

ABSTRACT
Increased T-regulatory cell activity drives tumor progression in the compound APCmin/+/enterotoxic Bacteroides fragilis colon cancer model. At the same time, how microbially-induced inflammation promotes T-regulatory cell expansion in the dysplastic intestine remains poorly described. Analysis of post-infection immune cell kinetics in the colon lamina propria revealed that CD4+ Foxp3+ cell numbers increased by 25-fold between days 3–14. Importantly, T-regulatory cell expansion was preceded by a 12-fold spike in lamina propria CD11b+ cell numbers between days 0–4; suggesting a link between the myeloid compartment and the T-regulatory cells. Consistent with this notion, in vitro co-culture studies utilizing sorted myeloid cell subsets and CD4+ T-cells demonstrated that the CD11b+CX3CR1+ but not the CD11b+CX3CR1− subset preferentially induced Foxp3 expression in CD4+ T-cells. Phenotypic analysis revealed that the CD11b+CX3CR1+ subset represented a homogenous CD64+CD24−CD103a− macrophage population. Global CX3CR1 knockout or conditional depletion of CX3CR1+ myeloid cells resulted in diminished CD4+Foxp3+ cell expansion and a 3 to 6-fold reduction in tumor burden establishing CX3CR1+ macrophages as a major driver of the T-regulatory cell-tumor axis. Quantitative analysis of CD11b+ myeloid cell subsets for IFNβ mRNA revealed that the CX3CR1+ macrophages expressed 15-fold higher levels of IFNβ in comparison to the CX3CR1− myeloid subset. Antibody mediated neutralization of IFNβ resulted in the suppression of CD4+Foxp3+ cell induction and tumor growth, demonstrating the central role of IFNβ in mediating CX3CR1+ macrophage-driven T-regulatory cell expansion. These studies shed new mechanistic light on the cellular ontogeny of pro-tumorigenic T-regulatory cells in the inflamed colon of the APCmin/+ mouse.
KEYWORDS: Colon cancer, T-regulatory cells, CX3CR1, macrophages, IFNβ
Introduction
APCmin/+/enterotoxic Bacteroides fragilis (ETBF) murine model of colon cancer1 has been used extensively to study the role of microbially-induced inflammation in colon tumorigenesis and tumor progression. Specifically, a series of elegant studies by the Sears and the Housseau laboratories have demonstrated the central role of type 17 T-cell immunity in tumor development and growth in this model; and delineated how ETBF promotes type 17 immunity via its pleiotropic activity on gut epithelium and the myeloid cell compartment.1–5 Separately, these and other studies, including those from our laboratory, revealed an equally critical role for T-regulatory cells (Treg) in driving tumor growth in the ETBF-colonized gut.6,7 Importantly, a recent report demonstrated that Treg can preferentially help to expand Th17 cells by acting as a sink for IL-2 in the above model, establishing a novel link between the two pathogenic subsets.6
While these reports illuminated the cellular origin and the pathogenic role of type 17 inflammation that accompanies and drives colon tumorigenesis, the ontogeny of the ETBF-driven pro-tumorigenic Treg response remains poorly understood. It is known that under steady-state conditions gut-resident CD11b+CX3CR1+ myeloid cells are critical to the maintenance of immune homeostasis in the GI tract8,9 and more specifically that of the lamina propria (LP) Treg compartment.10 However, the cellular mechanisms that regulate the generation and expansion of pro-tumorigenic Treg in the inflamed dysplastic colon have not been directly addressed.
Herein we report that in the APCmin/+/ETBF model Treg can drive tumor growth independent of Th17 cell activity and that the ontogeny of pro-tumorigenic Treg is linked to the CD11b+CD64+CD24−CD103a−CX3CR1+ gut-resident macrophages, which expand within 48 hours of bacterial colonization. We further show that the unique ability of these macrophages to rapidly expand Treg is associated, at least in part, with production of IFNβ that is required both for Treg induction and tumorigenesis.
Results
CD4+Foxp3+ Treg expansion is rapid and is required for tumor growth independent of Th17 cell activity
In order to obtain insight into the ontogeny of ETBF-induced Treg we first analyzed the Treg kinetics in the inflamed colon of the APCmin/+ mouse. Treg numbers in the colon LP were monitored between days 0 (un-infected mice) and day 21 post-colonization. The data shown in Figure 1a demonstrate that Treg expansion commenced between days 3 and 6 post-ETBF (8-fold), reached its peak on day 14 (25-fold) and declined thereafter but remained significantly above baseline (13-fold). To confirm that this dramatic increase drove tumor growth, we determined whether depletion of Treg affected tumor burden in APCmin/+ mice that were heterozygous for Foxp3DTR/+. Mice were administered diphtheria toxin (DT) starting on day 7 post-ETBF administration for 3 weeks and tumor burden was analyzed in control vs DT-treated animals. Figure 1b shows that depletion of Foxp3+ T-cells (Supplemental Figure 1A) resulted in a robust 5-fold reduction in tumor number confirming the critical role of Treg alone in tumor growth. Analysis of colon LP T-cell subsets revealed that Treg depletion resulted in global T-effector cell proliferation (Figure 1c). Specifically, all effector T-cell subset numbers, including Th1, Th17 and CD8+ T-cells increased by ~28, 7 and 55-fold, respectively. Suppression of tumor growth in Treg-depleted mice, despite continued Th17 cell expansion, suggested that Treg drove tumor growth independent of Th17 cells. Separately, in addition to dramatically-increased numbers, Th1 and CD8+ T-cells displayed enhanced activation as measured by IFNγ+ production (Figure 1c), establishing a link between Treg presence and cytotoxic T-effector function.
