

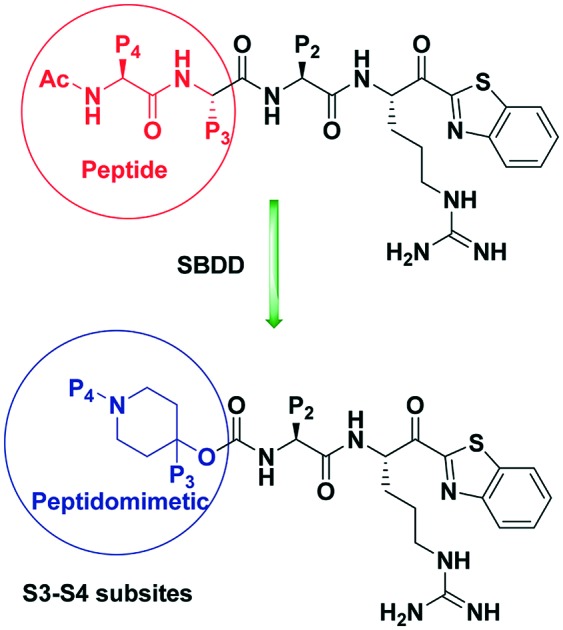
 A series of piperidine-based peptidomimetic inhibitors have been synthesized and evaluated their activity against the three serine proteases HGFA, matriptase, and hepsin. All analogs showed nanomolar activity against matriptase and hepsin.
A series of piperidine-based peptidomimetic inhibitors have been synthesized and evaluated their activity against the three serine proteases HGFA, matriptase, and hepsin. All analogs showed nanomolar activity against matriptase and hepsin.
Abstract
Matriptase and hepsin are type II transmembrane serine proteases (TTSPs). Along with related S1 trypsin like serine protease HGFA (hepatocyte growth factor activator), their unregulated proteolytic activity has been associated with cancer including tumor progression and metastasis. These three proteases have two substrates in common, hepatocyte growth factor (HGF) and macrophage stimulating protein (MSP), the ligands for MET and recepteur d'origine nantais (RON) receptor tyrosine kinases. Mechanism-based tetrapeptide and benzamidine inhibitors of these proteases have been shown to block HGF/MET and MSP/RON cancer cell signaling. Herein, we have rationally designed a new class of peptidomimetic hybrid small molecule piperidine carbamate dipeptide inhibitors comparable in potency to much larger tetrapeptides. We have identified multiple compounds which have potent activity against matriptase and hepsin and with excellent selectivity over the off-target serine proteases factor Xa and thrombin.
Introduction
Aberrant cell signaling via overexpression of cell surface receptors and the unregulated overproduction of their cytokines and growth factor ligands have been associated with tumor progression and metastasis in multiple tumor types.1–4 Hepatocyte growth factor (HGF) is the only known activating ligand for the receptor MET and likewise macrophage stimulating protein (MSP) is the only ligand for RON.5–7 MET and RON are structurally related receptor tyrosine kinases (RTKs) which stimulate intracellular kinases leading to multiple downstream effects including EMT (epithelial mesenchymal transition) in addition to cell proliferation, survival, and motility.8–13
The growth factors HGF and MSP are secreted as single-chain inactive zymogens, pro-HGF and pro-MSP which require post-translation proteolytic processing to produce a two-chain heterodimeric form14 to activate their respective kinase receptors, MET and RON.14–17 Several S1 trypsin-like serine proteases have been shown to proteolytically activated HGF and MSP however, the three proteases matriptase, hepsin, and HGFA (HFG-activator) are the three most efficient.7,18–20 This is reflected in the selectivity profile of the endogenous polypeptide inhibitors HAI-1 and HAI-2 which potently inhibit only these three proteases,20–23 Therefore, specifically targeting these three proteases with so-called ‘triplex’ inhibitors would block all HGF and MSP ligand-mediated tumor cell signaling of MET and RON.24–27
We have recently reported substrate-based22,28,29 covalent inhibitors of HGFA, matriptase, and hepsin containing electrophilic heterocyclic ketone warheads (Fig. 1) which react with the active-site serine reversibly. We have also developed a series of small molecule peptidomimetic benzamidine inhibitors.24 Others have reported on both peptide-based30–32 and small molecule benzamidine33–35 inhibitors of matriptase and hepsin including an interesting series of cyclic urea benzamidines exemplified by SRI-31215 (Fig. 1).33 In this present manuscript, we describe our continuing efforts to develop optimized mechanism-based α-ketobenzothiazole inhibitors (kbt) inhibitors (Fig. 1) with improved drug-like characteristics.
Fig. 1. Structures of tetrapeptide α-ketobenzothiazole (kbt) inhibitors based on the sequences of the pro-HGF (1a) and pro-MSP (1b) and peptidomimetic benzamidine serine protease inhibitors, SRI 31215, 1c, and 1d of HGFA, matriptase and hepsin.

Results and discussion
Rational design of P3–P4 hybrid piperidine carbamate dipeptide inhibitors48
There are several disadvantages associated with peptide-derived inhibitors as drugs, including high conformational flexibility, susceptibility to proteolytic degradation leading to high clearance and low half-life, and poor membrane permeability resulting is low oral bioavailability.36,37 The attributes of peptides leading to their poor drug-like properties stem from several reasons including their high molecular weight, large number of amide bonds susceptible to enzymatic hydrolysis also resulting in high polarity and multiple H-bond-donors and acceptors make it problematic for cell permeability. Reduction of peptidyl character of the drugs typically enhances the cellular permeability, proteolytic stability, and oral bioavailability.38,39 Thus, our goal in this present study is to rationally design novel inhibitors of HGFA, matriptase and hepsin which have much less peptide character. To that end, we introduced non-peptidyl functional groups into the P4 and P3 positions of the tetrapeptide inhibitors 1a and 1b, designed to make binding interaction in the corresponding S4 and S3 subsite pockets of the three proteases.28
The superposition of three protease structures: HGFA (PDB code 2WUC),40 matriptase (PDB code ; 2GV7)41 and hepsin (PDB code ; 1Z8G)42 and their ligands allows for the design of hybrid inhibitors (Fig. 2). SRI 31215, reported by Galemmo et al.43 is a non-peptide cyclic urea benzamidine (cub) inhibitor of matriptase and hepsin which binds the S1, S3 and S4 pockets but not the S2. When overlaid on 1b, the piperidine is positioned close to the P3 amino acid nitrogen suggesting that a piperidine ring attached through a two-atom linker such as a carbamate from the P2 position would place the piperidine in a similar position to that of SRI 31215. Our previous structure–activity relationship (SAR) studies and reported PS-SCL (positional scanning of substrate combinatorial libraries) studies on matriptase,20,29,44 hepsin22,44,45 and HGFA45 indicated that all three proteases require substrates with an Arg (R) at the P1 and prefer Leu (L) at the P2 position.28,29 We hypothesize that the low potency of SRI 31215 is partly reflected by the lack of binding in the S2 pocket (Leu of 1b). Based on this analysis and inspired by the SRI 31215 structure we designed novel hybrid dipeptide inhibitors with the preferred Leu (L) in the P2 position, but which contain the piperidine group of SRI 31215 in the P3 position installed via a carbamate from the P2 Leu as suggested by our model (Fig. 2). In another set of analogs, we created a P4 position library with alkyl or aryl sulfonyl substituents on the piperidine nitrogen to identify compounds to access the S4 pocket with optimal substitution for both potency and selectivity for the individual proteases.
