

ABSTRACT
Chimeric antigen receptor (CAR) T cells are in prime focus of current research in cancer immunotherapy. Facilitating CAR T cell generation is among the top goals. We have recently demonstrated direct in vivo generation of human CD19-CAR T cells by targeting CD8+ cells using lentiviral vectors (LVs). The anti-tumor potency of in vivo generated CAR T cells was assessed in human PBMC-transplanted NSG mice carrying i.v. injected CD19+ Nalm-6 tumor cells. A single injection of CD8-targeted LV delivering CD19-CAR was sufficient to completely eliminate the tumor cells from bone marrow and spleen, whereas control animals contained high levels of CD19+ cells. Tumor elimination was due to in vivo generated CAR+ cells. Notably, these were not only composed of T lymphocytes but also included CAR+ natural killer cells (NK and NKT). This is the first demonstration of tumor elimination by in vivo generated human CAR T cells.
KEYWORDS: Cell therapy, CAR T cells, in vivo gene delivery, receptor targeting, T-cell targeting
Introduction
Genetic modification of T cells with chimeric antigen receptors (CARs) recognizing surface antigens on tumor cells has emerged as an effective therapeutic treatment for patients with B cell hematological malignancies.1-44. Over 300 clinical trials are ongoing worldwide, focusing on various aspects such as improving CAR activity, expanding the approach to other tumor entities and facilitating the complex manufacturing process.5 By now, two products, namely Kymriah for pediatric acute lymphoblastic leukemia (ALL) and Yescarta for adult diffused large B-cell lymphomas (DLBCL), have received marketing authorization.6
CARs recognize tumor antigens by single‐chain variable fragments (scFvs) displayed on a hinge domain. T cell activation is mediated by one or more intracellular signaling domains, which usually include the CD3ζ chain domain. Other co‐stimulatory domains like CD28 and/or 4‐1BB are present in second- and third-generation CARs.7 CAR T cells are individualized cell therapy products requiring extensive and time-consuming manufacturing procedures. The process can potentially be simplified by using T cell-targeted vectors, which transfer the CAR coding sequence selectively into particular lymphocytes thus enabling direct in vivo CAR gene delivery. Proof of principle for this approach in human and mouse T lymphocytes has recently been published by our group and others.8,9 We have demonstrated that human CD19-CAR T cells can be generated directly in vivo using the lentiviral vector CD8-LV which uses the human CD8α chain as an entry receptor.10,11 CD8-LV specifically transduced human CD8+ cells in both human peripheral blood mononuclear cells (huPBMC) engrafted NOD-scid-IL2Rγnull (NSG) mice and mice humanized with human CD34+ hematopoietic stem cells. In vivo CAR T cell generation was accompanied by B lymphocyte elimination.8 Clear evidence for tumor cell clearance by in vivo generated CAR T cells was however missing.8 Here we report that in vivo generated CD19-reactive CD8+ CAR T cells resulted in the complete elimination of CD19+ Nalm-6 cells in vector-treated mice, whereas in control animals CD19+ tumor cells expanded in an uncontrolled way.
Material and methods
Vector production for in vivo CAR T cell generation
CD8-LV encoding the CD19-CAR was generated exactly as described previously using transient transfection of HEK-293T cells with plasmids pCAGGS-NiV-Gd34-CD8, pCAGGS-NiV-Fd22, pCMVdR8.9, and pS-CD19-CAR-W.8 The functional activity of vector stocks was determined by transducing Molt4.8 cells in serial fivefold dilutions. The number of transduced cells was determined after 4 d by flow cytometry to detect CD19-CAR surface expression via its myc tag. Particle numbers in vector stocks were determined by nanoparticle tracking analysis (Nanosight NS300, Malvern Panalytical). For in vivo CAR T cell generation 2.5 × 1011 particles diluted in a total volume of 200 µl PBS were injected into mice intravenously.
