

ABSTRACT
Immunotherapy with oncolytic herpes simplex virus-1 therapy offers an innovative, targeted, less-toxic approach for treating brain tumors. However, a major obstacle in maximizing oncolytic virotherapy is a lack of comprehensive understanding of the underlying mechanisms that unfold in CNS tumors/associated microenvironments after infusion of virus. We demonstrate that our multiplex biomarker screening platform comprehensively informs changes in both topographical location and functional states of resident/infiltrating immune cells that play a role in neuropathology after treatment with HSV G207 in a pediatric Phase 1 patient. Using this approach, we identified robust infiltration of CD8+ T cells suggesting activation of the immune response following virotherapy; however there was a corresponding upregulation of checkpoint proteins PD-1, PD-L1, CTLA-4, and IDO revealing a potential role for checkpoint inhibitors. Such work may ultimately lead to an understanding of the governing pathobiology of tumors, thereby fostering development of novel therapeutics tailored to produce optimal responses.
KEYWORDS: HSV, oncolytic virotherapy, glioma, multiplex immunofluorescence, checkpoint proteins
Introduction
Effective, less-toxic therapies for malignant pediatric brain tumors represent an unmet clinical need. The significant morbidity and mortality associated with childhood brain cancer is compounded by neurotoxicity within the developing brain caused by current therapies (e.g., hormone dysfunction, neurosensory and neurocognitive decline).1 Novel, targeted therapies directed toward tumor cells are needed to improve outcomes and reduce damaging side effects in children with brain cancer. Oncolytic herpes simplex virus (oHSV) therapy offers an innovative, targeted, less-toxic approach for treating pediatric brain tumors. Our institution has conducted 3 Phase 1 trials of a first-generation oHSV (G207), which has both copies of γ134.5 deleted and a lacZ insertion in the ribonucleotide reductase locus for added protection, given alone and with a single small dose of radiation in adults with recurrent high-grade glioma.2–4 These trials conclusively demonstrated safety of injecting high doses [up to 3 × 109 plaque-forming units (PFU)] directly into the tumor or surrounding brain tissue, and approximately 50% of patients had radiographic evidence of tumor response, including two long-term survivors (>5.5 years).
Our preclinical data indicate that children are likely the ideal candidates for oHSV; we recently reported that 10 pediatric patient-derived brain tumor xenografts were on average ~40-fold more sensitive to killing by oHSV than 8 adult patient-derived glioblastoma (GBM) xenografts.5 Moreover, pediatric medulloblastoma tumor cells (including the most resistant group 3 tumors) and chemo- and radio-resistant CD133+ or CD15+ cancer stem cells were highly sensitive to oHSV, and CD133+ glioma cells were likewise sensitive and had no inherent resistance to oHSV.6–8 Based on our preclinical findings, we have ongoing clinical trials of G207 alone or combined with a single 5 Gy dose of radiation in children with recurrent or progressive malignant supratentorial (NCT02457845) and cerebellar tumors (NCT03911388).9,10 The critical obstacle to maximizing oHSV and achieving more durable responses is developing a strategy to amplify and then maintain the anti-tumor immune response induced by the virus.11 Recent studies have shown that human brain tumors evade immune surveillance through “checkpoint” proteins which inhibit tumor infiltrating lymphocytes and prevent T cell activation.12 Higher expression of these proteins in brain tumors has correlated with worse patient outcomes and blocking these proteins with checkpoint inhibitors has resulted in dramatic responses in some human cancers.13,14 However, significant challenges to inciting and maintaining a potent anti-tumor immune response remain for the immunologically privileged site of the brain.
In an effort to maximize the anti-tumor immune response of oHSV, we have developed an innovative multiplex biomarker screening platform that is capable of evaluating changes in both the topographical location, architectural distribution and functional states of resident and infiltrating immune cell types that play a role in resultant tumor/immunopathology after infusion of G207. Recent advances in multiplex immunofluorescence techniques have allowed simultaneous visualization of a small set of antigens on formalin-fixed, paraffin-embedded tissue for disease diagnosis and translational research.15 Here, we describe our novel methodology that allows for simultaneous visualization of antigen labels that highlight the neuroinflammatory response, immune checkpoint state, tumor phenotype, and vascular niche. We applied this technique to study the treatment response of a patient with pediatric GBM following G207 virotherapy. The results garnered will inform the next series of pediatric virotherapy clinical trials via the identification of adjuvant targets (e.g. checkpoint proteins) to maximize efficacy.
Materials and methods
Patient/gross tissue pathology
Pre-treatment biopsy tissue from an 11-year-old female with a right parietal lobe glioblastoma was obtained to confirm recurrent tumor prior to treatment with G207 per protocol. Post-treatment HSV tissue from the same patient was obtained during tumor resection approximately 3 months after the infusion of G207.
Tumor biopsy/tissue processing
The University of Alabama at Birmingham Institutional Review Board reviewed and approved the trial and study (IRB-150319005); the studies described below have been performed in accordance with our Assurance of Compliance approved by the Department of Health and Human Services. Informed consent was obtained from the patient’s parent and assent was obtained from the patient prior to being screened for treatment. Biopsies were taken to confirm presence of tumor tissue prior to placement of catheters. Following frozen section demonstration of recurrent tumor, 3 silastic catheters were placed in stereotactically predefined coordinates of tumor.10
Both pre- and post-G207 treatment brain tumor tissue blocks were fixed in 10% neutral buffered formalin and underwent standard clinical processing into paraffin blocks. In brief, following serial dehydration in 70% (25 minutes), 80% (40 minutes), 90% (25 minutes), and 3 changes of absolute alcohol (25 minutes each) at 35°C, the tissue blocks were incubated in 2 changes of xylene solution (40 minutes each) at the same temperature and transferred through 4 separate paraffin baths (25 minutes each) at 58°C. Finally, the tissue blocks were embedded in paraffin and sectioned at 5 µm thickness for multiplex fluorescence immunohistochemistry.