Fig. 2. Computational model of tetrapeptide inhibitor Ac-KQLR-kbt (1b; yellow) bound to HGFA (white) matriptase (green) and hepsin (orange) and overlaid on peptidomimetic benzamidine inhibitor SRI 31215 (blue).

Synthesis of P2–P1 Leu-Arg-kbt dipeptides capped with substituted piperidine carbamates at the P3 and P4 positions
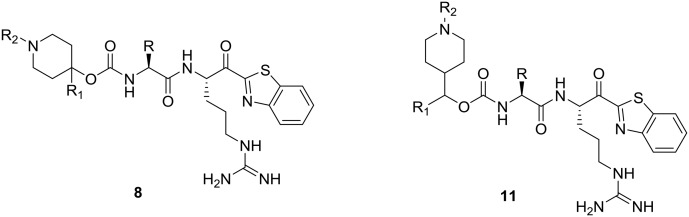
Construction of the target compounds which were selected based on our computational binding model of 1a, 1b and SRI 31215 is shown in Schemes 1 and 2. Shown in Scheme 1A, the leucine amino acid isocyanate is prepared by refluxing leucine methyl ester hydrochloride with trichloromethyl chloroformate (2a). The cyclohexyl alanine (Cha) isocyanate 2b is formed in a similar fashion. As seen in Scheme 1B, Grignard reactions with commercially available tert-butyl 4-oxopiperidine-1-carboxylate gives 1-Boc-4-piperidinol derivatives 3a–c and likewise reaction with tert-butyl 4-formylpiperidine-1-carboxylate gives 3d–e in good yield. Shown in Scheme 2A, leucine isocyanate 2 is then treated with piperidinol derivatives (3a–e) yielding the corresponding carbamate esters (4a–e) or 9a–b (Scheme 2B). Treatment of the carbamates 4a–e with dry HCl in dioxane followed by reaction with alkyl sulfonyl chloride gave the corresponding sulfonamides (5a–h). Hydrolysis of substituted piperidine esters 5a–h or 9a–b (Scheme 2B) with LiOH in aqueous THF provided the carboxylic acids (6a–m or 10a–b) which were then reacted with the Pbf side chain protected Arg-kbt (7)28 using standard amide coupling conditions (EDC/HOBt or HATU) to give piperidine dipeptides which were subjected to global side-chain deprotection with a cocktail of TFA/water/thioanisole. Reverse phase preparatory HPLC purification was then conducted to produce final target compounds (8a–m and 11a–b) in high purity.
Scheme 1. A) Synthesis of amino acid isocyanates 2a–b and B) piperidine alcohols 3a–e.

Scheme 2. Synthetic route for hybrid piperidine dipeptide ketobenzothiazole (kbt) inhibitors of matriptase, hepsin, and HGFA. *Synthesis of H-Arg(Pbf)-kbt-HCl 7 has been reported previously.28.

Using the fluorogenic protease substrates, Boc-QAR-AMC (matriptase and hepsin) or Boc-QLR-AMC (HGFA) in previously published kinetic enzyme assays.22 We tested the activity of all target compounds using eleven different concentrations of compound. Inhibitors were pre-incubated with protease followed by the addition of the substrate. Inhibition of substrate proteolysis derived fluorescence was monitored kinetically over a period of one hour. Shown in Table 1 are the experimentally determined IC50 values of each compound for their concentration-dependent inhibition of HGFA, matriptase, and hepsin.
Table 1. Biological activity and selectivity of hybrid piperidine dipeptide kbt HGFA, matriptase and hepsin serine protease inhibitors, 8a–m and 11a–b.
| ||||||||
| Compd | R | R1 | R2 | Matriptase IC50 a (μM) | Hepsin IC50 a (μM) | HGFA IC50 a (μM) | Factor Xa IC50 a (μM) | Thrombin IC50 a (μM) |
| 8a | Leu |
|
H | 0.09 | 0.005 | 0.78 | NT | >20 |
| 8b | Leu |
|
H | 0.03 | 0.0006 | 1.2 | 6.5 | >20 |
| 8c | Leu |
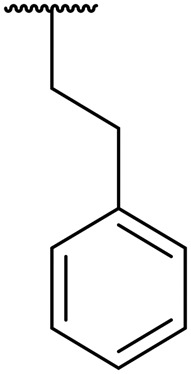
|
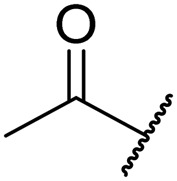
|
0.07 | 0.0005 | >20 | 2.0 | 7.7 |
| 8d | Cha |
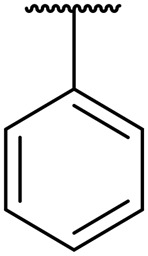
|
H | 2.1 | 0.09 | >20 | 9.0 | >20 |
| 8e | Leu | H |
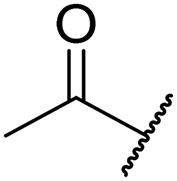
|
0.64 | 0.02 | >20 | NT | NT |
| 8f | Leu | H |
|
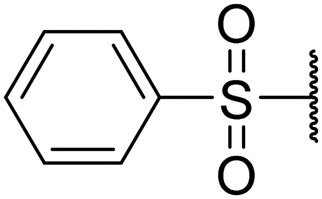
0.81 | 0.02 | 8.6 | >20 | >20 |
| 8g | Leu | H |
|
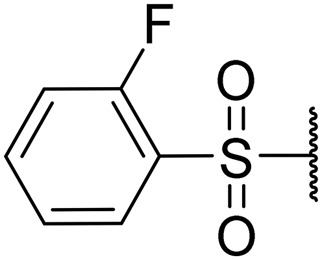
4.5 | 0.07 | 18 | NT | NT |
| 8h | Leu | H |
|
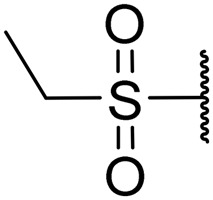
0.73 | 0.01 | >20 | >20 | >20 |
| 8i | Leu | H |
|
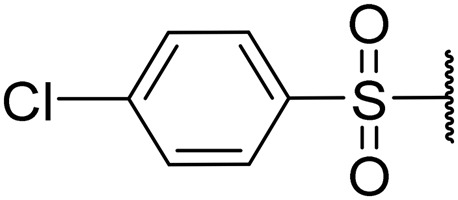
2.4 | 0.06 | 9.0 | NT | NT |
| 8j | Leu | H |
|
7.5 | 0.07 | >20 | NT | NT |
| 8k | Leu | H |
|
10 | 0.11 | >20 | NT | NT |
| 8l | Leu | H |
|
4.0 | 0.06 | 6.3 | NT | NT |
| 8m | Leu | H |
|
>20 | 0.11 | 8.8 | NT | NT |
| 11a | Leu |
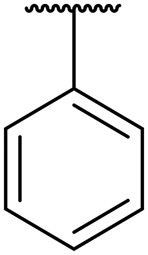
|
H | 1.1 | 0.008 | 14 | >20 | >20 |
| 11b | Leu |
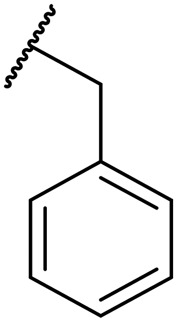
|
H | 0.02 | 0.002 | 6.0 | NT | NT |
aIC50 values are an average of three experiments.
