

ABSTRACT
Background: We have identified, in melanomas, a set of genes encoding proteins that mediate mechanical barrier function in normal skin (barrier molecule genes, BMGs) and whose overexpression is associated with decreased immune signatures and shorter patient survival. The most overexpressed of these, filaggrin (FLG), is expressed on chromosome 1q21.3, which also encodes genes of the epidermal differentiation complex (EDC). EDC genes may be regulated by the transcription factors (TFs) AHR and ARNT. We hypothesized that ARNT-related genes would be expressed concordantly with BMG and EDC genes, inversely associated with immune signatures, and enhanced by 1q21.3 copy gain.
Methods: Gene expression data from human melanomas in the Cancer Genome Atlas (TCGA), and a validation GEO dataset were evaluated, with copy number profiles from TCGA. Expression of Th1 immune genes and BMG/EDCs at 1q21.3 was visualized using clustered copy number and mRNA profiles. Associations of clusters and 1q21.3 copy number with patient survival and mRNA expression were assessed using Kaplan Meier curves, log-rank tests, and Wilcoxon rank sum tests.
Results: BMGs are concordantly expressed with EDC genes. Clustering divided tumors into 4 categories: (1) ImmuneHI, (2) BMG/EDCHI, (3) ARNTHI, (4) Mixed. Both ARNTHI and BMG/EDCHI tumors had low immune signatures and significantly shortened survival. KLF4 and FOXF2 are putative TFs that may regulate these genes.
Conclusions: ARNTHI tumors may represent another subset of tumors, in addition to BMG/EDCHI tumors, with barriers to immune infiltrates, likely with different mechanisms. These genes have prognostic significance and may be relevant targets for future therapy.
KEYWORDS: Barrier molecule genes, epidermal differentiation complex, melanoma, aryl hydrocarbon receptor, aryl hydrocarbon receptor nuclear translocator, chromosome 1q21.3 copy number gain
Introduction
Melanoma metastases lacking immune cell infiltrates are less responsive to immune therapy and are more likely to be lethal.1−5 Thus, there is a need to understand barriers to immune infiltration. Several such barriers have been proposed, including activation of WNT/β-catenin signaling6,7 and overexpression of endothelin receptor B (EDNRB).8,9 However, prior work did not demonstrate a survival effect when either of these gene sets were overexpressed. Additionally, we previously identified a set of 8 genes that are upregulated dramatically in a subset of melanomas lacking Th1 immune gene signatures: overexpression of these genes is also associated with poor survival.10 These genes encode proteins that mediate mechanical barrier functions in normal skin: filaggrin (FLG), dystonin (DST), plakophilin-3 (PKP3), desmocollin-3 (DSC-3), desmoplakin (DSP), trop-2 (TACSTD2), junction plakoglobin (JUP), and periplakin (PPL) and are expressed primarily in a mutually exclusive manner from WNT/β-catenin genes.10 Their gene products are key components of desmosomes or tight junctions, and these genes are referred to here as barrier molecule genes (BMGs). The mechanisms governing BMG overexpression may advance our understanding of their role in melanoma progression and whether their overexpression can be reversed therapeutically.
The most upregulated of these BMGs is FLG,10 which is encoded on chromosomal locus 1q21.3. Interestingly, 1q21.3 is a reported susceptibility locus in melanoma, based on single nucleotide polymorphisms in this region.11 Other genes on 1q21.3 code for proteins involved in terminal differentiation of keratinocytes and represent the epidermal differentiation complex (EDC).12–14 These are all co-expressed in keratinocytes, as are FLG and the other BMGs; however, we are not aware of any studies that have assessed whether they are concordantly expressed in melanomas. Interestingly, genomic analysis of melanomas by TCGA investigators defined 3 melanoma subgroups, one of which was a “keratin” subtype; however, that analysis did not address details of the EDC or the BMGs.4 We hypothesized that EDC genes would be concordantly upregulated with BMGs in a subset of human melanomas, and that they would thus similarly be inversely associated with lack of Th1 immune signatures in those tumors.
Concordant expression of the 8 BMGs that we originally reported10 suggests the possibility of global upregulation, possibly mediated by common transcription factors (TFs) or copy number amplification. In keratinocytes, upregulation of EDC gene expression during atopic dermatitis has been reported to be mediated by the aryl hydrocarbon receptor (AHR) and aryl hydrocarbon receptor nuclear translocator (ARNT),14,15 which heterodimerize and translocate, as an AHR:ARNT complex, to the nucleus.16 Also, there is evidence that EDC gene expression may be enhanced by other TFs, including krüppel-like factor 4 (KLF4), GATA3, GRHL3, AP1, and NRF2, among others.14 However, the effects of these TFs on BMGs outside 1q21.3 or on EDC genes in human melanomas is unclear. We hypothesized that ARNT would promote BMG/EDC expression in melanomas and would thus be overexpressed in BMG/EDCHi melanomas. Alternatively, we hypothesized that copy number variation in the 1q21.3 locus may result in EDC gene overexpression in some patients’ melanomas and may be associated with worse clinical outcomes. To address these hypotheses, we tested whether BMG, EDC, and AHR/ARNT genes are co-expressed in melanomas and are associated with clinical outcome. We also assessed copy number variation at chromosome 1q21.3 and enrichment for TF binding sites in the promoter regions of BMG, EDC, and ARNT associated genes that may explain concordant overexpression of those genes.