Figure 1.
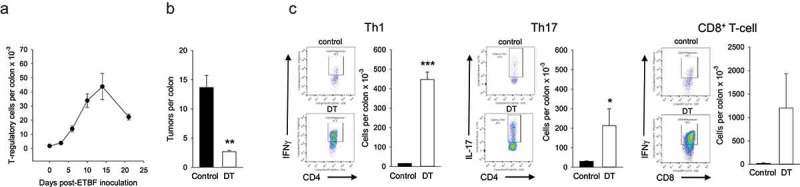
Effect of CD4+Foxp3+ Treg on tumorigenesis and T-effector cell activity.
Panel a. Kinetics of Treg expansion in ETBF-colonized colon. Colons were harvested on indicated days post-ETBF colonization and total LP Treg numbers were determined per colon. Error bars = SEM, n = 3/time point. Days 5–21 were significantly different from Day 0 (p < .01). Panel b. Colon tumor burden in the presence and absence of Treg. Tumor burden was quantified 3 weeks after ETBF administration in control and DT-administered mice. Error bars = SEM, n = 3/time point (** denotes p < .01). Panel c. The colons from control and DT-administered mice were harvested and LP T-effector subsets were quantified. Representative flow panels showing percent cytokine-producing cells and histograms depicting respective T-effector cell numbers/colon are shown. Error bars = SEM, n = 3/time point (* and *** denote p < .05 and 0.001, respectively). Data from one of two repeats with similar results are shown.
Treg expansion is driven by the CD11b+CX3CR1+ myeloid cell subset
The essential role of LP-resident CX3CR1+ myeloid cell-Treg axis in the maintenance of gut immune homeostasis is well-known.8–10 Separately, recent studies established that ETBF colonization results in major quantitative and qualitative changes in myeloid cell populations that are found in the colonic LP.4 We thus hypothesized that gut-resident CX3CR1+ myeloid cells may be critical to regulating post-infection Treg activity. To this end, we first analyzed the quantitative changes in CD11b+ myeloid cells in the colon during the first week of colonization. Figure 2a shows that ETBF induced rapid quantitative changes in both the CD11b+CX3CR1+ and the CD11b+CX3CR1− subsets. Specifically, CD11b+CX3CR1+ cell numbers increased by 6.5-fold on day 2 and peaked at 13-fold above background on day 4. CD11b+CX3CR1− subset prevalence increased with somewhat slower kinetics (~2-fold on day 2) but ultimately caught up (11-fold on day 4). These findings showed that the CD11b+CX3CR1+ subset expansion preceded the Treg response by ~2–3 days, consistent with a role for this subset in driving Treg ontogeny.
Figure 2.
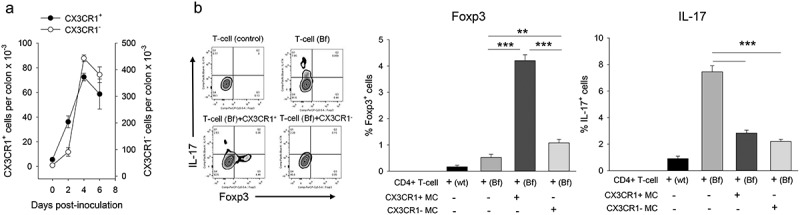
Post-ETBF myeloid cell subset kinetics and function.
Panel a. CD11b+ myeloid cell subset kinetics in the colon. LP CD11b+CX3CR1+ and CD11b+CX3CR1− myeloid cell numbers were quantified in post-ETBF mice. Error bars = SEM, n = 3-6/time point. Days 2–6 were significantly different than day 0 (p < .05). Panel b. In vitro co-culture assay. Sorted CD11b+ cell subsets were incubated with CD4+ T-cells as described in the Methods. Control T-cells were isolated from naïve APCmin/+ mice whereas experimental T-cells (Bf) were purified from ETBF-inoculated mice. Representative FACS panels are shown for control (T-cells only, top row) and experimental (T-cells + myeloid cells, bottom row). CD4+ T-cells were gated on and analyzed for Foxp3 vs IL-17 production. Quantitative data are shown for percent Foxp3+ and IL-17+ cells for each well. Error bars = SEM, n = 3 mice per group (** and *** denote p < .01 and < 0.001, respectively). Data from one of two repeats with similar results are shown.
We then focused on the ability of CX3CR1+ vs CX3CR1 – subsets to promote Foxp3 expression in CD4+ T-cells. For this we utilized an in vitro co-culture system similar to one we used previously.11 CD11b+CX3CR1+ and CD11b+CX3CR1− subsets were sorted and co-incubated with purified CD4+ T-cells in culture in the presence of IL-2 for 4 days and analyzed for Foxp3 vs IL-17 expression (corresponding to the two subsets that have been linked to tumorigenesis in this model). The data are shown in Figure 2b. The findings demonstrate that the CX3CR1+ subset preferentially induced Foxp3 expression in CD4+ T-cells that were sorted from ETBF-colonized mice (8-fold increase) vs the CX3CR1− subset (2-fold increase). In contrast, both myeloid cell subsets suppressed IL-17 expression in CD4+ T-cells, suggesting that post-ETBF Th17-cell expansion, which accompanied Treg expansion with similar kinetics (Supplemental Figure 1B) was likely driven by another cell type.
