

ABSTRACT
Biofluid-accessible extracellular vesicles (EVs) may represent a new means to improve the sensitivity and specificity of detecting disease. However, current methods to isolate EVs encounter challenges when they are used to select specific populations. Moreover, it has been difficult to comprehensively characterize heterogeneous EV populations at the single vesicle level. Here, we robustly assessed heterogeneous EV populations from cultured cell lines via nanoparticle tracking analysis, proteomics, transcriptomics, transmission electron microscopy, and quantitative single molecule localization microscopy (qSMLM). Using qSMLM, we quantified the size and biomarker content of individual EVs. We applied qSMLM to patient plasma samples and identified a pancreatic cancer-enriched EV population. Our goal is to advance single molecule characterization of EVs for early disease detection.
Abbreviations: EV: Extracellular Vesicle; qSMLM: quantitative Single Molecule Localization Microscopy; PDAC: Pancreatic Ductal Adenocarcinoma; EGFR: epidermal growth factor receptor 1; CA19-9: carbohydrate antigen 19-9; SEC: size exclusion chromatography; WGA: wheat germ agglutinin; AF647: Alexa Fluor 647; Ab: antibody; HPDEC: Healthy Pancreatic Ductal Epithelial Cell; TEM: Transmission Electron Microscopy.
KEYWORDS: Quantitative single molecule localization microscopy, extracellular vesicles, pancreatic cancer, translational medicine, imaging, diagnostics
Introduction
Since the symptoms of pancreatic ductal adenocarcinoma (PDAC) are often non-specific, patients are usually diagnosed at a late stage of the disease [1]. Early detection strategies for PDAC remain inadequate [2]. However, encouraging results were obtained when liquid biopsies were interrogated for extracellular vesicles (EVs) [3]. These membrane-encapsulated structures are continuously shed by both healthy and diseased cells. EVs originating from diverse biogenesis pathways, vary in size, and harbour distinct molecular cargo (proteins, lipids, and nucleic acids). For example, exosomes are typically 30–100 nm in diameter and originate from the endosomal pathway. Microvesicles, which are more variable, are typically 100–1000 nm in diameter, and originate from plasma membrane budding. EVs shed by healthy cells contain molecular regulators of normal intercellular communication [4] whereas EVs shed by cancer cells play a role in disease pathogenesis, such as tumour initiation and progression [4].
EVs are excellent biomarker sources. They can be collected frequently and noninvasively from accessible biofluids. However, patient biofluids contain complex mixtures of EVs originating from different healthy and diseased tissues. Two major challenges in leveraging their diagnostic potential have been to robustly isolate EVs from specific cell types and to characterize EV subpopulations at the level of individual vesicles [5]. Here we describe a new application of quantitative single molecule localization microscopy (qSMLM) to target plasma EV populations through highly expressed membrane glycoproteins and to comprehensively characterize individual EVs.
PDAC has been associated with high levels of specific receptors and altered forms of various membrane glycoproteins [6,7]. Two important examples are epidermal growth factor receptor 1 (EGFR) and carbohydrate antigen 19–9 (CA19-9). EGFR is both overexpressed in pancreatic tumours [7] and abundant in EVs shed from PDAC cells [3,8]. Likewise, CA19-9 is attached to a variety of membrane proteins highly expressed in PDAC [9,10] and is considered a marker for long-term PDAC prognosis [11]. However, CA19-9 plasma levels have limited diagnostic potential [12,13].
qSMLM is a sensitive fluorescence-based imaging method that can achieve single molecule sensitivity with ~10 nm precision. In previous cell culture applications, qSMLM was used to determine EV numbers and sizes [14] or the presence of proteins of interest [15,16]. Here, qSMLM was used to comprehensively assess EVs from both cell culture and patient plasma. We combined 1) affinity selection of EVs via EGFR or CA19-9, 2) robust molecular counting [17], and 3) fast data analysis. Using this strategy, we quantified both the size and membrane protein content of individual EVs. Importantly, we detected a PDAC-enriched EV subpopulation in patient plasma.
These single molecule measurements on both EGFR- and CA19-9-enriched EVs provide several important benefits. Since molecules within EVs are protected from the circulating milieu, they may represent a more accurate reflection of disease states [18]. Moreover, this methodology can also be used to define EV heterogeneity. In this context, both the molecular profile and size of individual EVs may provide biological insight into the communication mechanisms between cancer cells. Finally, low volumes of plasma are required, and the approach can be easily extended to many other membrane targets. Ultimately, our goal is to use qSMLM to characterize specific EV populations for diagnostics.
Results
EV isolation and reproducibility
EVs from PANC-1 cells were isolated using size exclusion chromatography (SEC). SEC fractions were analysed for the following: 1) EV concentration with nanoparticle tracking analysis (NanoSight, Figure 1(a), light grey), 2) protein concentration with NanoDrop (Figure 1(a), dark grey), and 3) amount of EGFR and EV markers, CD63 and TSG101, in EV fractions (Figure 1(a), top) via Western blots. SEC fraction 8 (F8) consistently exhibited the highest amount of EVs. We thus assessed the reproducibility of F8 with three independent EV preparations analysed by LC-MS/MS. The number of unique proteins identified in each replicate was between 180–193, with >95% of proteins annotated in ExoCarta and >95% overlap between all three replicates (Figure S1). Functional commonalities shared by EVs in F8 emerged from the functional enrichment analysis of this highly reproducible protein cargo. Unsurprisingly, terms associated with EVs were highly represented (Table S1). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD012660.
Figure 1.
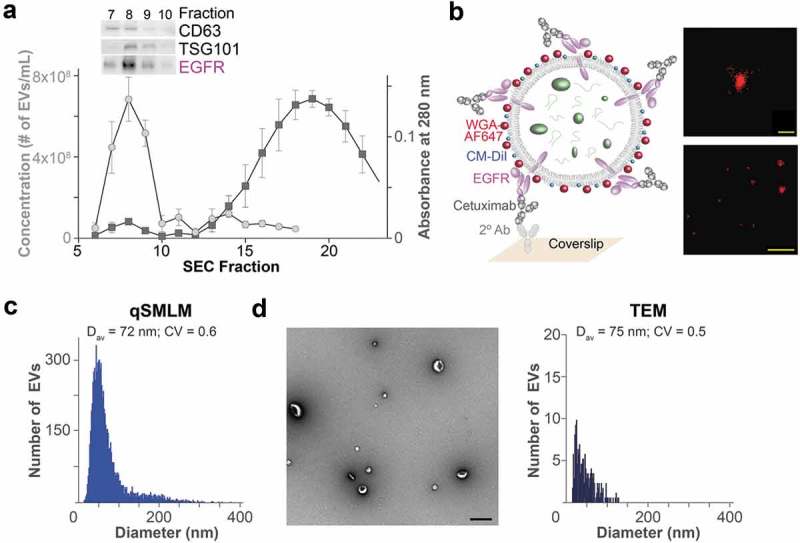
Quantification of EV sizes. (a) EVs from PANC-1 cells were isolated using SEC and characterized for EV and protein concentration. Error bars represent SEM; N = 3. Protein levels for the EV markers (CD63, TSG101) and EGFR were obtained for SEC fractions 7–10 with Western blots. (b) Left, scheme of an EV affinity isolated with cetuximab and labelled with WGA-AF647 as a reporter. Right, filtered dSTORM images of WGA-AF647 with localizations in red. A single EV (top; scale bar, 100 nm) and a larger field of view (bottom; scale bar, 1 µm) are shown. (c) qSMLM size distribution of EGFR-enriched EVs from PANC-1 cells (SEC F8) using WGA-AF647 as a reporter; N = 3, 15 ROI. (d) Representative TEM image (left; scale bar 200 nm) and TEM size distribution (right) of EVs from PANC-1 cells (SEC F8); N = 3.
