

Abstract
Fibroblast growth-factor receptor (FGFR) is a potential target for cancer therapy. We designed three novel series of FGFR1 inhibitors bearing indazole, benzothiazole, and 1H-1,2,4-triazole scaffold via fragment-based virtual screening. All the newly synthesised compounds were evaluated in vitro for their inhibitory activities against FGFR1. Compound 9d bearing an indazole scaffold was first identified as a hit compound, with excellent kinase inhibitory activity (IC50 = 15.0 nM) and modest anti-proliferative activity (IC50 = 785.8 nM). Through two rounds of optimisation, the indazole derivative 9 u stood out as the most potent FGFR1 inhibitors with the best enzyme inhibitory activity (IC50 = 3.3 nM) and cellular activity (IC50 = 468.2 nM). Moreover, 9 u also exhibited good kinase selectivity. In addition, molecular docking study was performed to investigate the binding mode between target compounds and FGFR1.
Keywords: Anticancer, FGFR1, FGFR1 inhibitor, Fragment-based virtual screening
Graphical Abstract

Introduction
Protein kinases constitute one of the largest protein families in humans1–5. The kinase enzymes in this family catalyse phosphorylation of serine, threonine, or tyrosine residues, which regulate the majority of signal transduction pathways in cell, and thus play an important role in cell growth, metabolism, differentiation and apoptosis consequently. Deregulation of protein kinases is implicated in a number of diseases including cancer, diabetes and inflammation. Targeted inhibition of protein kinases has thereby become an attractive therapeutic strategy in treatment of relevant diseases6,7.
Fibroblast growth-factor receptors (FGFRs) form a sub-family of the receptor tyrosine kinase (RTK) superfamily, which consists of four highly conserved functional members (FGFR 1–4)8,9. FGFR signalling is initiated by binding of extracellular FGF ligand which leads to receptor dimerisation and cross-phosphorylation of the kinase domains proceed to phosphorylate intracellular substrates such as FRS2, Gab1, PLCγ, and STAT1. Subsequent downstream signalling is complex and includes activation of the PI3K-Akt and the Ras/Raf/Mek/Erk pathways. In normal cells, FGFR plays fundamental roles in many physiologic processes, including embryogenesis, tissue homeostasis, tissue repairing, wound healing, and inflammation10–12. Therefore, the inhibition of FGFR signalling pathway presents a promising option for cancer therapeutics.
In the past few years, different strategies have been pursued to inhibit abnormal FGFR signalling pathways, among them several small-molecule FGFR inhibitors have advanced into clinical trial (Figure 1)13. While the pan-kinase inhibitors (e.g. dovitinib (TKI-258)14, nintedanib (BIBF-1120)15, and ponatinib (AP-24534)16) which were originally adopted for the treatment of cancers that harbour aberrant FGFR, severe side effects were observed due to inhibition of several off-target kinases such as EGFR, VEGFR, PDGFR, etc. More recently, there has been an increasing interest in developing selective FGFR inhibitors. Several selective FGFR inhibitors bear various scaffolds have been developed into clinical investigation (e.g. NVP-BGJ39817, LY-287445518, AZD454719, JNJ-4275649320, and Debio-134721).
Figure 1.

Structure of representative FGFR inhibitors.
In our previous studies, a series of indazole derivatives were discovered as potent FGFR1 inhibitors22. The most promising compound L1 exhibited good enzymatic inhibition against FGFR1 and modest cellular inhibition (Figure 1). In this study, in order to improve the cellular inhibition, discover novel scaffold and enrich the structure-activity relationship, further work has been putting into modification of indazole scaffold and the hydrophobic substituents. The goal of this compound modification was to increase the ligand-receptor interaction, and improve the physico-chemical property. As a result, we designed the novel FGFR1 inhibitors by the following two strategies: (a) according to the fragment-based drug design strategy, we introduced novel flat hetero aromatic ring which formed key H-bond interaction with hinge region; (b) further optimisation of the original indazole inhibitors via introduced halogen atoms, that were often used to improve permeability through the modulation of modulation of compound’s lipophilicity and halogen-protein interactions.
Herein, with the fragment-based virtual screening strategy, we designed three novel series of FGFR1 inhibitors bearing 1H-1,2,4-triazole, benzothiazole and indazole scaffold (Figure 2(b)). The indazole derivative 9 u stood out as the most potent FGFR1 inhibitor, and it also exhibited good kinase selectivity. Additionally, the docking studies were carried out to investigate the receptor-ligand interaction.
Figure 2.

(a) Binding model of AZD4547 and NVP-BGJ398 to the FGFR1 kinase domain (PDB ID: 4V05, 3TTO). (b) The fragment-based virtual screening protocol.
Materials and methods
Fragment-based virtual screening
Pharmacophore screening
The fragment library was derived from kinase hinge region directed library provided by enamine (https://enamine.net/index.php), which contained 11809 fragments. On the basis of the chemical features of FGFR1 inhibitors, these pharmacophore features, hydrogen bond acceptor (HBA), hydrogen bond donor (HBD), and hydrophobic (HY) were defined as a query for pharmacophore screening. The database screening was performed using the Ligand Pharmacophore Mapping protocol in DS 4.0. The fit values were calculated based on the chemical substructures map the location constraints of the pharmacophoric features and their distance deviation from the feature centres. Finally, only the compounds showed good fit values in pharmacophore model could enter the further molecular docking study.
Molecular docking study
Molecular docking of compounds into the three dimensional X-ray structure of FGFR1 (PDB ID: 4ZSA) was carried out using the surflex-dock module of the Sybyl-x 2.0 software package.The three-dimensional structure of compound was constructed using ChemBio 3 D Ultra software [Chemical Structure Drawing Standard; Cambridge corporation, USA 2010], then it was energetically minimised by using MMFF94 with 5000 iterations and minimum RMS gradient 0.05. The protein was prepared by the Protein preparation wizard of Sybyl-x 2.0. The waters were eliminated from the protein and the polar hydrogen was added. Receptor grids were generated using Receptor Grid Generation. The generated binding site was just the ATP-binding pocket of FGFR1, including several key amino acid residues: Glu562, Ala564.
Compound 9d was placed during the molecular docking procedure. Types of interactions of the docked protein with ligand were analysed after the end of molecular docking. The detailed structures and calculation results were shown in Supplementary Table S1.
Chemistry
Solvents were distilled under the positive pressure of dry argon before use and dried using standard methods. Chemicals were obtained from local suppliers and were used without further purification. All reactions were monitored by thin-layer chromatography (silica gel 60 F254 glass plates). NMR spectra were recorded on Bruker 400 MHz instruments, and the chemical shifts were presented in terms of parts per million with TMS as the internal reference. Electron-spray ionisation mass spectra in positive mode (ESI-MS) data were obtained with a Bruker Esquire 3000+ spectrometer. Flash column chromatography was performed on silica gel (200–300 mesh, Adamas, China).
General method for preparation of compounds 9a-9z (exemplified by 9d)
The detail experimental procedures of intermediate 2, 3, 4, 6, 7 and 8 are described as previous reference 22 (see in Supplementary Material).
4–(4-ethylpiperazin-1-yl)-N-(6–(3-methoxyphenyl)-1H-indazol-3-yl)benzamide (9d) The building block 8 (100.0 mg, 0.2 mmol) was dissolved in dioxane (2 ml), then followed by the addition of (3-methoxyphenyl)boronic acid (85.2 mg, 0.5 mmol), Pd(dppf)Cl2 (20 mg), 1 M Cs2CO3 (500 µL). The reaction mixture was heated at 120 °C. for 1 h, then it cooled to rt. After concentrated, the residue was dissolved in EtOAc (50 ml) and washed with H2O (10 ml ×2), and brine (10 ml ×2), dried over Na2SO4, concentrated in vacuo. The residue was purified by chromatography on silica gel DCM-MeOH (10:1) to give the 9d (45.6 mg, 44%). Mp 226.5–230.2 °C. 1H NMR (400 MHz, DMSO-d6) δ: 12.80 (brs, 1H), 10.53 (brs, 1H), 7.99 (d, J = 8.9 Hz, 2H), 7.78 (d, J = 8.6 Hz, 1H), 7.68 (s, 1H), 7.35–7.45 (m, 2H), 7.30 (d, J = 7.9 Hz, 1H), 7.24–7.26 (d, J = 2.0 Hz, 1H), 7.03 (d, J = 9.0 Hz, 2H), 6.97 (dd, J = 8.2, 2.0 Hz, 1H), 3.85 (s, 3H), 2.48–2.55 (m, 4H), 2.39 (q, J = 7.1 Hz, 2H), 1.05 (t, J = 7.2 Hz, 3H). 13 C NMR (100 MHz, DMSO-d6) δ: 165.5, 160.2, 153.6, 142.6, 142.2, 141.1, 138.9, 130.5, 129.8, 123.1, 123.0, 120.0, 119.8, 117.0, 113.9, 113.6, 113.1, 108.3, 55.6, 52.6, 52.1, 47.4, 12.4. ESI-MS (m/z): [M + H]+ = 456.7 (Calcd: 456.55). HRMS: calcd for C27H31N5O2 (M + H)+ 456.2400, found 456.2391. HPLC analysis: MeOH-H2O (80: 20), 6.85 min, 96.8% purity.
N-(6–(3,5-dimethoxyphenyl)-1H-indazol-3-yl)-4-((3R,5S)-3,5-dimethylpiperazin-1-yl)benzamide (9a) White solid: 29.2 mg, yield 38%; Mp 228.1–232.2 °C. 1H NMR (400 MHz, DMSO-d6) δ: 12.81 (brs, 1H), 10.53 (brs, 1H), 7.99 (d, J = 8.3 Hz, 2H), 7.77 (d, J = 8.0 Hz, 1H), 7.68 (s, 1H), 7.37 (dd, J = 8.6, 1.3 Hz, 1H), 7.04 (d, J = 9.0 Hz, 2H), 6.85 (d, J = 2.2 Hz, 2H), 6.54 (t, J = 2.1 Hz, 1H), 3.85 (s, 2H), 3.83 (s, 6H), 2.90–3.01 (m, 2H), 2.35–2.41 (m, 2H), 1.12 (s, 3H), 1.10 (s, 3H). 13 C NMR (100 MHz, DMSO-d6) δ: 165.7, 161.3, 153.2, 143.3, 142.1, 141.0, 139.0, 129.9, 122.9, 120.0, 117.1, 114.0, 108.4, 105.8, 99.9, 55.8, 53.1, 50.6, 18.7. ESI-MS (m/z): [M + H]+ = 486.1 (Calcd: 486.24). ESI-HRMS: calcd for C28H33N5O3 (M + H)+ 486.2505, found 486.2502. HPLC analysis: MeOH-H2O (80: 20), 7.36 min, 96.0% purity.