The majority of compounds tested showed good activity and excellent selectivity for matriptase and hepsin over HGFA (Table 1). The most potent inhibitors we identified are 8b and 8c which have IC50 values 0.6 and 0.5 nM for hepsin and 30 and 70 nM for matriptase, respectively. We also made compound 8d, which replaces the P2 Leu with the unnatural amino acid cyclohexyl alanine (Cha) but found that the activity lowers for all three proteases (Table 1). We found that inhibitors with substituted alkyl or aryl sulfonyl groups at R2/P4 position attached on the piperidine ring nitrogen generally exhibit better potency against hepsin relative to matriptase. As examples, ethyl sulfonyl (8h) and acetyl (8e) analogs showed the best activity of all aryl sulfonyl analogs at the R2/P4 position, clearly demonstrating that smaller functional groups are preferred in the S4 pocket of matriptase and hepsin.
To introduce more flexibility into the piperidine carbamate we made the two additional analogs 11a–b which have a methylene spacer between the piperidine ring and the carbamate linker to the Leu P2 position. In these analogs, the R1 substituent is now not a tertiary group on the piperidine ring but rather a secondary group on the spacer. We found that these two inhibitors exhibit excellent potency against both hepsin and matriptase with IC50 values 8 nM and 2 nM, respectively; and also found moderate effect in inhibiting HGFA (IC50 values 14 μM and 6 μM) but less active than the corresponding matched pairs 8a and 8b (IC50 values of 0.78 and 1.2 μM). Our SAR data also revealed that adding the methylene linker between the aryl and carbamate group decreases the activity against matriptase and hepsin. Similar to that found for 8a and 8b, the benzyl group on the R1 position of the piperidine having an additional methylene spacer is optimal to the phenyl group in 11a and 11b.
It is noteworthy that all hybrid piperidine dipeptides displayed only weak or no activity against HGFA with the analogs 8a and 8b containing phenyl and benzyl groups at the R1 position having the best IC50 values of 0.78 and 1.2 μM, respectively. Increasing the alkyl linker length between the phenyl and piperidine ring as in compound 8c results in loss of any activity up to 20 μM. Evaluation of the SAR derived from different sulfonyl and acyl groups at the R2 position showed only moderate effects on HGFA activity. Interestingly, when tested in our enzyme assays, we found that SRI 31215 only has weak activity for HGFA (IC50 20 μM). Furthermore, all other benzamidine inhibitors reported to data24,33 show either no or weak potency for HGFA as well.
To determine the target selectivity profile, a handful of compounds were tested against the similar trypsin-like serine proteases, factor Xa and thrombin. In general, we found that all compounds tested had good selectivity over both factor Xa and thrombin. However, we found that 8c with an acetyl on R2 and phenethyl group off of R1 had some inhibition of factor Xa (IC50 2.0 μM) and thrombin (IC50 7.7 μM). A notable piece of SAR is that that 8b which has no substitution at R2 and a shorter benzyl R1 showed no activity against thrombin (>20 μM) and a 4-fold higher IC50 relative to 8c.
Conclusion
In summary, we have developed a new class of hybrid piperidine carbamate dipeptidyl ketobenzothiazole serine protease inhibitors based on tetrapeptide inhibitors and benzamidine inhibitor SRI 31215 of matriptase and hepsin. These inhibitors replace the P4 and P3 amino acids with non-peptide piperidine carbamates which span the S3–S4 sidechain-bindings pockets. The piperidine moiety was appended with a variety of aryl and sulfonyl substituted groups off the nitrogen to interact more specifically in the S4 pockets of matriptase and hepsin. Using this strategy, we have identified multiple new potent inhibitors of matriptase and hepsin such as 8b, 8c, 8f, and 8h with excellent selectivity over factor Xa and thrombin. Excitingly, these new dipeptide inhibitors of HGFA matriptase and hepsin approach the activity level of the tetrapeptides 1a and 1b. Therefore, we were successful in our goal here of rationally designing novel inhibitors of the same potency but with much less peptide character. Current efforts are in progress to obtain X-ray crystal co-structures of these compounds to aid in the further structure-based optimization.
Experimental
General synthesis, purification, and analytical chemistry procedures
Starting materials, reagents, and solvents were purchased from commercial vendors unless otherwise noted. 1H NMR spectra were measured on a Varian 400 MHz NMR instrument. The chemical shifts were reported as δ ppm relative to TMS using residual solvent peak as the reference unless otherwise noted. The following abbreviations were used to express the multiplicities: s = singlet; d = doublet; t = triplet; q = quartet; m = multiplet; br = broad. High-performed liquid chromatography (HPLC) was carried out on GILSON GX-281 using Waters C18 5 μM, 4.6 × 50 mm and Waters Prep C18 5 μM, 19 × 150 mm reverse phase columns, eluted with a gradient system of 5 : 95 to 95 : 5 acetonitrile : water with a buffer consisting of 0.05% TFA. Mass spectra (MS) were performed on HPLC/MSD using electrospray ionization (ESI) for detection. All reactions were monitored by thin layer chromatography (TLC) carried out on Merck silica gel plates (0.25 mm thick, 60F254), visualized by using UV (254 nm) or dyes such as KMnO4, p-anisaldehyde and CAM (Cerium Ammonium Molybdate or Hanessian's Stain). Silica gel chromatography was carried out on a Teledyne ISCO CombiFlash purification system using pre-packed silica gel columns (12–330 g sizes). All compounds used for biological assays are greater than 95% purity based on NMR and HPLC by absorbance at 220 nm and 254 nm wavelengths.