Immunohistochemistry
Briefly, 4 µm sections were obtained from formalin-fixed, paraffin-embedded block preparations. The immunostaining was accomplished with a fully-automated immunostainer (Bond IHC stainer, Leica Biosystems, IL). Clinical immunohistochemistry was performed with ready-to-use antibodies from Leica Biosystems directed against CD3 (clone: LN10), CD4 (clone: 4B12), CD8 (clone: 4B11) and CD68 (clone: 514H12). Appropriate positive and negative control slides were prepared; the negative control slides consisted of tissue sections of each case processed without the addition of primary antibody.
Multiplex fluorescence immunohistochemistry
Multiplex fluorescence immunohistochemistry was performed on 5 µm-thick paraffin sections sourced from pre- and post-HSV treated GBM tissue using up to 4 iterative rounds of sequential immunostaining with select antibody panels (see Supplemental Table 1 for each panel) tailored for deep phenotyping of GBM cytoarchitecture and examination of the tumor microenvironment. Each round of antibody staining was imaged by fluorescence microscopy followed by antibody stripping and tissue restaining steps to repeat the cycle, each time using a different antibody panel, which ultimately culminated in screening 18 different validated biomarkers relevant to GBM phenotyping per each tissue section. Briefly, the sections were first deparaffinized using a standard xylene/ethanol rehydration protocol followed by antigen unmasking with a 10-minute heat mediated antigen retrieval step in 10 mM Tris/EDTA buffer pH 9.0 (Tris/EDTA buffer) using a 800W microwave set at 100% power. The sections were then incubated with Human BD Fc Blocking solution (BD Biosciences) to saturate endogenous Fc receptors, and then in True Black Reagent (Biotium) to quench intrinsic tissue autofluorescence. The sections were then immunoreacted for 1 hour at RT using 1–5 μg/ml cocktail mixture of immunocompatible Antibody Panel #1 targeting neuroinflammatory cell phenotypes. This step was followed by washing off excess primary antibodies in PBS supplemented with 1 mg/ml bovine serum albumin (BSA) and staining the sections using a 1 μg/ml cocktail mixture of the appropriately cross-adsorbed secondary antibodies (purchased from either Thermo Fisher, Jackson ImmunoResearch or Li-Cor Biosciences) conjugated to one of the following spectrally compatible fluorophores: Alexa Fluor 430, Alexa Fluor 546, Alexa Fluor 594, Alexa Fluor 647 or IRDye 800CW. After washing off excess secondary antibodies, sections were counterstained using 1 μg/ml DAPI (Thermo Fisher Scientific) for visualization of cell nuclei. Slides were then coverslipped using Immu-Mount medium (Thermo Fisher Scientific) and imaged using a multi-channel wide field epifluorescence microscope (see below). After imaging, tissue bound primary and secondary antibodies were both stripped off the slides after a 5-minute incubation at RT in NewBlot Nitro 5X Stripping buffer (Li-Cor Biosciences) followed by 1-minute additional heat mediated antigen retrieval step in Tris/EDTA buffer. The above processing cycle beginning with re-blocking of tissues in Human BD Fc Blocking solution was repeated and the same sections then incubated using a second mixture of antibodies (Antibody Panel #2) targeting select checkpoint proteins. The whole process was sequentially repeated two more times using Antibody Panels # 3 and 4 to characterize GBM phenotypes and the vascular niche, respectively.
Multiplex fluorescence immunohistochemistry image acquisition
Images were acquired from whole specimen sections using the Axio Imager.Z2 slide scanning fluorescence microscope (Zeiss) equipped with a 20X/0.8 Plan-Apochromat (Phase-2) non-immersion objective (Zeiss), a high resolution ORCA-Flash4.0 sCMOS digital camera (Hamamatsu), a 200W X-Cite 200DC broad band lamp source (Excelitas Technologies), and 7 customized filter sets (Semrock) optimized to detect the following fluorophores: DAPI, Alexa Fluor 430, Alexa Fluor 488, Alexa Fluor 546, Alexa Fluor 594, Alexa Fluor 647 and IRDye 800CW. Image tiles (600 × 600 μm viewing area) were individually captured at 0.325 micron/pixel spatial resolution, and the tiles seamlessly stitched into whole specimen images using the ZEN 2 image acquisition and analysis software program (Zeiss), with an appropriate color table having been applied to each image channel to either match its emission spectrum or to set a distinguishing color balance. Pseudocolored stitched images acquired from all 4 rounds of antibody staining and imaging were then exported to Adobe Photoshop and overlaid as individual layers to create multicolored merged composites as previously described.16
Quantification and statistical analysis
Quantification was performed on a total of n = 6 random regions of interest (ROI) by a clinically trained neuropathologist (RL). The number of nuclei, as determined via DAPI positive staining, was ≥100 per ROI. Data are expressed as percentage of positive cells for a given marker over the total number of DAPI positive cells; these data are presented via standard box-and-whiskers plots, in which the central-line represents the median and the upper and lower bounds of the boxes are min and max value. All statistical tests were performed in GraphPad Prism (version 5.00 for Mac, GraphPad Software). Data were tested for normality using a D’Agostino and Pearson omnibus normality test and subsequently assessed for homogeneity of variance. Data that passed both tests were further analyzed using a two-tailed unpaired Student’s t-test. For all statistical tests, p-value <0.05 were considered statistically significant.
Results
The complex host-tumor relationship is the result of interplay between tumor cells, normal parenchyma, stromal elements, and the immune system. Despite such acknowledged complexities, a comprehensive method capable of phenotyping clinically relevant tissue (e.g. pre- and post-treatment) has yet to emerge. Herein, we successfully demonstrate the ability to analyze clinical tissue samples with a plex of antibodies (18 in total). ROIs centered on hemorrhage as defined by glycophorin A (GPA) and the tumor vasculature (i.e. the relative presence or absence thereof) as defined by PECAM-1/CD31 and collagen IV serve as tissue landmarks in pre-treatment (Figure 1a,b) and post-treatment tissue (Figure 2a,b). Of note, quantification of the percentage of CD31 (4.1% ± 1.0% pre- vs 3.3% ± 0.5% post-, p-value = 0.511, Figure 3a) and collagen IV (3.7% ± 0.9% pre- vs 4.3% ± 0.5%, p-value = 0.596, Figure 3b) positive cells were not statistically different.
Figure 1.
