living organisms [Figure 2]. There is always an inherent variability in the case of complex biological molecules which are difficult to characterize and hence cannot be reproduced just as it, even between batches of the same product; either reference product or biosimilars. Nevertheless, biosimilars should have no clinically meaningful differences in terms of safety, purity, and potency from the reference product.
Table 2.
| Parameters | Biosimilars | Small molecule generics |
|---|---|---|
| Properties | ||
| Size | Large | Small |
| Structure | Complex | Simple |
| Stability | Unstable | Stable |
| Manufacturing | Genetically modified cell lines Complex fermentation and purification process Complex analytical characterization Difficult to make identical copies (results of physicochemical comparability analysis with innovator should fall within allowable range set by regulatory authorities) |
Chemical synthesis Standard analytics Identical copies can be prepared |
| Preclinical (tissue/animal) |
In vitro/in vivo bioassay Toxicity studies (requirement varies based on molecules and guidelines by different regulators) |
Generally, none |
| Clinical | Pharmacokinetic (PK)/pharmacodynamic (PD) studies Clinical efficacy and safety study Postmarketing surveillance |
Usually bioequivalence/bioavailability study |
Figure 2.
Chemical Drugs versus Biologicals
As defined by EMA, a biosimilar is a biological medicine that is highly like another biological medicine already approved in the EU (the so-called “reference medicine”). Since biosimilars are produced from living organisms, there may be some minor differences from the reference medicine. These minor differences are not clinically meaningful, i.e., no differences are expected in safety and efficacy. Natural variability is inherent to all biological medicines and strict controls are always in place to ensure that it does not affect the way the medicine works or its safety. Biosimilars are approved according to the same standards of pharmaceutical quality, safety, and efficacy that apply to all biological medicines approved in the EU. The aim of biosimilar development is to demonstrate biosimilarity - high similarity in terms of structure, biological activity and efficacy, safety and immunogenicity profile. Because of complexity in structure and high molecular weight in the development process of biosimilars, researchers experience similar challenges as innovator biologics. Complex and elaborate manufacturing and stringent approval process are required for both innovator biologics and biosimilars. Physicochemical and biological characterizations along with well-designed PK/PD study, phase 3 efficacy, and safety study are required to obtain regulatory approval of biosimilars.[4] However, for approval of generics, simple bioequivalence and bioavailability studies are required [Figure 3].
Figure 3.

Development pathway of biosimilars vs innovators: Physicochemical and biological characterization phases of the biosimilar development program are more comprehensive to generate a 'highly similar' product, whereas phase 1, 2, and 3 clinical development require more emphazis for regulatory approval of innovator biologics[9,10]
Figure 4.
Regulatory pathways for the approval of biosimilar insulin
Development pathway of biosimilar insulin: Comparability studies
Comparability studies are indispensable for the development of biosimilar. Comparability is devised as a stepwise process that is customised for each product. The knowledge acquired from the initial quality comparability studies ( first step) is further used to determine the extent and type of nonclinical (second step) and clinical studies (third step) which is subsequently required in the next step of development, always with the aim of ruling out differences in clinical performance between the biosimilar and the reference medicine. The first step is primarily comprised of the analytical as well as functional quality control studies. The physical and chemical properties along with biological activity of the biosimilar are extensively studied in this phase. In vitro studies are carried out to compare the protein structure and biological function with the help of sensitive techniques. These studies are much more sensitive than the clinical trials in detecting any minor differences of clinical relevance between the biosimilar and the reference product.
The second step comprises of nonclinical comparative studies that include in vitro pharmacodynamics studies exploring binding and activation of physiological targets as well as immediate physiological effects in cells. In vivo pharmacodynamics animal studies are recommended only in the absence of suitable in vitro models. Similarly in vivo toxicological studies are only required in certain cases viz. when the biosimilar is produced in a new type of cell or organism or usage of a new excipient in the formulation.
The third step is comprised of comparative clinical studies. The aim is not to establish the safety and efficacy in patients but to confirm biosimilarity and to address any unanswered question from the previous analytical or functional studies.