Materials and methods
RNA-seq, GISTIC copy number variation, and patient survival analysis
RNA-seq data from 471 patients with cutaneous melanoma were available from the Cancer Genome Atlas (TCGA) at cbioportal.org using the TCGA Provisional data set.17,18 RNA-seq data were downloaded for the genes in the WNT/β-catenin pathway, endothelin receptor B (EDNRB),8,19 EDC pathway,12–14 BMGs,10 ARNT-related genes, and Th1 immune genes, using our user-defined lists as inputs for gene query.1,10,20 WNT/β-catenin genes were curated from a publicly available list.6,21 Because ARNT was contained in cluster IV as shown in Figure 1, we named this gene set “ARNT-related genes”. Heatmaps were generated using unsupervised hierarchical clustering with Pearson correlation distance metric and Ward clustering method (“Ward.D” method using hclust in R). Affymetrix mRNA expression profiles from a cohort of 83 patients with melanoma were available from GEO with the GEO Series accession number GSE8401 and served as a validation study. The RNA expression profiles were converted to normalized Z-scores before being analyzed further.22 Only 54 of the 72 genes of interest were probed on the array in the validation study, and some genes had multiple probe sets. The genes that were not available in the validation data set, and therefore were excluded, were: ANXA8, C1ORF43, CD8B, FLG2, GRHL3, KPRP, LCE1C, LCE2A, LCE3A, LCE3C, LCE3D, LCE3E, PGLYRP3, RPTN, S100A7A, SPRR2E, SPRR2G, and SPRR4. Copy number profiles from 354 patients with metastatic melanoma were available from TCGA. Our gene set included BMG/EDC genes located on chromosome 1q21.3, as well as the 150 genes located on chromosome 1q21.3. Copy number gain was defined as greater than or equal to +1 (triploid), and a heatmap was generated using hierarchical clustering with a Euclidean distance metric and Ward clustering method. Associations of clusters with patient survival were assessed with Kaplan Meier curves and log-rank tests.
Figure 1.
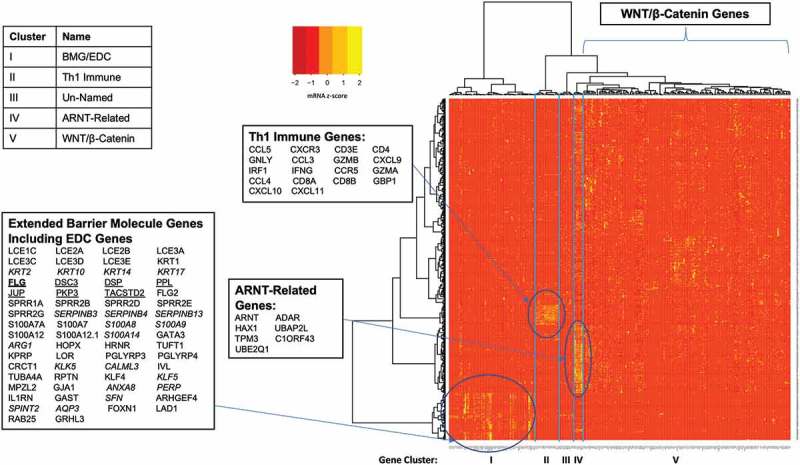
WNT/β-catenin pathway genes are expressed discordantly from barrier molecule and epidermal differentiation complex genes in melanomas. Heatmap of RNA-seq data from 471 patients with melanoma from the TCGA generated by hierarchical clustering of genes (x-axis) and patients (y-axis) identifies tumor clusters with high barrier molecule gene expression that is independent of WNT/β-catenin pathway gene expression.
Transcription factor binding site enrichment analysis
TF motif enrichment was performed by inputting BMG/EDC and ARNT-related genes into oPOSSUM Human Single Site Analysis package.23 The top 10 and bottom 10 TF results were selected for further analysis. Boxplots and Wilcoxon rank sum test were used to assess associations of 1q21.3 copy number gain with mRNA z-score. Box plots and scatterplots were generated in R. Linear regression was applied to fit the Z-score data shown in Figure 4(e).
Figure 4.
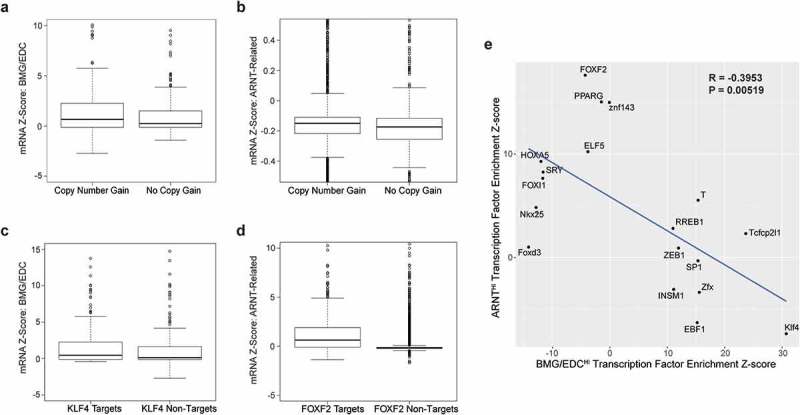
Chromosome 1q21.3 copy number gain, KLF4, and FOXF2 are potential mechanisms of upregulation of ARNT-related genes and BMG/EDC genes in melanoma. Boxplot of mRNA expression of BMG/EDC genes associated with no copy gain or copy number gain (a) or to KLF4 motif enrichment and non-enrichment (b). Boxplot of mRNA expression of ARNT-related genes associated with no copy gain or copy number gain (c) or to FOXF2 motif enrichment and non-enrichment (d). (e) Scatterplot of significantly enriched (Z-score > 10) or de-enriched (Z-score < −10) transcription factor motifs for BMG/EDC genes or ARNT-related genes identified using oPOSSUM.