Next, we wanted to determine whether the CX3CR1+ subset was essential to Treg expansion in vivo. For this we initially used a global CX3CR1 knockout model in the APCmin/+ background. APCmin/+ mice were backcrossed to CX3CR1flstopflDTR/flstopflDTR mice to obtain APCmin/+ mice homozygous for the modified DTR gene insertion downstream of the endogenous CX3CR1 promotor, which results in an inactive CX3CR1 gene (Supplemental Figure 2). These mice were then administered ETBF and tumor development was evaluated 4 weeks post-infection. Figure 3a demonstrates that global loss of CX3CR1 expression led to a 5.5-fold reduction in colon tumor burden, establishing the critical requirement for CX3CR1 expression during tumorigenesis. Moreover, analysis of colon LP T-cell subsets revealed a significant ~2-fold reduction in Treg accompanied with a 2-fold increase in IFNγ-producing CD8 + T-cells consistent with the Figure 1 data. In contrast, there was no significant effect on Th1/Th17 populations (Supplemental Figure 3A). These findings were consistent with our hypothesis that CX3CR1+ myeloid cells were important to Treg expansion and tumor growth in the above model.
Figure 3.
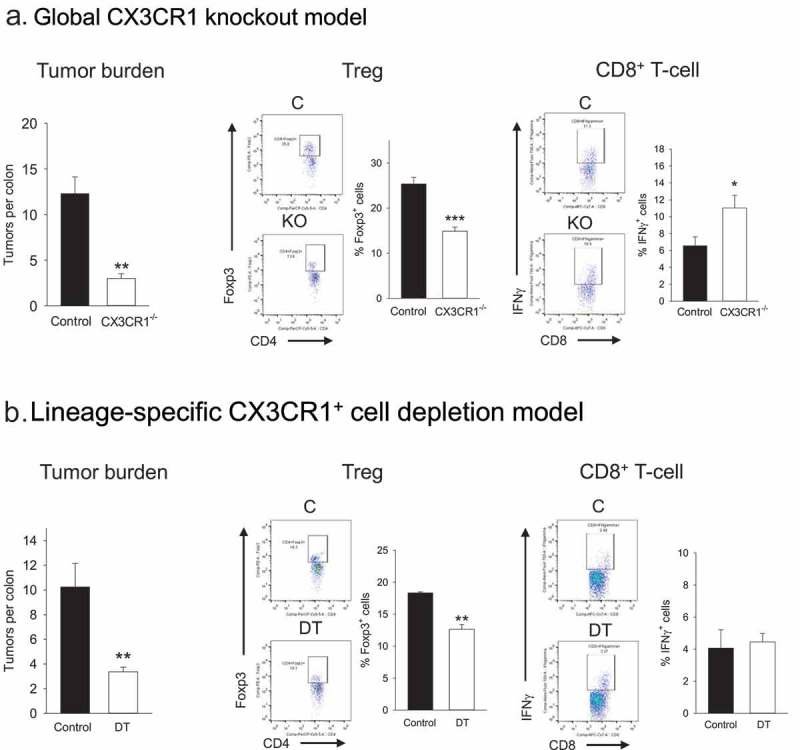
Role of CX3CR1+ myeloid cells in Treg induction and T-effector cell activity in vivo.
Panel a. Effect of global knockout of CX3CR1+ on tumor burden and T-cell activity. Control APCmin/+ or APCmin/+ CX3CR1 flstopflDTR/flstopflDTR mice were inoculated with ETBF and tumor burden/colon LP T-cell activity were analyzed 3 weeks after bacteria administration. Representative FACS panels and quantitative data for Treg and CD8+ T-cells are shown. Error bars = SEM, n = 5–6 mice per group (*, ** and *** denote p < .05, 0.01 and 0.001, respectively). Panel b. Effect of CX3CR1+ myeloid cell depletion on tumor burden and T-cell activity. Control or DT-administered LysMcre/+CX3CR1flstopflDTR/+APCmin/+ mouse colons were analyzed 3 weeks post-ETBF for tumor burden and T-cell activity. Representative FACS panels and quantitative data for Treg and CD8+ T-cells are shown. Error bars = SEM, n = 6 mice per group. ** denotes p < .01.