Cell culture
Human PBMC isolated from two donors were cultured in RPMI 1640 medium (Biowest) supplemented with 10% fetal bovine serum (FBS; Biochrom AG), 2 mM glutamine (Sigma-Aldrich), 0.5% penicillin/streptomycin, 25 mM HEPES (Sigma-Aldrich) and 50 IU/ml IL-2 (Miltenyi Biotech). B cells were depleted prior to activation by using anti-human CD19 microbeads (Miltenyi Biotech). For the activation of PBMC, plates were coated with 1 µg/ml anti-human CD3 mAb (clone: OKT3, Miltenyi Biotech) and 3 µg/ml anti-human CD28 mAb (clone: 15E8, Miltenyi Biotech) were added to the cell culture medium and incubated for 72 h at 37°C. Nalm-6 cells were cultivated in RPMI 1640 medium supplemented with 10% FBS and 2 mM glutamine. Their identity was confirmed by genetic phenotyping performed by the German cell culture collection (DSMZ).
Tumor mouse model
NSG mice (NOD.Cg.PrkdcscidIL2rgtmWjl/SzJ, Jackson Laboratory) were intravenously (i.v.) injected with 1 × 105 Nalm-6-luc cells, which stably express firefly luciferase.12 Two days prior to vector application, in vivo imaging (IVIS Spectrum, Perkin Elmer) was performed to arrange animals in two different groups based on luciferase signal intensities for unbiased outcomes. A day later 5 × 106 activated human PBMC from two donors were i.v. injected (mice 1–4 and 9–12 received PBMC from donor 1; the other eight mice received PBMC from donor 2). On the next day, 2.5 × 1011 particles of CD8-LV encoding CD19-CAR or PBS as control were i.v. administered. PBS was chosen as control to exclude any influence on tumor growth by the transplanted donor lymphocytes. To follow up tumor progression, IVIS imaging was performed on days 4, 7, 12, 14 and 17 post-vector application. For this purpose, mice were intraperitoneally injected with D-luciferin (Perkin Elmer) at 150 µg/g body weight. Imaging data were obtained 10 min after luciferin injection. Mice were regularly checked for their health status and tumor load by IVIS. They were sacrificed when termination criteria had been reached.
Animal experiments were performed in accordance with the regulations of the German animal protection law and the respective European Union guidelines after having obtained formal approval of the institutional and the national animal care committee. Human blood was provided by anonymous donors in accordance with the rules of the local ethics committee.
Preparation of single-cell suspension and flow cytometry analysis
Single-cell suspensions of spleen were obtained by mincing the organ through a 70 µm cell mesh. Erythrocytes were lysed by resuspending splenocytes in lysis buffer (BD Pharm Lyse™). Bone marrow single-cell suspensions were obtained by flushing the bones with a syringe containing RPMI medium. The cell suspension was passed through a 70 µm cell mesh and erythrocytes were lysed by resuspending cells in lysis buffer. FACS analysis was performed by LSRII or Fortessa (BD Bioscience) and data were analyzed using FACS Express V6 (De Novo Software) or FlowJo V10.1 (FlowJo). Antibodies specific for the following antigens were used for FACS staining: CD45 (2D1, BV510, BioLegend), CD3 (BW264/56, PerCP, Miltenyi Biotec), CD4 (RPA-T4, PE-CF594, BD), CD8 (BW135/80, FITC, Miltenyi Biotec), CD62L (DREG-56, BV605, BioLegend), CD45RA (HI100, BV421, BioLegend), CD19 (HIB19, AlexaFluor700, Thermo Fisher Scientific), myc-tag on the CD19-CAR (9B11, PE, Cell Signaling Technology) PD-1 (PD1.3.1.3, PE-Vio770, Miltenyi Biotec) Tim-3 (REA635, APC, Miltenyi Biotec), Lag-3 (REA351, VioBlue, Miltenyi Biotec) and CD56 (B159, AlexaFluor700, BD Bioscience). To determine the viability, the LIVE/DEAD Fixable Dead cell stain kit (eFluor 780, Thermo Fisher Scientific) was used.
Statistical analysis
Data were analyzed using the GraphPad Prism 7 software (Graph-Pad Software, USA). Statistical differences were assessed as indicated by using an unpaired t-test and two-way ANOVA test. Differences were considered significant at p < 0.05.