Few of the important studies are described below:
Physicochemical analysis: This comparability exercise characterises molecular mass, protein content, peptide mapping, peptide sequencing, disulphide linkage, glycosylation pattern, physicochemical integrity, stability, impurities, and additives compared to reference insulin[9,11]
Biological characterization: Comparative in vitro biological analysis for insulin includes assessment of metabolic potency, mitogenic activity, binding affinity to insulin receptors, insulin-like growth factor-1 (IGF-1) receptor binding assay etc[11,12]
Preclinical phase: One repeat-dose toxicology study in rats might generally be included in biosimilar insulin development program. Studies assessing safety pharmacology, reproduction toxicology, and carcinogenicity are not routinely required.[12]
Clinical studies
Phase 1 study (PK/PD): Demonstration of similar PK and PD profiles through insulin clamp study is considered the mainstay to confirm similar efficacy of biosimilar and reference insulin[11,12]
Phase 3 study: Comparative efficacy and safety studies in Type 1 diabetes mellitus (T1DM) and insulin naïve Type 2 diabetes mellitus (T2DM) are recommended.[11]
Clinical trials required to demonstrate comparability of biosimilar insulin to innovator
-
European Medicines Agency (EMA) [12]
- PK/PD study
- Phase 3 study in patients with T1DM (Duration: 6 months of safety study)
-
U.S. Food and Drug Administration (USFDA) [13]
- PK/PD study
- Phase 3 study in patients with T1DM
- (Duration: 12 months, determination of primary efficacy parameter at 6 months)
- Phase 3 study in insulin naïve patients with T2DM (Duration: 6 months)
- Interchangeability study (guidance pending)
-
Pharmaceutical and Medical Devices Agency (PMDA), Japan [14]
- PK/PD study
- Phase 3 study in T1DM
- (Duration: 12 months, determination of primary efficacy parameter at 6 months)
-
Central Drugs Standard Control Organization (CDSCO), India [6]
- PK/PD study
- Phase 3 study in T1DM
-
World health organization (WHO)[7]
- PK/PD study
- Phase 3 study.
Phase 2 study is not required for biosimilars as it typically finds dose range which is already decided by the innovator. Biosimilar must match the dose requirement of an innovator in the phase 3 study. Amongst these authorities, EMA provides insulin specific guideline that describes nonclinical and clinical requirements for demonstration of biosimilarity of similar biological medicinal products containing recombinant human insulin and insulin analogs.[12]
-
Requirements for PK/PD study:
- Study design: Double-blind, crossover, hyperinsulinemia euglycemic clamp study using single SC doses of test and reference insulins
- Study population: normal-weight healthy volunteers/patients with T1DM.[12]
-
Equivalence margin:
- PK/PD requires equivalence testing that compares the 90% or 95% confidence interval (CI) of the observed treatment difference between the biosimilar and reference product with a predefined equivalence margin
- For primary PK endpoints (AUC0-t, Cmax),90% confidence interval of the ratio test/reference should fall within the predefined equivalence margins of 80% to 125% (0.8–1.25), unless otherwise justified
- For primary PD parameters (AUCGIR0-t), the 95% confidence interval of the ratio test/reference should fall within the predefined equivalence margins (0.8–1.25).[12]
Phase 3 clinical study and immunogenicity of biosimilar insulins
The comparative phase 3 trial should be of equivalence or noninferiority design.[11] As per EMA, there is no anticipated need for specific efficacy studies as endpoints used, usually, HbA1c level, are not considered sensitive enough to detect clinically relevant differences between two insulins.[12]
As all biological medicines have large molecular size detectable by the immune system, they bear the potential to be spotted by the immune system of the body as “foreign” and have the underlying possibility to bring on undesired immune reactions due to their composition. Hence, appraisal of immunogenicity profile is an integral part of safety assessment with regards to all biological medicines. According to EMA, safety studies should be performed with a specific focus on immunogenicity in patients with T1DM.[12] It is well admitted that blinding of study participants is likely not feasible but, at minimal, the determination of the antidrug antibodies should be carried out in a blinded fashion. The primary outcome in the assessment of immunogenicity is the incidence and antibody titre of the biosimilar insulin with respect to the reference insulin. Any little imperfection in protein structure (particularly for insulin) may produce antibodies, which can neutralise the effect of insulin itself leading to loss of efficacy. Furthermore, an unwanted immune response may be induced by a variety of factors such as disease state; drug-related factors (process and product-related) and patient-related (age, gender, genetic background etc.), stratification by type of diabetes along with pre-existing anti-insulin antibodies is warranted where a mixed population is included in the data analysis. If any impact of antidrug antibodies on the glycemic control is detected, the investigations should include insulin dose requirement and safety parameters, particularly local and systemic hypersensitivity. If any other insulin is administered to the patient in addition to test insulin, the regimen and type of background insulin should be kept unchanged during the evaluation period. For a biosimilar manufacturer developing different insulin preparations with the same active ingredient, the preparation with the highest immunogenic potential should be assessed in the safety study. For a formulation containing excipients with limited data, the safety/immunogenicity of the formulation necessitates to be investigated.[12]
Since drug-induced antibodies are anticipated to uprise early-on, a 6-month study is advocated by EMA to compare the incidence and titres of antibodies to the test and reference medicinal products, whereas FDA and PMDA demand 12 months' duration to be completed before approval.[12] FDA had released the draft guidance on the development of therapeutic protein biosimilars in May 2019. However, we need to wait and watch for this guideline to get finalized.