Figure 3a data convincingly demonstrated an important role for CX3CR1 in Treg expansion/tumor growth. At the same time, since CX3CR1 can be expressed by multiple leukocyte subsets we could not exclude the possibility of contribution by CX3CR1+ cells that are non-myeloid in origin to Treg expansion/tumor progression. To address this issue directly, we generated a double transgenic mouse in the APCmin/+ background, i.e. LysMcre/+CX3CR1flstopflDTR/+APCmin/+ mice in which CX3CR1+ myeloid cells could be partially depleted in a lineage-specific manner by DT administration (Supplemental Figure 2B). These mice were administered ETBF and starting one week after bacterial inoculation were either treated with DT or PBS for 3 weeks. The colons were then analyzed for tumor burden and T-cell subset prevalence. Figure 3b data show that partial elimination of CX3CR1+ myeloid cells resulted in a 3-fold reduction of tumor burden confirming the specific role of this subset in tumorigenesis/tumor growth (Figure 3b). Furthermore, loss of CX3CR1+ myeloid cells resulted in a modest but significant decrease in Treg prevalence (Figure 3b) consistent with a central role for CD11b+CX3CR1+ myeloid subset in specifically driving the Treg-tumor axis. In this model, partial depletion of CX3CR1+ myeloid cells did not affect CD8+ T-cell activity (Figure 3b) or Th1/17 cell prevalence (Supplemental Figure 3B).
CD11b+CX3CR1+ subset represents a homogenous CD64+CD24−CD103a− macrophage population
The above data confirmed the critical involvement of gut-resident CD11b+CX3CR1+ myeloid cells in post-ETBF Treg expansion. However, whether this population represented a unique cell type, or a heterogenous mix of dendritic cell (DC) and/or macrophage subsets that are found in the inflamed gut,12,13 remained undetermined. To this end, further phenotypic analysis of CD11b+CX3CR1+ myeloid cells infiltrating the colonic LP in day 3 post-ETBF mice was undertaken. The data shown in Figure 4 demonstrate that this subset consisted overwhelmingly (≥90%) of CD64+CD24−CD103a− cells, consistent with macrophage lineage, as well as morphology.14,15 Further analysis confirmed that CD64, CX3CR1 and CD103a expression patterns clearly distinguished these macrophages from CD11c+MHCII+CD64−CD24+CD103a+ DC in both phenotype and morphology (Supplemental Figure 4). Separately, CX3CR1+ macrophages expressed high levels of CD11c and MHCII, again consistent with that of an activated LP-resident subset16 (Supplemental Figure 4).
Figure 4.
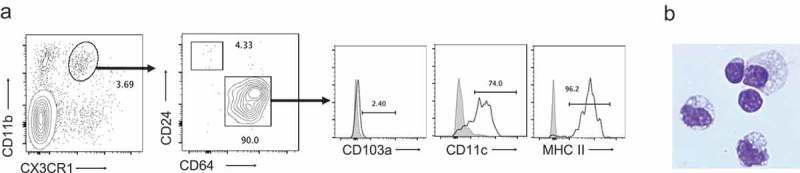
Phenotypic and morphological characterization of CD11b+CX3CR1+ myeloid cells.
Panel a. LP CD45.2+ cells that were CD11b+CX3CR1+ were analyzed for CD24 and CD64 expression. CD11b+CX3CR1+CD64+ macrophages were then assessed for CD103a, CD11c and MHCII (shaded histograms = fluorescence minus one control; open histograms = antibody staining). Panel b. CD11b+CX3CR1+ subset was sorted and analyzed by cytospin. Intact cells demonstrated abundant foamy cytoplasm with prominent cytoplasmic vacuoles typical of macrophage morphology. Magnification = 800x.
Preferential expansion of Treg by CX3CR1+ macrophages requires IFNβ
Whereas the above findings supported a major role for the CX3CR1+ macrophages in promoting the expansion of Treg in the LP of ETBF-inflamed colon, they did not provide insight into the molecular mechanism that mediates this expansion. A single recent study reported a requirement for IFNβ in the maintenance of Treg in the gut by resident CX3CR1+ myeloid cells.17 To determine whether a similar mechanism was responsible for the ontogeny of the Treg in the ETBF-infected colon, we first analyzed IFNβ expression in CD11b+ cell subsets. CX3CR1-positive and negative subsets were sorted and analyzed for IFNβ expression by qPCR. Figure 5a shows that CD11b+CX3CR1+ macrophages expressed significantly higher levels of IFNβ mRNA in comparison to CX3CR1− subset both prior to (~5-fold) and subsequent to (~15-fold) bacterial inoculation. In contrast, no significant differences were observed in the relative levels of TNFα mRNA between the two subsets. Importantly, treatment of mice with a neutralizing anti-IFNβ antibody not only reduced Treg prevalence in the colonic LP but also resulted in a significant (~2.5-fold) reduction in tumor burden (Figure 5b). The reduction in Treg/tumor burden was accompanied with increased CD8+ T-cell activity similar to that observed in Figure 1, again consistent with a link between IFNβ and Treg activity. IFNβ neutralization resulted in a tendency toward increased Th1 and Th17 activity, which did not reach significance (Supplemental Figure 5). Collectively, these findings strongly support a role, at least in part, for IFNβ in mediating the CX3CR1+ macrophage-mediated expansion of colonic Treg.
Figure 5.
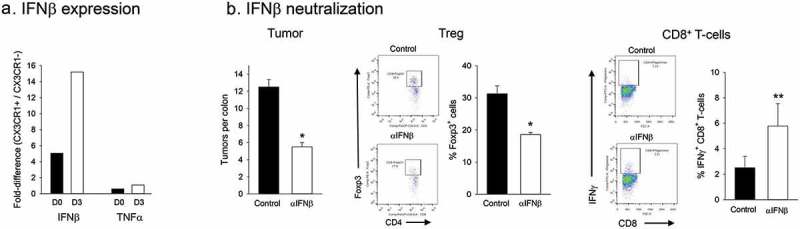
Role of IFNβ in CX3CR1+ macrophage-mediated Treg expansion.