Results and discussion
In our previous study on in vivo generated CAR T cells, CD19+ Raji cells had been injected intraperitoneally as CAR T cell target. In this mouse model, Raji cells did not efficiently spread to spleen, blood and bone marrow due to intraperitoneal injection and it was not possible to easily distinguish them from the B lymphocytes.8 In order to assess the anti-tumoral efficacy of in vivo generated CAR T cells, NSG mice transplanted systemically with Raji-luc cells were initially evaluated for the activity of CD8-LV delivering the CD19-CAR. While CAR T cells became readily detectable, there was no significant reduction in tumor burden detectable, with the exception of a tendency for reduced tumor cell load in the bone marrow of some mice.13 We next tested the anti-tumoral function of in vivo generated CAR T cells in the context of luciferase-encoding Nalm-6 tumor cells, which are often assessed in CAR T preclinical studies.14,15 Four days after i.v. injection of luciferase-encoding Nalm-6 cells into NSG mice, the animals were distributed to two groups based on the luciferase intensities measured by IVIS. After another day, activated human PBMC were i.v. injected, followed by i.v. administration of CD8-LV or PBS (control group) a day later (Figure 1(a)). At day 14 after vector injection, 4 out of 8 mice in the vector-treated group displayed complete tumor remission when analyzed by IVIS (Figure 1(b)). One mouse (#9), sacrificed on day 14, exhibited a stable signal compared to day 7 (Figure 1(b)). Three further mice reached complete tumor remission at day 17. Mouse #15 showed an isolated low luciferase signal in the right hind leg (Figure 1(b)). However, also in this mouse, the tumor burden was substantially reduced (about 12-fold) after vector application. In the control group, none of the mice showed any reduction in luciferase signals thus exhibiting a steady increase in tumor load over time (Figure 1(b,c)). Overall, statistically significant tumor remission was evident already by day 12 and became more pronounced by day 14 and 17 when comparing the total flux between the PBS-treated and vector-treated mice (Figure 1(c)).
Figure 1.

Tumor load over time.
(a) Experimental layout. 1 × 105 Nalm-6-Luc tumor cells were intravenously (i.v.) injected into NSG mice. Five days later 5 × 106 activated human PBMC from two donors (Donor 1 and Donor 2) were i.v. administered and finally, another day later mice were i.v. injected with either 2.5 × 1011 particles of CD8-LV encoding CD19-CAR or PBS. (b) Luciferase imaging. Ventral view of all mice included in the experiment recorded at the indicated time points after control (PBS) or vector (CD8-LV) administration. X in the day 17 panel indicates mice sacrificed at day 14. (c) Tumor load over time. Kinetics of quantified luciferase signals over time is shown as logarithm of the total flux (p/sec). The dotted line indicates the time point of vector administration. Data represent mean ± SEM for all groups (PBS, n = 8; CD8-LV, n = 8). Statistical significance was determined using two-way ANOVA.
At the cellular level, the tumor burden was quantified during final analysis based on high CD19 and low CD45 expression (Fig. S1). The reduction of CD19 positive cells in the vector-treated animals was well in agreement with the tumor burden quantified by IVIS measurements (Figure 2(a)). For mouse #9 a low frequency of tumor cells was detectable in bone marrow (2.69%), while in mouse #15 tumor burden in all analyzed organs was too low to be detected. In none of the other vector-treated animals CD19+ cells were detectable, neither in bone marrow, nor in spleen or blood, while the control group contained high levels of tumor cells especially in bone marrow and spleen (Figure 2(a)). Between 5% and 12% of human CD3+CD8+ cells isolated from bone marrow, spleen and blood were CAR-positive on day 14 of analysis (Figure 2(b)). No CAR+ cells were detected in the CD8− population thereby confirming the high selectivity of the vector. Interestingly, CAR levels were significantly lower in bone marrow and spleen at day 18 compared to day 14, which may be due to reduced CAR T cell proliferation after tumor cell clearance. This was further supported by the fact that the two mice with residual tumor (#9 and #15) contained by far higher CAR T cell levels than the tumor-free mice, especially in spleen and blood (Figure 2(b), labeled by arrowheads). These results clearly demonstrate the anti-tumoral potency of the in vivo generated CAR T cells.