Equivalence trial
In PK/PD study, the clinical resemblance between the biosimilar candidate and the reference product must be demonstrated based on equivalence testing. The aim of an equivalence trial is thereby based on the hypothesis and statistical inference which are dissimilar from that of a conventional superiority trial. The null hypothesis in an equivalence trial uses a two-sided test based on a prespecified range. The null hypothesis is that the biosimilar is either (1) inferior to the reference product or (2) superior to the reference product based on a prespecified equivalence margin. The equivalence margins are chosen to identify if there are any clinically meaningful differences in effectiveness between the biosimilar and reference product at 90% or 95% confidence interval. The upper (superiority) and the lower (inferiority) bounds of the equivalence margin generally will be the same. The goal in an equivalence design is to reject the null hypothesis of nonequivalence and accept the alternative hypothesis that the two treatments (in this case, the biosimilar and the reference product) are equivalent (i.e., the differences between the two are not clinically and statistically meaningful). The equivalence test equates the confidence interval (CI) of the observed treatment difference with a predefined equivalence margin. Choice of equivalence margin is supported by statistical assessment based on historical data from the reference product and clinical relevance.[10]
The Marvel Lifesciences (UK) in 2007 had earlier withdrawn its European Union (EU) marketing authorization application (MAA) for different variants of its biosimilar human insulin failing to demonstrate equivalence in PKPD testing. Although the data of its rapid insulin had a total area under concentration (AUC) curve well within the classical interval of 80–125%, the glucose infusion AUC was 27% lower for its long marvel product (NPH insulin) with reference to Humulin I and 23% higher for its mix marvel (premix human insulin 30/70). Furthermore, AUCs calculated up to 2 hours after dosing were substantially higher, while the elimination half-life was significantly shorter for marvel soluble insulin. Thereby, marvel soluble insulin demonstrated faster absorption, more potent effect, and rapid elimination than the reference product. The Committee for Medicinal Products for Human Use (CHMP) had estimated marvel soluble insulin to substantially induce 45% greater glucose-lowering effect within the first hour of dosing as compared to the reference biological product and rejected its MAA.[15] Insugen (Biocon, India) has been available in India since 2004 and in Nigeria since 2010, despite the fact that even a phase III study (CTRI/2010/091/000627) in type 1 diabetes is being conducted now to assess the safety and efficacy of Insugen R and N with Actrapid and Insulatard (Novo Nordisk, Denmark), respectively. Also, Insuman (Sanofi) and Wosulin (Wockhardt, India) are available for more than a decade but lack head-on comparison with innovator human insulin. However, Insugen (Biocon, India) in a real-world study, although not compared with innovator human insulin, has given promising results.[16] Moreover, these human insulins follow a robust pharmacovigilance plan (discussed in next section) and DCGI that has never issued any warning in use of these products and there are no reports on any issue related to efficacy, safety, and immunogenicity available in public domain. No other data is currently available in the literature for other biosimilar insulin glargine available in India.
Switching/interchangeability study
USFDA has released the guidelines on the interchangeability of biosimilar product with reference biological product in May 2019. Interchangeability means the biologic product may be substituted without the intervention of the prescriber. The guidelines state that a biosimilar can be designated as interchangeable with reference biological product when biosimilar can be expected to produce the same clinical result as the reference biological product in any given patient. The primary objective of a switching study or studies is to demonstrate the risk in terms of safety or diminished efficacy of switching between use of the proposed biosimilar insulin and the reference insulin is not greater than the risk of using the reference insulin without such alternation or switch. However, USFDA does not mandate to conduct such studies.[17] Till date, no biosimilar insulin approved has been granted the interchangeable designation by USFDA.