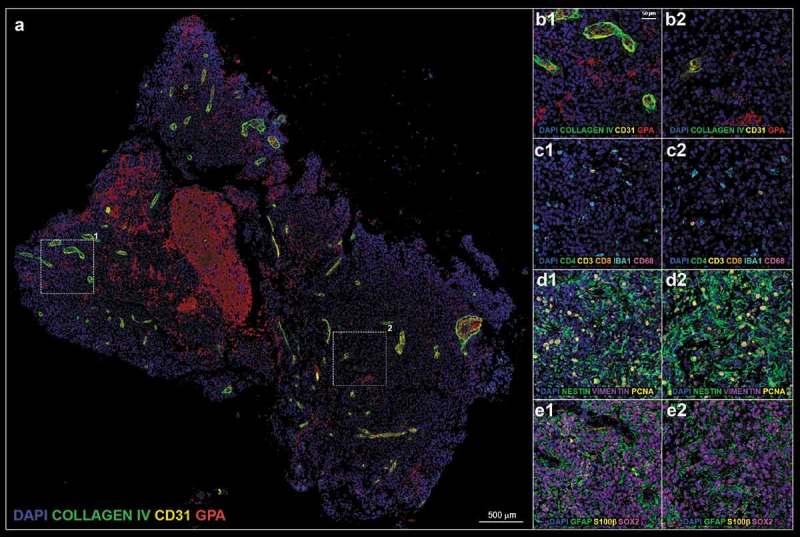
(a). Pre-treatment tissue biopsy; white boxes define two representative regions of interest (ROI) 1 and 2 enlarged in A1–E1 and A2–E2, respectively. (b). vascular panel; blue: DAPI, green: collagen IV, yellow: CD31, red: glycophorin A (GPA). (c). inflammatory panel; blue: DAPI, green: CD4, yellow: CD3, orange: CD8, teal: IBA1, magenta: CD68. (d). glia stem panel; blue: DAPI, green: nestin, purple: vimentin, yellow: proliferation cell nuclear antigen (PCNA). (e). glia differentiation panel; blue: DAPI, green: glial fibrillary acidic protein (GFAP), yellow: S100β, magenta: SOX2.
Figure 2.
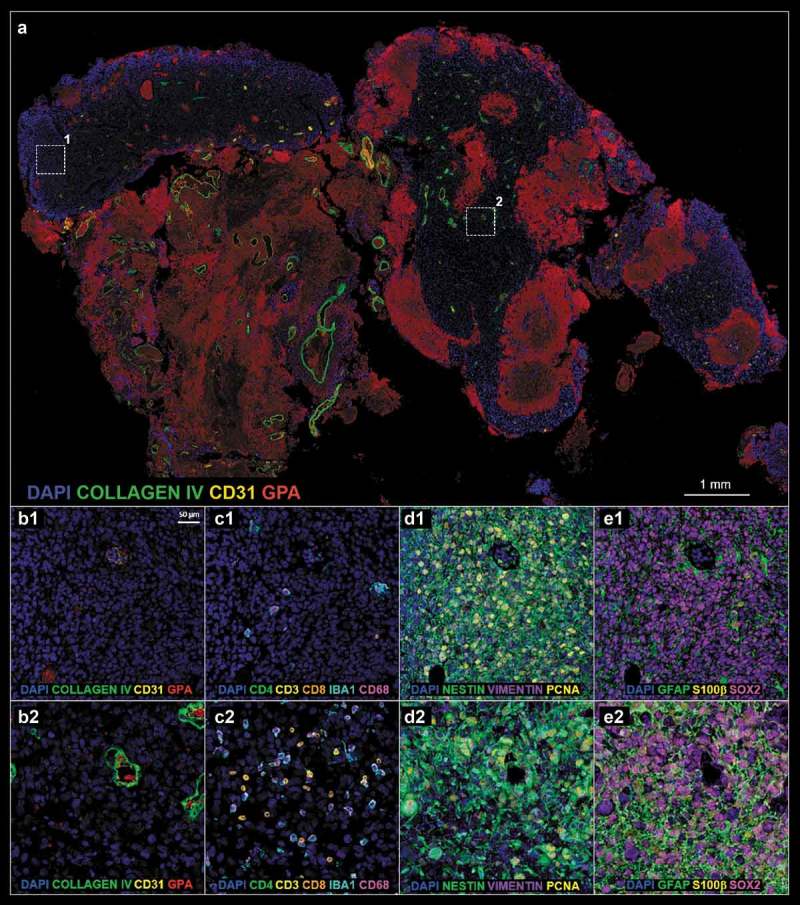
(a). Post-treatment tissue biopsy; white boxes define two representative ROIs 1 and 2 enlarged in A1–E1 and A2–E2, respectively. (b–e). Refer to Figure 1 legend for biomarker definition.
Figure 3.
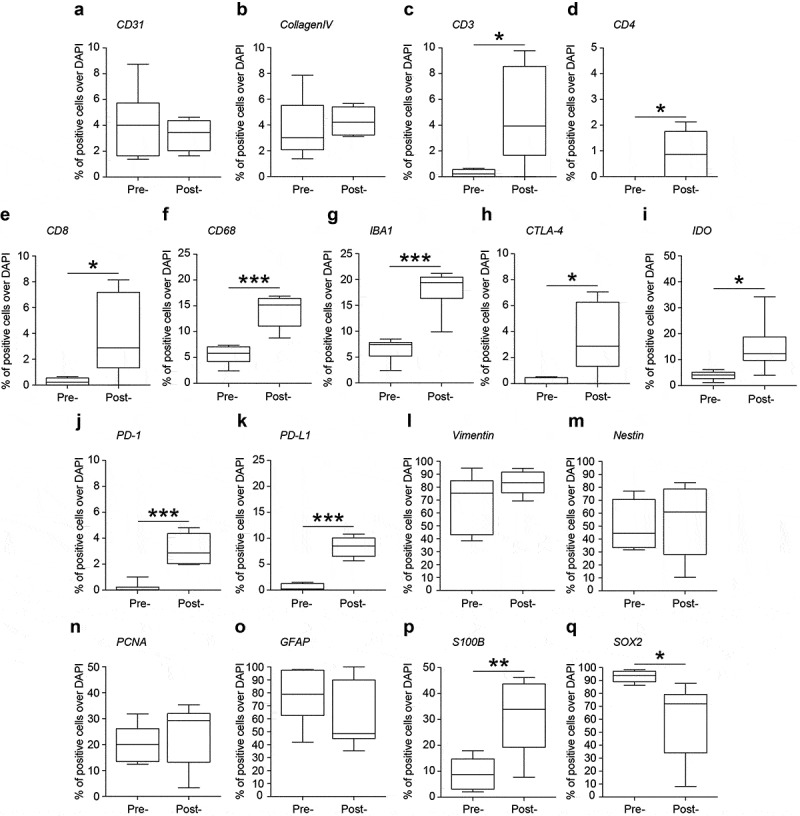
Quantification of the percentage of positive cells over DAPI positive cells in pre-treatment (pre-) and post-treatment (post-) samples. (a). CD31; (b). Collagen IV; (c). CD3; (d). CD4; (e). CD8; (f). CD68; (g). IBA1; (h). CTLA-4; (i). IDO; (j). PD-1; (k). PD-L1; (l). Vimentin; (m). Nestin; (n). PCNA; (o). GFAP; (p). S100β; (q). SOX2. Data are percentage of positive cells over DAPI and shown via standard box-and-whiskers plot in which the central-line represents the median and the upper and lower bounds of the boxes are min and max values. *: significant at a threshold of p < .05. **: significant at a threshold of p < .01. ***: significant at a threshold of p < .001.