Figure 2.

Tumor load and CAR expression at the cellular level.
Analysis of cells in CD8-LV (black symbols) and PBS (gray symbols) injected animals were evaluated by flow cytometry. Representative density plots are shown for bone marrow cells for one mouse from each group sacrificed at day 14. The gating strategies are represented in Fig. S1. Cells isolated from bone marrow, spleen and blood were evaluated by flow cytometry for the percentage of (a) CD19+ tumor cells within the fraction of human CD45+ cells and (b) CAR+ cells in the CD3+CD8+ population at 14 and 18 days post-vector administration. Donor 1 and donor 2 are represented as open and filled circles, respectively. Black arrowheads point to values obtained for mouse #9 at day 14 and mouse #15 at day 18. Data represent mean ± SEM for all groups. Statistical significance was determined using multiple t-test.
Approaches to optimize the conventional CAR T cell product currently focus on the identification of conditions that favor the enrichment of specific T cell phenotypes such as central memory T cells (TCM) or effector memory T cells (TEM) in order to obtain a product exhibiting long-lasting anti-tumor efficacy.16-1818. Since most of the anti-tumoral activities take place in the bone marrow we evaluated the phenotype of in vivo generated CAR T cells in bone marrow based on CD45RA and CD62L expression. In vivo generated CD8-targeted CAR T cells displayed predominantly effector T cell (Teff) or effector memory T cell (TEM) phenotype in a donor-dependent manner (Figure 3(a)). In donor 1, there were substantially more TEM cells in the vector-treated animals than in the control animals (Figure 3(a–b)). Animals transplanted with donor 2 cells, in contrast, showed similar high levels of Teff cells in both groups. Irrespective of the differences in the phenotypes, CAR T cells from both donors had efficiently cleared the tumor. These results suggest that in vivo generated effector CAR TEM and Teff cells are equally potent in clearing tumor cells. This is well in line with ex vivo generated CAR T cells engrafted into NSG/Nalm6 mice exhibiting mainly Teff phenotype.19 Repetitive antigen stimulation often drives CAR T cells into an exhaustion phenotype thereby losing their anti-tumor potency.20 PD-1 expression on CD8+ T cells ranged between 85% and 95% in the control groups at day 14 (Figure 3(c)). This high level was likely due to ex vivo plate-bound activation of the engrafted PBMC. Yet, PD-1 expression was even higher in the vector-treated animals, in CD8+CAR− as well as CD8+CAR+ populations. This effect was especially pronounced on day 14 thus being well in line with this day showing the main anti-tumoral activity. For Lag-3 and Tim-3, there were no significant differences between the control and the vector-treated mice (Figure 3(c)). At day 18, however, the vector-treated group displayed an increased dual expression of Lag-3 and Tim-3 compared to the control mice (Figure 3(d)). While these data demonstrate activation-induced exhaustion of the CAR T cells, it has to be kept in mind that exhaustion in this mouse model is strongly influenced by the xenogeneic activation of the human T cells. Final conclusions about the exhaustion of in vivo generated CAR T cells will require studies in syngeneic settings.
Figure 3.

Memory and exhaustion phenotype.
(a) Graphic representation of memory T cell phenotypes in CD8+CAR− fractions in control (PBS) and CD8+CAR+ fraction in vector (CD8-LV) treated mice at 18 d post-vector administration from two donors (D1 and D2) are shown. Cells isolated from the bone marrow were evaluated by flow cytometry for the percentage of (b) memory phenotype TSCM/Naïve, Teff, TEM and TCM, and (c) inhibitory receptors PD-1, Tim-3 and Lag-3 within the CD3+CD8+ fraction in mice treated with PBS (PBS CAR−; gray circles) or CD8-LV (CD8-LV CAR−; black circles and CD8-LV CAR+; black triangles). Donor 1 and donor 2 are represented as open and filled circles/triangles, respectively. Data represent mean ± SEM for all groups. Statistical significance was determined using two-way ANOVA. (d) Graphic representation of inhibitory receptor expression in CD8+CAR− fraction in control (PBS) and CD8+CAR+ fraction in vector (CD8-LV) treated mice analyzed at 14 and 18 d post-vector administration.
