

ABSTRACT
In Parkinson disease (PD), there is a preferential degeneration of neurons that contain the dark-brown cytoplasmic pigment neuromelanin, in particular dopaminergic neurons of the substantia nigra (SN), the loss of which leads to the typical motor symptoms of the disease and constitutes the cardinal pathological diagnostic criterion for PD. Neuromelanin is generally considered a byproduct of dopamine oxidative metabolism and, in humans, it is first detected in early childhood and accumulates progressively with age until occupying most of the neuronal cytoplasm, as neurons apparently lack the means to degrade or eliminate this pigment. Aging is the main risk factor for developing PD, but the molecular substrate underlying this link remains unknown. Despite the close and long-established association between neuromelanin and PD, the potential contribution of neuromelanin to PD pathogenesis has remained elusive because, in contrast to humans, common laboratory animal species, such as rodents, lack neuromelanin. To overcome this major limitation, we have recently generated the first experimental in vivo rodent model exhibiting age-dependent production and accumulation of human-like neuromelanin within PD-vulnerable dopaminergic nigral neurons, at levels up to those reached in elderly humans.
KEYWORDS: Alpha-synuclein, autophagy, cellular trafficking, dopamine, Lewy bodies, neurodegeneration, neuromelanin, Parkinson’s disease
Our model is based on the unilateral viral vector-mediated expression in the rat SN of human (Hs) TYR (tyrosinase), the rate-limiting enzyme for the synthesis of peripheral melanins (e.g. skin and hair) that is also present at very low levels in the human SN, where it might play a putative role in neuromelanin synthesis. Using this unique neuromelanin-producing rat model, we found that progressive intracellular neuromelanin accumulation above a specific threshold ultimately compromises neuronal function and triggers, in an age-dependent manner, pathological features typical of PD, including impaired dopamine release, motor deficits, Lewy body (LB)-like inclusion formation and nigrostriatal neurodegeneration [1]. Relevant to humans, intracellular neuromelanin levels reach this pathogenic threshold in both PD patients and pre-symptomatic PD subjects (i.e. incidental Lewy body disease) but not in aged-matched elderly control brains. In parallel to dopaminergic cell death, HsTYR-overexpressing rodents also exhibit other neuropathological features typical of PD, such as extracellular neuromelanin, released from dying neurons, surrounded by, or within, activated microglia (a process known as neuronophagia, which is indicative of ongoing neurodegeneration).
In humans, intracellular neuromelanin is known to continuously accumulate over a lifetime within undegraded or partly degraded macroautophagic/autophagic structures, probably in an (unsuccessful) attempt by the cell to degrade this insoluble pigment. In agreement with this, neuromelanin from HsTYR-overexpressing rodents appears enclosed within single-membrane-delimited mature lysosomes/autolysosomes or inside double-membrane-delimited autophagosomes that accumulated with age until occupying most of the neuronal cytoplasm. Consistent with the autophagic nature of neuromelanin granules, lysosomal and autophagosomal markers increase in parallel with neuromelanin production and colocalize with neuromelanin granules in HsTYR-expressing neurons. The continuous buildup of neuromelanin within autophagic compartments in these cells is associated with a parallel decrease in lysosomal-mediated proteolysis and a marked reduction in ubiquitin-proteasome system (UPS) activity. Relevant to humans, both autophagy and the UPS have been reported to be impaired in neuromelanin-laden but not in non-melanized regions from post-mortem PD brains. Consistent with a failure in autophagy and UPS-mediated proteolysis, neuromelanin-laden cells exhibit a marked accumulation of SQSTM1/p62, a common component of PD-associated neuropathological inclusions, and an extensive deposition of SNCA/α-synuclein oligomers, as directly visualized by an SNCA proximity ligation assay. In HsTYR-overexpressing rodents, cytoplasmic inclusions emerge within neuromelanin-filled areas, either overlapping with or displacing neuromelanin granules, and often appear as early multiple punctate small aggregates immunopositive for SQSTM1 that seem to progressively coalesce into more compacted LB-like spherical structures, as similarly reported in post-mortem PD brains. PD-type inclusion formation in HsTYR-overexpressing rodents peaks at the time of early neuronal dysfunction and is substantially reduced once neurodegeneration is established, suggesting that inclusion-containing neurons are those that become dysfunctional and preferentially degenerate in these animals. In line with this, the number of neuronal inclusions in PD brains at advanced stages of the disease is much lower than that observed in early PD cases. Remarkably, while some of the PD-type inclusions from HsTYR-overexpressing rodents are immunopositive for SNCA, the latter is dispensable for inclusion formation and nigrostriatal degeneration in these animals, as attested by experiments combining HsTYR-induced neuromelanin production with SNCA genetic ablation. This observation suggests that SNCA, which is currently regarded as a major pathogenic factor in PD, might not be in fact required for LB formation and PD-linked neurodegeneration.
The accumulation of SQSTM1-immunopositive inclusions in HsTYR-overexpressing rodents suggests a general failure of cellular proteostasis, well beyond SNCA, in neuromelanin-laden neurons. Consistent with a late-stage proteostasis failure in which cell function and survival may become compromised, neuromelanin-producing cells also exhibit accumulation of dysfunctional mitochondria and increased production of reactive oxygen species, 2 pathogenic events that have been consistently linked to PD. Confirming a pivotal pathogenic role for impaired proteostasis in neuromelanin-laden neurons, enhancement of lysosomal-mediated proteolysis in HsTYR-overexpressing rodents by overexpression of the master autophagy regulator TFEB (transcription factor EB) reduces intracellular neuromelanin to levels below the pathogenic threshold, attenuates PD-like inclusion formation, prevents nigrostriatal neurodegeneration and reverses motor deficits in these animals. Part of TFEB’s effects at reducing intracellular neuromelanin levels are attributed to TFEB-induced exocytosis of neuromelanin-filled lysosomes outside the cell, thereby demonstrating the feasibility and therapeutic potential of modulating intracellular neuromelanin levels in vivo.