Panel a. Relative IFNβ expression in CD11b+ myeloid cell subsets. CD11b+ CX3CR1+ or CD11b+CX3CR1− cells from the colon LP were sorted, RNA extracted and IFNβ mRNA levels were quantified by qPCR. TNFα levels were also analyzed as an independent variable. Relative ratios of cytokine levels (CD11b+ CX3CR1+/CD11b+CX3CR1−) prior to and 3 days after ETBF colonization are shown. Average of 3 technical replicates from pooled samples (5 mice/group) were calculated for CX3CR1+ and CX3CR1− subsets and relative proportion was determined. Panel b. In vivo IFNβ neutralization. Effect of anti-IFNβ antibody on tumor burden and T-cell activity in ETBF colonized mice was determined. Representative FACS panels and quantitative data (% Foxp3+ CD4 T-cells and number of IFNγ+ CD8 T-cells per colon) are shown. Error bars = SEM, n = 4 mice per group. * and ** denote p < .05 and 0.01.
Discussion
Our studies, for the first time, demonstrate that the expansion of Treg in the colonic LP of ETBF-colonized APCmin/+ mice is driven by CX3CR1+ tissue-resident macrophages. The findings also establish that Treg expansion requires the production of IFNβ by these macrophages. Additionally, our data suggest that the Treg, independent of Th17 cells, are essential to tumor growth in this model. Collectively, this information sheds significant new light on the post-ETBF Treg ontogeny in the APCmin/+ mouse colon.
The finding that in the inflamed colon CX3CR1+ macrophages promote Treg activity in an IFNβ-dependent manner is consistent with the literature on the central role of the CX3CR1+ myeloid cell-Treg axis in maintaining steady-state tolerance in the gut.8–10 Our data confirm and extend the physiological relevance of this pathway to the regulation of microbially-induced inflammation in the dysplastic colon. Whereas we identified gut-resident CX3CR1+ macrophages as the primary driver of Treg expansion, our data cannot distinguish between the ontologically and functionally distinct subsets within this population.13 Therefore, whether a unique CX3CR1+ macrophage subpopulation is directly responsible for the activity observed here remains to be determined.
In addition to driving Treg expansion, CX3CR1+ macrophages actively inhibited IL-17 production in CD4+ T-cells. This observation suggests that co-expansion of the Th17 compartment in ETBF-colonized gut is driven by another cell type such as the recently-described inflammatory DC-like antigen-presenting cells that arise from Ly6Chi monocytes.18–20 In this study we did not attempt to identify the cell type that promotes the expansion of Th17 cells or the mechanism by which CX3CR1+ macrophages suppress IL-17. One potential candidate for the latter is IFNβ, which has been reported to suppress IL-17 production in both murine and human CD4+ T-cells.21,22
Previous studies showed that Treg can enhance Th17 expansion by acting as a sink for IL-26. Our data show that Th17 expansion persisted in the absence of Treg, and thus could occur independent of Treg help. One possible explanation for this seeming contradiction is that Treg act as a secondary enhancer, rather than the primary driver, of Th17 expansion. Alternatively, the timing of the Treg elimination may be important. That is, in the previous report Treg depletion was initiated prior to ETBF administration and performed for one week followed by analysis of the colon for Th17 activity. In contrast, we initiated depletion one week after ETBF colonization and administered DT for 3 weeks (starting DT prior to ETBF and continuing for 4 weeks resulted in significant mortality). It is therefore possible that the one-week period between ETBF infection and Treg depletion was sufficient for Treg to enhance Th17 cell polarization, which then expanded without further Treg help.
In this study, we did not address the mechanism that mediates tumor suppression in Treg-depleted mice. Our data showed that global elimination of CX3CR1 resulted in potent CTL activation, which would be expected to suppress tumor growth. Of note, this effect was strong enough to counter expanding pro-tumorigenic Th17 cell activity, suggesting that the functional balance between CTL and Th17 is important to outcome in this model. Consistent with this notion, we previously reported that suppression of colon tumorigenesis by orally-administered IL-10 formulations was associated with the paradoxical ability of IL-10 to suppress Th17 cell activity while enhancing CD8 + T-cell cytotoxicity in the ETBF-colonized gut.7
Finally, our findings may have broader implications for the treatment of colon cancer, which (with the exception of a small microsatellite unstable subset) is unresponsive to immune therapy.23 Specifically, as ETBF has been associated with colorectal cancer (CRC) as well as familial adenomatous polyposis (FAP), IFNβ and IL-17 may represent relevant immunological targets in CRC and/or FAP patients.