Quantifying properties of fluorescent reporters
Quantification of SMLM images requires thorough characterization of fluorescently labelled probes. Here, primary antibodies (Abs) were labelled with approximately one Alexa Fluor 647 (AF647) dye (see Methods). However, small labelling heterogeneity is expected even with optimized protocols. To minimize this effect, we calculated the average number of appearances for fluorescently labelled Abs using a surface assay for molecular isolation (SAMI) [17], Figure S2. This approach allowed us to directly test photophysical properties of labelled Abs using the same optical setup and imaging conditions as those used for detecting EVs. Concentrations of fluorescently labelled Abs were also assessed with dSTORM using PANC-1 EVs. Compared to staining with 13 nM of Ab (2 μg/mL), staining with 1 nM of Ab produced less signal and isolated fewer EVs, whereas staining with 30 nM of Ab produced more noise. Additionally, similar localization densities were obtained on EVs stained with 13 nM and 30 nM of Abs. While every effort was made to properly quantify molecular content, reported densities represents estimates for given processing conditions. Thus, we report detected densities, rather than absolute molecular densities.
qSMLM on cell line EVs
We assessed PANC-1 EVs from SEC F8. After staining (see Methods), EVs were affinity isolated onto coverslips using the clinical Ab cetuximab against EGFR (Figure 1(b), left). EVs were subsequently immobilized via fixation (see Methods). This immunocapture method facilitated efficient SMLM imaging. Simulations were performed thereafter to identify optimal NIS-Elements processing filters for the qSMLM data (Figure S3A,B), helping to remove signal unassociated with EVs (e.g. fluorescent molecules not bound to EVs, Figure S3B). Rapid Voronoi tessellation was subsequently used to extract details on EV diameter and the number of molecules per EV (Figure S3C,D); see Methods). Additionally, we showed that Brownian fluctuations were largely negligible in our characterization of isolated EVs, Figure S4.
Figure 1(b), right, shows qSMLM images obtained when cetuximab isolated EVs were stained with AF647 coupled to a membrane binding lectin called wheat germ agglutinin (WGA-AF647); additional EV images are shown in Figure S5. qSMLM was next used to establish the size of isolated EVs (Figure 1(c)). The average EV diameter was 72 nm with a coefficient of variation (CV) of 0.6. These measurements were validated with transmission electron microscopy (TEM). Largely intact and unaggregated EVs were observed (Figure 1(d), left) with an average EV diameter of 75 nm and CV of 0.5 (Figure 1(d), right). We also examined the EVs eluted in fraction 10 (Figure S5B, Figure S6). As expected from the SEC elution profile, both qSMLM and TEM detected fewer EVs with smaller diameters in fraction 10.
To examine EVs for EGFR content, we used AF647 coupled to cetuximab (cetuximab-AF647). Images are shown in Figures S5C–G and a scheme in Figure S7A. The qSMLM characterization is shown in Figure 2(a). Each black dot represents one EV and provides two corresponding values: the number of detected EGFR receptors on the membrane surface (y-axis) and the apparent EV diameter using cetuximab-AF647 as a reporter (x-axis). On average, EVs had 28 detected EGFRs and a diameter of 51 nm. Measurements of replicates were highly reproducible (Figure S8A). Further, simulated EVs were generated from a random sampling of experimental data to demonstrate that these nanoscale details are associated with a distinct distribution of EVs (Figure S7B). As expected, the average EV size obtained from qSMLM with cetuximab-AF647 decreased from SEC fraction 6 to SEC fraction 10 (Figure S7C).
Figure 2.
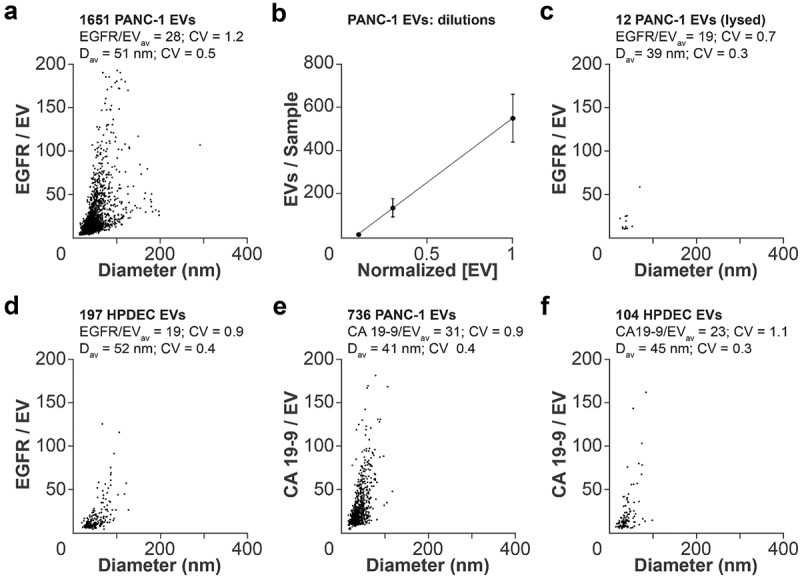
Quantification of EV content. (a) qSMLM quantification of EGFR-enriched EVs from PANC-1 cells using cetuximab-AF647 as a reporter. (b) Average number of detected EVs from PANC-1 cells using different dilutions. (c) qSMLM quantification of EGFR-enriched EVs from PANC-1 cells permeabilized with Triton-X 100 using cetuximab-AF647 as a reporter. (d) qSMLM quantification of EGFR-enriched EVs from HPDEC cells using cetuximab-AF647 as a reporter. (e) qSMLM quantification of CA19-9-enriched EVs from PANC-1 cells using anti CA19-9 Ab-AF647 as a reporter. (f) qSMLM quantification of CA19-9-enriched EVs from HPDEC cells using anti CA19-9 Ab-AF647 as a reporter. In all cases, SEC F8 was used; N = 3, 15 ROI.