4-((3R,5S)-3,5-dimethylpiperazin-1-yl)-N-(6–(3-methoxyphenyl)-1H-indazol-3-yl)benzamide (9 b) White solid: 24.8 mg, yield 30%; Mp 224.8–227.4 °C. 1H NMR (400 MHz, DMSO-d6) δ: 12.81 (brs, 1H), 10.52 (brs, 1H), 7.98 (d, J = 8.9 Hz, 2H), 7.78 (d, J = 8.1 Hz, 1H), 7.68 (s, 1H), 7.37–7.44 (m, 2H), 7.30 (d, J = 7.9 Hz, 1H), 7.26 (s, 1H), 7.03 (d, J = 8.2 Hz, 2H), 6.97 (dd, J = 8.1, 2.2 Hz, 1H), 3.85 (s, 3H), 3.79–3.81 (m, 2H), 2.85–2.93 (m, 2H), 2.28–2.34 (m, 2H), 1.09 (s, 3H), 1.07 (s, 3H). 13 C NMR (100 MHz, DMSO-d6) δ: 165.7, 160.2, 153.1, 142.5, 142.2, 141.0, 139.0, 130.6, 129.9, 123.0, 119.9, 117.0, 114.1, 113.6, 113.1, 108.3, 55.6, 52.8, 50.7, 18.4. ESI-MS (m/z): [M + H]+ = 456.1 (Calcd: 456.23). ESI-HRMS: calcd for C27H31N5O2 (M + H)+ 456.2400, found 456.2390. HPLC analysis: MeOH-H2O (80: 20), 7.27 min, 95.9% purity.
N-(6–(3-methoxyphenyl)-1H-indazol-3-yl)-4–(4-methylpiperazin-1-yl)benzamide (9c) White solid: 40.2 mg, yield 45%; Mp 224.8–227.4 °C. 1H NMR (400 MHz, DMSO-d6) δ: 12.82 (brs, 1H), 10.55 (brs, 1H), 8.00 (d, J = 8.6 Hz, 2H), 7.80 (d, J = 8.8 Hz, 1H), 7.65 (s, 1H), 7.35–7.45 (m, 2H), 7.29 (d, J = 8.0 Hz, 1H), 7.25 (d, J = 2.0 Hz, 1H), 7.05 (d, J = 8.8 Hz, 2H), 6.95 (dd, J = 8.2, 1.9 Hz, 1H), 3.83 (s, 3H), 2.48–2.55 (m, 4H), 2.39 (q, J = 7.1 Hz, 2H), 1.05 (t, J = 7.2 Hz, 3H). 13 C NMR (100 MHz, DMSO-d6) δ: 165.6, 160.2, 153.7, 142.6, 141.1, 139.0, 130.5, 129.8, 123.1, 122.9, 120.0, 119.9, 117.0, 113.9, 113.6, 113.1, 108.3, 56.2, 52.6, 52.1, 46.2. ESI-MS (m/z): [M + H]+ = 442.2 (Calcd: 442.21). ESI-HRMS: calcd for C26H28N5O2 (M + H)+ 442.2198, found 442.2190. HPLC analysis: MeOH-H2O (80: 20), 7.38 min, 96.5% purity.
N-(6–(3-ethoxyphenyl)-1H-indazol-3-yl)-4–(4-ethylpiperazin-1-yl)benzamide (9e) White solid: 22.7 mg, yield 39%; Mp 223.6–226.0 °C. 1H NMR (400 MHz, DMSO-d6) δ: 12.80 (brs, 1H), 10.54 (brs, 1H), 8.00 (d, J = 8.9 Hz, 2H), 7.78 (d, J = 8.5 Hz, 1H), 7.66 (s, 1H), 7.35–7.43 (m, 2H), 7.29 (d, J = 7.8 Hz, 1H), 7.24 (s, 1H), 7.03 (d, J = 8.9 Hz, 2H), 6.96 (dd, J = 8.1, 1.9 Hz, 1H), 4.13 (q, J = 6.9 Hz, 2H), 3.26–3.40 (m, 4H), 2.51–2.62 (m, 4H), 2.34–2.45 (m, 2H), 1.37 (t, J = 8.0 Hz, 3H), 1.06 (t, J = 7.1 Hz, 3H). 13 C NMR (100 MHz, DMSO-d6) δ: 165.6, 159.5, 153.6, 142.6, 142.2, 141.1, 139.0, 130.5, 129.8, 123.1, 123.0, 119.9, 117.0, 114.0, 113.9, 113.6, 108.3, 63.6, 52.5, 52.1, 47.4, 15.2, 12.3. ESI-MS (m/z): [M + H]+ = 470.2 (Calcd: 470.25). ESI-HRMS: calcd for C28H33N5O2 (M + H)+ 470.2556, found 470.2550. HPLC analysis: MeOH-H2O (80: 20), 7.42 min, 96.9% purity.
4–(4-ethylpiperazin-1-yl)-N-(6–(3-isopropoxyphenyl)-1H-indazol-3-yl)benzamide (9f) White solid: 21.3 mg, yield 39%; Mp 227.9–231.7 °C. 1H NMR (400 MHz, DMSO-d6) δ: 12.77 (brs, 1H), 10.52 (brs, 1H), 8.00 (d, J = 8.7 Hz, 2H), 7.78 (d, J = 8.5 Hz, 1H), 7.65 (s, 1H), 7.35–7.41 (m, 2H), 7.27 (d, J = 7.7 Hz, 1H), 7.22 (s, 1H), 7.03 (d, J = 8.0 Hz, 2H), 6.92 (dd, J = 8.3, 1.9 Hz, 1H), 4.72–4.78 (m, 1H), 3.27–3.38 (m, 4H), 2.40–2.58 (m, 4H), 2.34 (q, J = 7.1 Hz, 2H), 1.32 (s, 3H), 1.31 (s, 3H), 1.06 (t, J = 7.1 Hz, 3H). 13 C NMR (100 MHz, DMSO-d6) δ: 166.6, 158.4, 153.6, 142.7, 142.2, 141.1, 139.0, 130.6, 129.8, 123.1, 119.8, 117.0, 115.0, 114.9, 114.0, 69.7, 52.5, 52.0, 47.3, 22.4, 12.3. ESI-MS (m/z): [M + H]+ = 484.3 (Calcd: 484.27). ESI-HRMS: calcd for C29H35N5O2 (M + H)+ 484.2713, found 484.2706. HPLC analysis: MeOH-H2O (80: 20), 7.22 min, 97.1% purity.
N-(6–(3-(sec-butoxy)phenyl)-1H-indazol-3-yl)-4–(4-ethylpiperazin-1-yl)benzamide (9 g) White solid: 23.9 mg, yield 47%; Mp 225.9–290.0 °C. 1H NMR (400 MHz, DMSO-d6) δ: 12.79 (brs, 1H), 10.54 (brs, 1H), 8.00 (d, J = 8.9 Hz, 2H), 7.78 (d, J = 8.5 Hz, 1H), 7.67 (s, 1H), 7.37–7.42 (m, 2H), 7.29 (d, J = 7.9 Hz, 1H), 7.24–7.26 (m, 1H), 7.03 (d, J = 9.0 Hz, 2H), 6.96 (dd, J = 8.1, 1.8 Hz, 1H), 3.86 (s, 1H), 3.84 (s, 1H), 3.26–3.38 (m, 4H), 2.51–2.60 (m, 4H), 2.40–2.46 (m, 2H), 2.03–2.09 (m, 1H), 1.06 (t, J = 7.1 Hz, 3H), 1.03 (s, 3H), 1.01 (s, 3H). 13 C NMR (100 MHz, DMSO-d6) δ: 166.6, 159.8, 153.6, 142.6, 142.2, 141.1, 139.0, 130.5, 129.8, 123.1, 119.9, 117.0, 114.0, 113.6, 108.3, 74.3, 52.5, 52.0, 47.3, 31.2, 28.3, 19.6, 12.3. ESI-MS (m/z): [M + H]+ = 498.3 (Calcd: 498.29). ESI-HRMS: calcd for C30H37N5O2 (M + H)+ 498.2869, found 498.2875. HPLC analysis: MeOH-H2O (80: 20), 7.30 min, 97.0% purity.
methyl 3–(3-(4–(4-ethylpiperazin-1-yl)benzamido)-1H-indazol-6-yl)benzoate (9 h) White solid: 27.5 mg, yield 45%; Mp 228.7–233.4 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.91 (brs, 1H), 10.61 (brs, 1H), 8.27 (s, 1H), 7.98–8.06 (m, 4H), 7.84 (d, J = 8.5 Hz, 1H), 7.74 (s, 1H), 7.65–7.69 (m, 1H), 7.42 (d, J = 8.6 Hz, 1H), 7.08 (d, J = 8.5 Hz, 2H), 3.91 (s, 3H), 3.42–3.48 (m, 4H), 2.80–2.90 (m, 4H), 2.48–2.52 (q, J = 7.6 Hz, 2H), 1.19 (t, J = 7.1 Hz, 3H). 13 C NMR (100 MHz, DMSO-d6) δ: 166.7, 165.6, 153.0, 142.2, 141.5, 141.1, 137.9, 132.5, 130.9, 130.1, 129.9, 128.6, 128.0, 123.4, 119.6, 117.2, 114.3, 108.5, 52.8, 51.6, 46.1, 29.5, 10.8. ESI-MS (m/z): [M + H]+ = 484.3 (Calcd: 484.24). ESI-HRMS: calcd for C28H31N5O3 (M + H)+ 484.2349, found 484.2358. HPLC analysis: MeOH-H2O (80: 20), 7.05 min, 96.5% purity.
4–(4-ethylpiperazin-1-yl)-N-(6–(3-methoxy-4-methylphenyl)-1H-indazol-3-yl) benzamide (9i) White solid: 27.7 mg, yield 41%; Mp 227.3–231.2 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.83 (brs, 1H), 10.54 (brs, 1H), 8.01 (d, J = 8.7 Hz, 2H), 7.75 (d, J = 8.5 Hz, 1H), 7.60 (s, 1H), 7.54 (s, 2H), 7.33 (dd, J = 8.6, 1.1 Hz, 1H), 7.05 (d, J = 9.1 Hz, 3H), 3.84 (s, 3H), 3.33–3.38 (m, 4H), 2.59–2.64 (m, 4H), 2.46 (q, J = 7.3 Hz, 2H), 2.25 (s, 3H), 1.09 (t, J = 7.7 Hz, 3H). 13 C NMR (100 MHz, DMSO-d6) δ: 165.5, 157.6, 142.4, 141.0, 138.9, 132.9, 129.9, 126.5, 126.1, 122.9, 119.5, 116.4, 114.1, 111.2, 107.3, 56.5, 55.9, 51.8, 46.6, 19.0, 16.6. ESI-MS (m/z): [M + H]+ = 470.3 (Calcd: 470.26). ESI-HRMS: calcd for C28H33N5O2 (M + H)+ 470.2556, found 470.2551. HPLC analysis: MeOH-H2O (80: 20), 7.14 min, 96.3% purity.