Synthesis of amino acid methyl ester isocyanates 2a–b. General procedure
The leucine amino acid methyl ester hydrochloride (4.55 g, 25 mmol) was placed in a dry RB flask and then dried overnight on the vacuum pump. The flask was flushed with nitrogen and dry dioxane (60 mL) was added followed by trichloromethyl chloroformate (7.42 g, 37.5 mmol). After refluxing for 14 h, the solvent was removed on the rotary evaporator to yield pure isocyanates 2a–b as colorless oils.46
Synthesis of compounds 3a–e. General procedure
To a 250 mL round bottom flask kept under nitrogen atmosphere was added a 2.0 M solution (1.40 mL, 2.76 mmol) of the appropriate Grignard reagent in THF and the solution was cooled to 0–5 °C. A solution of the appropriate aldehyde (0.5 g, 2.50 mmol) in dry THF (5 mL) was added dropwise to the cooled Grignard solution over ∼20 min. The reaction mixture was allowed to warm up to room temperature and stirred for 5 h under nitrogen. The disappearance of the aldehyde was monitored by TLC. The reaction mixture was cooled to 0–5 °C and acidified to pH ∼3.0 using 5% aqueous hydrochloric acid. The organic solvent was evaporated off and the residue was extracted with ethyl acetate (2 × 50 mL). The combined organic extracts were washed with brine (10 mL) dried over anhydrous Na2SO4, filtered, and concentrated. The crude product was purified by column chromatography to yield alcohols 3a–c and 3d–e as oils.46
Synthesis of carbamates 4a–d and 9a–b. General procedure
A solution of compound Boc-(4-hydroxy) piperidine (3) (2.02 g, 10 mmol) in dry acetonitrile (20 mL) was treated with trimethylamine (2.02 g; 20 mmol) followed by an appropriate amino acid methyl ester isocyanate 2 (11 mmol). The resulting reaction mixture was refluxed for 3 h and then allowed to cool to room temperature. The disappearance of the alcohol was monitored by TLC. The solvent was evaporated, and the residue was taken up in ethyl acetate (100 mL) and the organic layer was washed with 5% aqueous HCl (2 × 20 mL) and saturated NaCl (20 mL). The organic layer was dried over anhydrous sulfate, filtered, and concentrated to yield an oily product. Purification by flash chromatography yielded 4a–d and 9a–b esters as colorless oils/solids. Dissolved the above purified compound 4d in dry DCM (5 mL) and added a solution of 4 M HCl in dioxane (15 mL) with stirring. The reaction mixture was stirred for 2 h at room temperature. The disappearance of the starting material was monitored by TLC. The solvent was evaporated under reduced pressure and compounds 4e was used in the next step without further purification.46,47
Synthesis of sulfonyl compounds 5a–h. General procedure
A solution of compound 4e (100 mg, 0.32 mmol) in dry THF (3 mL) treated with triethyl amine (0.13 mL, 0.64 mmol) followed by added appropriate sulfonyl chloride (63 mg, 0.32 mmol) while stirring and continue the reaction for 12 h. Residue was dissolved in ethyl acetate (50 mL) and washed with 5% HCl (2 × 20 mL) and saturated NaCl (20 mL), dried over sodium sulfate, filtered and concentrated to yield crude product, which is purified by column chromatography yield the corresponding esters 5a–h as colorless oils.48
Synthesis of acids 6a–m and 10a–b. General procedure
A solution of ester 4 or 5/9 (0.28 g, 0.557 mmol) in tetrahydrofuran (2 mL) was treated with 1 M aqueous LiOH (2 mL). The reaction mixture was stirred for 3 h at room temperature while monitoring the disappearance of the ester by TLC. Most of the solvent was evaporated off and the solution was acidified to pH ∼3 using 5% hydrochloric acid (2 mL). The aqueous layer was extracted with ethyl acetate (2 × 25 mL) and the combined organic layer was washed with brine (10 mL). The organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated to yield compounds 6a–m and 10a–b.
Synthesis of compounds 8a–m and 11a–b. General procedure
EDCI (0.28 mmol) and HOBt (0.28 mmol) were added to a solution of compound 6 or 10 (100 mg, 0.222 mmol) in dry DMF (2 mL) and the mixture was stirred for 30 min at room temperature and cooled the reaction to 0–5 °C and added the Arg (Pbf)-kbt HCl (7) (129 mg, 0.222 mmol) followed by DIEA (115 mg, 0.888 mmol) and stirred for 15 min. Allowed the reaction to room temperature and stirred for 12 h at RT, the reaction was concentrated under reduced pressure and the residue was partitioned between ethyl acetate (25 mL) and 10% citric acid (2 × 10 mL). The layers were separated, and the ethyl acetate was further washed with aqueous NaHCO3 (10 mL), saturated NaCl solution (10 mL). The organic layer was dried over Na2SO4, filtered, and concentrated. The deprotection of the crude product was accomplished by stirring in 1.0 mL of a TFA–thioanisole–water mixture (95 : 2.5 : 2.5) for 2–3 h. After concentrating in vacuo, the crude material was dissolved in DMSO and purified using reverse phase HPLC (0.05% TFA/acetonitrile/water gradient). The pure fractions were pooled, frozen and lyophilized to give the pure dipeptides 8a–m and 11a–b as white powders.
4-Phenylpiperidin-4-yl ((S)-1-(((S)-1-(benzo[d]thiazol-2-yl)-5-guanidino-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (8a)
White solid, yield (65%), 1H NMR (400 MHz, CD3OD) δ ppm = 8.23–8.20 (m, 1 H), 8.15–8.11 (m, 1 H), 7.68–7.59 (m, 1 H), 7.34 (d, J = 3.9 Hz, 3 H), 7.29–7.18 (m, 4 H), 5.71–5.64 (m, 1 H), 3.88–3.79 (m, 1 H), 3.23–3.18 (m, 3 H), 3.12 (s, 1 H), 2.68–2.63 (m, 3 H), 2.44 (s, 2 H), 1.90–1.77 (m, 2 H), 1.60–1.49 (m, 1 H), 1.26 (d, J = 6.7 Hz, 2 H), 1.07–0.88 (m, 6 H). LCMS (ESI+) expected m/z 607.30, found 608.5 (M + H+).
4-Benzylpiperidin-4-yl ((S)-1-(((S)-1-(benzo[d]thiazol-2-yl)-5-guanidino-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (8b)
White solid, yield (68%), 1H NMR (400 MHz, CDCl3) δ ppm = 8.17 (d, J = 7.8 Hz, 1 H), 7.96 (d, J = 7.4 Hz, 1 H), 7.65–7.50 (m, 4 H), 6.94 (br. s., 2 H), 5.79 (br. s., 1 H), 4.76 (dd, J = 4.9, 10.8 Hz, 1 H), 4.15 (d, J = 5.5 Hz, 1 H), 3.45 (br. s., 2 H), 3.16 (br. s., 2 H), 2.81 (br. s., 4 H), 2.37–2.06 (m, 3 H), 1.93–1.58 (m, 8 H), 1.61–1.38 (m, 3 H), 0.98–0.85 (m, 6 H). LCMS (ESI+) expected m/z 621.31, found 622.4 (M + H+).
1-Acetyl-4-phenethylpiperidin-4-yl ((S)-1-(((S)-1-(benzo[d]thiazol-2-yl)-5-guanidino-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (8c)
White solid, yield (55%), 1H NMR (400 MHz, CHLOROFORM-d) δ ppm = 8.19 (d, J = 7.04 Hz, 1 H), 7.92–8.06 (m, 2 H), 7.43–7.76 (m, 7 H), 6.95 (br. s., 1 H), 5.79 (d, J = 7.83 Hz, 2 H), 4.71–4.85 (m, 1 H), 4.04–4.26 (m, 2 H), 3.16 (br. s., 1 H), 2.11–2.49 (m, 8 H), 1.40–1.95 (m, 10 H), 0.83–1.03 (m, 6 H). LCMS (ESI+) expected m/z 677.34, found 587.5 (M – Bn+).