Influx of CD8+ effector T-lymphocytes after oncolytic HSV G207 therapy
Using the antibody panel 1 (Supplemental Table 1, neuroinflammatory cell phenotyping), we demonstrated a massive influx of T-lymphocytes after treatment with oHSV G207 as evidenced by CD3, CD4, and CD8 expression. Prior to HSV therapy, there was a dearth of adaptive immunity noted throughout the pre-treatment tissue and within the examined ROIs (Figure 1c and 4). Examination of post-treatment tissue revealed a massive influx in CD3+CD8+ T cells throughout the tissue (Figure 2c and 5) and a significant increase in the proportion of CD3 (0.3% ± 0.1% pre- vs 4.7% ± 1.5% post-, p-value = 0.016, Figure 3c), CD4 (0.0% ± 0.0% pre- vs 0.9% ± 0.4% post-, p-value = 0.031, Figure 3d), and CD8 (0.3% ± 0.1% pre- vs 3.8% ± 1.3% post-, p-value = 0.021, Figure 3e) positive cells within examined ROIs; while CD3+CD4+ T cells did increase post-treatment, the vast majority of CD3+ cells were effector CD8+ T cells (Figure 5). In addition, a greater proportion of macrophages (i.e. CD68 positive cells) and microglia (i.e. IBA1 positive cells) were observed in post-treatment tissue (Figure 2c and 5) compared to pre-treatment tissue (Figure 1c and 4), both throughout the tissue and within the examined ROIs. These findings were further validated using standard clinical immunohistochemistry on representative tissue blocks (Figure S1). Of note, about a 3 fold-increase in the percentage of CD68 (5.5% ± 0.7% pre- vs 14.0% ± 1.3% post-, p-value = 0.002, Figure 3f) and IBA1 (6.6% ± 0.9% pre- vs 18.0% ± 1.7% post-, p-value <0.0001, Figure 3g) positive cells were found. Beyond oncolysis, these data suggest that oHSV therapy increases and/or amplifies the anti-tumor immune response.
Figure 4.
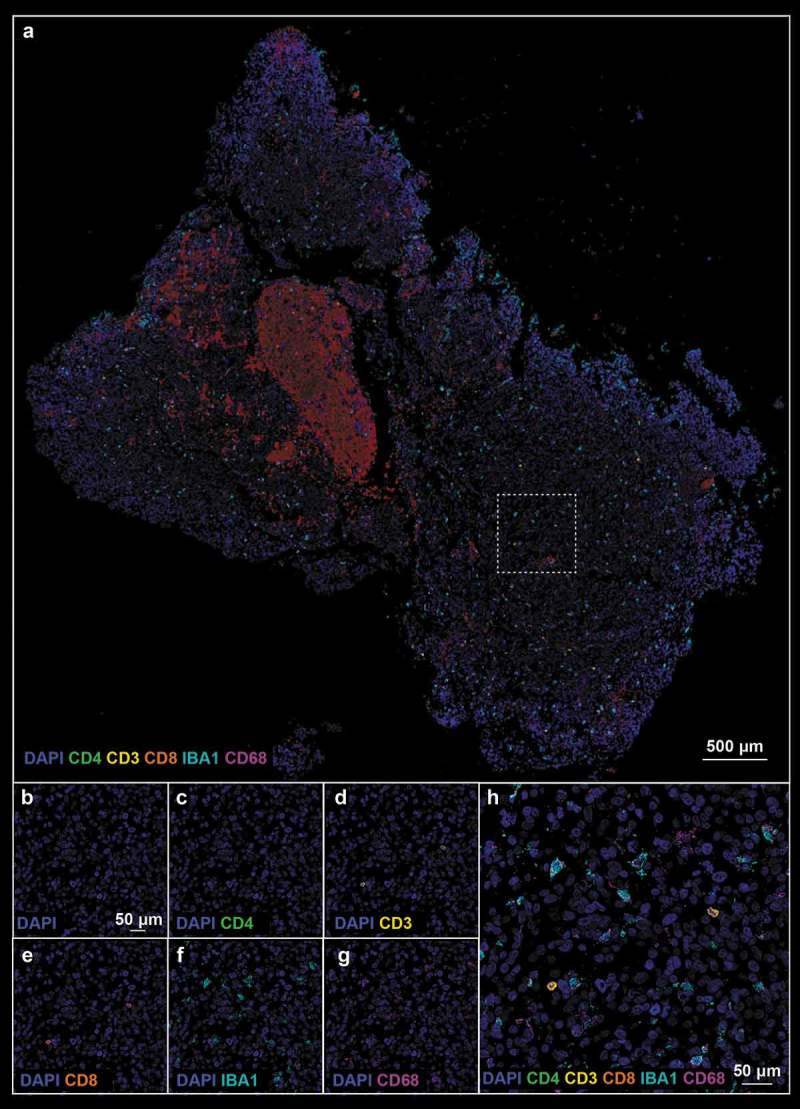
(a). Pre-treatment tissue biopsy stained with neuroinflammatory cell phenotyping (Panel 1 from Supplemental Table 1) antibodies; white boxes define the region of interest. (b–h). Single marker pictures (B–G) and merge (H) of the ROI indicated in (A). Blue: DAPI, green: CD4, yellow: CD3, orange: CD8, teal: IBA1, magenta: CD68.