Results
Screening for genes upregulated in tumors without immune signatures
In a prior analysis of gene expression in human melanomas, we screened 200 genes with greatest variability across samples24 and calculated mean levels for each, across each of three tumor subgroups.10 Group 1 tumors had high immune signatures, while groups 2 and 3 had low expression of immune genes. Expression in group 2, which lacked immune signatures, was divided by mean expression of 200 genes with greatest variance in the other groups to calculate fold increase. Only group 2 had significant patterns of upregulation of variably expressed genes, while groups 1 and 3 did not. Genes with 4.5-fold or greater expression in group 2 were selected for further study and are listed in Supplemental Table 1. These extend beyond our initial report on 8 BMGs.10 We refer to this larger dataset as the “extended BMGs.”
WNT/β-catenin pathway genes are discordantly expressed from BMGs in melanomas
In our prior study, BMG overexpression was largely discordant from WNT/β-catenin gene overexpression.10 For the present study, we evaluated the extended BMGs (n = 57, Supplemental Table 1), EDC genes on 1q21.3 (n = 50, Supplemental Table 2), EDNRB, Th1 immune genes (n = 18), and a large set of WNT/β-catenin genes (n = 131, Supplemental Table 3) for concordant or discordant expression in melanomas, using TCGA data (n = 471 patients). We hypothesized that extended BMGs and EDC genes would be (a) co-expressed with the 8 BMGs, (b) upregulated in melanomas lacking Th1 immune gene signatures, and (c) discordantly expressed from WNT/β-catenin pathway genes, suggesting different mechanisms of barriers to immune cell exclusion. We found that a subset of tumors had high Th1 immune gene expression and low expression of EDC, BMG, and WNT/β-catenin genes (Figure 1, Cluster II). Another subset concordantly upregulated both extended BMG and EDC genes, and lacked Th1 immune signatures (Cluster I). WNT/β-catenin genes were sporadically overexpressed in some other tumors lacking a Th1 immune gene signature and included AHR (Cluster V). Another cluster of 7 EDC genes from 1q21.3 (cluster IV) had striking concordant upregulation in yet another subset of melanomas lacking immune signatures. The Cluster IV genes include ARNT, so we refer to this cluster as “ARNT-related genes” (Figure 1). Further, we found that the extended BMGHI patient cluster (BMGHI ImmuneLO) had significantly shorter overall survival than ImmuneHI BMGLO tumors (Supplemental Figure 1).
Discordance of ARNT and BMG/EDC upregulation and survival associations
WNT/β-catenin genes were not concordantly overexpressed in any subset. Rather, among the Cluster V tumors, small numbers of WNT/β-catenin genes were overexpressed in different tumors. Since these expression patterns were sporadic, we repeated unsupervised hierarchical clustering of RNA-seq data from TCGA melanomas with genes representing clusters I, II, and IV. This divided tumors into 4 clusters: (1) ImmuneHI, (2) BMG/EDCHI, (3) ARNTHI, and (4) Mixed (Figure 2(a)). Seven ARNT-related genes clustered with DST, FGF4, GCG, and SPRR4, where SPRR4 is on 1q21.3. The remaining BMG/EDC genes were concordantly overexpressed, but ARNT-related genes clustered separately from them. As seen in the larger hierarchical clustering (Figure 1), ARNTHI melanomas and BMG/EDCHI melanomas again had low Th1 immune signatures, and ImmuneHI tumors had low expression of both BMG/EDC and ARNT-related genes (Figure 2(a,b)). This suggests independent mechanisms of immune exclusion in the BMG/EDCHI and ARNTHI patients and that ARNT may not regulate BMG/EDC gene expression in these melanomas.
Figure 2.
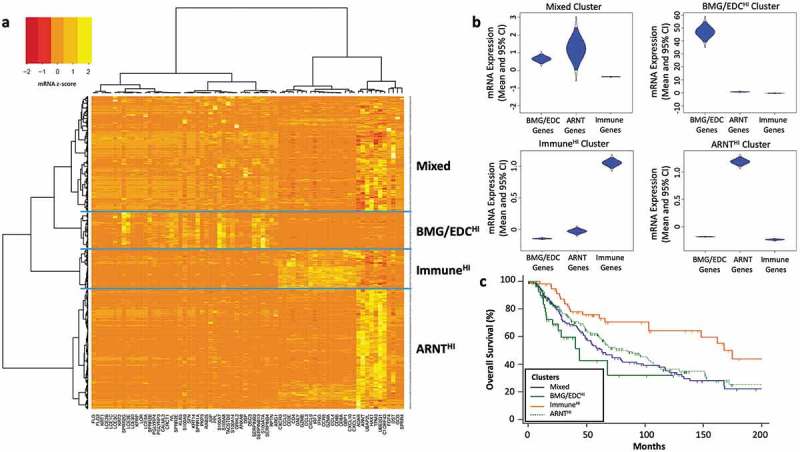
Barrier molecule and epidermal differentiation complex genes cluster together but are expressed discordantly from ARNT-related genes in melanomas. (a) Heatmap of RNA-seq data from 471 patients with metastatic melanoma from the TCGA generated by hierarchical clustering of genes (x-axis) and patients (y-axis) identifies 4 tumor clusters based on mRNA expression in gene subsets: Mixed (n = 174), BMG/EDCHI (n = 56), ImmuneHI (n = 62), and ARNTHI (n = 180). (b) Violin plots show distributions of mRNA expression data (c) and Kaplan Meier curves with survival data for patients across 4 clusters of tumors. Differences were assessed by log rank tests.