N-(6–(3,5-bis(trifluoromethyl)phenyl)-1H-indazol-3-yl)-4–(4-ethylpiperazin-1-yl)benzamide (9j) White solid: 33.6 mg, yield 44%; Mp 227.5–213.3 °C. 1H NMR (400 MHz, DMSO-d6) δ: 12.95 (brs, 1H), 10.59 (brs, 1H), 8.42 (s, 2H), 8.13 (s, 1H), 8.00 (d, J = 8.9 Hz, 2H), 7.94 (s, 1H), 7.87 (d, J = 8.6 Hz, 1H), 7.54 (d, J = 8.2 Hz, 1H), 7.04 (d, J = 8.0 Hz, 2H), 3.27–3.38 (m, 4H), 2.44–2.52 (m, 4H), 2.40 (q, J = 7.1 Hz, 2H), 1.05 (t, J = 7.2 Hz, 3H). 13 C NMR (100 MHz, DMSO-d6) δ: 165.6, 153.6, 143.7, 141.9, 141.3, 135.7, 131.5, 131.2, 129.9, 128.4, 125.0, 123.6, 122.9, 122.8, 119.7, 117.6, 113.9, 109.7, 52.6, 52.1, 47.4, 12.4. ESI-MS (m/z): [M + H]+ = 562.2 (Calcd: 562.20). ESI-HRMS: calcd for C28H27F6N5O (M + H)+ 562.2042, found 562.2049. HPLC analysis: MeOH-H2O (80: 20), 7.08 min, 95.9% purity.
N-(6–(3,5-dichlorophenyl)-1H-indazol-3-yl)-4–(4-ethylpiperazin-1-yl)benzamide (9k) White solid: 28.9 mg, yield 41%; Mp 226.4–228.7 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.90 (brs, 1H), 10.58 (brs, 1H), 7.99 (t, J = 9.4 Hz, 2H), 7.81 (dd, J = 10.2, 5.0 Hz, 3H), 7.67–7.73 (m, 1H), 7.63 (t, J = 1.7 Hz, 1H), 7.16–7.47 (m, 1H), 7.03 (dd, J = 8.8, 4.5 Hz, 2H), 3.34 (s, 4H), 2.46–2.67 (m, 4H), 2.41 (s, 2H), 1.06 (t, J = 7.0 Hz, 3H). 13 C NMR (100 MHz, DMSO-d6) δ: 165.5, 153.6, 144.6, 141.9, 136.0, 135.1, 129.8, 127.3, 126.4, 124.7, 123.5, 123.1, 120.2, 119.5, 117.5, 113.9, 113.1, 109.1, 52.5, 52.0, 47.3, 12.3. ESI-MS (m/z): [M + H]+ = 494.2 (Calcd: 494.15). ESI-HRMS: calcd for C26H27Cl2N5O (M + H)+ 494.1514, found 494.1508. HPLC analysis: MeOH-H2O (80: 20), 7.23 min, 96.5% purity.
N-(6–(2,5-dichlorophenyl)-1H-indazol-3-yl)-4–(4-ethylpiperazin-1-yl)benzamide (9 l) White solid: 30.3 mg, yield 39%; Mp 231.5–234.6 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.93 (brs, 1H), 10.59 (brs, 1H), 8.01 (d, J = 8.8 Hz, 2H), 7.79 (d, J = 8.5 Hz, 1H), 7.64 (d, J = 8.6 Hz, 1H), 7.58 (d, J = 2.5 Hz, 1H), 7.52 (dd, J = 8.3, 2.8 Hz, 2H), 7.09–7.15 (m, 1H), 7.05 (d, J = 8.8 Hz, 2H), 3.36–3.40 (m, 4H), 2.61–2.68 (m, 4H), 2.46–2.50 (q, J = 7.2 Hz, 2H), 1.10 (t, J = 6.8 Hz, 3H). 13 C NMR (100 MHz, DMSO-d6) δ: 165.7, 153.4, 142.3, 141.3, 141.1, 136.0, 132.4, 131.9, 131.6, 130.9, 128.9, 129.5, 123.2, 122.5, 121.6, 117.1, 114.1, 111.2, 52.1, 51.9, 46.8, 11.6. ESI-MS (m/z): [M + H]+ = 494.2 (Calcd: 494.15). ESI-HRMS: calcd for C26H27Cl2N5O (M + H)+ 494.1514, found 494.1510. HPLC analysis: MeOH-H2O (80: 20), 7.36 min, 96.7% purity.
N-(6–(2,3-dichlorophenyl)-1H-indazol-3-yl)-4–(4-ethylpiperazin-1-yl)benzamide (9 m) White solid: 22.1 mg, yield 36%; Mp 220.1–222.4 °C. 1H NMR (400 MHz, DMSO-d6) δ: 12.89 (brs, 1H), 10.58 (brs, 1H), 8.00 (d, J = 8.4 Hz, 2H), 7.78 (d, J = 8.0 Hz, 1H), 7.70–7.72 (m, 1H), 7.46–7.50 (m, 3H), 7.10 (d, J = 7.8 Hz, 1H), 7.04 (d, J = 8.4 Hz, 2H), 3.26–3.40 (m, 4H), 2.51–2.55 (m, 4H), 2.42 (q, J = 7.1 Hz, 2H), 1.05 (t, J = 7.2 Hz, 3H). 13 C NMR (100 MHz, DMSO-d6) δ: 165.6, 153.5, 143.1, 141.3, 141.2, 137.1, 132.8, 130.8, 130.4, 130.2, 129.8, 128.8, 123.0, 122.5, 121.6, 117.0, 114.0, 111.0, 52.4, 52.0, 47.2, 12.2. ESI-MS (m/z): [M + H]+ = 494.2 (Calcd: 494.15). ESI-HRMS: calcd for C26H27Cl2N5O (M + H)+ 494.1514, found 494.1509. HPLC analysis: MeOH-H2O (80: 20), 6.88 min, 95.5% purity.
N-(6–(3,4-dichlorophenyl)-1H-indazol-3-yl)-4–(4-ethylpiperazin-1-yl)benzamide (9n) White solid: 21.8 mg, yield 38%; Mp 224.9–228.0 °C. 1H NMR (400 MHz, DMSO-d6) δ: 12.91 (brs, 1H), 10.57 (brs, 1H), 7.99–8.03 (m, 3H), 7.73–7.81 (m, 3H), 7.53–7.60 (m, 1H), 7.40–7.47 (m, 1H), 7.04 (d, J = 8.4 Hz, 2H), 3.26–3.40 (m, 4H), 2.51–2.55 (m, 4H), 2.41 (q, J = 7.1 Hz, 2H), 1.06 (t, J = 7.2 Hz, 3H). 13 C NMR (100 MHz, DMSO-d6) δ: 165.5, 153.6, 142.0, 141.7, 141.2, 136.3, 132.2, 131.5, 130.7, 129.8, 129.4, 127.9, 123.5, 122.9, 119.4, 117.3, 113.9, 108.7, 52.6, 52.1, 47.4, 12.4. ESI-MS (m/z): [M + H]+ = 494.2 (Calcd: 494.15). ESI-HRMS: calcd for C26H27Cl2N5O (M + H)+ 494.1514, found 494.1514. HPLC analysis: MeOH-H2O (80: 20), 7.05 min, 96.5% purity.
4–(4-ethylpiperazin-1-yl)-N-(6–(3-fluoro-5-methoxyphenyl)-1H-indazol-3-yl)benzamide (9o) White solid: 22.1 mg, yield 34%; Mp 225.5–227.0 °C. 1H NMR (400 MHz, DMSO-d6) δ: 12.86 (brs, 1H), 10.55 (brs, 1H), 8.00 (d, J = 8.9 Hz, 2H), 7.80 (d, J = 8.5 Hz, 1H), 7.72 (s, 1H), 7.40 (dd, J = 8.6, 1.3 Hz, 1H), 7.12–7.21 (m, 2H), 7.03 (d, J = 9.0 Hz, 2H), 6.85–6.89 (m, 1H), 3.87 (s, 3H), 3.26–3.41 (m, 4H), 2.51–2.57 (m, 4H), 2.39 (q, J = 7.1 Hz, 2H), 1.05 (t, J = 7.2 Hz, 3H). 13 C NMR (100 MHz, DMSO-d6) δ: 165.0, 162.2, 161.1, 143.5, 141.5, 140.7, 137.2, 129.3, 122.8, 122.4, 119.2, 116.8, 113.4, 109.1, 108.1, 106.2, 106.0, 55.7, 52.1, 51.6, 46.9, 11.9. HRMS: calcd for C27H30FN5O2 (M + H)+ 474.2305, found 474.2296. HPLC analysis: MeOH-H2O (80: 20), 7.24 min, 96.7% purity.
N-(6–(2-chloro-4-(trifluoromethyl)phenyl)-1H-indazol-3-yl)-4–(4-ethylpiperazin-1-yl)benzamide (9p) White solid: 19.9 mg, yield 35%; Mp 227.9–232.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.92 (brs, 1H), 10.58 (brs, 1H), 7.97–8.04 (m, 3H), 7.80–7.85 (, 2H), 7.74 (d, J = 8.0 Hz, 1H), 7.55 (s, 1H), 7.14 (dd, J = 8.5, 1.2 Hz, 1H), 7.03 (d, J = 8.9 Hz, 2H), 3.33–3.40 (m, 4H), 2.48–2.52 (m, 4H), 2.45 (s, 2H), 1.06 (t, J = 7.1 Hz, 3H). 13 C NMR (100 MHz, DMSO-d6) δ: 165.7, 153.5, 144.8, 141.3, 136.0, 133.2, 133.0, 129.9, 127.2, 124.8, 123.0, 122.7, 121.4, 117.2, 114.0, 111.2, 52.4, 52.0, 47.1, 12.1. ESI-MS (m/z): [M + H]+ = 528.1 (Calcd: 528.17). ESI-HRMS: calcd for C27H27ClF3N5O (M + H)+ 528.1778, found 528.1777. HPLC analysis: MeOH-H2O (80: 20), 7.18 min, 96.9% purity.
N-(6–(2,6-dimethylphenyl)-1H-indazol-3-yl)-4–(4-ethylpiperazin-1-yl)benzamide (9q) White solid: 30.9 mg, yield 40%; Mp 235.7–239.2 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.75 (brs, 1H), 10.56 (brs, 1H), 8.02 (d, J = 8.7 Hz, 2H), 7.77 (d, J = 8.3 Hz, 1H), 7.18 (d, J = 8.4 Hz, 2H), 7.14 (d, J = 8.2 Hz, 2H), 7.07 (d, J = 8.6 Hz, 2H), 6.82 (d, J = 8.3 Hz, 1H), 3.37–3.43 (m, 4H), 2.54–2.68 (m, 4H), 2.35 (q, J = 7.2 Hz, 2H), 2.01 (s, 6H), 1.14 (t, J = 7.0 Hz, 3H). 13 C NMR (100 MHz, DMSO-d6) δ: 165.6, 153.0, 142.0, 141.1, 139.1, 135.8, 129.9, 127.8, 127.5, 122.8, 121.5, 116.5, 114.3, 110.2, 51.7, 51.6, 46.4, 21.0, 12.2. ESI-MS (m/z): [M + H]+ = 454.3 (Calcd: 454.26). ESI-HRMS: calcd for C26H28ClN5O (M + H)+ 454.2607, found 454.2606. HPLC analysis: MeOH-H2O (80: 20), 7.30 min, 95.7% purity.