Apart from T cells, CD8α is expressed on natural killer cells (NK), natural killer T cells (NKT), dendritic cells (DCs) and macrophages. During ex vivo activation, macrophages and DCs were eliminated from the PBMC due to applied cultivation conditions. Therefore, we evaluated CAR expression in NK and NKT cells present in the animals. Remarkably, we observed CAR expression on CD56+CD8+ cells in both CD3+ (NKT cells) and CD3− (NK cells) fractions, on day 14 and day 18 in bone marrow (Figure 4(b,c)). Their frequencies decreased from day 14 to day 18 (Figure 4(e,f)). This correlated well with the decrease in CD19+ cells and reflects what was observed for CAR T cells (Figure 4(d)). Overall, substantially higher frequencies of CD8+ NK and NKT cells were observed in the vector-treated groups compared to the control groups at day 14. Interestingly, CAR+ NKT and NK cells were also detected in spleen at day 14 (Fig. S3). Hence suggesting that all types of CAR+ cells induced by CD8-LV had proliferated through antigen stimulation.
Figure 4.
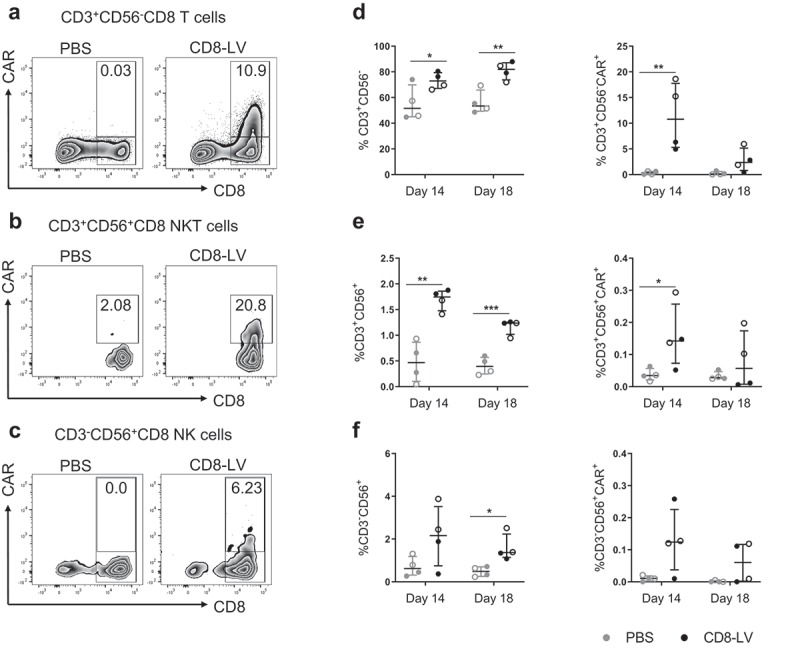
CAR expression on NK and NKT cells.
Cells isolated from the bone marrow of CD8-LV (black symbols) or PBS (gray symbols) injected animals were evaluated by flow cytometry. Frequencies of CAR+ cells within T cells (a), NKT cells (b), NK cells (c) and their percentage of total CD8 cells (d–f) are shown. (a–c) Exemplary density plots showing different populations in the bone marrow cells of two representative mice at day 14 post-vector administration. The gating strategy is represented in Fig. S2. (d–f) Frequencies of individual populations are calculated as count of individual cell populations normalized to total CD8 cell count. Percentages of (d) CD3+CD56− T cells and T cell-derived CAR+ cells, (e) CD3+CD56+(NKT cells) and NKT-derived CAR+ cells as well as (f) CD3−CD56+ (NK cells) and NK-derived CAR+ cells at 14 and 18 d post-vector administration are shown. Donor 1 and donor 2 are represented as open and filled circles, respectively. Data represent mean ± SEM for all groups. Statistical significance was determined using multiple t-test.