Our method to detect EGFR-enriched EVs was sensitive. The number of detected EVs correlated with the amount of EVs incubated onto coverslips across a wide concentration range (Figure 2(b), Figure S5C,D). Several controls were performed to validate the method. A negligible number of EVs were detected under the following conditions: 1) EV membranes were disrupted with Triton-X 100 (Figure 2(c), Figure S5E); 2) EVs were incubated with cetuximab-AF647 and then placed onto coverslips coated with either PEG (Figure S5F, Figure S9A) or anti-rabbit secondary Ab (Figure S5G, Figure S9B); and 3) EVs labelled only with CM-DiI were incubated onto coverslips coated with anti-human Fc secondary Ab (Figure S9C).
We also assessed EVs from normal human pancreatic ductal epithelial cells (HPDEC) purified by SEC. F8 was assessed for both EGFR and EV marker content (Figure S9D). Using qSMLM, we detected fewer EVs with lower EGFR content from HPDECs compared to PANC-1 cells (Figure 2(d), Figure S5H). No significant difference in EV diameter was observed between the two cell lines. qSMLM replicates were also reproducible (Figure S8B).
Additionally, we characterized CA19-9 content in SEC F8 EVs from PANC-1 cells and HPDECs (Figure S5I). The Ab against CA19-9 is well characterized, extremely selective, and has been extensively used in biomarker discovery [10]. Affinity isolation with the anti CA19-9 Ab-AF647 led to a narrower EV size range for both cell lines. Compared to HPDECs, PANC-1 cells yielded significantly more EVs with higher CA19-9 content (Figure 2(e,f)). While the detected target densities depend on experimental conditions (e.g. optical setup, Ab quality, and labelling efficiency), these numbers are reproducible and can be used to compare samples (Figure S8C,D).
RNASeq
To further assess EV content, RNA was isolated from SEC fractions 7–9 of PANC-1 and HPDEC cells and quantified using Ribogreen. The RNA content was higher in PANC-1 fractions, and the RNA in F8 was consistently high in both cell lines (Table S2). Raw sequence data can be found on the exRNA Atlas (exrna-atlas.org, accession number: EXR-KJENS1PANCANC1-AN). Sequencing depth was similar across all samples (Table S3). F8 EVs from PANC-1 cells had the greatest number of detected transcripts identified in all three replicates (Figure S10). A list of differentially expressed genes for F8 can be found in Table S4.
qSMLM on plasma EVs
SEC was used to isolate EVs from ~200 μL of plasma from 6 healthy subjects and 5 PDAC patients. We characterized the EV concentration of F8 using nanoparticle tracking analysis and adjusted the starting concentration of EVs for qSMLM to be identical for each patient sample (diluted to 7 × 108 EVs/150 μL). Samples were affinity isolated with either cetuximab-AF647 or anti CA19-9 Ab-AF647 (Figure 3(a–c), Figure S5J,K, and Figure S11). Significantly more EVs were isolated from PDAC patients compared to healthy subjects. An average of 5- and 15-fold increases in EV numbers were observed for EGFR and CA19-9, respectively (Figure S11E). EVs from the plasma of PDAC patients showed complex and heterogeneous distributions. We detected multiple populations, one resembling that found in healthy subjects and an additional PDAC-associated population. We quantified this PDAC-associated population by negative gating: fewer than 5% of the total healthy subject EVs were contained within the gated region (grey polygons, Figure 3(c)). The PDAC-enriched area contained EVs larger in diameter but with fewer target molecules, possibly indicating a microvesicle enrichment. In this region, significantly more EVs were isolated from PDAC patients compared to healthy subjects. An average of 35- and 130-fold increases in EV numbers were observed for EGFR and CA19-9, respectively (Figure 3(d)). Importantly, our results are in agreement with patient characteristics. Taken altogether, our method appears to be sensitive for PDAC detection (Figure S11G).
Figure 3.
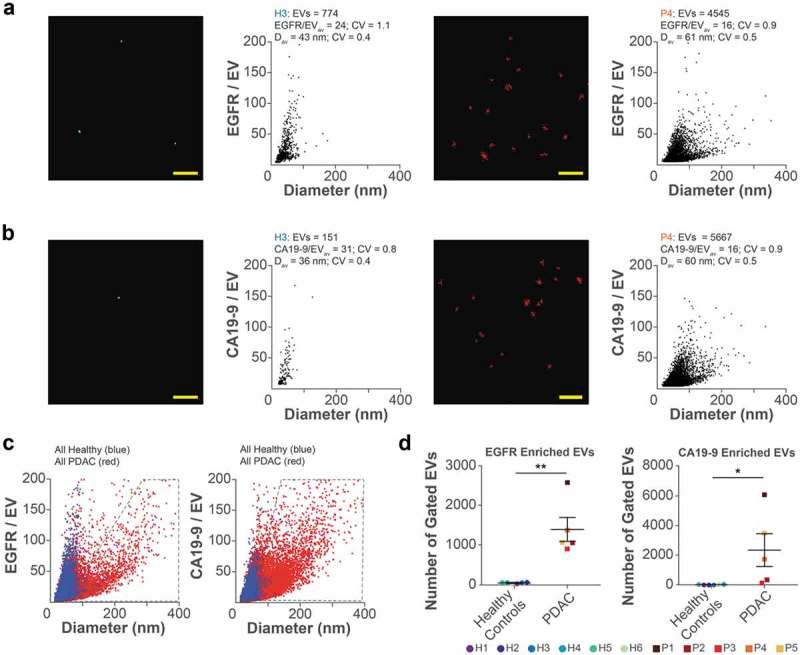
Quantification of EVs from patient plasma. (a) Filtered dSTORM images and qSMLM quantification of EGFR-enriched EVs from plasma of healthy subject 3 (H3, left) and PDAC patient 4 (P4, right) using cetuximab-AF647 as a reporter (N = 3, 15 ROI). (b) Filtered dSTORM images and qSMLM quantification of CA19-9-enriched EVs from plasma of healthy subject 3 (H3, left) and PDAC patient 4 (P4, right) using anti CA19-9 Ab-AF647 as a reporter (N = 2, 15 ROI). Each dot in the dSTORM images represents a localization from the corresponding fluorescent reporter. Scale bars, 1 µm. (c) Combined EGFR-enriched (left) and CA19-9-enriched (right) EVs from healthy subjects (blue) and PDAC patients (red). (d) Number of EVs in gated area (grey polygons, panel c). In all cases, SEC F8 was used.
Discussion
EVs play critical roles in both normal and pathogenic intercellular communication. In cancer, EVs can mediate tumour development, vascularization, and metastasis [4]. Since EVs are a source of cell-specific bioactive molecules, they represent ideal candidates for biomedical and therapeutic applications. However, it has been challenging to assess individual, cell-specific EVs from complex patient biofluids. Here, we extensively characterized EVs from cell lines using nanoparticle tracking analysis, proteomics, transcriptomics, TEM, and qSMLM. We extended our methodology to patient EVs. We determined EV concentration, size distribution, and molecular content of EGFR and CA19-9. Using these parameters, we defined a pancreatic cancer-enriched EV population.