4–(4-ethylpiperazin-1-yl)-N-(6–(2-(trifluoromethoxy)phenyl)-1H-indazol-3-yl)benzamide (9r) White solid: 28.2 mg, yield 38%; Mp 230.9–233.5 °C. 1H NMR (400 MHz, DMSO-d6) δ: 12.87 (brs, 1H), 10.57 (brs, 1H), 8.00 (d, J = 8.4 Hz, 2H), 7.79 (d, J = 7.8 Hz, 1H), 7.62–7.64 (m, 1H), 7.53–7.57 (m, 4H), 7.16 (d, J = 8.0 Hz, 1H), 7.04 (d, J = 8.8 Hz, 2H), 3.28–3.43 (m, 4H), 2.47–2.53 (m, 4H), 2.41 (q, J = 7.1 Hz, 2H), 1.06 (t, J = 7.2 Hz, 3H). 13 C NMR (100 MHz, DMSO-d6) δ: 165.6, 153.6, 145.9, 141.6, 141.2, 135.5, 134.7, 132.4, 129.9, 128.5, 123.0, 122.6, 121.5, 117.0, 113.9, 110.8, 52.5, 52.0, 47.4, 12.3. ESI-MS (m/z): [M + H]+ = 510.2 (Calcd: 510.21). ESI-HRMS: calcd for C27H28F3N5O2 (M + H)+ 510.2117, found 510.2119. HPLC analysis: MeOH-H2O (80: 20), 7.18 min, 96.9% purity.
N-(6–(4-cyanophenyl)-1H-indazol-3-yl)-4–(4-ethylpiperazin-1-yl)benzamide (9 s) White solid: 15.8 mg, yield 38%; Mp 226.4–230.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.94 (brs, 1H), 10.52 (brs, 1H), 7.93–8.08 (m, 6H), 7.85 (d, J = 8.5 Hz, 1H), 7.78 (s, 1H), 7.44 (d, J = 8.6 Hz, 1H), 7.03 (d, J = 8.6 Hz, 2H), 3.28–3.39 (m, 4H), 2.51–2.59 (m, 4H), 2.40 (q, J = 6.7 Hz, 2H), 1.06 (t, J = 7.1 Hz, 3H). 13 C NMR (100 MHz, DMSO-d6) δ: 165.6, 153.7, 145.6, 142.1, 141.2, 137.1, 133.3, 129.9, 128.5, 123.6, 122.9, 119.6, 117.5, 113.9, 110.5, 109.1, 52.5, 52.1, 47.4, 12.3. ESI-MS (m/z): [M + H]+ = 451.2 (Calcd: 451.22). ESI-HRMS: calcd for C27H28N6O (M + H)+ 451.2246, found 451.2238. HPLC analysis: MeOH-H2O (80: 20), 7.40 min, 96.3% purity.
4–(4-ethylpiperazin-1-yl)-N-(6–(1-methyl-1H-pyrazol-3-yl)-1H-indazol-3-yl) benzamide (9t) White solid: 18.7 mg, yield 35%; Mp 221.6–224.2 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.67 (brs, 1H), 10.46 (brs, 1H), 8.23 (s, 1H), 7.98 (d, J = 8.8 Hz, 2H), 7.94 (s, 1H), 7.68 (d, J = 8.5 Hz, 1H), 7.57 (s, 1H), 7.30 (d, J = 8.5 Hz, 1H), 7.02 (d, J = 8.8 Hz, 2H), 3.89 (s, 3H), 3.37–3.45 (m, 4H), 2.52–2.60 (m, 4H), 2.41 (q, J = 6.7 Hz, 2H), 1.06 (t, J = 7.2 Hz, 3H). 13 C NMR (100 MHz, DMSO-d6) δ: 165.6, 153.6, 142.4, 141.1, 136.8, 131.3, 129.8, 128.7, 123.0, 122.7, 118.6, 116.1, 113.9, 105.5, 52.5, 52.0, 47.3, 19.0, 12.3. ESI-MS (m/z): [M + H]+ = 430.2 (Calcd: 430.24). ESI-HRMS: calcd for C24H29N7O (M + H)+ 430.2355, found 430.2357. HPLC analysis: MeOH-H2O (80: 20), 7.11 min, 95.5% purity.
4–(4-(dimethylamino)piperidin-1-yl)-N-(6–(3-fluoro-5-methoxyphenyl)-1H-indazol-3-yl)benzamide (9 u) White solid: 14.3 mg, yield 46%; Mp 223.8–227.4 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.87 (brs, 1H), 10.54 (brs, 1H), 7.99 (d, J = 8.7 Hz, 2H), 7.79 (d, J = 8.5 Hz, 1H), 7.72 (s, 1H), 7.40 (d, J = 8.8 Hz, 1H), 7.11–7.22 (m, 2H), 7.04 (d, J = 8.8 Hz, 2H), 6.87 (d, J = 10.9 Hz, 1H), 3.98 (d, J = 12.2 Hz, 2H), 3.87 (s, 3H), 2.82–2.90 (m, 2H), 2.59 (s, 1H), 2.35 (s, 6H), 1.92 (s, 2H), 1.51 (d, J = 10.8 Hz, 2H). 13 C NMR (100 MHz, DMSO-d6) δ: 165.5, 164.8, 162.9, 161.6, 153.2, 143.9, 142.0, 141.2, 137.7, 129.9, 123.3, 122.5, 119.6, 117.3, 114.1, 109.6, 108.6, 106.7, 100.9, 62.1, 56.2, 46.9, 41.3, 27.4, 21.6. ESI-MS (m/z): [M + H]+ = 488.3 (Calcd: 488.26). HRMS: calcd for C27H30FN5O2 (M + H)+ 488.2462, found 488.2459. HPLC analysis: MeOH-H2O (80: 20), 7.22 min, 97.5% purity.
4-((3R,5S)-3,5-dimethylpiperazin-1-yl)-N-(6–(3-fluoro-5-methoxyphenyl)-1H-indazol-3-yl)benzamide (9v) White solid: 26.5 mg, yield 38%; Mp 229.6–233.4 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.87 (brs, 1H), 10.56 (brs, 1H), 7.99 (d, J = 8.8 Hz, 2H), 7.79 (d, J = 8.6 Hz, 1H), 7.72 (s, 1H), 7.40 (d, J = 8.5 Hz, 1H), 7.14–7.18 (m, 2H), 7.04 (d, J = 9.0 Hz, 2H), 6.87 (d, J = 10.9 Hz, 1H), 4.47 (brs, 1H), 3.87 (s, 3H), 3.82 (s, 2H), 2.96 (s, 2H), 2.36 (d, J = 10.7 Hz, 2H), 1.11 (d, J = 6.1 Hz, 6H). 13 C NMR (100 MHz, DMSO-d6) δ: 165.5, 164.8, 162.9, 161.6, 153.2, 143.9, 142.0, 141.1, 137.7, 129.9, 126.7, 123.3, 119.6, 117.3, 113.9, 109.6, 108.6, 106.7, 100.9, 63.3, 56.2, 53.3, 50.6, 19.0. ESI-MS (m/z): [M + H]+ = 474.2 (Calcd: 474.23). ESI-HRMS: calcd for C27H30FN5O2 (M + H)+ 474.2305, found 474.2290. HPLC analysis: MeOH-H2O (80: 20), 7.11 min, 95.5% purity.
N-(6–(3-fluoro-5-methoxyphenyl)-1H-indazol-3-yl)-4–(4-methyl-1,4-diazepan-1-yl)benzamide (9w) White solid: 30.1 mg, yield 33%; Mp 230.4–234.6 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.83 (brs, 1H), 10.44 (brs, 1H), 7.97 (d, J = 8.4 Hz, 2H), 7.79 (d, J = 8.0 Hz, 1H), 7.71 (s, 1H), 7.39 (d, J = 8.1 Hz, 1H), 7.14–7.18 (m, 2H), 6.85–6.89 (m, 1H), 6.80 (d, J = 7.9 Hz, 2H), 3.87 (s, 7H), 3.63–3.65 (m, 2H), 3.53 (t, J = 6.8 Hz, 2H), 2.70–2.78 (m, 2H), 2.55–2.62 (m, 2H), 2.48–2.51 (m, 4H), 2.36 (s, 3H). 13 C NMR (100 MHz, DMSO-d6) δ: 165.6, 164.8, 162.9, 161.6, 151.7, 144.0, 142.0, 137.6, 130.16, 123.3, 120.2, 119.6, 117.3, 110.8, 109.6, 108.6, 106.7, 101.1, 57.2, 56.6, 56.2, 48.1, 46.1, 26.8. ESI-MS (m/z): [M + H]+ = 474.0(Calcd: 473.22). ESI-HRMS: calcd for C27H30FN5O2 (M + H)+ 474.2305, found 474.2297. HPLC analysis: MeOH-H2O (80: 20), 7.15 min, 96.2% purity.
N-(6–(3-fluoro-5-methoxyphenyl)-1H-indazol-3-yl)-4-((4-methylpiperazin-1-yl)methyl)benzamide (9x) White solid: 20.1 mg, yield 44%; Mp 240.3–242.5 °C. 1H NMR (400 MHz, DMSO-d6) δ: 13.01 (brs, 1H), 10.84 (brs, 1H), 8.06 (d, J = 8.2 Hz, 2H), 7.80 (d, J = 8.2 Hz, 1H), 7.74 (s, 1H), 7.46 (d, J = 8.0 Hz, 2H), 7.41 (dd, J = 8.0, 1.4 Hz, 1H), 7.14–7.18 (m, 2H), 6.85–6.89 (m, 1H), 3.87 (s, 1H), 3.55 (s, 2H), 2.33–2.46 (m, 8H), 2.16 (s, 3H). 13 C NMR (100 MHz, DMSO-d6) δ: 165.9, 164.8, 162.9, 161.6, 144.0, 143.0, 142.0, 140.6, 137.7, 132.9, 130.1, 129.1, 128.4, 123.0, 119.8, 117.1, 109.6, 108.7, 106.7, 100.9, 62.1, 56.2, 55.2, 53.1, 46.2. ESI-MS (m/z): [M + H]+ = 474.2 (Calcd: 474.23). ESI-HRMS: calcd for C27H30FN5O2 (M + H)+ 474.2305, found 474.2298. HPLC analysis: MeOH-H2O (80: 20), 7.11 min, 95.5% purity.