4-Phenylpiperidin-4-yl ((S)-1-(((S)-1-(benzo[d]thiazol-2-yl)-5-guanidino-1-oxopentan-2-yl)amino)-3-cyclohexyl-1-oxopropan-2-yl)carbamate (8d)
White solid, yield (60%), 1H NMR (400 MHz, CDCl3) δ ppm = 8.20 (d, J = 7.83 Hz, 2 H), 7.92–8.04 (m, 2 H), 7.75 (br. s., 1 H), 7.37–7.68 (m, 9 H), 6.97 (br. s., 2 H), 5.81 (br. s., 2 H), 4.70–4.88 (m, 2 H), 4.08–4.29 (m, 2 H), 3.41–3.63 (m, 1 H), 3.17 (br. s., 1 H), 2.51 (br. s., 2 H), 2.10–2.39 (m, 4 H), 1.43–1.93 (m, 8 H), 0.79–1.36 (m, 11 H). LCMS (ESI+) expected m/z 646.33, found 647.5 (M + H+).
1-Acetylpiperidin-4-yl ((S)-1-(((S)-1-(benzo[d]thiazol-2-yl)-5-guanidino-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (8e)
White solid, yield (65%), 1H NMR (400 MHz, DMSO-d6) δ = 8.57 (d, J = 5.5 Hz, 1 H), 8.30–8.21 (m, 1 H), 7.68 (quin, J = 6.5 Hz, 1 H), 7.51 (br. s., 1 H), 7.25 (d, J = 8.2 Hz, 1 H), 5.41 (br. s., 1 H), 4.65 (br. s., 1 H), 4.08 (d, J = 6.7 Hz, 1 H), 3.84 (d, J = 13.3 Hz, 1 H), 3.59 (br. s., 1 H), 3.24 (br. s., 1 H), 3.15 (d, J = 5.5 Hz, 2 H), 1.99 (s, 3 H), 1.75 (br. s., 2 H), 1.58 (dd, J = 6.3, 14.5 Hz, 3 H), 1.41–1.31 (m, 3 H), 0.82 (d, J = 6.7 Hz, 6 H). LCMS (ESI+) expected m/z 573.28, found 574.5 (M + H+).
1-(Phenylsulfonyl)piperidin-4-yl ((S)-1-(((S)-1-(benzo[d]thiazol-2-yl)-5-guanidino-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (8f)
White solid, yield (55%), 1H NMR (400 MHz, CDCl3) δ = 8.17 (d, J = 6.7 Hz, 1 H), 8.03–7.92 (m, 2 H), 7.83 (br. s., 1 H), 7.79–7.66 (m, 3 H), 7.64–7.50 (m, 4 H), 7.46 (d, J = 7.4 Hz, 1 H), 6.98 (br. s., 1 H), 5.83–5.71 (m, 1 H), 5.52 (d, J = 5.5 Hz, 1 H), 5.33 (br. s., 1 H), 4.64 (br. s., 2 H), 4.16 (br. s., 1 H), 3.34–3.07 (m, 2 H), 2.88 (br. s., 2 H), 2.17 (br. s., 1 H), 1.91 (br. s., 3 H), 1.75 (br. s., 4 H), 1.57 (dt, J = 7.0, 15.7 Hz, 4 H), 0.89 (d, J = 6.7 Hz, 6 H). LCMS (ESI+) expected m/z 671.26, found 672.4 (M + H+).
1-((2-Fluorophenyl)sulfonyl)piperidin-4-yl ((S)-1-(((S)-1-(benzo[d]thiazol-2-yl)-5-guanidino-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (8g)
White solid, yield (60%), 1H NMR (400 MHz, CDCl3) δ ppm = 8.16 (d, J = 7.83 Hz, 1 H), 7.97 (d, J = 7.83 Hz, 1 H), 7.86 (br. s., 1 H), 7.70 (d, J = 7.83 Hz, 1 H), 7.50–7.64 (m, 4 H), 7.45 (d, J = 6.65 Hz, 1 H), 7.29–7.37 (m, 1 H), 6.88 (br. s., 1 H), 5.74 (br. s., 1 H), 5.33 (d, J = 6.65 Hz, 1 H), 4.68 (br. s., 1 H), 3.84–4.28 (m, 5 H), 3.44–3.62 (m, 2 H), 3.24 (br. s., 4 H), 2.90–3.04 (m, 2 H), 2.17 (br. s., 2 H), 1.93 (m, 3 H), 1.76 (m, 2 H), 1.45–1.68 (m, 2 H), 0.77–1.01 (m, 6 H). LCMS (ESI+) expected m/z 689.25, found 690.4 (M + H+).
1-(Ethylsulfonyl)piperidin-4-yl ((S)-1-(((S)-1-(benzo[d]thiazol-2-yl)-5-guanidino-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (8h)
White solid, yield (62%), 1H NMR (400 MHz, CDCl3) δ ppm = 8.18 (d, J = 7.43 Hz, 1 H), 7.97 (d, J = 7.83 Hz, 1 H), 7.73 (d, J = 7.43 Hz, 1 H), 7.46–7.65 (m, 4 H), 5.75 (br. s., 2 H), 5.55 (br. s., 1 H), 4.82 (br. s., 1 H), 4.22 (br. s., 1 H), 3.45 (br. s., 2 H), 3.05–3.31 (m, 4 H), 2.96 (q, J = 7.30 Hz, 2 H), 2.10–2.28 (m, 3 H), 1.50–2.03 (m, 7 H), 1.34 (t, J = 7.43 Hz, 3 H), 0.94 (d, J = 7.04 Hz, 6 H). LCMS (ESI+) expected m/z 623.26, found 624.4 (M + H+).
1-((4-Chlorophenyl)sulfonyl)piperidin-4-yl ((S)-1-(((S)-1-(benzo[d]thiazol-2-yl)-5-guanidino-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (8i)
White solid, yield (65%), 1H NMR (400 MHz, CDCl3) δ ppm = 8.19 (d, J = 7.43 Hz, 1 H), 8.00 (d, J = 7.04 Hz, 1 H), 7.44–7.75 (m, 5 H), 7.38 (d, J = 7.04 Hz, 2 H), 6.93 (br. s., 1 H), 5.83 (br. s., 1 H), 5.42 (d, J = 7.43 Hz, 1 H), 4.66 (br. s., 1 H), 4.15 (br. s., 1 H), 3.17–3.41 (m, 6 H), 2.94 (br. s., 4 H), 1.93 (m, 2 H), 1.76 (m., 4 H), 1.19–1.37 (m, 1 H), 0.75–0.93 (m, 6 H). LCMS (ESI+) expected m/z 705.22, found 706.4 (M + H+).