Median overall survival was 160, 90, 68, and 46 months, respectively, for ImmuneHI ARNTHI, Mixed, and BMG/EDCHI tumor clusters, and was significantly shorter for BMG/EDCHI patients than ImmuneHI (p = .0005) and ARNTHI (p = .0392, Figure 2(c)). Patient survival was also significantly worse for ARNTHI than ImmuneHI tumors (p = .0003).
This analysis was repeated using publicly available data from GEO with RNA expression profiles for 83 patients with melanoma. Similar clustering occurred in tumors, dividing them into 4 groups: (1) ImmuneHI, (2) BMG/EDCHI, (3) ARNTHI, and (4) Mixed (Supplemental Figure 2a). The genes also demonstrated nearly identical clustering patterns with few exceptions (four additional genes clustering with ARNT-related genes: TACSTD2, CD4.1, SERPINB13.3, and CALML3). A low Th1 immune signature was characteristic of BMG/EDCHI and ARNTHI melanomas (Supplemental Figure 2b). Overall survival was significantly shorter for Mixed (p = .0094) and ARNThi (p = .0192) tumors versus ImmuneHI, but not in BMG/EDCHI compared to ImmuneHI tumors (Supplemental Figure 2c).
Copy number gain at 1q21.3 locus contributes to upregulation of BMG/EDC and ARNT-related genes in metastatic melanoma
The association of chromosome 1q with copy number amplifications in melanomas25-28 and identification of BMG and EDC genes on chromosome 1q21.3, led us to hypothesize that copy number gain at this 1q21.3 may be a mechanism of upregulation of BMG/EDC and/or ARNT genes in patients with metastatic melanoma. Of 328 patients with metastatic melanoma, 52% had copy number gain encompassing the entirety of chromosome 1q21.3 (Figure 3(a); Supplemental Table 4), while 4% (n = 14) had deep deletions, and 44% (n = 143) were diploid. Among patients with 1q21.3 copy gains, 94% (n = 179) were triploid and 6.3% (n = 12) were tetraploid. Survival was significantly decreased for tumors with four copies of 1q21.3 compared to diploid (Figure 3(b), p = .0198). However, there was no significant survival difference between triploid and diploid cases. This suggests that marked amplification of chromosome 1q21.3 may be a marker of poor prognosis in melanoma.
Figure 3.
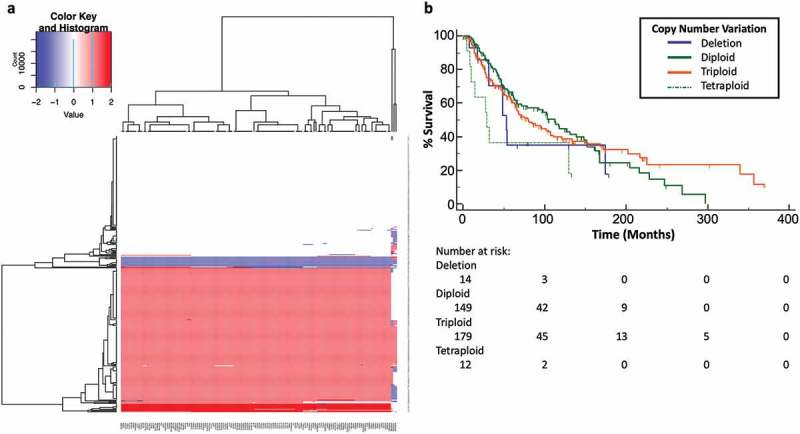
Chromosome 1q21.3 copy number gain contributes to global upregulation of ARNT-related genes and BMG/EDCs. (a) Heatmap of copy number profiles across genes at chromosome 1q21.3 (x-axis) and patients (y-axis) generated by hierarchical clustering in patients with metastatic melanomas from the TCGA. Deep deletion (−1) is indicated with dark blue, diploid (0) with white, triploidy (1) with light red, and tetraploidy (2) with dark red. (b) Kaplan Meier curves with survival data for patients with deep deletion (−1), diploidy (0), triploidy (+1), or tetraploidy (+2). Differences were assessed by log rank tests.
Of 43 genes that are concordantly upregulated in the BMG/EDCHI cluster, 4 of 21 extended BMGs and 20 of the 22 EDC genes are located at chromosome 1q21.3. We tested whether copy number gain of 1q21.3 explains upregulation of BMG/EDC genes in BMG/EDCHI tumors. Amplification of 1q21.3 was associated with higher mRNA expression of BMG/EDC genes on 1q21.3 in 61% of patients (Figure 4(a), p = .0469). Similarly, copy number gain at 1q21.3 was associated with increased expression of ARNT-related genes (Figure 4(b), p = 1.2e-07). Thus, copy number gain may be a mechanism for increased expression of both ARNT-related genes and BMG/EDC genes.