Overall, our results reveal a novel pathogenic scenario in PD in which the continuous, age-dependent buildup of neuromelanin within autophagic compartments may ultimately exhaust the vesicular storage capacity of the cell, interfere with lysosomal proteases and other degradative pathways, impair intracellular vesicular trafficking and alter endocytic/secretory tasks, creating a cellular traffic jam that interferes with intracellular communication and protein synthesis/degradation, eventually leading to Lewy pathology and PD-linked neuronal dysfunction/degeneration (Figure 1). Accordingly, strategies to modulate intracellular neuromelanin levels may provide unprecedented therapeutic opportunities to prevent, halt or delay neuronal dysfunction and degeneration linked to PD and brain aging.
Figure 1.
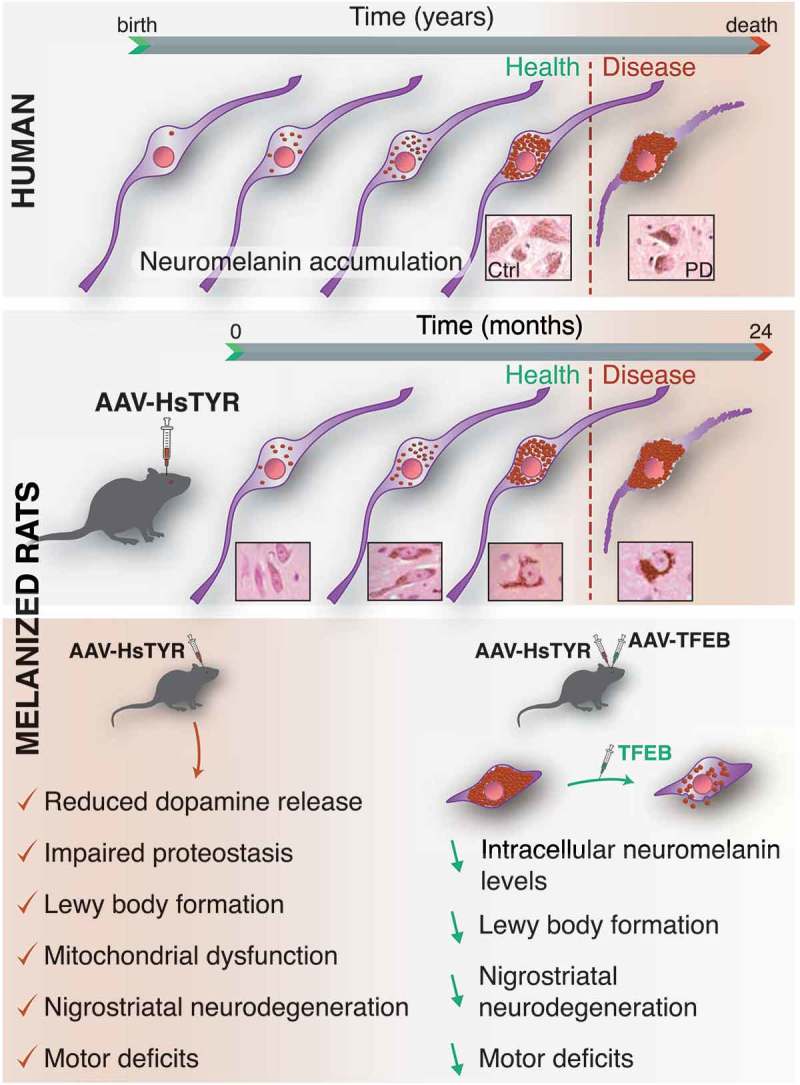
Proposed pathogenic effects of age-dependent intracellular neuromelanin accumulation in humans (top) and HsTYR-overexpressing rats (bottom). The continuous, age-dependent intracellular buildup of neuromelanin within undegraded autophagic compartments above a specific pathogenic threshold of accumulation (dotted red line) is associated with a general failure of cellular proteostasis concomitant with lysosomal/autophagy dysfunction, UPS impairment, Lewy body-type inclusion formation, reduced mitochondrial respiration, increased production of reactive oxygen species, impaired neurotransmission, nigrostriatal neurodegeneration and neuroinflammation, all of which are major pathological features of PD. Enhancement of lysosomal-mediated proteolysis with TFEB reduces intracellular neuromelanin to levels below the pathogenic threshold, attenuates PD-like inclusion formation, prevents nigrostriatal neurodegeneration and reverses motor impairment in HsTYR-overexpressing rodents. Part of TFEB’s effects are attributed to enhanced exocytosis of neuromelanin-filled lysosomes outside the cell, thus supporting the feasibility and therapeutic potential of modulating intracellular neuromelanin levels in vivo.
Funding Statement
This work was supported by the Michael J. Fox Foundation for Parkinson’s Research [TAP 15291]; Ministerio de Economía y Competitividad [SAF2015-73997-JIN]; Ministerio de Economía y Competitividad [SAF2016-77541-R]; “la Caixa” Foundation [LCF/PR/HR17/52150003].
Disclosure statement
No potential conflict of interest was reported by the authors.
Reference
- [1].Carballo-Carbajal I, Laguna A, Romero-Gimenez J, et al. Brain tyrosinase overexpression implicates age-dependent neuromelanin production in Parkinson’s disease pathogenesis. Nat Commun. 2019;10(1):973 PMID: 30846695. [DOI] [PMC free article] [PubMed] [Google Scholar]