Materials and methods
Mice and the tumor model
C57BL/6 (B6), C57BL/6J-ApcMin/J (APCmin/+), B6.129(Cg)-Foxp3tm3(DTR/GFP)Ayr/J (Foxp3DTR/DTR), B6N.129P2-Cx3cr1tm3(DTR)Litt/J (CX3CR1 flstopflDTR/flstopflDTR) and B6.129P2-Lyz2tm1(cre)Ifo/J (LysMcre) mice were purchased from Jackson Laboratory. Enterotoxic B fragilis strain 86-5443-2-2 (ETBF) which secretes B. fragilis toxin 2 (BFT-2) was a kind gift from Dr. Cynthia L Sears (Johns Hopkins University School of Medicine, Baltimore, Maryland). ETBF was grown under anaerobic conditions at 37ºC overnight prior to administration to mice. For colonization with ETBF, 5–6 week old APCmin/+ mice (or transgenic mice in APCmin/+ background) were administered clindamycin (0.1g/L) and streptomycin (5g/L) for 3–5 days before oral gavage (~5 × 107 bacteria in PBS) essentially as described.1 All experiments were conducted in accordance with guidelines set forth by the Institutional Animal Care and Use Committees at the University of Louisville (Louisville, KY).
Genetic models
For APCmin/+Foxp3DTR/DTR mice, APCmin/+ male mice were crossed to Foxp3DTR/DTR female mice to generate APCmin+Foxp3DTR/+ male mice. Then APCmin+Foxp3DTR/+ male mice were crossed to Foxp3DTR/DTR female mice for the second time to generate APCmin/+Foxp3DTR/DTR mice. For APCmin/+CX3CR1 flstopflDTR/+LysMcre/+ mice, APCmin/+ male mice were initially crossed to CX3CR1flstopflDTR/flstopflDTR mice to establish APCMin/+CX3CR1 flstopflDTR/+ male mice. Then APCMin/+CX3CR1 flstopflDTR/+ male mice were crossed to LysMcre female mice to establish APCmin/+CX3CR1 flstopflDTR/+LysMcre/+ mice. Genotyping for each strain was performed using the primer sequences and PCR conditions recommended by the supplier (Jackson Laboratories, Bar Harbor, ME).
Flow cytometry
MLN were processed into single cell suspensions and colons were digested and LP lymphocytes were isolated as described.24 For experiments requiring detection of intracellular antigens, cell suspensions were cultured in the presence of Golgistop (5 μL/mL; BD), phorbol myristate acetate (50 ng/mL; Sigma) and ionomycin (1mg/mL Sigma). Cells were then permeabilized and fixed using an intracellular staining kit (eBioscience). The following antibodies were used: CD4 (RM4-5, eBioscience), CD8α (53–6.72, Biolegend), CD11b (M1/70, Biolegend), CD45R/B220 (RA3-6B2, BD Pharmingen), CX3CR1 (SA011F11, Biolegend), CD11c (HL3, BD), MHCII (M5/114, Biolegend), CD24 (M1/69, Biolegend), CD64 (X54-5/7.1, Biolegend), CD103a (2E7, Biolegend), IL-17A (TC11-18H10, Biolegend), Foxp3 (FJK-16s, eBioscience) and IFNγ (XMG1.2, BD Pharmingen).
Cytospin analysis
CD11b+CX3CR1+ or CD11c+CD24+CD64− cells were sorted to near homogeneity (>93%, FACSARIA III) from colonic lamina propria of day 3 APCmin/+/B fragilis mice, were stained with Wright-Giemsa, and were evaluated by light microscopy (800x magnification).
Diphtheria toxin (DT) administration
For Treg depletion, APCmin/+Foxp3DTR/DTR mice received 1μg of DT (Sigma) dissolved in 100μl of Dulbecco PBS by intraperitoneal injection on days 7, 14, and 21 after ETBF inoculation.25 APCmin/+CX3CR1flstopflDTR/+ LysMcre/+ mice also received 1μg of DT dissolved in 100μl of Dulbecco PBS by intraperitoneal injection on days 7, 14, and 21 after ETBF inoculation for lineage-specific knockdown of CX3CR1+ myeloid cells.
IFNβ neutralization
Anti-mIFN-β mAb (HDβ-4A7, Leinco Technologies) was given intraperitoneally to APCmin+ mice (0.25mg in 0.25ml PBS, three times per week for 3 weeks) to neutralize IFN-β (PBS as control). The above treatment was initiated 1 week after ETBF inoculations.
Quantitative real-time PCR
Total RNA was extracted using Trizol (Invitrogen/ThermoFisher). The Trizol solution was then added to the Zymo-Spin IIC column for further purification per the manufacturer’s instructions (Zymo Research, Irvine, CA). DNA was removed by adding rDNase I (Ambion/ThermoFisher) to the RNA solution. RNA was then eluted in DNase/RNase free water and immediately processed for reverse transcription. Complementary DNA (cDNA) was synthesized by using TaqMan reverse Transcription reagents (Applied Biosystems/ThermoFisher). The prepared cDNAs were amplified using iTaq Universal SYBR Green supermix kit (Bio-Rad, Hercules, CA) using the Stratagene Mx3005P system with the following primers: IFN-β Fwd: 5ʹ-CCAGCTCCAAGAAAGGACGA −3ʹ, Rev: 5ʹ-CGCCCTGTAGGTGAG GTTGAT −3ʹ; TNFα Fwd 5ʹ- GAA CTG GCA GAA GAG GCA CT −3ʹ and Rev 5ʹ- AGG GTC TGG GCC ATA GAA CT −3ʹ; β-actin Fwd 5ʹ- TCA CCC ACA CTG GCC CAT CTA CGA −3ʹ and Rev 5ʹ- TGG TGA AGC TGT AGC CAC GCT −3ʹ. Relative quantitative measurement of target gene levels was performed using the 2−ΔΔΔΔCt method. β-actin was used as the endogenous housekeeping control gene.