KLF4 is a putative transcriptional regulator of BMG/EDC genes
The discordant expression of ARNT-related genes and of AHR from BMG/EDC genes did not support AHR or ARNT as regulatory TFs for BMG/EDC genes in a subset of patients (Figure 2(a)). Additionally, we performed functional studies with AHR agonists/antagonists to test whether they may regulate the BMG/EDC genes in vitro. However, preliminary experiments did not support the role of AHR as a regulatory TF (data not shown). Thus, we explored whether other TFs may explain upregulation of BMG/EDC genes. TF motif enrichment analysis was performed on BMG/EDCHI tumors (n = 55) and identified KLF4 as a putative regulatory TF (Table 1) based on significantly increased prevalence of binding sites in 26 (61%) of those genes. mRNA z-scores of KLF4 motif-enriched genes BMG/EDC (n = 26) were upregulated compared to non-enriched genes (Figure 4(c), p = 8.6e-15). Other TFs with increased binding sites in genes in the BMG/EDCHI cluster were identified (Table 1). Interestingly, binding sites for ARNT and IRF1 were highly de-enriched (i.e., occurring below background frequencies) in promoter regions of BMG/EDC genes (Table 1). KLF4 co-expression with representative genes from each gene cluster was assessed. KLF4 expression was not significantly correlated with CD8A (Immune) or ARNT (Figure 5(a,b)); however, it correlated significantly with FLG expression (Figure 5(c)). These findings raise the possibility that KLF4 may enhance BMG/EDC gene expression.
Table 1.
Enriched and de-enriched transcription factors in promoter regions for BMG/EDCHI and ARNTHI melanomas.
| BMG/EDC Genes |
ARNT-Related Genes |
|||
|---|---|---|---|---|
| Transcription Factor | Z-Score | Transcription Factor | Z-Score | |
| Enriched | Klf4 | 30.733 | FOXF2 | 17.618 |
| Tcfcp2l1 | 23.684 | PPARG | 15.037 | |
| Zfx | 15.586 | znf143 | 14.986 | |
| T | 15.379 | ELF5 | 10.225 | |
| SP1 | 15.361 | NFATC2 | 9.617 | |
| EBF1 | 15.224 | HOXA5 | 9.282 | |
| ZEB1 | 11.961 | NFE2L2 | 9.243 | |
| INSM1 | 11.123 | FOXD1 | 9.148 | |
| RREB1 | 11.003 | FOXA1 | 9.073 | |
| Tal1:Gata1 Dimer | 9.961 | TLX1:NFIC Dimer | 8.684 | |
| De-Enriched | Foxd3 | −14.152 | Klf4 | −7.374 |
| Nkx2-5 | −12.848 | Ddit3:Cebpa Dimer | −6.313 | |
| HOXA5 | −11.994 | EBF1 | −6.3 | |
| FOXI1 | −11.684 | RORA_2 | −5.884 | |
| SRY | −11.627 | NR2F1 | −5.877 | |
| ARID3A | −9.869 | IRF1 | −5.798 | |
| NKX3-1 | −8.772 | NFYA | −5.776 | |
| Arnt | −7.571 | HIF1A:ARNT Dimer | −5.449 | |
| Pou5f1 | −7.328 | MIZF | −5.4 | |
| IRF1 | −7.225 | HNF4A | −4.775 | |
Figure 5.
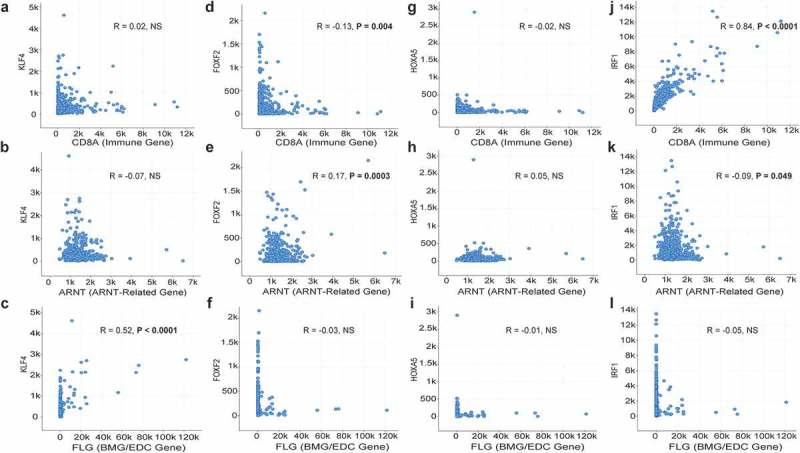
Co-expression of KLF4, FOXF2, HOXA5, and IRF1 with representative genes from Th1 immune, BMG/EDC, and ARNT-related gene clusters. RNA-seq data from 471 patients with metastatic melanoma from the TCGA with mRNA expression of: KLF4 compared to CD8A (a), ARNT (b), and FLG (c); FOXF2 compared to CD8A (d), ARNT (e), and FLG (f); HOXA5 compared to CD8A (g), ARNT (h), and FLG (i); and IRF1 compared to CD8A (j), ARNT (k), and FLG (l). R values are Pearson correlation values.
FOXF2 is a putative transcriptional regulator of ARNT-related genes
To identify, in silico, possible TFs supporting upregulation of ARNT-related genes, TF motif enrichment was repeated for ARNTHI tumors (n = 180). FOXF2 was identified as a putative regulatory TF, along with other TFs (Table 1). To assess whether FOXF2 may contribute to upregulation of ARNT-related genes and the 4 other genes that clustered with them in the ARNTHI cluster, mRNA z-scores of FOXF2 motif-enriched genes (6 or 54.5% of ARNT-related genes) to non-enriched genes (5 or 45.5% of ARNT-related genes) were compared. FOXF2 motif-enriched genes were significantly upregulated compared to others (Figure 4(d), p = 2.2e-16), suggesting that FOXF2 may promote expression of some ARNT-related genes. Binding sites for KLF4, IRF1, HIF1A:ARNT dimer, and others were highly de-enriched in promoter regions of ARNT-related genes (Table 1). Interestingly, FOXF2 expression was significantly anti-correlated with CD8A and positively correlated with ARNT (Figure 5(d,e)). FOXF2 expression did not correlate with FLG expression (Figure 5(f)). These findings suggest that FOXF2 may enhance expression of ARNT-related genes, but not immune or BMG/EDC genes.