Pharmacovigilance plan for approved biosimilar
It is a must for all biologics including biosimilars to have a risk management plan including immunogenicity assessment. The aim of the plan should be to accumulate additional information early on to characterize the risk profile and provide information on the safety and efficacy of the product. The EMA warrants submission of a comprehensive pharmacovigilance plan as an integral part of the original approval plan. The plan should allow identification of immunogenicity risk during the product development stage and should anticipate future risks if any. The assessment of immunogenicity should incorporate immune response case definitions, infrastructure programmes for processing patient samples as well as support for clinicians to report ADRs easily.[12]
Issue on biosimilars are “similar” to innovator biologics, but not “identical”
-
Variability of biologics:
- Inherent variability exists for all biological products as those are large in size, complex in structure and produced from ”nature's factory” using living cell lines
- Irrespective of the manufacturing process (same or different), variation can occur in both reference and biosimilar products
- Even the different batches of the innovator or reference product, derived from the same manufacturing process, will exhibit a certain level of variation (microheterogeneity).[18]
-
Acceptance range for biosimilar set by regulators:
- A biosimilar and the respective originator product will not be entirely identical
- Based on the variability of different batches of innovator, regulatory authorities set a specification limit or acceptable range for each test
- The values of biosimilar products must be inside the prespecified acceptance range set by the regulators on the basis of the range generated through the values of different batches of innovators
- To obtain regulatory approval, this variation must be manifested to be “clinically irrelevant” and have no impact on efficacy and safety
- Comprehensive physiochemical testing, biological characterisation, PK/PD, efficacy, and immunogenicity testing compared to innovator's product will ensure a high level of similarity that forms the basis of regulatory approval.[18]
Impact of biosimilar insulin on healthcare
Economic impact of diabetes: Diabetes imposes a large economic burden on individuals, families, and national health systems. High cost and suboptimal access to medicines contribute significantly to the burden of the disease. In India, 80% of pharmaceutical spend is out of pocket. Approximately 70% of the country's population is rural and the cost of therapy is having a direct impact on their acceptance of therapy. Accessibility, affordability, and awareness are the key factors[19,20]
Biosimilar and competition: Introduction of genuine competition which will drive development, advancement, and modernization in the field of biologics. Availability of biosimilar insulins can potentially lead to lower insulin cost and increased access for patients with diabetes, worldwide
Approved quality with state-of-the-art technology: Biosimilar insulins approved by the stringent regulator will surely endorse the quality. Biosimilar manufacturers are using state-of-the-art technologies. Technology that has evolved since the launch of the innovator biologics may offer additional conveniences to patients and healthcare providers[21]
Substantial cost-benefit: Cost of biosimilar products is still relatively high unlike, generics. Developing a biosimilar product is expensive and time-consuming process (approximately 8–10 years to introduce a biosimilar in the market). The extensive research and development lifecycle more closely resemble next-generation drugs rather than inexpensive generics. This can surely lower out of the pocket annual expenditure. The chronic nature of the use of insulin can lead to significant absolute cost savings
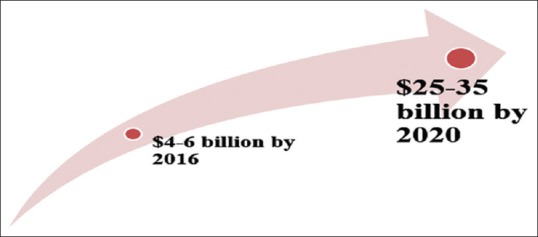
An exciting new reality: Worldwide biosimilar market is projected to be worth USD 25–35 billion by 2025. Globally more than 450 biosimilars are being developed and more than 250 are in pipeline. Innovator companies, too, have started manufacturing biosimilars. Till now, USFDA has approved five biosimilars: Sandoz's Zarxio is biosimilar to Amgen's Neupogen (filgrastim), Celltrion's Inflectra and Samsung Bioepis's Renflexis is biosimilar to Janssen's Remicade (infliximab), Sandoz's Erelzi is biosimilar to Amgen's Enbrel (etanercept), Amgen's Amjevita is biosimilar to AbbVie's Humira (adalimumab). Lilly and Boehringer's insulin glargine have been approved in Europe, USA and Japan. Merck and Samsung Bioepis's and Biocon/Mylan's insulin glargine has completed Phase III trials.[4,22] Recently Sanofi has initiated a phase 1 trial aiming to demonstrate bioequivalence of a concentrated U200 formulation of its biosimilar lispro (SAR342434) to the U100 formulation, currently marketed as Insulin Lispro Sanofi. The crossover euglycemic clamp study investigates pharmacokinetics, pharmacodynamics, and tolerability of SAR342434 U200 vs Insulin Lispro Sanofi (U100) in adult T1D patients (n = 90). Primary endpoints are insulin Cmax and total AUC (0–10 hours). Secondary endpoints include PD parameters (0–8 hours). The study is planned to be completed in October 2019.[23]