N-(6–(3-fluoro-5-methoxyphenyl)-1H-indazol-3-yl)-4–(4-methylpiperazin-1-yl)benzamide (9 y) White solid: 17.1 mg, yield 30%; Mp 237.4–241.6 °C. 1H NMR (400 MHz, DMSO-d6) δ: 13.01 (brs, 1H), 10.84 (brs, 1H), 8.06 (d, J = 8.2 Hz, 2H), 7.80 (d, J = 8.2 Hz, 1H), 7.74 (s, 1H), 7.46 (d, J = 8.0 Hz, 2H), 7.41 (dd, J = 8.0, 1.4 Hz, 1H), 7.14–7.18 (m, 2H), 6.85–6.89 (m, 1H), 3.87 (s, 1H), 3.24–3.39 (s, 4H), 2.44–2.52 (s, 4H), 2.16 (s, 3H). 13 C NMR (100 MHz, DMSO-d6) δ: 165.9, 164.7, 163.1, 161.6, 144.0, 143.0, 142.0, 140.7, 137.7, 132.9, 129.1, 128.4, 123.0, 119.8, 117.2, 109.6, 108.7, 106.7, 101.1, 62.1, 56.2, 55.2, 53.1, 46.2. ESI-MS (m/z): [M + H]+ = 460.2 (Calcd: 460.21). ESI-HRMS: calcd for C27H30FN5O2 (M + H)+ 460.2149, found 460.2155. HPLC analysis: MeOH-H2O (80: 20), 7.42 min, 96.4% purity.
N-(6–(3-fluoro-5-methoxyphenyl)-1H-indazol-3-yl)-5–(4-methylpiperazin-1-yl)pyrazine-2-carboxamide (9z) White solid: 19.8 mg, yield 35%; Mp 237.7–240.3 °C. 1H NMR (400 MHz, DMSO-d6) δ: 12.95 (brs, 1H), 10.42 (brs, 1H), 8.80 (s, 1H), 8.44 (s, 1H), 7.94 (d, J = 8.4 Hz, 1H), 7.74 (s, 1H), 7.42 (d, J = 8.3 Hz, 1H), 7.05–7.25 (m, 2H), 6.87 (d, J = 10.9 Hz, 1H), 3.87 (s, 7H), 2.70 (s, 4H), 2.42 (s, 3H). ESI-MS (m/z): [M + H]+=462.2 (Calcd: 462.21). 13 C NMR (100 MHz, DMSO-d6) δ: 164.8, 162.8, 161.6, 155.6, 142.8 142.1, 140.1, 137.8, 132.4, 130.1, 129.5, 123.2, 119.8, 116.7 109.6, 108.6, 106.7, 101.0, 56.2, 54.5, 46.0, 44.2. ESI-MS (m/z): [M + H]+ = 462.2 (Calcd: 462.20). HRMS: calcd for C24H25FN7O2 (M + H)+ 462.2054, found 462.2038. HPLC analysis: MeOH-H2O (80: 20), 7.11 min, 95.5% purity.
General method for preparation of compounds 12a-12d (exemplified by 12a)
N-(6–(3-methoxyphenyl)benzo[d]thiazol-2-yl)-4–(4-methylpiperazin-1-yl) benzamide (12a) white solid: 73.7 mg, yield 53%. Mp 230.5–233.2 °C. 1H NMR (500 MHz, DMSO-d6) δ: 12.57 (brs, 1H), 8.34 (s, 1H), 8.07 (d, J = 8.8 Hz, 2H), 7.81 (d, J = 8.4 Hz, 1H), 7.76 (d, J = 8.3 Hz, 1H), 7.40 (t, J = 7.9 Hz, 1H), 7.31 (d, J = 7.7 Hz, 1H), 7.28 (s, 1H), 7.03 (d, J = 8.9 Hz, 2H), 6.95 (d, J = 8.0 Hz, 1H), 3.85 (s, 3H), 2.44 (s, 4H), 2.23 (s, 3H). 13 C NMR (125 MHz, DMSO-d6) δ 165.57, 160.24, 160.06, 154.19, 148.63, 141.98, 135.97, 132.96, 130.46, 130.43, 125.64, 120.78, 120.29, 120.19, 119.59, 113.71, 113.34, 112.70, 55.62, 54.77, 46.94, 46.18. HRMS: calcd for C26H27N4O2S (M + H)+ 459.1776, found 459.1708. HPLC analysis: MeOH-H2O (85: 15), 7.50 min, 97.1% purity.
N-(6–(3,5-dimethoxyphenyl)benzo[d]thiazol-2-yl)-4–(4-methylpiperazin-1-yl) benzamide (12 b) White solid 56.7 mg, yield 49%. Mp 234.6–238.2 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.58 (brs, 1H), 8.34 (s, 1H), 8.06 (d, J = 8.3 Hz, 2H), 7.73–7.84 (m, 2H), 7.03 (d, J = 8.3 Hz, 2H), 6.88 (s, 2H), 6.51 (s, 1H), 3.84 (s, 6H), 2.44 (s, 4H), 2.23 (s, 3H). 13 C NMR (125 MHz, DMSO-d6) δ 165.57, 161.33, 160.09, 154.19, 148.72, 142.62, 136.02, 132.89, 130.43, 125.67, 120.69, 120.29, 120.25, 113.70, 105.39, 99.68, 55.77, 54.77, 46.94, 46.17. HRMS: calcd for C27H29N4O3S (M + H)+ 489.1882, found 489.1852. HPLC analysis: MeOH-H2O (85: 15), 7.70 min, 96.4% purity.
4–(4-methylpiperazin-1-yl)-N-(6–(3,4,5-trimethoxyphenyl)benzo[d]thiazol-2-yl)benzamide (12c) White solid 50.5 mg, yield 47%. Mp 236.4–238.3 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.59 (brs, 1H), 8.17 (s, 1H), 8.07 (d, J = 8.8 Hz, 2H), 7.83 (d, J = 8.4 Hz, 1H), 7.60 (d, J = 8.3 Hz, 1H), 7.24 (t, J = 7.9 Hz, 1H), 7.19 (t, J = 7.5 Hz, 1H), 7.12 (t, J = 6.6 Hz, 1H), 7.04 (d, J = 8.9 Hz, 2H), 3.90 (s, 3H), 2.45 (s, 4H), 2.23 (s, 3H). 13 C NMR (125 MHz, DMSO-d6) δ 165.57, 159.85, 154.19, 153.68, 148.40, 137.43, 136.32, 136.26, 132.86, 130.43, 125.67, 120.63, 120.30, 120.08, 113.71, 104.76, 60.54, 56.46, 54.77, 46.94, 46.18. HRMS: calcd for C28H31N4O4S (M + H)+ 519.1988, found 519.2007. HPLC analysis: MeOH-H2O (85: 15), 7.35 min, 96.8% purity.
N-(6–(2-fluoro-3-methoxyphenyl)benzo[d]thiazol-2-yl)-4–(4-methylpiperazin-1-yl)benzamide (12d) White solid 64.8 mg, yield 52%. Mp 232.5–235.2 °C. 1H NMR (500 MHz, DMSO-d6) δ 12.59 (brs, 1H), 8.17 (s, 1H), 8.07 (d, J = 8.8 Hz, 2H), 7.83 (d, J = 8.4 Hz, 1H), 7.60 (d, J = 8.3 Hz, 1H), 7.24 (t, J = 7.9 Hz, 1H), 7.19 (t, J = 7.5 Hz, 1H), 7.12 (t, J = 6.6 Hz, 1H), 7.04 (d, J = 8.9 Hz, 2H), 3.90 (s, 3H), 2.45 (s, 4H), 2.23 (s, 3H). 13 C NMR (125 MHz, DMSO-d6) δ 165.59, 160.39, 154.19, 150.12, 148.69, 148.32, 148.23, 148.17, 132.55, 130.65, 130.44, 129.38, 129.29, 127.56, 127.54, 125.01, 124.97, 122.36, 122.28, 120.49, 120.24, 113.70, 113.24, 56.58, 54.75, 46.92, 46.15. HRMS: calcd for C26H26FN4O2S (M + H)+ 477.1682, found 477.1653. HPLC analysis: MeOH-H2O (85: 15), 7.58 min, 95.5% purity.
General method for preparation of compounds 18a-18c (exemplified by 18a)
4–(4-methylpiperazin-1-yl)-N-(5-phenethyl-1H-1,2,4-triazol-3-yl)benzamide (18a) white solid 145.4 mg, yield 32%. Mp 218.1–222.5 °C. 1H NMR (500 MHz, DMSO-d6) δ 13.14 (brs, 1H), 11.59 (brs, 1H), 7.96 (d, J = 7.7 Hz, 2H), 7.24–7.28 (m, 4H), 7.18 (t, J = 7.1 Hz, 1H), 7.00 (d, J = 8.9 Hz, 2H), 3.31 (s, 4H), 3.00 (t, J = 7.7 Hz, 2H), 2.88 (s, 2H), 2.37–2.48 (m, 4H), 2.22 (s, 3H). 13 C NMR (125 MHz, DMSO-d6) δ 165.27, 160.15, 153.93, 130.06, 128.75, 128.71, 126.38, 121.02, 113.77, 111.75, 54.80, 47.09, 46.19, 34.09, 30.25. HRMS: calcd for C24H31N6O3 (M + H)+ 391.2168, found 391.2083. HPLC analysis: MeOH-H2O (85: 15), 9.05 min, 97.5% purity.
N-(5–(3-methoxyphenethyl)-1H-1,2,4-triazol-3-yl)-4–(4-methylpiperazin-1-yl) benzamide (18 b) White solid 82.1 mg, yield 31%. Mp 216.4–219.5 °C. 1H NMR (500 MHz, DMSO-d6) δ 13.09 (brs, 1H), 11.55 (brs, 1H), 7.96 (d, J = 5.2 Hz, 2H), 7.19 (t, J = 7.8 Hz, 1H), 7.00 (d, J = 8.9 Hz, 2H), 6.81 (d, J = 7.6 Hz, 2H), 6.75 (d, J = 8.4 Hz, 1H), 3.72 (s, 3H), 3.31 (s, 4H), 2.97 (t, J = 7.5 Hz, 2H), 2.88 (d, J = 8.6 Hz, 2H), 2.37–2.47 (m, 4H), 2.23 (s, 3H). 13 C NMR (125 MHz, DMSO-d6) δ 165.29, 159.70, 153.93, 130.05, 129.74, 120.96, 114.42, 113.77, 111.80, 55.33, 54.80, 47.11, 46.21, 34.05. HRMS: calcd for C23H29N6O2 (M + H)+ 421.2274, found 421.2215. HPLC analysis: MeOH-H2O (85: 15), 8.80 min, 97.2% purity.