Reciprocal regulation of BMG/EDC genes and ARNT-related genes
To explore whether anticorrelation of TF motif enrichment or de-enrichment could account for discordant expression of the BMG/EDC genes and the ARNT-related genes, a scatterplot was generated for enriched and de-enriched TF motif Z-scores, with Z-scores greater than 10 or less than −10 for genes expressed in BMG/EDCHI and ARNTHI clusters, respectively. There was significant anticorrelation between the motif Z-scores associated with genes expressed in the BMG/EDCHI and ARNTHI clusters (Figure 4(e)). There were significant associations of IRF1 with CD8A, and of KLF4 with FLG, and negative associations of FOXF2 vs. CD8A, and of IRF1 vs. FLG (Figure 5). These findings suggest that BMG/EDC genes and ARNT-related genes may have reciprocal regulation, which may be mediated by KLF4 and other TFs.
Discussion
We have found that EDC and extended BMGs are co-expressed together and upregulated in the absence of an immune gene signature in melanoma in two independent studies. There was an absence of a survival effect in the BMG/EDCHI group in the validation data set, which may be due to the small study size or inability to control for other prognostic factors. However, our other findings were validated. Many BMGs and EDC genes are important for terminal differentiation of keratinocytes and barrier function in normal skin and are located at chromosomal locus 1q21.3. Copy number amplification of 1q21.3 is weakly associated with upregulation of 1q21.3 genes, which includes some BMG/EDC and ARNT-related genes. Tetraploidy at this locus is a marker of poor prognosis. Interestingly, 1q21.3 amplification has been identified in breast cancers at higher risk of recurrence and may be actionable therapeutically.27
The absence of a survival effect in the BMG/EDCHI group in the validation data set was explored further. Interestingly, when metastatic melanomas were separated from primary melanomas, median survival was shorter for BMG/EDCHI metastases, but was not significant (P = .19), which can be attributed to the small sample size (n = 3). Separation of the small subset of tumors in the BMG/EDCHI group with a high immune signature from those with a low immune signature demonstrated greater median survival for BMG/EDCHI tumors with a high immune signature and a survival curve that also overlapped with ImmuneHI tumors. These findings may partially be due to the small sample size, but the BMG/EDCHI tumors with high immune signature are also all primary tumors, which also may explain some of the survival differences. Limitations of our study include small sample size of the validation study, differences in tumor composition (primary versus metastasis), inability to control for other prognostic factors, use of external data sets, and lack of confirmation of gene expression findings by immunohistochemical staining of tumors.
Although AHR and ARNT have been reported to induce BMG/EDC gene expression in keratinocytes,15,16 our in vitro studies did not confirm that, and the discordant expression of BMG/EDC genes from AHR and ARNT in melanomas suggests that BMG/EDC expression is largely ARNT-independent. Regardless, another novel finding is that ARNTHI melanomas represent a large subset that lack a Th1 immune signature, highlighting another subset of tumors, in addition to BMG/EDCHI tumors, where barriers to immune infiltration exist that may differ between these groups (Figure 2(a,b)). It will be important for future studies to assess whether BMG, EDC, or ARNT-related genes are mechanistically involved in barriers to immune cell infiltrates or in tumor progression.
Understanding TFs responsible for upregulating these genes will support these future studies and may enable therapeutic interventions. Our in silico studies suggest that KLF4 may upregulate BMG/EDC overexpression. KLF4 has not been well studied in melanomas; however, what little is known suggests a pro-tumorigenic role.29,30 Other studies support roles of KLF4 in decreasing apoptosis,29,31 and supporting melanoma cell stemness.32 Additionally, KLF4 knockout is associated with loss of barrier function and aberrations in keratinocyte differentiation in the skin and cornea.33–35 Thus, targeting KLF4 activation is a promising therapeutic approach for melanomas with high BMG/EDC gene expression to reduce barriers to immune infiltrates and also to mitigate the survival disadvantage conferred in this subgroup of patients. Interestingly, KLF4 and FOXF2 may have a potential antagonistic role toward β-catenin,36–38 and thus may contribute to the discordant expression of BMG/EDC genes and WNT/β-catenin genes.
This study identifies two distinct subsets of patients characterized by high expression of BMG/EDC or ARNT-related genes, each with a survival disadvantage and immune signature exclusion in melanomas. It also identifies 1q21.3 amplification as a contributory mechanism of upregulation of these genes and poor prognostic factor, and potential regulatory TFs KLF4 and FOXF2 that may act on these genes. Future studies will define how BMG/EDC genes and ARNT-related genes may interfere with immune cell infiltration and patient survival in melanoma and other cancers.
Funding Statement
This work was supported by the Cancer Research Institute [Clinical Laboratory Integration Project];Cancer Research Institute [Clinical Laboratory Integration Project];National Institutes of Health [R01 CA057653];National Institutes of Health [T32 CA163177].
Acknowledgments
Support was provided by United States Public Health Services Research grants R01 CA057653 (CL Slingluff), United States Public Health Services (USPHS) Training grant in Surgical Oncology T32 CA163177 (KM Leick), and Cancer Research Institute Clinical Laboratory Integration Project (CL Slingluff, KM Leick). We acknowledge Rebeka Eki, Mouadh Benamar, and Tarek Abbas for their assistance with preliminary experiments using AHR/ARNT agonists and antagonists, the results of which were not shown. Ashana Weeraratna provided guidance with regards to the WNT/β-catenin pathway.