Few methods comprehensively assess EVs at the single vesicle level. Electron microscopy quantifies EV size [19,20], nanoparticle tracking analysis measures both EV size and numbers, tunable resistive pulse sensing measures EV size, numbers and charge, and high-resolution flow cytometry assesses EV numbers and membrane protein expression levels [20,21]. Studies have provided conflicting data regarding absolute quantification of EVs [5,22]. Here we provide an approach that can efficiently assess EV numbers, size, and molecular content from a limited amount of sample material. The present study advances qSMLM to assess pancreatic cancer-enriched EV populations from both cell lines and human plasma. Importantly, our approach can be extended to other cell- or tissue-specific EV populations for diagnostic applications.
To reproducibly isolate EVs, we used SEC. Per recent guidelines [23], we verified that EV fractions contained EV specific markers, CD63 and TSG101. Using TEM, we confirmed EVs were largely unaggregated, intact, and efficiently separated from soluble factors in media (Figure 1). The SEC isolation method was shown to be highly reproducible through proteomic and transcriptomic analyses. Additionally, using the information on EV cargo obtained through these types of analyses, other molecular candidates can be selected in the future for isolation of PDAC-enriched EVs.
We utilized surface glycoproteins enriched in PDAC to affinity isolate vesicles and established an analysis pipeline to quantify EV size and membrane content. Fluorescently labelled Abs were used to detect EVs isolated from PANC-1 cells (pancreatic cancer cells) and HPDECs (normal pancreatic ductal epithelial cells) (Figure 2). We detected a greater number of EVs with higher EGFR and CA19-9 content from PANC-1 cells compared to HPDECs. Our independent repeats provided reproducible results (Figure S8). Thus, the spread in size and receptor content is likely due to inherent EV heterogeneity. Our data in cell lines indicate that qSMLM can identify signatures associated with particular EV populations.
Interestingly, staining with cetuximab-AF647, compared to staining with WGA-AF647, yielded a smaller apparent EV diameter. We speculate this may be caused by a lower EGFR density on larger (>100 nm diameter) EVs and non-uniform EGFR distributions (Figure S3C). Simulations guided by experimental results (Figure S7B) suggest that the distribution of EGFR on membranes (i.e. membrane areas without detected EGFR) may contribute to the slight underestimation of sizes.
We extended our methodology to more complex clinical samples. EVs from both the plasma of healthy subjects and PDAC patients were assessed for the PDAC-relevant markers, EGFR and CA19-9. EVs from the plasma of PDAC patients showed complex and heterogeneous distributions. The PDAC-associated population included larger EVs with fewer receptors (Figure 3). This result could indicate that more abundant shedding of EGFR- and CA19-9-enriched microvesicles is characteristic of PDAC tumours. This is consistent with a previous study indicating that EGFR can transfer between cell populations through microvesicles, increasing oncogenic activity [24]. Our blinded qSMLM analysis indicated that P1 and P4 had highly elevated numbers of EGFR- and CA19-9-enriched EVs in PDAC-associated EV populations. P3, however, had minimally elevated numbers of EGFR- and CA19-9-enriched EVs in PDAC-associated EV populations. Clinical data for P1 and P4 displayed elevated serum CA19-9 and these patients ultimately succumbed to the disease. P3 did not show elevated serum CA19-9 at the start of the trial when plasma was collected. Thus, the comparison of qSMLM results to clinical data demonstrates that qSMLM has diagnostic potential and that our overall approach may be sensitive for effective PDAC detection.
Conclusion
The advancement presented here allowed us to characterize distinct populations of EVs by quantifying the size and detected receptor content of individual vesicles. Furthermore, we differentiated PDAC-enriched EVs from other EV populations within complex biofluids. In conclusion, our qSMLM approach can be extended to the study of EVs from other diseases and a range of molecular EV targets broadly used in research and medicine.
Methods
Cell culture
PANC-1 cells (ATCC) were cultured in RPMI-1640 phenol red-free media supplemented with 10% (v/v) EV depleted FBS (Gibco; A2720801), 1% (v/v) penicillin-streptomycin (Thermo; 15140148), 2 mM glutamax (Gibco; 35030061), and 1 mM sodium pyruvate (Gibco; 11360070). Human immortalized pancreatic ductal epithelial cells (HPDEC, AddexBio, San Diego, CA) were grown in Keratinocyte SFM medium with defined Keratinocyte SFM supplements (Life Technologies; 17005042 and S0015) per supplier suggestion and subsequently cultured in phenol red–free Epilife media supplemented with calcium chloride and Epilife supplements (Life Technologies; MEPICFPRF500 and S0125). EVs from PANC-1 cells were collected between passages 8–15 and EVs from HPDECs were collected between passages 3–7.
EV purification from cells
Three 150 mm plates (Corning; 430599) were each plated with 6 × 106 cells in 40 mL of culture media, cells reached 80% confluency three days after seeding. 40 mL of conditioned media was collected from each plate after three days in culture. The resulting 120 mL of conditioned media was spun at 300xG for 10 minutes at room temperature to remove cell debris. Then the total volume of conditioned media was concentrated to 400 μL using a Vivaspin 20 100 kDa concentrator (VWR; 95056–134) by centrifuging at 1000xG in 10 minute increments. Between each spin, dilute media was added and pipetted to homogenize the concentrating solution, ensuring the EVs remained soluble. For EV purification from cell line conditioned media, the resulting 400 μL of concentrated media was loaded onto a qEV column (iZON; qEVoriginal) equilibrated at room temperature with PBS, according to manufacturer instructions.
EV purification from plasma
For EV purification from PDAC patients or healthy subject plasma, ~200 μL of plasma was loaded onto a PBS-equilibrated qEVoriginal column. 500 μL fractions 6–22 were collected. F8 was analysed by NanoSight for EV concentration.
Nanoparticle tracking analysis (NTA)
A NS300 Nanosight (Nanosight, Malvern) instrument was used to analyse EVs. To determine the concentration of EVs in qEV fractions, purified EVs were diluted 1:20 in PBS. Automatic settings were applied for the blur and minimum track length. For capture settings, screen gain was set at 1 and camera level was set at 15 or 16. For analysis settings, screen gain was set at 8–10 and detection threshold was set at 5. Two movies of 60 seconds were captured at 30 frames per second for each sample, and the determined concentrations were averaged.
Negative staining transmission electron microscopy
4 μL of EVs were absorbed to glow discharged, carbon-coated 200 mesh EM grids. Grids were washed in Milli-Q water three times for 20 seconds followed by conventional negative staining with 1% (w/v) uranyl acetate. Images were collected using an FEI Tecnai 12 transmission electron microscope (Thermo Fisher Scientific) equipped with a LaB6 filament and operated at an acceleration voltage of 120 kV. Images were recorded with a Gatan 2 k × 2 k CCD camera (Gatan, Inc., Pleasanton, CA, USA) at a magnification of 11,000X and a defocus value of ~1.5 μm.