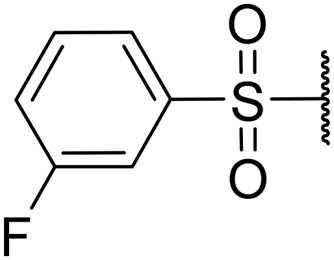
1-((3-Fluorophenyl)sulfonyl)piperidin-4-yl ((S)-1-(((S)-1-(benzo[d]thiazol-2-yl)-5-guanidino-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (8j)
White solid, yield (65%), 1H NMR (400 MHz, CDCl3) δ ppm = 8.16 (d, J = 7.83 Hz, 1 H), 7.97 (d, J = 7.83 Hz, 1 H), 7.86 (br. s., 1 H), 7.70 (d, J = 7.83 Hz, 1 H), 7.50–7.64 (m, 4 H), 7.45 (d, J = 6.65 Hz, 1 H), 7.29–7.37 (m, 1 H), 6.88 (br. s., 1 H), 5.74 (br. s., 1 H), 5.33 (d, J = 6.65 Hz, 1 H), 4.68 (br. s., 1 H), 3.84–4.28 (m, 5 H), 3.44–3.62 (m, 2 H), 3.24 (br. s., 4 H), 2.90–3.04 (m, 2 H), 2.17 (br. s., 2 H), 1.93 (m, 3 H), 1.76 (m, 2 H), 1.45–1.68 (m, 2 H), 0.77–1.01 (m, 6 H). LCMS (ESI+) expected m/z 689.25, found 690.5 (M + H+).
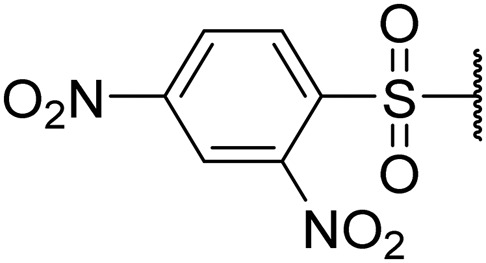
1-((2,4-Dinitrophenyl)sulfonyl)piperidin-4-yl ((S)-1-(((S)-1-(benzo[d]thiazol-2-yl)-5-guanidino-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (8k)
Off-white solid, yield (60%), 1H NMR (400 MHz, CDCl3) δ ppm = 8.75–8.66 (m, 1 H), 8.48–8.45 (m, 1 H), 8.19 (s, 3 H), 8.03–7.98 (m, 2 H), 7.67–7.53 (m, 5 H), 7.16–7.06 (m, 1 H), 5.33–5.27 (m, 1 H), 3.95–3.88 (m, 1 H), 3.52–3.47 (m, 1 H), 2.68 (s, 2 H), 1.87 (br. s., 52 H), 1.02–0.87 (m, 6 H). LCMS (ESI+) expected m/z 761.23, found 762.5 (M + H+).
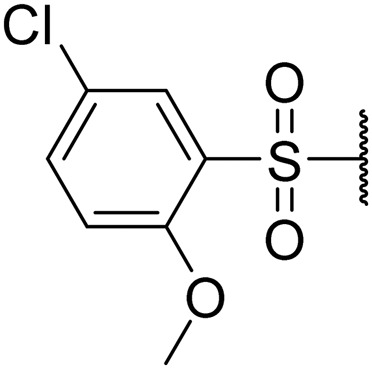
1-((5-Chloro-2-methoxyphenyl)sulfonyl)piperidin-4-yl ((S)-1-(((S)-1-(benzo[d]thiazol-2-yl)-5-guanidino-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (8l)
White solid, yield (63%), 1H NMR (400 MHz, CDCl3) δ ppm = 8.12–8.24 (m, 1 H), 7.71–8.02 (m, 4 H), 7.40–7.65 (m, 4 H), 6.95 (d, J = 8.61 Hz, 1 H), 5.60–5.85 (m, 1 H), 5.46 (d, J = 7.04 Hz, 1 H), 4.74 (br. s., 1 H), 3.96–4.28 (m, 7 H), 3.90 (s, 2 H), 3.47 (d, J = 4.70 Hz, 2 H), 3.13 (m, 2 H), 2.18 (br. s., 2 H), 1.49–2.00 (m, 8 H), 0.91 (d, J = 6.78 Hz, 6 H). LCMS (ESI+) expected m/z 735.24, found 736.4 (M + H+).
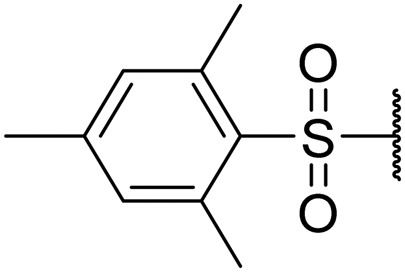
1-(Mesitylsulfonyl)piperidin-4-yl ((S)-1-(((S)-1-(benzo[d]thiazol-2-yl)-5-guanidino-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (8m)
White solid, yield (64%), 1H NMR (400 MHz, CDCl3) δ ppm = 8.17 (d, J = 7.04 Hz, 1 H), 7.97 (t, J = 6.26 Hz, 2 H), 7.76 (d, J = 5.09 Hz, 1 H), 7.50–7.65 (m, 4 H), 6.94 (s, 2 H), 5.68–5.86 (m, 1 H), 4.77 (br. s., 1 H), 4.21 (br. s., 1 H), 3.01–3.57 (m, 6 H), 2.58 (s, 3 H), 2.30 (s, 3 H), 2.19 (s, 2 H), 1.50–1.94 (m, 6 H), 0.91 (d, J = 6.39 Hz, 6 H). LCMS (ESI+) expected m/z 713.31, found 714.5 (M + H+).
Phenyl(piperidin-4-yl)methyl ((S)-1-(((S)-1-(benzo[d]thiazol-2-yl)-5-guanidino-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (11a)
White solid, yield (50%), 1H NMR (400 MHz, CD3OD) δ ppm = 8.23–8.20 (m, 1 H), 8.15–8.11 (m, 1 H), 7.68–7.59 (m, 1 H), 7.34 (d, J = 3.9 Hz, 3 H), 7.29–7.18 (m, 4 H), 5.71–5.64 (m, 1 H), 3.88–3.79 (m, 1 H), 3.23–3.18 (m, 3 H), 3.12 (s, 1 H), 2.68–2.63 (m, 3 H), 2.44 (s, 2 H), 1.90–1.77 (m, 2 H), 1.60–1.49 (m, 1 H), 1.26 (d, J = 6.7 Hz, 2 H), 1.07–0.88 (m, 6 H). LCMS (ESI+) expected m/z 621.32, found 622.5 (M + H+).