N-(5–(3,5-dimethoxyphenethyl)-1H-1,2,4-triazol-3-yl)-4–(4-methylpiperazin-1-yl)benzamide (18c). White solid 77.8 mg, yield 31%. Mp 220.9–223.3 °C. 1H NMR (500 MHz, DMSO-d6) δ 13.11 (brs, 1H), 11.60 (brs, 1H), 7.96 (s, 2H), 6.94–7.05 (m, 2H), 6.38–6.41 (m, 2H), 6.31 (s, 1H), 3.71 (s, 6H), 3.31 (s, 4H), 2.79–2.97 (m, 4H), 2.40–2.47 (m, 4H), 2.22 (s, 3H). HRMS: calcd for C24H31N6O3 (M + H)+ 451.2379, found 451.2351. HPLC analysis: MeOH-H2O (85: 15), 8.72 min, 96.9% purity.
Biological evaluation
Kinase profiling
The kinase profiling of 9 u was conducted using Elisa kinase assay and the Eurofins Kinase Profiler Selectivity Testing Service.
Elisa kinase assay. The effects of the indicated compounds on the activities of receptor tyrosine kinases were determined using enzyme-linked immunosorbent assays (ELISAs) with purified recombinant proteins. Briefly, 20 μg/mL poly (Glu,Tyr)4:1 (Sigma, St Louis, MO, USA) was pre-coated in 96-well plates as a substrate. A 50 μL aliquot of 10 μmol/L ATP solution diluted in kinase reaction buffer (50 mmol/L HEPES [pH 7.4], 50 mmol/L MgCl2, 0.5 mmol/L MnCl2, 0.2 mmol/L Na3VO4, and 1 mmol/L DTT) was added to each well; 1 μL of different compounds with indicated concentration diluted in 1% DMSO (v/v) (Sigma, St Louis, MO, USA) were then added to each reaction well. DMSO (1%, v/v) was used as the negative control. The kinase reaction was initiated by the addition of purified tyrosine kinase proteins (FGFR1, FGFR2, FGFR3, FGFR4, ALK, Bcr-Abl, EPH-A2, Flt-1, RET, c-Src, IGF1R, c-Met, EGFR, ErbB2) diluted in 49 μL of kinase reaction buffer solution. After incubation for 60 min at 37 °C, the plate was washed three times with phosphate-buffered saline (PBS) containing 0.1% Tween 20 (T-PBS). Anti-phosphotyrosine (PY99) antibody (100 μL; 1:500, diluted in 5 mg/mL BSA T-PBS) was then added. After a 30 min incubation at 37 °C, the plate was washed three times, and 100 μL horseradish peroxidase-conjugated goat anti-mouse IgG (1:2000, diluted in 5 mg/mL BSA T-PBS) was added. The plate was then incubated at 37 °C for 30 min and washed 3 times. A 100 μL aliquot of a solution containing 0.03% H2O2 and 2 mg/mL o-phenylenediamine in 0.1 mol/l citrate buffer (pH 5.5) was added. The reaction was terminated by the addition of 50 μL of 2 mol/l H2SO4 as the colour changed, and the plate was analysed using a multi-well spectrophotometer (SpectraMAX 190, Molecular Devices, Sunnyvale, CA, USA) at 490 nm. The inhibition rate (%) was calculated using the following equation: [1-(A490/A490 control)] × 100%. The IC50 values were calculated from the inhibition curves in two separate experiments.
Cell proliferation assay
Human gastric cancer cell line SNU-16 was purchased from American Type Culture Collection (ATCC, Manssas, VA, USA). All the cell lines were routinely maintained in complete medium according to the suppliers’ recommendations.
Cells were seeded in 96-well tissue culture plates. On the day when seeding, the cells were exposed to various concentrations of compounds and further cultured for 72 h at 37 °C. Finally, cell proliferation was determined using Cell Counting Kit (CCK-8) or the thiazolyl blue tetrazolium bromide (MTT, from Sigma-Aldrich, St. Louis, MO, USA) assay. The IC50 values were calculated by concentration–response curve fitting using a SoftMax pro-based four-parameter method.
Results and discussion
Rational drug design
Most selective FGFR1 inhibitors are ATP-competitive inhibitors that bind to the ATP-binding pocket of FGFR. The ATP binding pocket of the FGFR1 consists of five regions3,23,24: (i) the adenine region, (ii) hydrophobic region I, (iii) hydrophobic region II, (iv) the nucleotide domain, (v) and the phosphate region. The adenine region is considered as the major binding site in which heterocycle scaffolds are anchored through several H-bonds with a hinge region of the kinase (Figure 1, red colour). Meanwhile, the hydrophobic region I (Figure 1, blue colour) and hydrophobic region II (Figure 1, pink colour) are the other two important sites, which plays key factor in binding small molecules by interacting with lipophilic moieties hydrophobically, and forms van der Waals interactions and H-bonds.
Accordingly, the inhibitors are commonly involving the following pharmacophore features. (i) The core structure of most inhibitors consists of a flat hetero aromatic ring system that contains at least one H-bond acceptor and H-bond donor, then it can form hydrogen bonds with the key amino acid residues: Glu562 and Ala564. (ii) Terminal hydrophobic head often interacts with the hydrophobic region I, which is a phenyl ring with different extra hydrophobic substitutions. (iii) Another hydrophobic scaffold directly linked to the flat hetero aromatic ring system which occupies the hydrophobic region II. Herein, we initially pay attention to explore the novel hetero aromatic ring, which was the core structure of FGFR1 inhibitor, formed the key H-bond interaction with hinge region, and connected the hydrophobic region I and II.
Furthermore, we superimposed the two FGFR1 crystal structures of 4V05 and 3TTO, which were complexed with AZD454725 and NVP-BGJ 39817, respectively. The results revealed that AZD4547 and NVP-BGJ 398 were similar in their binding mode with the target were similarly, when there was a significant spatial distance constraint between the pharmacophore features. The spatial distance of the hydrophobic head and the heterocycle ring is between 5.9 Å and 6.5 Å (Figure 2(a) and Figure S2). Therefore, the FGFR1 inhibitors might contain the corresponding pharmacophore features, and these pharmacophore features were also consisted to a space distance constraint.
The fragment library was derived from kinase hinge region directed library, which contained 11809 scaffolds. Subsequently, with the built pharmacophore model and molecular docking, seven hit fragments were obtained through serval round filtration (Supplementary Table S1 and Figure S4). One of them was indazole scaffold, which was proved to be an efficiency FGFR1 inhibitor scaffold22, which indicated the accuracy of virtual screening protocol. Then, in view of the difficulties in synthesis, only 1H-1,2,4-triazole, benzothiazole and indazole scaffold were selected for further modification (Figure 2(b)). The novel 1H-1,2,4-triazole and benzothiazole scaffolds were selected for the study of FGFR1 inhibitors research. Furthermore, we also continued to optimise the original indazole derivatives, improve the physicochemical property and enrich the structure-activity relationship of indazole derivatives. Finally, we synthesised several indazole derivatives through introduced halogen substituents at various positions. As a result, with the fragment-based virtual screening strategy, novel derivatives bearing 1H-1,2,4-triazole, benzothiazole and indazole scaffold were designed, synthesised and evaluated.
Chemistry
The preparation of target compounds 9a-t, 12a-d and 18a-c was described in Schemes 1–3. Compounds 9a-t were synthesised from starting material 4-fluorobenzoate (1) and 4-bromo-2-fluorobenzonitrile (2) through seven steps. The nucleophilic substitution reactions of 4-fluorobenzoate with various piperazines afforded the methyl ester 2 intermediate, which then could hydrolysis to carboxylic acid 3. Subsequently, the carboxylic acid 3 was condensed with HATU to provide the activated ester reagent 4. Meanwhile, the intermediate 5 could be condensed by 4-bromo-2-fluorobenzonitrile 1 with hydrazine. Then nitrogen atom at the 1-position of 1H-indazol-3-amine scaffold 5 was protected by BOC group. The key intermediate 8 was obtained by condensation of 4 and 7 in the presence of NaH. Treatment of the intermediate 8 with various substituted-phenylboronic acid under Suzuki coupling condition26 of Pd(dppf)Cl2 and Cs2CO3 in refluxing dioxane/water gave target compounds. According to the similar synthetic route, compounds 12a-d were synthesised from the starting material 6-bromobenzo[d]thiazol-2-amine (10) through two steps.
Scheme 1.

Synthesis of indazole derivatives. Reagents and conditions: (a) K2CO3, DMSO, 120 °C, 85–90%; (b) NaOH, MeOH, H2O, 80 °C, 93–96%; (c) HATU, K2CO3, DMF, rt, 62–70%; (d) hydrazine hydrate, n-butanol, 120 °C, 86%; (e) Boc2O, DMAP, THF, 89%; (f) NaH, THF, 60 °C, 52–61%; (g) TFA, CH2Cl2, 0 °C, 70%; (h) R-B(OH)2, Cs2CO3, Pd(dppf)Cl2, dioxane, 120 °C, 30–48%.
Scheme 2.

Synthesis of benzothiazole derivatives. Reagents and conditions: (a) 4, NaH, THF, 60 °C, 51–62%; (b) R-B(OH)2, Cs2CO3, Pd(dppf)Cl2, dioxane, 120 °C, 47–53%.
Scheme 3.

Synthesis of 1H-1,2,4-triazole derivatives. Reagents and conditions: (a) propanedioic acid, pyridine, piperidine, 105 °C, 77–82%; (b) Pd/C, H2, EtOH, 88–93%; (c) SO2Cl2, reflux; (d) amino guanidine hydrochloride, 140 °C, 49–53%; (f) 4, NaH, THF, 60 °C, 54–62%; (g) TFA, CH2Cl2, 0 °C, 70%.
The synthesis of the other 1H-1,2,4-triazole derivatives was shown in Scheme 3. The starting material substituted-benzaldehyde (13) was condensed with propanedioic acid in the presence of pyridine and piperidine at 105 °C via Aldol condensation, which the intermediate 14 was obtained. After hydrogenation of 14 released the corresponding aliphatic acid 15, which was connected with the amino guanidine hydrochloride giving triazole scaffold 16 in 49.1% yield. Then nitrogen atom at the 1-position of the 1H-1,2,4-triazole heterocycle 16 was protected by BOC group. The target compounds 18a-c were prepared in similar way by following condensation and Suzuki-coupling procedures.
Biological evaluation
Biological evaluation for various scaffold derivatives 9a-d, 12a-d, 18a-c
Initially, we designed and synthesised the indazole (9a-d), benzothiazole (12a-d) and 1H-1,2,4-triazole (18a-c) derivatives, with the goal of analysing the effects of different scaffolds on biological activity (Table 1). All these compounds were evaluated against FGFR1 enzyme and SNU-16 cell line. The results indicated that when the heterocycle was substituted by a 1H-1,2,4-triazole scaffold, it would result in almost no enzyme inhibitory activities and cell anti-proliferative activities. Furthermore, the corresponding benzothiazole derivatives 12a-d were also revealed poor inhibitory activity against FGFR1 enzyme (IC50 >1000 nM), thus led to slight reduction cellular anti-proliferative activities.
Table 1.
Structures and activities of various derivatives 9a-d, 12a-d, and 18a-c.