Images from three independent preparations were processed using custom MATLAB code to extract EV sizes from TEM images. Images were first binarized with an intensity threshold adjusted to account for the brightness of each image. Detected objects smaller than 800 pixels and those that were not roughly circular were removed. Remaining objects were then morphologically closed and filled. EV area was determined by integrating the number of pixels, given a 0.95 nm pixel size. Diameters were taken to be 2√(Area/π).
Western and dot blot
For western blots, 20 μL of purified EV fractions were added to 5 μL of sample buffer and boiled for 3 minutes. Samples were spun at 16000xG for 3 minutes at 4°C. 10 μL of sample was loaded and separated by SDS-PAGE using 4–20% gradient gels (Bio-Rad; 456–1096). Following electrophoresis, proteins were transferred to a nitrocellulose membrane (Bio-Rad; 1620115), using the Bio-Rad Trans-Blot Turbo transfer system. For dot blots, 0.1 μg of HPDEC or PANC-1 cell lysate was diluted into 20 μL of PBS. 5 μL of diluted cell lysate or F8 EVs were dotted onto nitrocellulose membranes, and allowed to dry as previously described [14]. Membranes were blocked with 5% BSA Fraction V (RPI; A30075-100) in TBS-T for 30 minutes and then incubated with primary Ab overnight at 4°C. Primary Abs used for these experiments were anti-TSG101 (1:1000, Sigma; T5701-200UL), anti-CD63 (1:250, Abcam; ab134045), and anti-EGFR (1:1000, Abcam; ab52894). After several washes with TBS-T, membranes were incubated with either goat anti-rabbit (1:5000, Abcam; ab97051) or goat anti-mouse (1:5000, Abcam; ab97023) HRP-conjugated secondary Ab for 1 hour at room temperature. Protein detection was performed using the Bio-Rad ChemiDoc Touch imaging system.
Surface assay
The following molecules, primary Abs, and secondary Abs were used for the specific isolation and detection of EVs: WGA-AF647 (Invitrogen; W32466), cetuximab (Bristol-Myers Squibb), mouse anti CA19-9 (US Biologics; C0075-03A), goat anti-human (Jackson Laboratory; 109-005-098), goat anti-mouse (Millipore; Ap124), and goat anti-rabbit (Abcam; ab6702).
The membrane label Cell Tracker CM-DiI (Cell Tracker, Invitrogen; C7000) stock was prepared in DMSO, as per manufacturer instructions. Where applicable, primary Abs were fluorescently labelled with AF647 N-hydroxysuccinimidyl (NHS) ester (Thermo; A20006). We used optimized Ab labelling conditions [25] to obtain approximately one dye per Ab. The degree of labelling was calculated with a NanoDrop for each batch of labelled Abs. To minimize the effects of labelling heterogeneity on molecular counting, we defined the average number of detected localizations using the SAMI assay [17]. To calculate the photophysical properties, surfaces of sparsely attached reporters were prepared. Briefly, 25-mm #1.5 coverslips (Warner Instruments, Hamden, CT) were cleaned, flame dried, stored, then activated for covalent protein attachment as described in detail previously [26]. The soluble extracellular fragment of EGFR (R&D systems; 344-ER) was attached to coverslips (30 nM final concentration in 150 µL of PBS) and detected with cetuximab-AF647 (30 nM final concentration in 150 µL of PBS). Anti CA19-9 Ab-AF647 was directly attached to coverslips (30 nM final concentration in 150 µL of PBS). Surfaces were blocked after initial protein attachment with PEG-His6 (50 μM final concentration in 150 μL of PBS). These surfaces were imaged under the same conditions as surfaces with EVs, described below. The resulting localizations were analysed to determine the average number of fluorophore appearances per molecule as described [17]. Average values of 2 appearances per molecule were determined for both cetuximab-AF647 and anti CA19-9 Ab-AF647 (Figure S2).
For EV affinity isolation, secondary Ab (1 μM final concentration in 150 μL of PBS) followed by PEG-His6 (50 μM final concentration in 150 μL of PBS) was covalently attached to the surface of activated coverslips as previously described [17]. Concurrently, 7 × 108 EVs were diluted in EV blocking buffer (PBS with 0.5% BSA and 0.01% Tween-20) to a final volume of 150 μL. Primary Ab or WGA was added to the 150 μL diluted EV sample (final concentration of 2 µg/mL) and rotated for 1 hour at room temperature. CM-DiI was added during the last 15 minutes of this incubation (final concentration of 2 µg/mL). Excess primary Ab, WGA, and CM-DiI were partially removed by centrifugation in a 300 kDa concentrator (VWR; 29300–626). Specifically, EV samples were washed in 400 μL of blocking buffer and centrifuged at max speed (15000xG) for 30 seconds (repeated three times). Subsequently, labelled EVs were incubated on secondary Ab or PEG coated surfaces, as indicated, for 30 minutes at room temperature. Surfaces were washed with EV blocking buffer three times then PBS three times, fixed with 4% paraformaldehyde (VWR; 102091–918) and 0.2% glutaraldehyde (VWR; 100505–010) in PBS for 30 minutes, and quenched for 10 minutes with 25 mM glycine. Fixative was removed by washing with PBS three times, then coverslips were loaded into Attofluor cell chambers (Life Technologies; A7816) with PBS until ready for imaging. Important controls included surfaces prepared with PEG-His6 alone (50 μM final concentration in 150 μL of PBS) and lysed EVs. For the lysed EV control, EVs were incubated with EV lysis buffer (50 mM TRIS pH 7.5, 150 mM NaCl, 1% Triton-X 100, 5% glycerol, 1 mM DTT) for 30 minutes at room temperature. Lysed EVs were then buffer exchanged into EV blocking buffer with a 100 kDa concentrator (VWR; 29300–624) followed by incubation with cetuximab-AF647 and CM-DiI as described above.
dSTORM imaging
EVs were localized in TIRF using fluorescent signal from the membrane dye CM-DiI. Surfaces were imaged immediately after preparation in dSTORM imaging buffer (50 mM Tris pH 8.0, 10 mM NaCl, 10% glucose, 100 mM mercaptoethylamine, and GLOX (10% v/v)) as previously described [27]. Imaging was performed on a 3D N-STORM super-resolution microscope (Nikon). The N-STORM system is a fully automatic Ti-E inverted microscope with a piezo stage on a vibration isolation table. This system includes a 100 × 1.49 NA TIRF objective (Apo), N-STORM lens, λ/4 plate, and Quad cube C-NSTORM (97355 Chroma). To maintain imaging at the appropriate focal plane, the microscope has a Perfect Focus Motor. A MLC-MBP-ND laser launch included 405, 488, 561, and 647 nm lasers (Agilent). Images are captured with an EM-CCD camera iXon DU897-Ultra (Andor Technology, South Windsor, CT).
Using NIS-Elements 4.3 Software (Nikon) dSTORM images of 41 × 41 μm were collected with an exposure time of 10 ms. 10,000 frames were acquired for each field of view. To activate/excite AF647, the 647 nm laser power was set to 146 mW. TIRF images were collected using NIS-Elements 4.3 Software and CM-DiI was excited using the 561 nm laser with a power of 0.615–0.123 mW.