Figure 5.
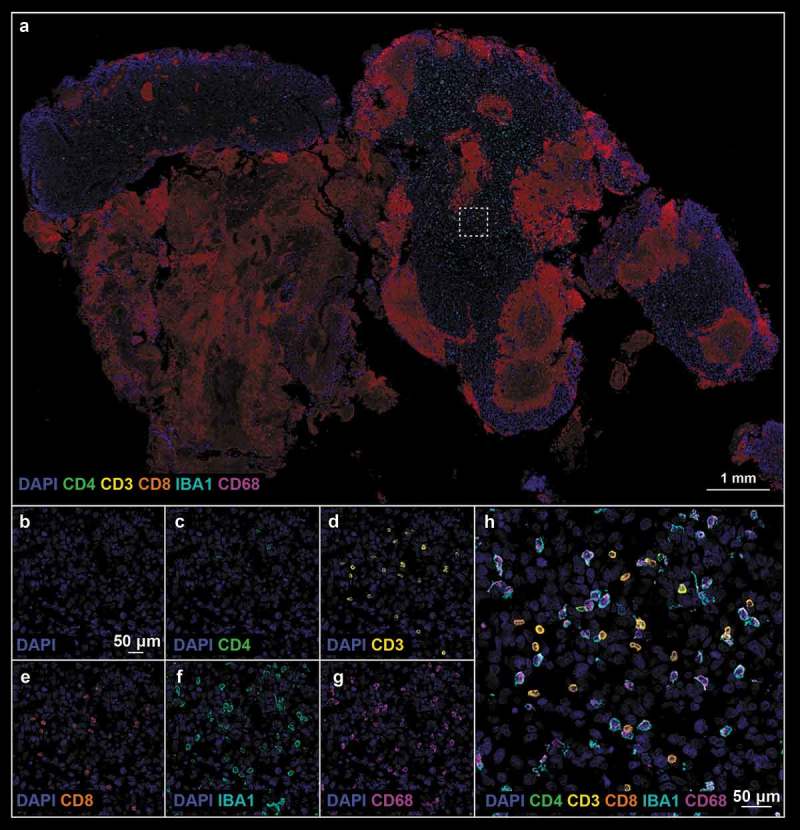
(a). Post-treatment tissue biopsy stained with neuroinflammatory cell phenotyping (Panel 1) antibodies; white boxes define the region of interest. (b–h). Refer to Figure 4 legend for biomarker definition.
Upregulation of checkpoint proteins cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4), programmed cell death protein 1 (PD-1), programmed death ligand 1 (PD-L1), and indoleamine 2,3-dioxygenase (IDO) after treatment with oncolytic HSV
The analysis of antibody panel 2 (Supplemental Table 1, checkpoint proteins) allowed us to further characterize the immunoregulatory state of our pre- and post-treatment tissue biopsies. Given the noted influx of T-lymphocytes described above we sought to characterize their expression of CTLA-4 and PD-1, checkpoint proteins that hold relevant clinical/prognostic value and in so doing found a robust increase in the proportion of CTLA-4 (0.2% ± 0.1% pre- vs 3.4% ± 1.1% post-, p-value = 0.013, Figure 3h) and PD-1 positive lymphocytes after treatment (0.2% ± 0.2% pre- vs 3.1% ± 0.5% post-, p-value = 0.002; Figure 3j). We noted that the few CD8+ T-lymphocytes in pre-treatment samples expressed CTLA-4, albeit weakly (Figure 6d), whereas CTLA-4 was ubiquitously expressed on CD8+ cells post-treatment (Figure 7d). Furthermore PD-1 was highly expressed on both CD4+ and CD8+ cells post-treatment (Figure 7e) In addition to examining lymphocyte expression of checkpoint proteins, we examined tumor cells for expression of PD-L1 and IDO. PD-L1 expression was significantly increased with 8.4% ± 0.9% cells expressing PD-L1 after treatment with G207 (Figures 3k and 7f) compared to 0.5% ± 0.3% cells pre-treatment (p-value = 0.001; Figure 6f). While faint IDO expression was seen in both pre- and post-treatment tissue (Figures 6c and 7c), there were significantly more cells with strong expression of IDO after treatment (4.0% ± 0.7% pre- vs 15.3% ± 3.7% post-, p-value = 0.0105 Figure 3i). These data suggest that while there is a vigorous immune cell infiltration of the tumor after oHSV, there is a concurrent increase in checkpoint protein expression in both tumor cells and immune cells.
Figure 6.

(a). Pre-treatment tissue biopsy stained with checkpoint protein (Panel 2 from Suplemental Table 1) antibodies; white boxes define the region of interest. (b–k). Single marker pictures (b–f), PD-1 and PD-L1 (g), merge (h), and neuroinflammatory cell markers (i–k) of the ROI indicated in A. Blue: DAPI, green: IDO, cyan: CTLA-4, yellow: PD-1, red: PD-L1.
Figure 7.