The IC50 values are shown as the average values from two separate experiments.
SNU-16 is FGFR2-amplified cell type.
“NA” means IC50 > 5 μM.
Fortunately, compound 9a with an indazole scaffold exhibited good FGFR1 enzyme inhibitory activity (IC50 = 30.6 nM) and modest cellular inhibition (IC50 = 1195.7 nM). While the 3,5-dimethoxy phenyl substituent was replaced with the 3-methoxy phenyl group, the enzyme inhibition of 9 b was relatively lower than 9a. However it was more potent in cellular assay (9a vs 9 b). Besides, co-crystal structure analysis revealed that the R1 substituent is at the solvent region of the target protein, which might be helpful to improve membrane permeability and enhance cellular potencies. Then the other two derivatives 9c and 9d with N-methyl piperazine and N-ethyl piperazine substituent at the R1 position, respectively, were synthesised and evaluated. As a result, the 9d demonstrated the best enzyme inhibitory (IC50 = 15.0 nM) and cellular anti-proliferative activities (IC50 = 785.8 nM). The above results demonstrated that the heterocycle could significantly affect the enzyme and cell growth inhibitory activity.
The structure-activity relationship of indazole derivatives 9e-z
The hydrophobic group I located in the hydrophobic cavity of the active site and formed strong hydrophobic interaction with the target. To increase the enzyme inhibitory activity, we initially optimised the hydrophobic group I (Table 2). It was indicated that 3-methoxy phenyl (9d, IC50 = 15.0 nM) was substituted by the 3-ethoxy phenyl (9e, IC50 = 13.2 nM), 3-isopropoxy phenyl (9f, IC50 = 9.8 nM) caused increase of activity. On the contrary, the compound 9 h with 3-s-butoxy phenyl exhibited low enzymatic inhibitory activity (IC50 > 1000 nM). Meanwhile, the compound with 3-COOMe-phenyl (9 h, IC50 = 77.7 nM), 3,5-dichloro-phenyl (9k, IC50 = 67.2 nM), 2,5-dichloro-phenyl (9 l, IC50 = 50.6 nM), 2,3-dichloro-phenyl (9 m, IC50 = 44.8 nM), 2,6-methyl-phenyl (9q, IC50 = 41.7 nM), shown moderate FGFR1 inhibitory activity. On the other hand, compounds containing a substituent either the electron-withdrawing or electron-donating group at para-position, were significant decreasing their activities (9d vs 9i; 9k, 9 l and 9 m vs 9n; 9 m vs 9p). Furthermore, the additional halogen atoms on phenyl might be helpful to improve physicochemical properties and enhance the cell anti-proliferative activities. Thus, the compound 9o (IC50 = 5.5 nM) containing a fluorine atom at the para-position of phenyl showed better inhibitory activity than their counterparts lack of these atoms (9d, IC50 = 15.0 nM).
Table 2.
Structures and activities of indazole derivatives 9e–t.
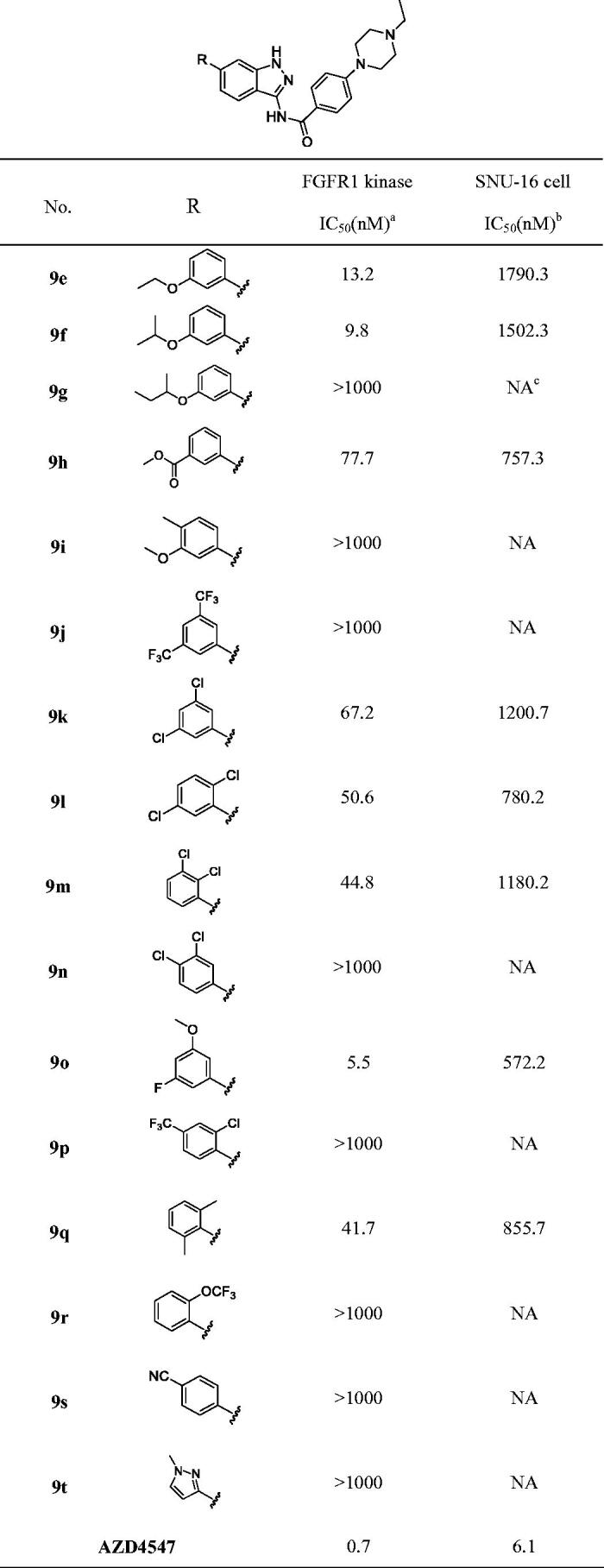
The IC50 values are shown as the average values from two separate experiments.
SNU-16 is FGFR2-amplified cell type.
“NA” means IC50 > 5 μM.
The active compounds 9e, 9f, 9 h, 9k, 9 l, 9 m, 9o and 9q were also selected for further evaluation of their cellular potencies (SNU-16 cell). Although, the FGFR1 enzyme inhibitory activities of compound 9 h and 9j were higher than that of 9d, while the cell anti-proliferative activities were similar. Besides, compound 9o bearing a fluorine atom at meta-position of phenyl resulted in 1.4-fold enhancement of the cellular inhibition (9o vs 9d), which exhibited the best enzyme and cellular inhibitory activity of these indazole compounds 9e-t.
As the above reported, R1 group located at the solvent region of the target protein, which might be help to improve membrane permeability and enhance cell anti-proliferative activities. Another six compounds 9 u-z were designed, prepared and evaluated for their biological activities (Table 3). The results of inhibition of FGFR1 assay revealed that (2S, 6 R)-2,6-dimethyl-1-phenylpiperazine (9v, IC50 = 13.2 nM), and N-methyl-4-phenylpiperazine (9 y, IC50 = 10.2 nM) led to slight reduction potency comparing to N-ethyl-4-phenylpiperazine (9o, IC50 = 5.5 nM), while introducing of a 1-benzyl-4-methylpiperazine group (9x, IC50 > 1000 nM) demonstrated poor inhibitory activity against FGFR1 enzyme. Meanwhile, the FGFR1 inhibitory activities of compounds 9 u, 9w and 9z were enhanced, and the lead compound 9 u was 1.5-fold enhancement. More importantly, the cellular potencies were improved by introducing various R1 group (9 u, 9v and 9w vs 9o), and among them 9 u exhibited the good FGFR1 inhibitory activity and cell anti-proliferative activity. Therefore, we selected 9 u as the lead compound for further pharmacological research.
Table 3.
Structures and activities of indazole derivatives 9 u–z.
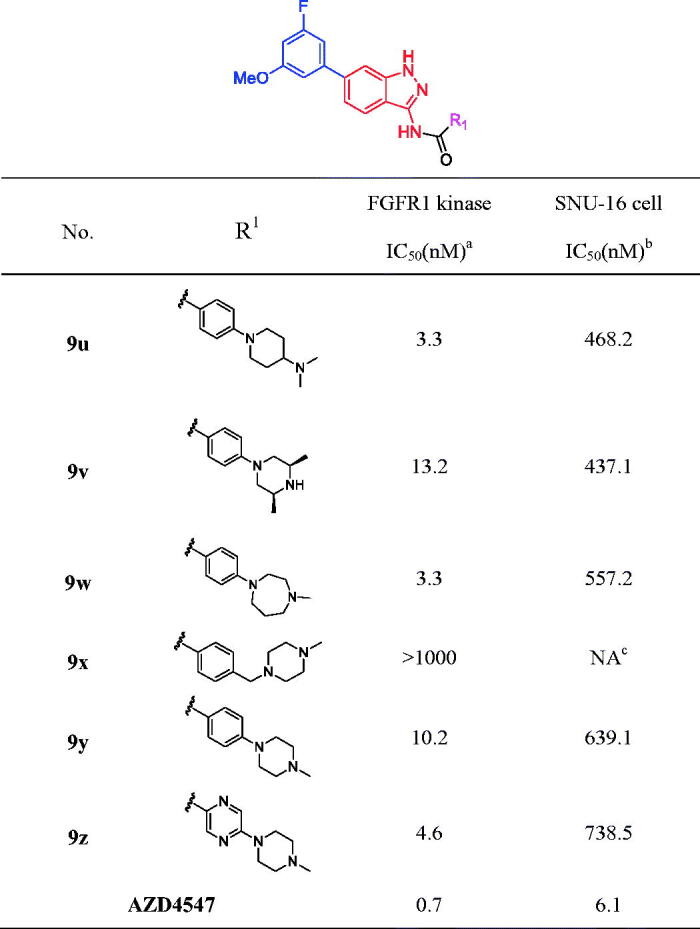
The IC50 values are shown as the average values from two separate experiments.
SNU-16 is FGFR2-amplified cell type.
“NA” means IC50 > 5 μM.
The kinase selectivity profile of compound 9 u
To identify the kinase selectivity of the lead compound 9 u, the pharmacological experiment was first evaluated by testing it against our in-house panel of tyrosine kinases. The results were summarised in Table 4, 9 u effectively inhibited the potent activities of FGFR1-3 with IC50 values of 3.3 nM, 5.2 nM and 12.2 nM, respectively. However, 9 u exhibited low enzymatic inhibitory activity (IC50 > 1000 nM), which indicated that 9 u is a selective FGFR1-3 inhibitor. The reason was that the sequence of FGFR1-3 was very similar, while the sequence of FGFR4 was quite different. Therefore, the binding mode of FGFR1-3 inhibitor was different from FGFR4 inhibitor, and the pharmacophore of FGFR4 inhibitor usually contained an acrylamide group [27–29]. Furthermore, IC50 values for most of kinases were greater than 1 μM except for Flt-1, RET and c-Src, which were inhibited at sub-micromolar concentrations of the compound. As a result, compound 9 u exhibited good kinase selectivity.