In vitro cell culture
Lymphocytes (CD45+CD4+, CD45+CD11b+CX3CR1+ and CD45+CD11b+CX3CR1− cells) were sorted to >90% purity from MLN of APCmin/+ mice 3 days after ETBF inoculation on a FACSAria III (BD Pharmingen). Foxp3-induction assays were performed by in vitro co-culture of 1 × 105/ml CD45+CD11b+CX3CR1+ or CD45+CD11b+CX3CR1− cells together with 1 × 106/ml CD45+CD4+ T cells and 40U/ml IL-2 (Peprotech, Inc) for 4 days.
Statistical analysis
Statistical calculations were performed using Student t test in pairwise comparisons of groups. In experiments with multiple groups homogeneity of inter-group variance was analyzed by one-way ANOVA. P values of 0.05 or less were considered statistically significant.
Funding Statement
The work was supported by the University of Louisville start-up funds (N.K.E.).
Abbreviations
| ANOVA | Analysis of variance |
| APCmin/+ | adenomatous polyposis coli multiple intestinal neoplasia |
| CD | Cluster of differentiation |
| CTL | Cytotoxic T-lymphocyte |
| CX3CR1 | CX3C chemokine receptor 1 |
| DT | Diphtheria toxin |
| ETBF | Enteroroxic Bacteroides fragilis |
| Foxp3 | Forkhead box P3 |
| IEL | Intra-epithelial lymphocyte |
| IFNβ | Interferon beta |
| IFNγ | Interferon gamma |
| IL | Interleukin |
| LP | Lamina propria |
| Ly6C | lymphocyte antigen 6 complex, locus C |
| MLN | Mesenteric lymph node |
| PBS | Phophate-buffered saline |
| qPCR | Quantitative polymerase chain reaction |
| SEM | Standard error of the mean |
| Th | T-helper |
| TNFα | Tumor necrosis factor alpha |
| Treg | T-regulatory cell |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15:1–7. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Housseau F, Sears CL.. Enterotoxigenic bacteroides fragilis (ETBF)-mediated colitis in Min (Apc±) mice: a human commensal-based murine model of colon carcinogenesis. Cell Cycle. 2010;9:3–5. doi: 10.4161/cc.9.1.10352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Housseau F, Wu S, Wick EC, Fan H, Wu X, Llosa NJ, Smith KN, Tam A, Ganguly S, Wanyiri JW, et al. Redundant innate and adaptive sources of IL17 production drive colon tumorigenesis. Cancer Res. 2016;76:2115–2124. doi: 10.1158/0008-5472.CAN-15-0749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thiele Orberg E, Fan H, Tam AJ, Dejea CM, Destefano Shields CE, Wu S, Chung L, Finard BB, Wu X, Fathi P, et al. The myeloid immune signature of enterotoxigenic bacteroides fragilis-induced murine colon tumorigenesis. Mucosal Immunol. 2017;10:421–433. doi: 10.1038/mi.2016.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chung L, Thiele Orberg E, Geis AL, Chan JL, Fu K, DeStefano Shields CE, Dejea CM, Fathi P, Chen J, Finard BB, et al. Bacteroides fragilis toxin coordinates a pro-carcinogenic inflammatory cascade via targeting of colonic epithelial cells. Cell Host Microbe. 2018;23:203–214 e205. doi: 10.1016/j.chom.2018.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geis AL, Fan H, Wu X, Wu S, Huso DL, Wolfe JL, Sears CL, Pardoll DM, Housseau F.. Regulatory T-cell response to enterotoxigenic bacteroides fragilis colonization triggers IL17-dependent colon carcinogenesis. Cancer Discov. 2015;5:1098–1109. doi: 10.1158/2159-8290.CD-15-0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gu T, De Jesus M, Gallagher HC, Burris TP, Egilmez NK. Oral IL-10 suppresses colon carcinogenesis via elimination of pathogenicCD4(+) T-cells and induction of antitumor CD8(+) T-cell activity. Oncoimmunology. 2017;6:e1319027. doi: 10.1080/2162402X.2017.1319027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shouval DS, Biswas A, Goettel JA, McCann K, Conaway E, Redhu NS, Mascanfroni ID, Al Adham Z, Lavoie S, Ibourk M, et al. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity. 2014;40:706–719. doi: 10.1016/j.immuni.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zigmond E, Bernshtein B, Friedlander G, Walker CR, Yona S, Kim KW, Brenner O, Krauthgamer R, Varol C, Muller W, et al. Macrophage-restricted interleukin-10 receptor deficiency, but not IL-10 deficiency, causes severe spontaneous colitis. Immunity. 2014;40:720–733. doi: 10.1016/j.immuni.2014.03.012. [DOI] [PubMed] [Google Scholar]