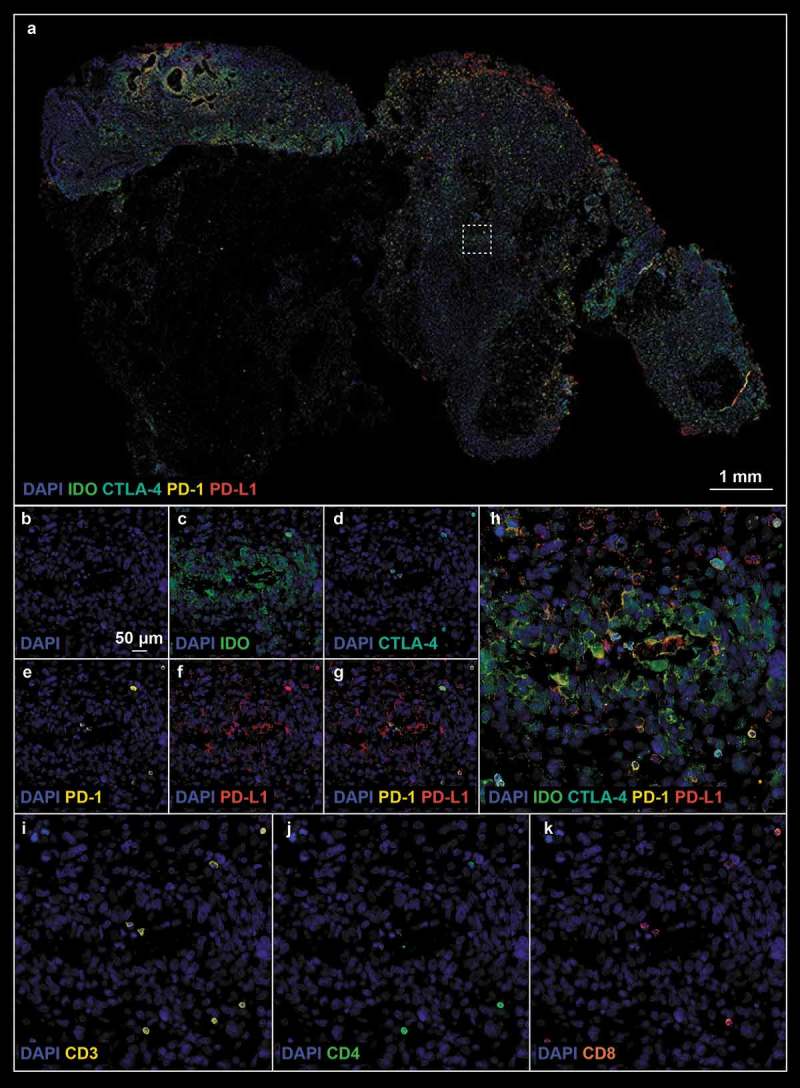
(a). Post- treatment tissue biopsy stained with checkpoint protein (Panel 2) antibodies; white boxes define the region of interest. (b–k). Refer to Figure 6 legend for biomarker definition.
Markers of stemness (Nestin/Sox2)/glia (GFAP/Vimentin/s100β) and replication (PCNA) demonstrate complex tumor heterogeneity
We sought to characterize tumor cell phenotype and global replication by means of antibody panel 3 (GBM cell phenotyping). Intensity of vimentin/nestin staining appeared relatively stable pre- (Figure 1d and S2) and post-HSV treatment (Figure 2d and S3) with no detectable differences in the percentage of vimentin (68.4% ± 9.1% pre- vs 83.2% ± 3.8% post-, p-value = 0.165, Figure 3l) or nestin (50.0% ± 7.6% pre- vs 54.5% ± 11.6% post-, p-value = 0.751, Figure 3m) positive cells. Furthermore, we did not detect any significant difference in the proportion of Proliferating Cell Nuclear Antigen (PCNA) positive cells in the pre-treatment (Figure S2) versus post-treatment tissue (Figure S3; 20.4% ± 2.9% pre- vs 24.1% ± 4.9% post-, p-value = 0.531, Figure 3n). Similarly, there was not a significant difference in expression of GFAP in pre-treatment (Figure S4) and post–treatment (Figure S5) tissue (77.4% ± 8.5% pre- vs 61.2% ± 10.5% post-, p-value = 0.256, Figure 3o). Finally, we quantified the proportion of S100β and SOX2 positive cells (Figures S4 and S5) and found a 3-fold increase in the percentage of S100β cells (9.1% ± 2.7% pre- vs 31.3% ± 6.0% post-, p-value = 0.008, Figure 3p) and a significant reduction of SOX2 positive cells (93.1% ± 1.9% pre- vs 59.9% ± 12.0% post-, p-value = 0.020, Figure 3q) post-treatment compared to pre-treatment.
Discussion
Despite the appeal of precision medicine, targeted therapeutics capable of successfully treating progressive pediatric high-grade gliomas based on unique features of a patient’s individual tumor have yet to materialize. The paucity of therapeutic options stands in stark contrast to the multitude of research efforts and clinical trials. The limited success of such a massive research investment demands a reevaluation of the pathobiology of pediatric brain tumors and a subsequent refinement of experimental/therapeutic approaches. Emerging immunofluorescence approaches have promised to provide a new perspective for understanding the tumor microenvironment, bringing the spatial relationships between immune cells and tumor cells into view.17 Accordingly, herein we have validated our novel in situ multiplex immunofluorescence biomarker screening approach and effectively demonstrated its use for the architectural assessment of tumor immunopathology and response to virotherapy. We simultaneously identified 18 relevant biomarkers that are reflective of the vascular niche, inflammation, replication, stemness and glial differentiation in an effort to fully characterize the pathogenesis/biology of such brain tumors and ultimately develop treatment modalities; for example, the high expression of checkpoint proteins after oHSV treatment identifies a rich target for checkpoint protein inhibitor therapy to maintain an anti-tumor immune response.