Table 4.
Selectivity of 9 u in a panel of kinases.
| Tyrosine kinase | IC50 (nM) | Tyrosine kinase | IC50 (nM) |
|---|---|---|---|
| FGFR1 | 3.3 | Flt-1 | >100 |
| FGFR2 | 5.2 | RET | >100 |
| FGFR3 | 12.2 | c-Src | >100 |
| FGFR4 | >1000 | IGF1R | >1000 |
| ALK | >1000 | c-Met | >1000 |
| Bcr-Abl | >1000 | EGFR | >1000 |
| EPH-A2 | >1000 | ErbB2 | >1000 |
Molecular docking studies
The docking simulation was performed to elucidate the binding mode of the various scaffold derivatives 9d, 12a and 18 b, the potent indazole inhibitor 9e and 9f, the inactive inhibitor 9 g with FGFR1. These compounds were docked into the ATP-binding pocket of FGFR1 (4ZSA).
As a result, the core indazole ring of compound 9d was entered the adenine region by forming the essential H-bond interaction with hinge residue Glu562 and Ala564 (docking score: 8.523), while the N-H of indazole scaffold formed H-bond with Glu562, and the nitrogen atom of indazole scaffold and N-H of amide formed another two hydrogen bond with Ala564 (Figure 3(a)). Meanwhile, the 3-methoxy phenyl group occupied the hydrophobic area I, and hydrophobic interact with the lipophilic residue Leu547, Val561 and Phe 642. However, while the hetero aromatic rings were converted to 1H-1,2,4-triazole or benzothiazole, there was weakly H-bond interaction with Ala564, and no H-bond interaction with Glu562 (Figure 3(b,c)). Besides, the hydrophobic group I was deviated from hydrophobic pocket. The above docking results might explain why the triazole and benzothiazole derivatives lost their biological activities (12a docking score: 6.380, 18 b docking score: 6.588).
Figure 3.

The docking mode of compound 9d (a), 12a (b), and 18 b (c). (d) Superposed docking poses of 9e (white), 9f (yellow), and 9 g (green).
Additionally, the methoxy group of 9d was inserted into a small hydrophobic cavity of the hydrophobic area I. While this cavity could contain a certain size of lipophilic group, thus the ethyloxy substituent of 9e (docking score: 8.983) and the iso-propyxoy substituent of 9f (docking score: 9.451) were more appropriate to this hydrophobic cavity, meanwhile 9e and 9f shown more potent FGFR1 inhibitory activity than 9d. However, the cavity could not accommodate large volume groups, including the sec-butoxy group (9 g docking score: 5.668, IC50 > 1000 nM). The molecular simulation results may further confirm the inhibitory potency of 9e and 9f against FGFR1, and the 9 g was inactive (Figure 3(d)).
Conclusion
In summary, we designed three novel series of FGFR1 inhibitors bearing indazole, benzothiazole, and 1H-1,2,4-triazole scaffold via fragment-based virtual screening. Interestingly, 33 new compounds were synthesised and evaluated for their inhibitory activity against FGFR1. Initially, the indazole derivative 9d was identified as a promising FGFR1 inhibitor, with the good enzymatic inhibition (IC50 = 15.0 nM) and modest anti-proliferative activity (IC50 = 785.8 nM). Then, the hit 9d was further optimised, through two rounds of optimisation, the compound 9 u stood out as the most potent FGFR1 inhibitors with the best enzyme inhibitory (IC50 = 3.3 nM) and cellular activity (IC50 = 468.2 nM). Moreover, 9 u also exhibited good kinase selectivity. Meanwhile, the docking study was performed to investigate the putative interaction mechanism with the FGFR1 target. Further studies on the structural optimisation and biological evaluation of 9 u are currently underway in our laboratory. Our study would provide a basis for discovering novel FGFR1 inhibitors.
Supplementary Material
Funding Statement
This work was supported by the National Natural Science Foundation of China (81703342, 81473110, 81773596), Natural Science Foundation of Jiangsu Higher Education Institutions (17KJA360004, 16KJB350003), Natural Science Foundation of Jiangsu (BK2016105), Postgraduate Research & Practice Innovation Program of Jiangsu Province (SJCX18_0448, KYCX18_1614).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- 1.Anand P, Kunnumakara A, Sundaram C, et al. Cancer is a preventable disease that requires major lifestyle changes. Pharm Res 2008;25:2097–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siemann DW, Chaplin DJ, Horsman MR.. Vascular-targeting therapies for treatment of malignant disease. Cancer 2004;100:2491–9. [DOI] [PubMed] [Google Scholar]
- 3.Traxler P, Furet P.. Strategies toward the design of novel and selective protein tyrosine kinase inhibitors. Pharmacol Ther 1999;82:195–206. [DOI] [PubMed] [Google Scholar]
- 4.Sharma PS, Sharma R, Tyagi T.. Receptor tyrosine kinase inhibitors as potent weapons in war against cancers. Curr Pharm Des 2009;15:758–76. [DOI] [PubMed] [Google Scholar]
- 5.Ocana A, Serrano R, Calero R, Pandiella A.. Novel tyrosine kinase inhibitors in the treatment of cancer. Curr Drug Targets 2009;10:575–6. [DOI] [PubMed] [Google Scholar]
- 6.Huang M, Shen A, Ding J, Geng M.. Molecularly targeted cancer therapy: some lessons from the past decade. Trends Pharmacol Sci 2014;35:41–50. [DOI] [PubMed] [Google Scholar]
- 7.Clark JD, Flanagan ME, Telliez JB.. Discovery and development of Janus Kinase (JAK) inhibitors for inflammatory diseases: miniperspective. J Med Chem 2014;57:5023–38. [DOI] [PubMed] [Google Scholar]
- 8.Touat M, Ileana E, Postel-Vinay S, et al. Targeting FGFR signaling in cancer. Clin Cancer Res 2015;21:2684–94. [DOI] [PubMed] [Google Scholar]
- 9.Carter EP, Fearon AE, Grose RP.. Careless talk costs lives: fibroblast growth factor receptor signalling and the consequences of pathway malfunction. Trends Cell Biol 2015;25:221–33. [DOI] [PubMed] [Google Scholar]
- 10.Dieci MV, Arnedos M, Andre F, Soria JC.. Fibroblast growth factor receptor inhibitors as a cancer treatment: from a biologic rationale to medical perspectives. Cancer Discovery 2013;3:264–79. [DOI] [PubMed] [Google Scholar]
- 11.Singh D, Chan JM, Zoppoli P, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012;337:1231–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu YM, Su F, Kalyana-Sundaram S, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discovery 2013;3:636–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Helsten T, Elkin S, Arthur E, et al. The FGFR landscape in cancer: analysis of 4,853 tumors by next-generation sequencing. Clin Cancer Res 2016;22:259–67. [DOI] [PubMed] [Google Scholar]
- 14.Hilberg F, Roth GJ, Krssak M, et al. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res 2008;68:4774–82. [DOI] [PubMed] [Google Scholar]
- 15.Roth GJ, Heckel A, Colbatzky F, et al. Design, synthesis, and evaluation of indolinones as triple angiokinase inhibitors and the discovery of a highly specific 6-methoxycarbonyl-substituted indolinone (BIBF 1120). J Med Chem 2009;52:4466–80. [DOI] [PubMed] [Google Scholar]
- 16.Gozgit JM, Wong MJ, Moran L, et al. Ponatinib (AP24534), a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models. Mol Cancer Ther 2012;11:690–9. [DOI] [PubMed] [Google Scholar]
- 17.Guagnano V, Furet P, Spanka C, et al. Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-ylphenylamino] -pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J Med Chem 2011;54:7066–83. [DOI] [PubMed] [Google Scholar]
- 18.Zhao GS, Li WY, Chen DH, et al. A novel, selective inhibitor of fibroblast growth factor receptors that shows a potent broad spectrum of antitumor activity in several tumor xenograft models. Mol Cancer Ther 2011;10:2200–10. [DOI] [PubMed] [Google Scholar]
- 19.Gavine PR, Mooney L, Kilgour E, et al. AZD4547: an orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res 2012;72:2045–56. [DOI] [PubMed] [Google Scholar]
- 20.Perera TPS, Jovcheva E, Mevellec L, et al. Discovery and pharmacological characterization of JNJ-42756493 (erdafitinib), a functionally selective smallmolecule FGFR family inhibitor. Mol Cancer Ther 2017;16:1010–20. [DOI] [PubMed] [Google Scholar]
- 21.Nakanishi Y, Akiyama N, Tsukaguchi T, et al. The fibroblast growth factor receptor genetic status as a potential predictor of the sensitivity to CH5183284/Debio 1347, a novel selective FGFR inhibitor. Mol Cancer Ther 2014;13:2547–58. [DOI] [PubMed] [Google Scholar]
- 22.Liu J, Peng X, Dai Y, et al. Design, synthesis and biological evaluation of novel FGFR inhibitors bearing indazole scaffold. Org Biomol Chem 2015;13:7643–54. [DOI] [PubMed] [Google Scholar]
- 23.Yosaatmadja Y, Patterson AV, Smaill JB, Squire CJ.. The 1.65 Å resolution structure of the complex of AZD4547 with the kinase domain of FGFR1 displays exquisite molecular recognition. Acta Crystallogr 2015;71:525–33. [DOI] [PubMed] [Google Scholar]
- 24.Zhang J, Yang PL, Gray NS.. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 2009;9:28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tucker JA, Klein T, Breed J, et al. Structural insights into FGFR kinase isoform selectivity: diverse binding modes of AZD4547 and ponatinib in complex with FGFR1 and FGFR4. Structure 2014;22:1764–74. [DOI] [PubMed] [Google Scholar]
- 26.Miyaura N, Suzuki A.. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem Rev 1995;95:2457–85. [Google Scholar]
- 27.Ken AB, Timothy DO, Erik V, et al. Discovery of the irreversible covalent FGFR inhibitor 8-(3-(4-acryloylpiperazin-1-yl)propyl)-6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methylamino)pyrido[2,3-d]pyrimidin-7(8H)‑one (PRN1371) for the treatment of solid tumors. J Med Chem 2017;60:6516–27. [DOI] [PubMed] [Google Scholar]
- 28.Li X, Guise CP, Taghipouran R, et al. 2-Oxo-3, 4-dihydropyrimido[4, 5-d]pyrimidinyl derivatives as new irreversible pan fibroblast growth factor receptor (FGFR) inhibitors. Eur J Med Chem 2017;135:531–43. [DOI] [PubMed] [Google Scholar]
- 29.Lu X, Chen H, Patterson AV, et al. Fibroblast growth factor receptor 4 (FGFR4) selective inhibitors as hepatocellular carcinoma therapy: advances and prospects. J Med Chem 2019;62:2905–15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.