In this study, we provide the first evidence for anti-tumoral activity mediated by in vivo generated human CAR T cells. Such CAR T cells were able to completely eliminate CD19+ Nalm-6 tumor cells in T cell transplanted NSG mice, a model frequently applied in the preclinical assessment of CAR T cells. While the activity against Nalm-6 cells was impressive, our approach failed in a similar mouse model with Raji tumor cells, 13 possibly due to their more aggressive and rapid disseminating nature. The question if in vivo generated CAR T cells could be as effective as ex vivo generated CAR T cells will have to be further investigated. Standardizing the dose of the administered vector particles versus the number of injected CAR T cells will be a particular challenge in such experiments. Beyond that, further preclinical testing, especially in large animal models, will be required to move this approach further toward translation.
An unexpected outcome of our study was the detection of CAR-positive NK and NKT cells in vector-injected mice. Due to their expression of the CD8α chain, which is the target antigen of the displayed single-chain antibody on the vector particles, these cells were on-target transductions. However, since NK cells are much harder to transduce ex vivo than T lymphocytes, their in vivo generation was unexpected. Although they made up only 1-2% of all CAR+ cells at the given time point, the presence of NK and NKT cells in the bone marrow at the site of tumor cell expansion, especially in the vector-treated group, suggests that they likely contributed to tumor cell clearance. Thus, CD8α-chain targeted LVs provide the potential of generating both, CAR+ T cells and NK cells, directly in vivo upon systemic administration. Since Her2-CAR expressing NK cells have been shown to induce T cell immunity against cancer cells in syngeneic mouse tumor models, the generation of CAR-NK cells could be another advantage of the in vivo approach used here.21
Funding Statement
This work was supportedby grants from the Deutsche Krebshilfe (70112578) and from the German government through the Federal Ministry of Health (03292364) to CJB.
Acknowledgments
The authors would like to thank Manuela Gallet and Gundula Braun for excellent help with animal experiments. Nalm-6-luc cells were kindly provided by Aditi Dey and Adele Fielding (UCL Cancer Institute, London).
Conflict of interest
CJB is listed as an inventor on a patent covering the CD8-targeted lentiviral vector. All the other authors declare that they have no conflict of interest.
Author contributions
SA planned and performed the experiments, analyzed data and drafted the manuscript. FBT contributed to experimental designs and supervised work. TW produced and evaluatedvector particles. CJB initiated and supervised the project, acquired grants and contributed to writing of the manuscript.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC.. CAR T cell immunotherapy for human cancer. Science. 2018;359:1–7. doi: 10.1126/science.aar6711. [DOI] [PubMed] [Google Scholar]
- 2.June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379:64–73. doi: 10.1056/NEJMra1706169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lichtman EI, Dotti G. Chimeric antigen receptor T-cells for B-cell malignancies. Transl Res. 2017;187:59–82. doi: 10.1016/j.trsl.2017.06.011. [DOI] [PubMed] [Google Scholar]
- 4.Miliotou AN, Papadopoulou LC. CAR T-cell therapy. a new era in cancer immunotherapy. Curr Pharm Biotechnol. 2018;19:5–18. doi: 10.2174/1389201019666181022115405. [DOI] [PubMed] [Google Scholar]
- 5.Hartmann J, Schüßler‐Lenz M, Bondanza A, Buchholz CJ. Clinical development of CAR T cells – challenges and opportunities in translating innovative treatment concepts. EMBO Mol Med. 2017;9:1183–1197. doi: 10.15252/emmm.201607485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barkholt L, Voltz-Girolt C, Raine J, Salmonson T, Schüssler-Lenz M. Regulatory watch. European regulatory experience with advanced therapy medicinal products. Nat Rev Drug Discov. 2018;18:8–9. doi: 10.1038/nrd.2018.200. [DOI] [PubMed] [Google Scholar]