Data analysis
Fluorophore localizations (above 700 photons) were extracted from raw image data using NIS-Elements. The NIS-Elements density filter with a 70 nm distance and 30 count threshold was used to remove background localizations (e.g. single fluorescent Ab molecules that were not removed during the 300 kDa filter dilution step; see filter optimization below). We performed drift correction and then used custom MATLAB code to remove artifact puncta (e.g. impurities that persistently fluoresce) via a filter removing all points within a 100 nm radius of any region where at least 400 frames registered a localization within a 1000 frame window.
Next, Voronoi tessellation was used to segment localizations into “clusters” and extract EV diameters and the number of molecules per EV. EVs with a radius below 7 nm or with fewer than 2 molecules were removed. Tessellation polygons were considered clustered if their aggregate area was smaller than 600 nm2. The Voronoi tessellation method was based on ClusterViSu [28], but modified to improve processing speed. To obtain the number of detected molecules, we divided the total number of localizations within a tessellated EV cluster by the average number of localizations for a given reporter (see Figure S2 and [17]).
Simulations to characterize filters and thresholds
To test the effect of the NIS-Elements thresholds used to remove signal unassociated with EVs, we simulated the localizations from individual EVs and background molecules. We varied the simulated EV size and molecules per EV to understand which EV parameters were detectable. Figure S3A shows the results of these simulations. Each grid cell corresponds to a simulated image with a particular EV size and molecules per EV. In Figure S3A (left) the cell colour indicates the ratio of the number of detected EVs to simulated EVs. Figure S3A (centre) shows the ratio of detected cluster diameter to simulated diameter. Figure S3A (right) shows the ratio of detected molecules per EV to simulated molecules per EV. Underestimations (dark blue) appear to occur in cases where there are less than 15 molecules per EV and large EVs with a low number of molecules per EV. Overestimations (yellow) appear to occur in cases where the diameters of EVs are below 40 nm, likely due to localization uncertainty (simulated to be 10 nm). Filters should be optimized by users for different preparation/imaging conditions.
EV movement over the course of dSTORM imaging was measured to confirm insignificant effects on the calculation of EV size. Individual EV centroids were first extracted from Voronoi tessellation results. Using these centroids and cross-correlation [29], with a bin size of 500 frames, the average Brownian motion was tracked across the total image acquisition. The root mean squared error (RMSE) was then calculated for the overall drift and included with the drift plots shown in Figure S4.
Simulations to characterize differences between EGFR-enriched EV populations detected with WGA-AF647 or cetuximab-AF647
Synthetic data was generated in MATLAB to validate the apparent differences in diameter between EVs detected with WGA and cetuximab. Experimental data (Figure S7B, top row) for EV diameters (WGA mean with SD: 72 ± 46 nm; cetuximab mean with SD: 51 ± 25 nm) and localization numbers (WGA mean with SD: 99 ± 117; cetuximab mean with SD: 56 ± 66) were used to inform the simulations (Figure S7B, middle row). Specifically, a spherical coordinate system was established to place localizations, in three dimensions, for an individual EV. The radial distance, polar angle, and azimuthal angle were all given levels of randomization (MATLAB rand function) within the space characterized by possible EV diameters, given a defined number of localizations. Features were also incorporated in the simulations to ensure that the majority of EVs were not perfectly spherical in shape. This included a second round of randomization on any one of the three dimensions to either extend or shorten the distance by the average localization precision (predetermined experimentally to be ~8 nm). In addition to the assigned random spherical coordinate, each localization was further provided with some average spatial error (<8 nm) to simulate placement on the “uneven” membrane surface of a given EV. The total number of localizations for each EV was provided by randomly sampling experimental data (approximately an Inverse-gamma distribution). Different fractions of this total number of localizations were allocated into clusters on the EV surface (Figure S7B, bottom row), and, in the case of using cetuximab as a reporter, the majority were encouraged to form clusters. This entire process was used to randomly place EVs across a simulated field of view (FOV; ~1600 μm2) for a total number of EVs mimicking experimental data averages, 550 or 110 EVs per FOV for WGA or cetuximab, respectively. Thus, a range of EV sizes and compositions were prepared in 100 simulated FOVs for each molecular target. The two-dimensional information was collected for each FOV and subsequently analysed using Voronoi tessellation (Figure S7B, middle row).
Proteomics on PANC-1 EVs
EV fractions were sonicated in lysis buffer (2% sodium deoxycholate (DOC), 25 mM Tris-HCl pH 7.0, 2X Thermo HALT). Proteins were reduced with 5 mM TCEP (30 minutes, 60°C) and alkylated with 10 mM iodoacetamide (30 minutes, dark) and digested overnight with trypsin at 1:50 enzyme to substrate ratio. DOC was removed by acid-precipitation. Peptide clean-up was performed using a Waters Sep-Pak C18 96-well plate. For LC-MS/MS analysis, peptides were reconstituted in buffer (98% water, 2% acetonitrile, 0.1% formic acid) containing 5 fmol/µL Pierce Retention Time Calibration mix.
Data was acquired on an Orbitrap Fusion Lumos (ThermoFisher Scientific, San Jose, CA) coupled to an Ultimate 3000 UHPLC system (ThermoFisher Scientific, San Jose, CA) operated in direct injection mode. Each sample was loaded on a C18 analytical column (45°C, PepMap RSLC C18, 75 µm ID * 25 cm, 2 µm particle size, 100 Å pore size) and eluted at a flow rate of 300 nL/minute using the following 120 minute method: 2% to 19% B in 80 minutes, 19% to 30% B in 20 minutes, 30% to 98% B in 5 minutes, remain at 98% B for 2 minutes followed by return to initial conditions in 1 minute and re-equilibration for 12 minutes. The Orbitrap mass spectrometer was operated in data-dependent mode (3 second duty cycle, top-speed mode, spray voltage of 1900 V, ion transfer tube temperature of 275°C, survey scan in the Orbitrap at a resolution of 120 K at 200 m/z, scan range of 400–1500 m/z, AGC target of 4E5 maximum ion injection time of 50 ms). Most abundant precursor ions with charge states between 2–7 were taken up for MS2 scan using High Energy Collision (HCD) dissociation and detection in the iontrap with the following settings: quadrupole isolation mode enabled, isolation window at 1.6 m/z, AGC target of 5E3 with maximum ion injection time of 35 ms and HCD collision energy of 35%. To avoid resampling of the same peaks, dynamic exclusion was set to 60 seconds.
Mass spectra were searched using Proteome Discoverer 2.2 (Thermo Fisher Scientific) and Mascot 2.6.0 (Matrix Science) against a SwissProt/UniprotKB Homo sapiens database (downloaded Jan 2017), allowing for tryptic rules and up to 2 missed cleavages, fixed cysteine carbamidomethylation, variable methionine oxidation, and N-terminal acetylation. Exosomal proteins were annotated using ExoCarta [30]. Enriched biological processes were determined using ToppFun [31], from proteins identified in 3 out of 3 replicates.