- 10.Kim M, Galan C, Hill AA, Wu WJ, Fehlner-Peach H, Song HW, Schady D, Bettini ML, Simpson KW, Longman RS, et al. Critical role for the microbiota in CX3CR1(+) intestinal mononuclear phagocyte regulation of intestinal T cell responses. Immunity. 2018;49:151–163 e155. doi: 10.1016/j.immuni.2018.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Q, Anderson CD, Egilmez NK. Inhaled IL-10 suppresses lung tumorigenesis via abrogation of inflammatory macrophage-Th17 cell axis. J Immunol. 2018;201:2842–2850. doi: 10.4049/jimmunol.1800141. [DOI] [PubMed] [Google Scholar]
- 12.Muzaki ARBM, Tetlak P, Sheng J, Loh SC, Setiagani YA, Poidinger M, Zolezzi F, Karjalainen K, Ruedl C. Intestinal CD103+CD11b− dendritic cells restrain colitis via IFN-γ-induced anti-inflammatory response in epithelial cells. Mucosal Immunol. 2016;9(2):336–351. doi: 10.1038/mi.2015.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Schepper S, Verheijden S, Aguilera-Lizarraga J, Viola MF, Boesmans W, Stakenborg N, Voytyuk I, Schmidt I, Boeckx B, Dierckx de Casterle I, et al. Self-maintaining gut macrophages are essential for intestinal homeostasis. Cell. 2018;175:400–415 e413. doi: 10.1016/j.cell.2018.07.048. [DOI] [PubMed] [Google Scholar]
- 14.Tamoutounour S, Henri S, Lellouard H, de Bovis B, de Haar C, van der Woude CJ, Woltman AM, Reyal Y, Bonnet D, Sichien D, et al. CD64 distinguishes macrophages from dendritic cells in the gut and reveals the Th1-inducing role of mesenteric lymph node macrophages during colitis. Eur J Immunol. 2012;42:3150–3166. doi: 10.1002/eji.201242847. [DOI] [PubMed] [Google Scholar]
- 15.Hilligan KL, Connor LM, Schmidt AJ, Ronchese F. Activation-induced TIM-4 expression identifies differential responsiveness of intestinal CD103+CD11b+ dendritic cells to a mucosal adjuvant. PLoS One. 2016;11(7):e0158775. doi: 10.1371/journal.pone.0158775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bain CC, Schridde A. Origin, differentiation, and function of intestinal macrophages. Front Immunol. 2018;9:2733. doi: 10.3389/fimmu.2018.02733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakahashi-Oda C, Udayanga KG, Nakamura Y, Nakazawa Y, Totsuka N, Miki H, Iino S, Tahara-Hanaoka S, Honda S, Shibuya K, et al. Apoptotic epithelial cells control the abundance of Treg cells at barrier surfaces. Nat Immunol. 2016;17:441–450. doi: 10.1038/ni.3345. [DOI] [PubMed] [Google Scholar]
- 18.Rivollier A, He J, Kole A, Valatas V, Kelsall BL. Inflammation switches the differentiation program of Ly6Chi monocytes from antiinflammatory macrophages to inflammatory dendritic cells in the colon. J Exp Med. 2012;209:139–155. doi: 10.1084/jem.20101387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zigmond E, Varol C, Farache J, Elmaliah E, Satpathy AT, Friedlander G, Mack M, Shpigel N, Boneca IG, Murphy KM, et al. Ly6C hi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity. 2012;37:1076–1090. doi: 10.1016/j.immuni.2012.08.026. [DOI] [PubMed] [Google Scholar]
- 20.Panea C, Farkas AM, Goto Y, Abdollahi-Roodsaz S, Lee C, Koscso B, Gowda K, Hohl TM, Bogunovic M, Ivanov II. Intestinal monocyte-derived macrophages control commensal-specific Th17 responses. Cell Rep. 2015;12:1314–1324. doi: 10.1016/j.celrep.2015.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang L, Yuan S, Cheng G, Guo B. Type I IFN promotes IL-10 production from T cells to suppress Th17 cells and Th17-associated autoimmune inflammation. PLoS One. 2011;6:e28432. doi: 10.1371/journal.pone.0028432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tao Y, Zhang X, Chopra M, Kim MJ, Buch KR, Kong D, Jin J, Tang Y, Zhu H, Jewells V, et al. The role of endogenous IFN-beta in the regulation of Th17 responses in patients with relapsing-remitting multiple sclerosis. J Immunol. 2014;192:5610–5617. doi: 10.4049/jimmunol.1302580. [DOI] [PubMed] [Google Scholar]
- 23.Boland PM, Ma WW. Immunotherapy for colorectal cancer. Cancers (Basel). 2017. May 11;9(5):pii: E50. doi: 10.3390/cancers9050050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weigmann B, Tubbe I, Seidel D, Nicolaev A, Becker C, Neurath MF. Isolation and subsequent analysis of murine lamina propria mononuclear cells from colonic tissue. Nat Protoc. 2007;2:2307–2311. doi: 10.1038/nprot.2007.315. [DOI] [PubMed] [Google Scholar]
- 25.Chung AY, Li Q, Blair SJ, De Jesus M, Dennis KL, LeVea C, Yao J, Sun Y, Conway TF, Virtuoso LP, et al. Oral interleukin-10 alleviates polyposis via neutralization of pathogenic T-regulatory cells. Cancer Res. 2014;74:5377–5385. doi: 10.1158/0008-5472.CAN-14-0918. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.