2-Phenyl-1-(piperidin-4-yl)ethyl ((S)-1-(((S)-1-(benzo[d]thiazol-2-yl)-5-guanidino-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (11b)
White solid, yield (52%), 1H NMR (400 MHz, DMSO-d6) δ = 8.65–8.56 (m, 1 H), 8.41 (s, 1 H), 8.36 (d, J = 4.7 Hz, 1 H), 8.30–8.23 (m, 2 H), 7.68 (dd, J = 2.3, 5.1 Hz, 2 H), 7.53–7.44 (m, 1 H), 7.29 (d, J = 8.2 Hz, 1 H), 7.18 (d, J = 8.2 Hz, 1 H), 5.52–5.35 (m, 1 H), 4.55–4.45 (m, 1 H), 4.06–3.96 (m, 1 H), 3.56 (d, J = 11.0 Hz, 1 H), 3.18–3.11 (m, 2 H), 2.42 (s, 2 H), 1.97 (d, J = 5.1 Hz, 1 H), 1.74 (dd, J = 3.9, 9.4 Hz, 3 H), 1.65–1.51 (m, 3 H), 1.40–1.29 (m, 2 H), 0.86 (d, J = 2.3 Hz, 6 H). LCMS (ESI+) expected m/z 635.33, found 544.4 (M – Bn+).
Computational methods
The published crystal structures of HGFA (PDB code 2WUC),40 matriptase (PDB code ; 2GV7)41 and hepsin (PDB code ; 1Z8G)42 were superimposed based on sequence alignment in MOE (Chemical Computing Group, v2014.09). The HGFA and hepsin structures include the peptide-based inhibitor Ace-KQLR-chloromethylketone. The kbt-peptide inhibitors were separately modeled into the binding sites of each protease based on the above structures, with energy minimization using the AMBER12 forcefield in MOE. The benzamidine 1d structure bound to matriptase is taken from the crystal structure. Inhibitors 8a–m and 11a–b were constructed in the binding site based on the above structure superposition. In each case, the inhibitors were minimized initially with the protease atom positions fixed, followed by minimization of the protein-ligand complex, again using the AMBER 12 forcefield.
Conflicts of interest
The authors declare no conflicts of interest associate with this manuscript.
Supplementary Material
Acknowledgments
Funding for this work was generously provided by the Alvin J. Siteman Cancer Research Fund (Washington University School of Medicine) #16-FY18-02, Susan G. Komen for the Cure Foundation grant CCR499051.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c9md00234k
References
- Gherardi E., Birchmeier W., Birchmeier C., Woude G. V. Nat. Rev. Cancer. 2012;12:89. doi: 10.1038/nrc3205. [DOI] [PubMed] [Google Scholar]
- Smyth E. C., Sclafani F., Cunningham D. OncoTargets Ther. 2014;7:1001–1014. doi: 10.2147/OTT.S44941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Otin C., Bond J. S. J. Biol. Chem. 2008;283:30433–30437. doi: 10.1074/jbc.R800035200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedosejevs E. T., Feil R., Lunn J. E., Plaxton W. C. FEBS Lett. 2018;592:2525–2532. doi: 10.1002/1873-3468.13197. [DOI] [PubMed] [Google Scholar]
- Sakai K., Aoki S., Matsumoto K. J. Biochem. 2015;157:271–284. doi: 10.1093/jb/mvv027. [DOI] [PubMed] [Google Scholar]
- Kataoka H., Itoh H., Hamasuna R., Meng J. Y., Koono M. Hum. Cell. 2001;14:83–93. [PubMed] [Google Scholar]
- Ganesan R., Kolumam G. A., Lin S. J., Xie M.-H., Santell L., Wu T. D., Lazarus R. A., Chaudhuri A., Kirchhofer D. Mol. Cancer Res. 2011;9:1175–1186. doi: 10.1158/1541-7786.MCR-11-0004. [DOI] [PubMed] [Google Scholar]
- Gaudino G., Follenzi A., Naldini L., Collesi C., Santoro M., Gallo K. A., Godowski P. J., Comoglio P. M. EMBO J. 1994;13:3524–3532. doi: 10.1002/j.1460-2075.1994.tb06659.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Organ S. L., Tsao M. S. Ther. Adv. Med. Oncol. 2011;3:S7–S19. doi: 10.1177/1758834011422556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M. H., Wang D., Chen Y. Q. Carcinogenesis. 2003;24:1291–1300. doi: 10.1093/carcin/bgg089. [DOI] [PubMed] [Google Scholar]
- Liu X., Newton R. C., Scherle P. A. Expert Opin. Invest. Drugs. 2011;20:1225–1241. doi: 10.1517/13543784.2011.600687. [DOI] [PubMed] [Google Scholar]
- Sakai K., Aoki S., Matsumoto K. J. Biochem. 2015;157:271–284. doi: 10.1093/jb/mvv027. [DOI] [PubMed] [Google Scholar]
- Yao H. P., Zhou Y. Q., Zhang R., Wang M. H. Nat. Rev. Cancer. 2013;13:466–481. doi: 10.1038/nrc3545. [DOI] [PubMed] [Google Scholar]
- Owen K. A., Qiu D., Alves J., Schumacher A. M., Kilpatrick L. M., Li J., Harris J. L., Ellis V. Biochem. J. 2010;426:219–228. doi: 10.1042/BJ20091448. [DOI] [PubMed] [Google Scholar]
- Naka D., Ishii T., Yoshiyama Y., Miyazawa K., Hara H., Hishida T., Kidamura N. J. Biol. Chem. 1992;267:20114–20119. [PubMed] [Google Scholar]
- Gak E., Taylor W. G., Chan A. M., Rubin J. S. FEBS Lett. 1992;311:17–21. doi: 10.1016/0014-5793(92)81356-q. [DOI] [PubMed] [Google Scholar]
- Wang M. H., Gonias S. L., Skeel A., Wolf B. B., Yoshimura T., Leonard E. J. J. Biol. Chem. 1994;269:13806–13810. [PubMed] [Google Scholar]
- Kawaguchi M., Orikawa H., Baba T., Fukushima T., Kataoka H. Febs J. 2009;276:3481–3490. doi: 10.1111/j.1742-4658.2009.07070.x. [DOI] [PubMed] [Google Scholar]
- Parr C., Sanders A. J., Jiang W. G. Anti-Cancer Agents Med. Chem. 2010;10:47–57. doi: 10.2174/1871520611009010047. [DOI] [PubMed] [Google Scholar]
- Kataoka H., Miyata S., Uchinokura S., Itoh H. Cancer Metastasis Rev. 2003;22:223–236. doi: 10.1023/a:1023051500010. [DOI] [PubMed] [Google Scholar]
- Friis S., Sales K. U., Schafer J. M., Vogel L. K., Kataoka H., Bugge T. H. J. Biol. Chem. 2014;289:22319–22332. doi: 10.1074/jbc.M114.574400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z., Harris P. K. W., Jones D. E., Chugani R., Kim T., Agarwal M., Shen W., Wildman S. A., Janetka J. W. ACS Med. Chem. Lett. 2014;5:1219–1224. doi: 10.1021/ml500254r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberst M. D., Johnson M. D., Dickson R. B., Lin C.-Y., Singh B., Stewart M., Williams A., Al-Nafussi A., Smyth J. F., Gabra H., Sellar G. C. Clin. Cancer Res. 2002;8:1101–1107. [PubMed] [Google Scholar]