- 7.Daniyan AF, Brentjens RJ. CARs of the future. Am J Hematol. 2019;49:S55–S58. doi: 10.1002/ajh.25416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pfeiffer A, Thalheimer FB, Hartmann S, Frank AM, Bender RR, Danisch S, Costa C, Wels WS, Modlich U, Stripecke R, et al. In vivo generation of human CD19-CAR T cells results in B-cell depletion and signs of cytokine release syndrome. EMBO Mol Med. 2018;10:e9158. doi: 10.15252/emmm.201809158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith TT, Stephan SB, Moffett HF, McKnight LE, Ji W, Reiman D, Bonagofski E, Wohlfahrt ME, Pillai SPS, Stephan MT. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers In Nat Nanotechnol. 2017;813–820. doi: 10.1038/nnano.2017.57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bender RR, Muth A, Schneider IC, Friedel T, Hartmann J, Plückthun A, Maisner A, Buchholz CJ. Receptor-targeted nipah virus glycoproteins improve cell-type selective gene delivery and reveal a preference for membrane-proximal cell attachment. PLoS Pathog. 2016;12:e1005641. doi: 10.1371/journal.ppat.1005641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou Q, Schneider IC, Edes I, Honegger A, Bach P, Schönfeld K, Schambach A, Wels WS, Kneissl S, Uckert W, et al. T-cell receptor gene transfer exclusively to human CD8(+) cells enhances tumor cell killing. Blood. 2012;120:4334–4342. doi: 10.1182/blood-2012-02-412973. [DOI] [PubMed] [Google Scholar]
- 12.Dey A, Zhang Y, Castleton AZ, Bailey K, Beaton B, Patel B, Fielding AK. The role of neutrophils in measles virus-mediated oncolysis differs between b-cell malignancies and is not always enhanced by GCSF. Mol Ther Nucleic Acids. 2016;24:184–192. doi: 10.1038/mt.2015.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pfeiffer A. CD8 Receptor-targeted lentiviral vectors – an approach for the in vivo generation of chimeric antigen receptor (CAR) T cells. Doctoral Thesis: Technical University Darmstadt; September 2018. [Google Scholar]
- 14.Brentjens RJ, Santos E, Nikhamin Y, Yeh R, Matsushita M, La Perle K, Quintás-Cardama A, Larson SM, Sadelain M. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin. Cancer Res. 2007;13:5426–5435. doi: 10.1158/1078-0432.CCR-07-0674. [DOI] [PubMed] [Google Scholar]
- 15.Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. 2018;24:7312–738. doi: 10.1038/s41591-018-0041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blaeschke F, Stenger D, Kaeuferle T, Willier S, Lotfi R, Kaiser AD, Assenmacher M, Döring M, Feucht J, Feuchtinger T. Induction of a central memory and stem cell memory phenotype in functionally active CD4+ and CD8+ CAR T cells produced in an automated good manufacturing practice system for the treatment of CD19+ acute lymphoblastic leukemia. Cancer Immunol. Immunother. 2018;67:1053–1066. doi: 10.1007/s00262-018-2155-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Casucci M, Falcone L, Camisa B, Norelli M, Porcellini S, Stornaiuolo A, Ciceri F, Traversari C, Bordignon C, Bonini C, et al. Extracellular NGFR spacers allow efficient tracking and enrichment of fully functional CAR-T cells co-expressing a suicide gene. Front Immunol. 2018;9:507. doi: 10.3389/fimmu.2018.00507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Popplewell LL, Wagner JR, Naranjo A, Blanchard MS, Mott MR, Norris AP, Wong CW, Urak RZ, Chang W-C, et al. Phase 1 studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with B-cell NHL. Blood. 2016;127:2980–2990. doi: 10.1182/blood-2015-12-686725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao Y, Rodgers DT, Du J, Ahmad I, Hampton EN, Ma JSY, Mazagova M, Choi S-H, Yun HY, Xiao H, et al. Design of switchable chimeric antigen receptor T cells targeting breast cancer. Angew Chem Int Ed Engl. 2016;55:7520–7524. doi: 10.1002/anie.201601902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, Sadelain M, Adusumilli PS. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126:3130–3144. doi: 10.1172/JCI83092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang C, Oberoi P, Oelsner S, Waldmann A, Lindner A, Tonn T, Wels WS. Chimeric antigen receptor-engineered NK-92 cells. An off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front Immunol. 2017;8:533. doi: 10.3389/fimmu.2017.00533. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.