Disclosures
The following disclosures apply to Dr. Slingluff, but are not related to this work: Research support to the University of Virginia from Celldex (funding, drug), Glaxo-Smith Kline (funding), Merck (funding, drug), 3M (drug), Theraclion (device staff support); Funding to the University of Virginia for Scientific Advisory Boards for Immatics, CureVac, Celldex. Also Dr. Slingluff receives license fee payments through the UVA Licensing and Ventures Group for patents for peptides used in cancer vaccines.
Synopsis
Melanomas with high ARNT-related gene expression represent a subset of tumors, in addition to those with high barrier molecule or epidermal differentiation complex gene expression, where immune barriers exist, suggesting that barriers to immune infiltrates may differ between these groups.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
References
- 1.Erdag G, Schaefer JT, Smolkin ME, Deacon DH, Shea SM, Dengel LT, Patterson JW, Slingluff CL.. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res. 2012;72(5):1. doi: 10.1158/0008-5472.CAN-11-3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bogunovic D, O’Neill DW, Belitskaya-Levy I, Vacic V, Yu Y-L, Adams S, Darvishian F, Berman R, Shapiro R, Pavlick AC, et al. Immune profile and mitotic index of metastatic melanoma lesions enhance clinical staging in predicting patient survival. Proc Natl Acad Sci U S A. 2009;106(48):20429–8. doi: 10.1073/pnas.0905139106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mihm MC Jr., Clemente CG, Cascinelli N.. Tumor-infiltrating lymphocytes in lymph node melanoma metastases: a histopathologic prognostic indicator and an expression of local immune response. Lab Invest. 1996;74:43–47. [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas, Network Genomic Classification of Cutaneous Melanoma. Cell. 2015;161(7):1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Edwards J, Wilmott JS, Madore J, Gide TN, Quek C, Tasker A, Ferguson A, Chen J, Hewavisenti R, Hershey P, et al. CD103+ tumor-resident CD8+ T cells are associated with improved survival in immunotherapy naive melanoma patients and expand significantly during anti-PD1 treatment. Clin Cancer Res. 2018. doi: 10.1158/1078-0432.CCR-17-2257. [DOI] [PubMed] [Google Scholar]
- 6.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523(7559):231–235. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 7.Spranger S, Zha YY, Gajewski T. Effect of melanoma intrinsic beta-catenin signaling on immune exclusion and resistance to immunotherapies. J Clin Oncol. 2015;33(15):1. doi: 10.1200/jco.2015.33.15_suppl.9014.25332246 [DOI] [Google Scholar]
- 8.Kandalaft LE, Facciabene A, Buckanovich RJ, Coukos G. Endothelin B receptor, a new target in cancer immune therapy. Clin Cancer Res. 2009;15(14):4521–4528. doi: 10.1158/1078-0432.CCR-08-0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, Katsaros D, O’Brien-Jenkins A, Gimotty PA, Coukos G. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med. 2008;14(1):28–36. doi: 10.1038/nm1699. [DOI] [PubMed] [Google Scholar]
- 10.Salerno EP, Bedognetti D, Mauldin IS, Deacon DH, Shea SM, Pinczewski J, Obeid JM, Coukos G, Wang E, Gajewski TF, et al. Human melanomas and ovarian cancers overexpressing mechanical barrier molecule genes lack immune signatures and have increased patient mortality risk. Oncoimmunology. 2016;5(12):e1240857. doi: 10.1080/2162402X.2016.1240857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Macgregor S, Montgomery GW, Liu JZ, Zhao ZZ, Henders AK, Stark M, Schmid H, Holland EA, Duffy DL, Zhang M, et al. Genome-wide association study identifies a new melanoma susceptibility locus at 1q21.3. Nat Genet. 2011;43(11):1114–1118. doi: 10.1038/ng.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mischke D, Korge BP, Marenholz I, Volz A, Ziegler A. Genes encoding structural proteins of epidermal cornification and S100 calcium-binding proteins form a gene complex (“epidermal differentiation complex”) on human chromosome 1q21. J Invest Dermatol. 1996;106(5):989–992. doi: 10.1111/1523-1747.ep12338501. [DOI] [PubMed] [Google Scholar]
- 13.Marenholz I, Zirra M, Fischer DF, Backendorf C, Ziegler A, Mischke D. Identification of human epidermal differentiation complex (EDC)-encoded genes by subtractive hybridization of entire YACs to a gridded keratinocyte cDNA library. Genome Res. 2001;11(3):341–355. doi: 10.1101/gr.114801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kypriotou M, Huber M, Hohl D. The human epidermal differentiation complex: cornified envelope precursors, S100 proteins and the ‘fused genes’ family. Exp Dermatol. 2012;21(9):643–649. doi: 10.1111/j.1600-0625.2012.01472.x. [DOI] [PubMed] [Google Scholar]
- 15.Furue M, Uchi H, Mitoma C, Hashimoto-Hachiya A, Chiba T, Ito T, Nakahara T, Tsuji G. Antioxidants for healthy skin: the emerging role of aryl hydrocarbon receptors and Nuclear factor-erythroid 2-related factor-2. Nutrients. 2017;9(3):223. doi: 10.3390/nu9030223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Furue M, Tsuji G, Mitoma C, Nakahara T, Chiba T, Morino-Koga S, Uchi H. Gene regulation of filaggrin and other skin barrier proteins via aryl hydrocarbon receptor. J Dermatol Sci. 2015;80(2):83–88. doi: 10.1016/j.jdermsci.2015.07.011. [DOI] [PubMed] [Google Scholar]
- 17.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, Katsaros D, O'Brien-Jenkins A, Gimotty PA, Coukos G. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. NatMed. 2008;14(1):28. [DOI] [PubMed] [Google Scholar]
- 20.Galon J, Angell HK, Bedognetti D, Marincola FM. The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity. 2013;39(1):11. doi: 10.1016/j.immuni.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 21.Nusse R, Lim X.. The wnt homepage. [accessed April1 2017] http://web.stanford.edu/~rnusse/pathways/targets.html.