In addition to selective tumor cell killing, oHSV may trigger anti-tumor immune responses. This may occur by several mechanisms: (1) the release of tumor neoantigens following oncolytic cell death; (2) stimulation of antiviral immune responses; (3) suppression of tumor immune evasion mechanisms; and (4) expression of engineered proinflammatory cytokines. In this pilot study, we directly visualized the spatiotemporal properties of the activated cytotoxic T cell response due to oHSV treatment in a pediatric patient with GBM. We show robust tumor infiltration at ~90 days post-treatment in focal regions of interest near the vascular niche. Additionally, we saw a significant increase in microglia and macrophages. This may be related to the increase in S100β which has been shown to act as a chemoattractant for macrophages.18 Importantly, we identified robust infiltration of a CTLA-4+ PD-1+ T cell population, and tumor cells demonstrated upregulation of PD-L1 and IDO. Cancer immunotherapy strategies, including CTLA-4 blockade (e.g. ipilimumab), PD-1/PD-L1 inhibition (e.g. nivolumab; pembrolizumab), or IDO inhibition (e.g. indoximod; epacadostat) activate the cell-mediated immune response against tumor cells. The expression of CTLA-4 and PD-1 on cytotoxic T cells and PD-L1 and IDO on tumor cells after treatment with oHSV suggests the potential for synergy between oncolytic virus treatment and checkpoint inhibitors.19–21
Such work sets the stage for the comprehensive description of CNS tumor pathobiology and may ultimately be extended to other diseases/disorders or insults to the CNS that require a comprehensive profile of tissue response to damage or disease. It is prudent to note that iterative rounds of multiplex IHC staining may ultimately increase the number of screened biomarkers to hundreds or even thousands per tissue section, which may become feasible for broader clinical-scale use with improvements in automation. Currently, we are in the process of expanding our biomarker panels by developing novel combinations of validated antibodies capable of fully characterizing the complex response of brain tumor tissue and the response to experimental therapeutics. Further, computational solutions are being devised to leverage this technology in order to comprehensively and robustly quantify the histologic changes mediating complex cellular processes in brain tumors and other neuropathological disorders using existing quantitative algorithms for image segmentation and analysis (e.g. the FARSIGHT toolkit).22 More advanced computational solutions are also being developed that will employ machine learning algorithms customized to our imaging paradigm, ultimately enabling easily scalable image data analysis to support systems biology and translational research into devising optimal treatment strategies to reduce the burden of neurological disease/disorders and/or traumatic insults.23 Computational advances, in combination with future mechanical automation of the sample preparation, can improve the scalability of this methodology so that it may be routinely performed on patients monitored for treatment response after virotherapy or other immunotherapies.24 Increased application of our novel in situ multiplex immunofluorescence will also promote an improved understanding of the range of immune and tumor cell response to virotherapy, and with clinical correlation will provide biological insights in patient stratification and monitoring response to treatment. Immediate implications from this work suggest that adjuvant immunotherapy with agents such as ipilimumab (an inhibitor of CTLA-4), pembrolizumab or nivolumab (inhibitors of PD-1), atezolizumab or durvalumab (inhibitors of PD-L1), or epacadostat or indoximod (inhibitors of IDO) may be beneficial in an effort to sustain the secondary immune response initiated by virotherapy.
Conclusion
In summary, our novel in situ multiplex immunofluorescence screening approach and imaging platform is a useful modality capable of providing a comprehensive analysis of the complex changes that occur in tumor cytoarchitecture specifically after immunologically centered therapies (e.g. virotherapy). Taken together, the results demonstrate that our multiplex biomarker screening platform lays the groundwork for the comprehensive characterization of changes that occur within the brain before and after treatment of a CNS neoplasm.
Funding Statement
The content is solely the responsibility of the authors and does not necessarily represent the official views of the U.S. Food and Drug Administration. This work was supported by the Assistant Secretary of Defense for Health Affairs through the Peer Reviewed Cancer Research Program. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense
Acknowledgments
This research was supported in part by a grant from the U.S. Food and Drug Administration and the National Institutes of Health (R01FD005379), the Department of Defense (W81XWH-15-1-0108), the Rally Foundation for Childhood Cancer Research, Hyundai Hope on Wheels, and Cannonball Kids cancer Foundation to GKF. JDB was supported by the University of Alabama at Birmingham Medical Scientist Training Program.
Conflicts of Interest
Drs. Markert and Gillespie are founders of and own stock and stock options (<8% interest) in Aettis, Inc., a biotech company that has licensed M032 HSV from The Board of Trustees of the University of Alabama for the University of Alabama at Birmingham. Dr. Gillespie currently serves as one of five unpaid members of the Board of Directors for Aettis, Inc. Dr. Gillespie is a founder of and owns stock and stock options (<10%) in Maji Therapeutics, which is developing other HSVs that are not the subject of the current investigation. Drs Markert and Gillespie were also founders of and owned stock and stock options (<8%) in Catherex Inc., a biotechnology company that had licensed additional intellectual property related to oHSV. Catherex, Inc., was sold to Amgen, Inc., on December 18, 2015, and they no longer participate in any decision making or have any control of any aspect of Catherex or Amgen, although they did receive proceeds from the sale of the company. Dr. Gillespie has served as a paid advisor to the Program Project at the Ohio State University that seeks to find improved methods for application of distinct oHSV to treat localized and metastatic cancers. This is generally, but not specifically, related to the subject matter of this investigation. Dr. Bernstock has positions/equity in CITC Ltd and Avidea Technologies and is member of the POCKiT Diagnostics Board of Scientific Advisors.