- Franco F. M., Jones D. E., Harris P. K., Han Z., Wildman S. A., Jarvis C. M., Janetka J. W. Bioorg. Med. Chem. 2015;23:2328–2343. doi: 10.1016/j.bmc.2015.03.072. [DOI] [PubMed] [Google Scholar]
- Hammami M., Rühmann E., Maurer E., Heine A., Gütschow M., Klebe G., Steinmetzer T. MedChemComm. 2012;3:807–813. [Google Scholar]
- Goswami R., Mukherjee S., Wohlfahrt G., Ghadiyaram C., Nagaraj J., Chandra B. R., Sistla R. K., Satyam L. K., Samiulla D. S., Moilanen A., Subramanya H. S., Ramachandra M. ACS Med. Chem. Lett. 2013;4:1152–1157. doi: 10.1021/ml400213v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venukadasula P. K. M., Owusu B. Y., Bansal N., Ross L. J., Hobrath J. V., Bao D., Truss J. W., Stackhouse M., Messick T. E., Klampfer L., Galemmo R. A. ACS Med. Chem. Lett. 2016;7:177–181. doi: 10.1021/acsmedchemlett.5b00357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z., Harris P. K., Karmakar P., Kim T., Owusu B. Y., Wildman S. A., Klampfer L., Janetka J. W. ChemMedChem. 2016;11:585–599. doi: 10.1002/cmdc.201500600. [DOI] [PubMed] [Google Scholar]
- Damalanka V. C., Han Z., Karmakar P., O'Donoghue A. J., La Greca F., Kim T., Pant S. M., Helander J., Klefström J., Craik C. S., Janetka J. W. J. Med. Chem. 2019;62:480–490. doi: 10.1021/acs.jmedchem.8b01536. [DOI] [PubMed] [Google Scholar]
- Colombo E., Désilets A., Duchêne D., Chagnon F., Najmanovich R., Leduc R., Marsault E. ACS Med. Chem. Lett. 2012;3:530–534. doi: 10.1021/ml3000534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damalanka V. C., Janetka J. W. Future Med. Chem. 2019;11:743–769. doi: 10.4155/fmc-2018-0446. [DOI] [PubMed] [Google Scholar]
- Li P., Jiang S., Lee S.-L., Lin C. Y., Johnson M. D., Dickson R. B., Michejda C. J., Roller P. P. J. Med. Chem. 2007;50:5976–5983. doi: 10.1021/jm0704898. [DOI] [PubMed] [Google Scholar]
- Venukadasula P. K. M., Owusu B. Y., Bansal N., Ross L. J., Hobrath J. V., Bao D., Truss J. W., Stackhouse M., Messick T. E., Klampfer L., Galemmo Jr. R. A. ACS Med. Chem. Lett. 2015;7:177–181. doi: 10.1021/acsmedchemlett.5b00357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant S. M., Mukonoweshuro A., Desai B., Ramjee M. K., Selway C. N., Tarver G. J., Wright A. G., Birchall K., Chapman T. M., Tervonen T. A., Klefström J. J. Med. Chem. 2018;61:4335–4347. doi: 10.1021/acs.jmedchem.7b01698. [DOI] [PubMed] [Google Scholar]
- Enyedy I. J., Lee S.-L., Kuo A. H., Dickson R. B., Lin C.-Y., Wang S. J. Med. Chem. 2001;44:1349–1355. doi: 10.1021/jm000395x. [DOI] [PubMed] [Google Scholar]
- Lin J. H. Curr. Drug Metab. 2009;10:661–691. doi: 10.2174/138920009789895499. [DOI] [PubMed] [Google Scholar]
- Craik D. J., Fairlie D. P., Liras S., Price D. Chem. Biol. Drug Des. 2013;81:136–147. doi: 10.1111/cbdd.12055. [DOI] [PubMed] [Google Scholar]
- Giordanetto F., Kihlberg J. J. Med. Chem. 2014;57:278–295. doi: 10.1021/jm400887j. [DOI] [PubMed] [Google Scholar]
- Mathiowetz A. M., Leung S. S. F. and Jacobson M. P., Optimizing the Permeability and Oral Bioavailability of Macrocycles, The Royal Society of Chemistry, 2015, ch. 10, pp. 367–397. [Google Scholar]
- Ganesan R., Eigenbrot C., Wu Y., Liang W.-C., Shia S., Lipari M. T., Kirchhofer D. Structure. 2009;17:1614–1624. doi: 10.1016/j.str.2009.09.014. [DOI] [PubMed] [Google Scholar]
- Steinmetzer T., Schweinitz A., Stürzebecher A., Dönnecke D., Uhland K., Schuster O., Steinmetzer P., Müller F., Friedrich R., Than M. E., Bode W., Stürzebecher J. J. Med. Chem. 2006;49:4116–4126. doi: 10.1021/jm051272l. [DOI] [PubMed] [Google Scholar]
- Herter S., Piper D. E., Aaron W., Gabriele T., Cutler G., Cao P., Bhatt A. S., Choe Y., Craik C. S., Walker N., Meininger D., Hoey T., Austin R. J. Biochem. J. 2005;390:125–136. doi: 10.1042/BJ20041955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owusu B. Y., Bansal N., Venukadasula P. K., Ross L. J., Messick T. E., Goel S., Galemmo R. A., Klampfer L. Oncotarget. 2016;7:29492–29506. doi: 10.18632/oncotarget.8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béliveau F., Désilets A., Leduc R. FEBS J. 2009;276:2213–2226. doi: 10.1111/j.1742-4658.2009.06950.x. [DOI] [PubMed] [Google Scholar]
- Herter S., Piper D. E., Aaron W., Gabriele T., Cutler G., Cao P., Bhatt A. S., Choe Y., Craik C. S., Walker N., Meininger D., Hoey T., Austin R. J. Biochem. J. 2005;390:125–136. doi: 10.1042/BJ20041955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damalanka V. C., Kim Y., Galasiti Kankanamalage A. C., Lushington G. H., Mehzabeen N., Battaile K. P., Lovell S., Chang K. O., Groutas W. C. Eur. J. Med. Chem. 2017;127:41–61. doi: 10.1016/j.ejmech.2016.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damalanka V. C., Kim Y., Galasiti Kankanamalage A. C., Rathnayake A. D., Mehzabeen N., Battaile K. P., Lovell S., Nguyen H. N., Lushington G. H., Chang K. O., Groutas W. C. Eur. J. Med. Chem. 2018;143:881–890. doi: 10.1016/j.ejmech.2017.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galasiti Kankanamalage A. C., Kim Y., Damalanka V. C., Rathnayake A. D., Fehr A. R., Mehzabeen N., Battaile K. P., Lovell S., Lushington G. H., Perlman S., Chang K. O., Groutas W. C. Eur. J. Med. Chem. 2018;150:334–346. doi: 10.1016/j.ejmech.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.