Increase in accessibility and availability with biosimilar insulins (optional if required)
Economic impact of diabetes
In addition to placing a large financial burden on individuals and their families due to the cost of insulin and other essential medicines, diabetes also has a substantial economic impact on the healthcare systems. Most countries spend between 5% and 20% of their total health expenditure on diabetes. There was a large disparity in health spending on diabetes between regions and countries. On an average, the estimated health spending due to diabetes was estimated at USD 5,374 to USD 9,641 per person with diabetes in high-income countries, compared to USD 401 to USD 688 in low- and middle-income countries. Only 19% of global health expenditure on diabetes was spent in low- and middle-income countries, where 75.4% of people with diabetes live. In some of the low-income countries, people with diabetes and their families bear almost the total cost of medical care. With such a high cost, the disease remains a significant challenge for healthcare systems and an obstacle to sustainable economic development. (Reference: IDF Atlas 7th ed.).
Situation in India
In India, a silent crisis in access to essential medicines confronts most patients who seek treatment of acute and chronic diseases. About 80% of pharmaceutical spend is out of pocket in our country. Approximately 70% of the country's population is rural and the cost of therapy is having a direct impact on their acceptance to therapy.
Diabetes related expenditure in India
According to IDF Diabetes Atlas, India, the country with the second-highest number of people living with diabetes, spent less than 3% of the global total (ID23 billion) expenditure on diabetes.[24]
High cost and suboptimal access to drugs contribute significantly to the burden of the disease and should be addressed through market shaping strategies. While hospitalization and complications are major components of the cost of diabetes, drug cost constitutes an important part of the expenses, often representing more than 50% of total direct costs for households. A study based on a large dataset found that drug cost accounted for 58% of out-of-pocket expenditure of diabetes.[19]
Few factors of the poor control are accessibility, affordability, and awareness amongst patients and physicians.
The issue of access to medicines
Price, affordability
Key to understanding the issue of access to medicines is the price (the amount of money needed to purchase goods or services) and affordability (being able to meet the expense of goods or services). Prices vary from country to country and affordability is impacted by purchasing power.
Availability
Availability of medicine is a key factor. In examining these two key concepts of affordability and availability, a variety of scenarios are possible, with medicines being available but not affordable, and affordable but not available. In an ideal scenario, medicines should be freely available and affordable to all.
CONCLUSION
Global burden of diabetes is significantly impacting health care expenditure. The huge unmet medical need for high quality and affordable biosimilars arise both in developing and developed countries. Biologics are highly complex, unstable, and high molecular weight substances. Biosimilars are a successor to a biologic medicine that has lost exclusivity or patent protection. Biosimilars cannot be considered “generic” equivalents of innovator products due to elaborate manufacturing and stringent approval process. The regulatory process of development of a biosimilar is well defined in Europe, USA and Japan. Development of biosimilars involves leveraging biological variability of the innovator's product. Rigorous physicochemical and biological characterizations with clinical studies are keys to demonstrate biosimilarity and quality.
Financial support and sponsorship
Nil.
Conflicts of interest
The co-authors Saptarshi Bose and Sandeep Gowda were employees of Biocon LTD.