RNA isolation and quantification
RNA was isolated from vesicle fractions 7, 8, and 9, each in triplicate, from the PANC-1 cancer cell line and HPDEC control cell line. Total RNA was extracted from 520 μL of isolated vesicles using Total Exosome RNA and Protein Isolation Kit (same as miRVana; Thermo Fisher, Cat. No. 4478545) with a second extraction as before [32]. Each isolated RNA sample was DNase treated with a TURBO DNA-free Kit (Thermo Fisher, Cat. No. AM1907), then cleaned and concentrated (RNA Clean & Concentrator, Zymo Research, Cat. No. R1016, following Appendix C in the kit protocol). The RNA was then quantified with Quant-iT Ribogreen RNA Assay (Thermo Fisher, Cat. No.: R11490).
Library preparation and whole transcriptome sequencing
Because there was enough RNA from PANC-1 EVs to examine both small and longer RNAs (requires two types of sample preparation) the sample was split in two with one half going into whole transcriptome library preparation and the other half going into small RNA library preparation. Input amounts were adjusted for the amount of RNA in that cell line and that fraction (see Table S2). There was not enough RNA from HPDEC EVs for both preparations, and only the whole transcriptome library preparation was made and sequenced.
Whole transcriptome: For each RNA sample, indexed, Illumina-compatible, double-stranded cDNA libraries were synthesized from total RNA with Takara Bio’s SMARTer Stranded Total RNA-Seq Kit v2 – Pico Input Mammalian kit (Takara Bio, Cat. No. 634411). Library preparation included chemical RNA fragmentation (94°C for 2 min), a 5-cycle indexing PCR, ribosomal cDNA depletion, and a 14-16-cycle enrichment PCR. Total RNA input was normalized to the lowest amount of RNA among technical triplicates within each vesicle fraction and each cell line, and the number of enrichment PCR cycles was dictated by the total RNA input according to manufacturer recommendations.
Each library was measured for size with Agilent’s High Sensitivity D1000 Screen Tape and reagents (Agilent, Cat. No. 5067–5584 & 5067–5603) and concentration with KAPA SYBR FAST Universal qPCR Kit (Kapa Biosystems, Cat. No. KK4824). Libraries were combined into two equimolar pools which were measured for size and concentration. Each pool was hybridized onto a paired-end flowcell (Illumina, Cat. No. PE-402-4002) with a 1% v/v PhiX Control v3 spike-in (Illumina, Cat. No. FC-110-3001) using Illumina’s HiSeq Rapid Duo cBot Sample Loading Kit (Illumina, Cat. No. CT-403-2001) on a cBot. Each template-hybridized flowcell was then clustered and sequenced on Illumina’s HiSeq 2500 with HiSeq Rapid v2 chemistry (Illumina, Cat. No. FC-402-4022). The first and second reads were each 83 bases.
Small RNA
For the PANC-1 cell line there was enough RNA to sequence both a small and a long RNA library for each replicate of fractions 7, 8, and 9, however, in the HPDEC cell line, the RNA was limited, and we sequenced only a long RNA library for each fraction and each replicate. For the PANC-1 EVs, small RNA libraries were generated using NEXTFlex Small RNA Library Prep Kit v2 (Cat #5132–03). The manufacturer instructions were followed through PCR amplification. The optional stop point was used after the RT reaction prior to clean up and PCR amplification. Samples underwent 18 cycles of PCR amplification. Following PCR amplification, the libraries were run on a 6% TBE gel for 30 minutes at 200 V, and the sample between 150 to 170 bp was excised. Samples were quantified with the Agilent High Sensitivity DNA Kit (5067–4626; Agilent). An equimolar pool of the samples was created, and the pool was denatured and clustered on a single read Illumina V3 flowcell (GD-401-3001; Illumina) and run on the Illumina HiSeq sequencing platform (HiSeq 2500; Illumina) for 50 cycles with a 7 cycle indexing read.
Fastq generation, genome alignment, and differential expression analysis
Fastqs were generated using bcl2fastq v2.19.1.403 using default parameters. 3 nt were trimmed from the reverse read, as suggested by the SMARTer Stranded Total RNA-Seq protocol, with cutadapt v1.17. Trimmed reads were then aligned to the GRCh38 genome with STAR v2.6.1d, and counted with featureCounts v1.6.3, (part of the subread package) using a non-redundant genome annotation combined from GENCODE 29 and LNCipedia5.2. Differential expression analysis was performed with DESeq 1.22.1.
Patient cohort
The qSMLM team was blinded to patient characteristics until all data acquisition and analyses was completed. All investigators were aware of healthy subject samples. Plasma samples were obtained under Institutional Review Board (IRB) number 015–196 (Baylor Scott & White Research Institute) and informed consent was obtained from all subjects. The patients analysed in this study were enrolled as part of the early-pancreatic cancer detection initiative at the Baylor Scott & White Research Institute, where plasma specimens were collected from patients with various stages of PDAC, pre-cancerous lesions and healthy subject controls. Analysed plasma samples and the clinical test for soluble serum CA19-9 were collected pre-neoadjuvant therapy. Additionally, plasma from two healthy subjects (H5, H6) was obtained under IRB 2013116 (TGen) and informed consent was obtained from all subjects.
Statistical information
All cell culture EV experiments were run in triplicates using separate cell culture preparations (biological replicates), with five fields of view (FOV, technical replicates). Each patient and healthy subject sample were run in triplicate, with five FOV (total FOV = 15), except for the CA19-9 analysis, which were run in duplicate with 7 or 8 FOV (total FOV = 15), due to a lack of sufficient sample volume for triplicate runs. Mean and SEM were determined using GraphPad Prism Software, Microsoft Excel, and MATLAB. One-tailed Student T-tests comparing two-samples of unequal variance were used to determine statistical significance. The coefficient of variation was used to describe the variance within EV populations. To determine the statistical significance of variation between the distributions of vesicles the data was divided into histograms and the SEM of each bin count was taken to be the square root of the bin count. A chi-squared test was then used to compare the histograms. Statistical significance was set at *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.0001. Scatter plots and histograms were generated in MATLAB, and graphs were made using GraphPad Prism Software (https://www.graphpad.com).