Supplementary Material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Diller L, Chow EJ, Gurney JG, Hudson MM, Kadin-Lottick NS, Kawashima TI, Leisenring WM, Meacham LR, Mertens AC, Mulrooney DA, et al. Chronic disease in the childhood cancer survivor study cohort: a review of published findings. J Clin Oncol. 2009;27(14):1–12. doi: 10.1200/JCO.2008.21.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Markert JM, Medlock MD, Rabkin SD, Gillespie GY, Todo T, Hunter WD, Palmer CA, Feigenbaum F, Tornatore C, Tufaro F, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7(10):867–874. doi: 10.1038/sj.gt.3301205. [DOI] [PubMed] [Google Scholar]
- 3.Markert JM, Liechty PG, Wang W, Gaston S, Braz E, Karrasch M, Nabors LB, Markiewicz M, Lakeman AD, Palmer CA, et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol Ther. 2009;17(1):199–207. doi: 10.1038/mt.2008.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Markert JM, Razdan SN, Kuo H-C, Cantor A, Knoll A, Karrasch M, Nabors LB, Markiewicz M, Agee BS, Coleman JM, et al. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol Ther. 2014;22(5):1048–1055. doi: 10.1038/mt.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedman GK, Bernstock JD, Chen D, Nan L, Moore BP, Kelly VM, Youngblood SL, Langford CP, Han X, Ring EK, et al. Enhanced sensitivity of patient-derived pediatric high-grade brain tumor xenografts to oncolytic HSV-1 virotherapy correlates with nectin-1 expression. Sci Rep. 2018;8(1):13930. doi: 10.1038/s41598-018-32353-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedman GK, Moore BP, Nan L, Kelly VM, Etminan T, Langford CP, Xu H, Han X, Markert JM, Beierle EA, et al. Pediatric medulloblastoma xenografts including molecular subgroup 3 and CD133+ and CD15+ cells are sensitive to killing by oncolytic herpes simplex viruses. Neuro Oncol. 2016;18(2):227–235. doi: 10.1093/neuonc/nov123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friedman GK, Langford CP, Coleman JM, Cassady KA, Parker JN, Markert JM, Yancey Gillespie G.. Engineered herpes simplex viruses efficiently infect and kill CD133+human glioma xenograft cells that express CD111. J Neuro Oncol. 2009;95(2):199–209. doi: 10.1007/s11060-009-9926-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernstock JD, Vicario N, Li R, Nan L, Totsch SK, Schlappi C, Gessler F, Han X, Parenti R, Beierle EA, et al. Safety and efficacy of oncolytic HSV-1 G207 inoculated into the cerebellum of mice. Cancer Gene Ther. 2019. doi: 10.1038/s41417-019-0091-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waters AM, Johnston JM, Reddy AT, Fiveash J, Madan-Swain A, Kachurak K, Bag AK, Gillespie GY, Markert JM, Friedman GK.. Rationale and design of a phase 1 clinical trial to evaluate HSV G207 alone or with a single radiation dose in children with progressive or recurrent malignant supratentorial brain tumors. Hum Gene Ther Clin Dev. 2017;28(1):7–16. doi: 10.1089/humc.2017.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bernstock JD, Wright Z, Bag AK, Gessler F, Gillespie GY, Markert JM, Friedman GK, Johnston JM.. Stereotactic placement of intratumoral catheters for continuous infusion delivery of HSV-1 G207 in pediatric malignant supratentorial brain tumors. World Neurosurg. 2018. doi: 10.1016/j.wneu.2018.11.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Totsch SK, Schlappi C, Kang K-D, Ishizuka AS, Lynn GM, Fox B, Beierle EA, Whitley RJ, Markert JM, Gillespie GY, et al. Oncolytic herpes simplex virus immunotherapy for brain tumors: current pitfalls and emerging strategies to overcome therapeutic resistance. Oncogene. 2019;38:6159–6171. doi: 10.1038/s41388-019-0870-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ring EK, Markert JM, Gillespie GY, Friedman GK. Checkpoint proteins in pediatric brain and extracranial solid tumors: opportunities for immunotherapy. Clin Cancer Res. 2017;23(2):342–350. doi: 10.1158/1078-0432.CCR-16-1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nduom EK, Wei J, Yaghi NK, Huang N, Kong LY, Gabrusiewicz K, Ling X, Zhou S, Ivan C, Chen JQ, et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol. 2016;18(2):195–205. doi: 10.1093/neuonc/nov172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tawbi HA, Forsyth PA, Algazi A, Hamid O, Hodi FS, Moschos SJ, Khushalani NI, Lewis K, Lao CD, Postow MA, et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N Engl J Med. 2018;379(8):722–730. doi: 10.1056/NEJMoa1805453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parra ER. novel platforms of multiplexed immunofluorescence for study of paraffin tumor tissues. J Cancer Treat Diagn. 2018;2:43–53. [Google Scholar]
- 16.Bernstock JD, Ye DG, Griffin A, Lee YJ, Lynch J, Latour LL, Friedman GK, Maric D, Hallenbeck JM. Cerebral ischemia increases small ubiquitin-like modifier conjugation within human penumbral tissue: radiological-pathological correlation. Front Neurol. 2017;8:738. doi: 10.3389/fneur.2017.00738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parra ER, Uraoka N, Jiang M, Cook P, Gibbons D, Forget M-A, Bernatchez C, Haymaker C, Wistuba II, Rodriguez-Canales J. Validation of multiplex immunofluorescence panels using multispectral microscopy for immune-profiling of formalin-fixed and paraffin-embedded human tumor tissues. Sci Rep. 2017;7(1):13380. doi: 10.1038/s41598-017-13942-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang H, Zhang L, Zhang IY, Chen X, Da Fonseca A, Wu S, Ren H, Badie S, Sadeghi S, Ouyang M, et al. S100B promotes glioma growth through chemoattraction of myeloid-derived macrophages. Clin Cancer Res. 2013;19(14):3764–3775. doi: 10.1158/1078-0432.CCR-12-3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen C-Y, Wang P-Y, Hutzen B, Sprague L, Swain HM, Love JK, Stanek JR, Boon L, Conner J, Cripe TP. Cooperation of oncolytic herpes virotherapy and PD-1 blockade in murine rhabdomyosarcoma models. Sci Rep. 2017;7(1):2396. doi: 10.1038/s41598-017-02503-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saha D, Martuza RL, Rabkin SD. Macrophage polarization contributes to glioblastoma eradication by combination immunovirotherapy and immune checkpoint blockade. Cancer Cell. 2017;32(2):253–67 e5. doi: 10.1016/j.ccell.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.LaRocca CJ, Warner SG. Oncolytic viruses and checkpoint inhibitors: combination therapy in clinical trials. Clin Transl Med. 2018;7(1):35. doi: 10.1186/s40169-018-0214-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bjornsson CS, Lin G, Al-Kofahi Y, Narayanaswamy A, Smith KL, Shain W, Roysam B. Associative image analysis: a method for automated quantification of 3D multi-parameter images of brain tissue. J Neurosci Methods. 2008;170(1):165–178. doi: 10.1016/j.jneumeth.2007.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bogoslovsky T, Bernstock JD, Bull G, Gouty S, Cox BM, Hallenbeck JM, Maric D.. Development of a systems-based in situ multiplex biomarker screening approach for the assessment of immunopathology and neural tissue plasticity in male rats after traumatic brain injury. J Neurosci Res. 2017. doi: 10.1002/jnr.24054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sotoudeh H, Shafaat O, Bernstock JD, Brooks MD, Elsayed GA, Chen JA, Szerip P, Chagoya G, Gessler F, Sotoudeh E, et al. Artificial intelligence in the management of glioma: era of personalized medicine. Front Oncol. 2019;9:768. doi: 10.3389/fonc.2019.00768. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.