- 22.Xu L, Shen SS, Hoshida Y, Subramanian A, Ross K, Brunet J-P, Wagner SN, Ramaswamy S, Mesirov JP, Hynes RO. Gene expression changes in an animal melanoma model correlate with aggressiveness of human melanoma metastases. Mol Cancer Res. 2008;6(5):760–769. doi: 10.1158/1541-7786.MCR-07-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ho Sui SJ, Mortimer JR, Arenillas DJ, Brumm J, Walsh CJ, Kennedy BP, Wasserman WW. oPOSSUM: identification of over-represented transcription factor binding sites in co-expressed genes. Nucleic Acids Res. 2005;33(10):3154–3164. doi: 10.1093/nar/gki624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, Gajewski TF. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009;69(7):3077–3085. doi: 10.1158/0008-5472.CAN-08-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Balazs M, Adam Z, Treszl A, Begany A, Hunyadi J, Adany R. Chromosomal imbalances in primary and metastatic melanomas revealed by comparative genomic hybridization. Cytometry. 2001;46:222–232. [DOI] [PubMed] [Google Scholar]
- 26.Chen D, Zhou D, Xu J, Zhou R, Ouyang J, Chen B. Prognostic value of 1q21 gain in multiple myeloma. Clin Lymphoma Myeloma Leuk. 2019. e159-e164;19(3):e159–e164. doi: 10.1016/j.clml.2018.12.002. [DOI] [PubMed] [Google Scholar]
- 27.Goh JY, Feng M, Wang W, Oguz G, Yatim SMJM, Lee PL, Bao Y, Lim TH, Wang P, Tam WL, et al. Chromosome 1q21.3 amplification is a trackable biomarker and actionable target for breast cancer recurrence. Nat Med. 2017;23(11):1319–1330. doi: 10.1038/nm.4405. [DOI] [PubMed] [Google Scholar]
- 28.Wu C, Yang T, Liu Y, Lu Y, Yang Y, Liu X, Liu X, Ye L, Sun Y, Wang X, et al. ARNT/HIF-1beta links high-risk 1q21 gain and microenvironmental hypoxia to drug resistance and poor prognosis in multiple myeloma. Cancer Med. 2018;7(8):3899–3911. doi: 10.1002/cam4.1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, Sun HJ, Li RG, Wang XM, Cheng ZQ, Lou N. Reprogramming factors induce proliferation and inhibit apoptosis of melanoma cells by changing the expression of particular genes. Mol Med Rep. 2019;19(2):967–973. doi: 10.3892/mmr.2018.9753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Riverso M, Montagnani V, Stecca B. KLF4 is regulated by RAS/RAF/MEK/ERK signaling through E2F1 and promotes melanoma cell growth. Oncogene. 2017;36(23):3322–3333. doi: 10.1038/onc.2016.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang D, Lin J, Chao Y, Zhang L, Jin L, Li N, He R, Ma B, Zhao W, Han C. Regulation of the adaptation to ER stress by KLF4 facilitates melanoma cell metastasis via upregulating NUCB2 expression. J Exp Clin Cancer Res. 2018;37(1):176. doi: 10.1186/s13046-018-0842-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y, Mou Y, Zhang H, Wang X, Li R, Cheng Z, Liu X. Reprogramming factors remodel melanoma cell phenotype by changing stat3 expression. Int J Med Sci. 2017;14(13):1402–1409. doi: 10.7150/ijms.21952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Segre JA, Bauer C, Fuchs E. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat Genet. 1999;22(4):356–360. doi: 10.1038/11926. [DOI] [PubMed] [Google Scholar]
- 34.Swamynathan SK, Davis J, Piatigorsky J. Identification of candidate Klf4 target genes reveals the molecular basis of the diverse regulatory roles of Klf4 in the mouse cornea. Invest Ophthalmol Vis Sci. 2008;49(8):3360–3370. doi: 10.1167/iovs.08-1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jaubert J, Cheng J, Segre JA. Ectopic expression of kruppel like factor 4 (Klf4) accelerates formation of the epidermal permeability barrier. Development. 2003;130(12):2767–2777. doi: 10.1242/dev.00477. [DOI] [PubMed] [Google Scholar]
- 36.Evans PM, Chen X, Zhang W, Liu C. KLF4 interacts with beta-catenin/TCF4 and blocks p300/CBP recruitment by beta-catenin. Mol Cell Biol. 2010;30(2):372–381. doi: 10.1128/MCB.00063-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ormestad M, Astorga J, Landgren H, Wang T, Johansson BR, Miura N, Carlsson P. Foxf1 and Foxf2 control murine gut development by limiting mesenchymal Wnt signaling and promoting extracellular matrix production. Development. 2006;133(5):833–843. doi: 10.1242/dev.02252. [DOI] [PubMed] [Google Scholar]
- 38.Zhang J, Zhang C, Sang L, Huang L, Du J, Zhao X. FOXF2 inhibits proliferation, migration, and invasion of hela cells by regulating Wnt signaling pathway. Biosci Rep. 2018;38(5). doi: 10.1042/BSR20180747. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Nusse R, Lim X.. The wnt homepage. [accessed April1 2017] http://web.stanford.edu/~rnusse/pathways/targets.html.