REFERENCES
- 1.DeVries JH, Gough SC, Kiljanski J, Heinemann L. Biosimilar insulins: A European perspective. Diabetes Obes Metab. 2015;17:445–51. doi: 10.1111/dom.12410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Epstein MS, Ehrenpreis ED, Kulkarni PM. FDA-Related Matters Committee of the American College of Gastroenterology. Biosimilars: The need, the challenge, the future: The FDA perspective. Am J Gastroenterol. 2014;109:1856–9. doi: 10.1038/ajg.2014.151. [DOI] [PubMed] [Google Scholar]
- 3.Mellstedt H. Clinical considerations for biosimilar antibodies. EJC Suppl. 2013;11:1–11. doi: 10.1016/S1359-6349(13)70001-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Konara CS, Barnard RT, Hine D, Siegel E, Ferro V. The Tortoise and the hare: Evolving regulatory landscapes for biosimilars. Trends Biotechnol. 2016;34:70–83. doi: 10.1016/j.tibtech.2015.10.009. [DOI] [PubMed] [Google Scholar]
- 5. [Last accessed on 2019 Jul 08]. Available from: https://www.pmda.go.jp/files/000163979.pdf .
- 6. [Last accessed on 2019 Jul 08]. Available from: http://dbtbiosafety.nic.in/DBT2016-17/CDSCO-DBT2016.pdf .
- 7. [Last accessed on 2019 Jul 08]. Available from: https://www.who.int/biologicals/publications/trs/areas/biological_therapeutics/TRS_977_Annex_2.pdf?ua=1 .
- 8.Misran A. Are biosimilars really generics? Expert Opin Biol Ther. 2010;10:489–94. doi: 10.1517/14712591003662615. [DOI] [PubMed] [Google Scholar]
- 9.McCamish M, Woollett G. Worldwide experience with biosimilar development. MAbs. 2011;3:209–17. doi: 10.4161/mabs.3.2.15005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alten R, Cronstein BN. Clinical trial development for biosimilars. Semin Arthritis Rheum. 2015;44(6 Suppl):S2–8. doi: 10.1016/j.semarthrit.2015.04.002. [DOI] [PubMed] [Google Scholar]
- 11.Minocha M, Gobburu J. Drug development and potential regulatory paths for insulin biosimilars. J Diabetes Sci Technol. 2014;8:14–9. doi: 10.1177/1932296813516954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. [Last accessed on 2019 Jul 08]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/03/WC500184161.pdf .
- 13. [Last accessed on 2019 Jul 08]. Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm397017.pdf .
- 14. [Last accessed on 2019 Jul 08]. Available from: https://www.pmda.go.jp/files/000153851.pdf .
- 15.Singh AK. Biosimilar insulins. In: Bajaj S, editor. RSSDI Update-2015. 1st ed. New Delhi: JAYPEE; 2016. pp. 240–7. (Marvel insulin) [Google Scholar]
- 16.Jabeen S, Pawar D, Shivaram VS, Raj P. Impact of insulin therapy assistance program, a patient support program, on 10,426 Indian T2DM patients to assess safety and efficacy with biosimilar insulin? Diabetes. 2018;67(Suppl 1) doi: 10.2337/db18-1059-P. [Google Scholar]
- 17. [Last accessed on 2019 Jul 08]. Available from: https://www.fda.gov/media/124907/download .
- 18.Polimeni G, Trifirò G, Ingrasciotta Y, Caputi AP. The advent of biosimilars for the treatment of diabetes: Current status and future directions. Acta Diabetol. 2015;52:423–31. doi: 10.1007/s00592-015-0771-7. [DOI] [PubMed] [Google Scholar]
- 19.Yesudian C, Grepstad M, Visintin E, Ferrario A. The economic burden of diabetes in India: A review of the literature. Global Health. 2014;10:80. doi: 10.1186/s12992-014-0080-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mohan D, Raj D, Shanthirani CS, Datta M, Unwin NC, Kapur A, et al. Awareness and knowledge of diabetes in Chennai--The Chennai Urban Rural Epidemiology Study [CURES-9] J Assoc Physicians India. 2005;53:283–7. [PubMed] [Google Scholar]
- 21.Schellekens H, Moors E. Clinical comparability and European biosimilar regulations. Nat Biotechnol. 2010;28:28–31. doi: 10.1038/nbt0110-28. [DOI] [PubMed] [Google Scholar]
- 22.Deloitte. Winning with biosimilars: Opportunities in global markets. 2016 [Google Scholar]
- 23. [Last accessed on 2019 Jul 08]. Available from: https://www.clinicaltrials.gov/ct2/show/NCT03903016 .
- 24.IDF Diabetes Atlas. 8th ed. Brussels, Belgium: International Diabetes Federation; 2017. International Diabetes Federation. [PubMed] [Google Scholar]