Funding Statement
This work was supported by the Hirshberg Foundation for Pancreatic Cancer Research, and the Beckman Research Institute of the City of Hope. We acknowledge the Dr. and Mrs. Roberts endowment from the Roberts Summer Academy at City of Hope. This work was also supported by [CA184792, CA202797 and CA214254] grants from the National Cancer Institute to Ajay Goel. Research reported in this publication included work performed in the Analytical Cytometry and Drug Discovery and Structural Biology Cores supported by the National Cancer Institute of the National Institutes of Health under award number [P30CA33572]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health
Acknowledgments
We thank Dr. D. Von Hoff for help obtaining patient samples. We thank Dr. Z. Li and R. Zerda at the City of Hope Electron Microscopy Core Facility for help with electron microscopy. We thank Dr. R. Sharma and V. David-Dirgo for their help with proteomic processing and data acquisition. We thank G. Choo, Y. Kalkum, and S. Hess for technical help and Dr. I. Talisman for manuscript editing.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. K.M.L. designed and performed experiments, analyzed and interpreted the data. D.L.W. developed codes, performed computational analysis, simulations, and interpreted the data. A.L.M. designed and performed TEM experiments and analyzed the data. M.S.B. developed codes, performed computational analysis, simulations, and interpreted the data. A.N.W. performed experiments and analyzed the data. K.G.M. analyzed the proteomics data. B.M., R.R., and E.H. performed RNA sequencing and subsequent data analysis. M.M.M. designed and guided TEM experiments and interpreted the data. A.G. recruited pancreatic patients with informed consent. P.P. designed and guided LC-MS/MS experiments, analyzed and interpreted the data. K.V.K.J. designed experiments, supervised RNA sequencing and data analysis, analyzed and interpreted the data. T.J.T. conceived and designed the project, designed experiments, guided qSMLM and simulation experiments, analyzed and interpreted the data.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Al-Sukhni W, Borgida A, Rothenmund H, et al. Screening for pancreatic cancer in a high-risk cohort: an eight-year experience. J Gastrointest Surg. 2012;16:771–783. [DOI] [PubMed] [Google Scholar]
- [3].Yang KS, Im H, Hong S, et al. Multiparametric plasma EV profiling facilitates diagnosis of pancreatic malignancy. Sci Transl Med. 2017;9:eaal3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].De Toro J, Herschlik L, Waldner C, et al. Emerging roles of exosomes in normal and pathological conditions: new insights for diagnosis and therapeutic applications. Front Immunol. 2015;6:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Maas SL, de Vrij J, van der Vlist EJ, et al. Possibilities and limitations of current technologies for quantification of biological extracellular vesicles and synthetic mimics. J Control Release. 2015;200:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Remmers N, Anderson JM, Linde EM, et al. Aberrant expression of mucin core proteins and o-linked glycans associated with progression of pancreatic cancer. Clin Cancer Res off J Am Assoc Cancer Res. 2013;19:1981–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Troiani T, Martinelli E, Capasso A, et al. Targeting EGFR in pancreatic cancer treatment. Curr Drug Targets. 2012;13:802–810. [DOI] [PubMed] [Google Scholar]
- [8].Adamczyk KA, Klein-Scory S, Tehrani MM, et al. Characterization of soluble and exosomal forms of the EGFR released from pancreatic cancer cells. Life Sci. 2011;89:304–312. [DOI] [PubMed] [Google Scholar]
- [9].Szajda SD, Waszkiewicz N, Chojnowska S, et al. Carbohydrate markers of pancreatic cancer. Biochem Soc Trans. 2011;39:340–343. [DOI] [PubMed] [Google Scholar]
- [10].Partyka K, Maupin KA, Brand RE, et al. Diverse monoclonal antibodies against the CA 19-9 antigen show variation in binding specificity with consequences for clinical interpretation. Proteomics. 2012;12:2212–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Humphris JL, Chang DK, Johns AL, et al. The prognostic and predictive value of serum CA19.9 in pancreatic cancer. Ann Oncol. 2012;23:1713–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Steinberg W. The clinical utility of the CA 19-9 tumor-associated antigen. Am J Gastroenterol. 1990;85:350–355. [PubMed] [Google Scholar]
- [13].Lamerz R. Role of tumour markers, cytogenetics. Ann Oncol. 1999;10(Suppl 4):145–149. [PubMed] [Google Scholar]
- [14].Nizamudeen Z, Markus R, Lodge R, et al. Rapid and accurate analysis of stem cell-derived extracellular vesicles with super resolution microscopy and live imaging. Biochim Biophys Acta Mol Cell Res. 2018;1865:1891–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lee K, Fraser K, Ghaddar B, et al. Multiplexed profiling of single extracellular vesicles. ACS Nano. 2018;12:494–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chen C, Zong S, Wang Z, et al. Imaging and intracellular tracking of cancer-derived exosomes using single-molecule localization-based super-resolution microscope. ACS Appl Mater Interfaces. 2016;8:25825–25833. [DOI] [PubMed] [Google Scholar]
- [17].Golfetto O, Wakefield DL, Cacao EE, et al. A platform to enhance quantitative single molecule localization microscopy. J Am Chem Soc. 2018;140:12785–12797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ebrahimkhani S, Vafaee F, Hallal S, et al. Deep sequencing of circulating exosomal microRNA allows non-invasive glioblastoma diagnosis. NPJ Precis Oncol. 2018;2:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Baran J, Baj-Krzyworzeka M, Weglarczyk K, et al. Circulating tumour-derived microvesicles in plasma of gastric cancer patients. Cancer Immunol Immunother. 2010;59:841–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Momen-Heravi F, Balaj L, Alian S, et al. Alternative methods for characterization of extracellular vesicles. Front Physiol. 2012;3:354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Vergauwen G, Dhondt B, Van Deun J, et al. Confounding factors of ultrafiltration and protein analysis in extracellular vesicle research. Sci Rep. 2017;7:2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].van der Pol E, Coumans FA, Grootemaat AE, et al. Particle size distribution of exosomes and microvesicles determined by transmission electron microscopy, flow cytometry, nanoparticle tracking analysis, and resistive pulse sensing. J Thromb Haemost. 2014;12:1182–1192. [DOI] [PubMed] [Google Scholar]
- [23].Thery C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the international society for extracellular vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7:1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Al-Nedawi K, Meehan B, Micallef J, et al. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol. 2008;10:619–624. [DOI] [PubMed] [Google Scholar]
- [25].Tobin SJ, Wakefield DL, Liu X, et al. Single molecule localization microscopy coupled with touch preparation for the quantification of trastuzumab-bound HER2. Sci Rep. 2018;11:15154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jorand R, Biswas S, Wakefield DL, et al. Molecular signatures of mu opioid receptor and somatostatin receptor 2 in pancreatic cancer. Mol Biol Cell. 2016;27:3659–3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dempsey GT, Vaughan JC, Chen KH, et al. Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat Methods. 2011;8:1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Andronov L, Orlov I, Lutz Y, et al. ClusterViSu, a method for clustering of protein complexes by Voronoi tessellation in super-resolution microscopy. Sci Rep. 2016;6:24084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang Y, Schnitzbauer J Hu Z, et al. Localization events-based sample drift correction for localization microscopy with redundant cross-correlation algorithm. Opt Express. 2014;22:15982––15991.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Keerthikumar S, Chisanga D, Ariyaratne D, et al. ExoCarta: a web-based compendium of exosomal cargo. J Mol Biol. 2016;428:688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chen J, Bardes EE, Aronow BJ, et al. ToppGene suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37:W305–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Burgos KL, Javaherian A, Bomprezzi R, et al. Identification of extracellular miRNA in human cerebrospinal fluid by next-generation sequencing. RNA. (New York, NY) 2013;19:712–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.