

ABSTRACT
Macroautophagy (hereafter referred to as autophagy) involves a lysosomal degradation pathway and plays a context-dependent role in promoting either cell survival or cell death during stress; excessive or impaired autophagy is implicated in various types of cell death. In particular, lipid peroxidation-associated ferroptosis has recently been recognized as a type of autophagy-dependent cell death, but the mechanisms involved remain largely obscure. Our recent findings demonstrate that clockophagy, namely the selective autophagic degradation of the circadian clock regulator ARNTL/BMAL1, promotes ferroptotic cancer cell death in vitro and in vivo. Mechanically, the cargo receptor SQSTM1/p62 is responsible for the autophagic degradation of ARNTL in response to type 2 ferroptosis inducers (e.g., RSL3 and FIN56), but not type 1 ferroptosis inducers (e.g., erastin, sulfasalazine, and sorafenib). Consequently, clockophagy-mediated ARNTL degradation promotes lipid peroxidation and subsequent ferroptosis through blocking HIF1A-dependent fatty acid uptake and lipid storage. These findings highlight a novel type of selective autophagy in regulated cell death.
KEYWORDS: Autophagy, circadian rhythm, cargo receptor, cell death, fatty acid uptake, ferroptosis, hypoxia, lipid droplets, lipid peroxidation, lipid storage
Graphical Abstract
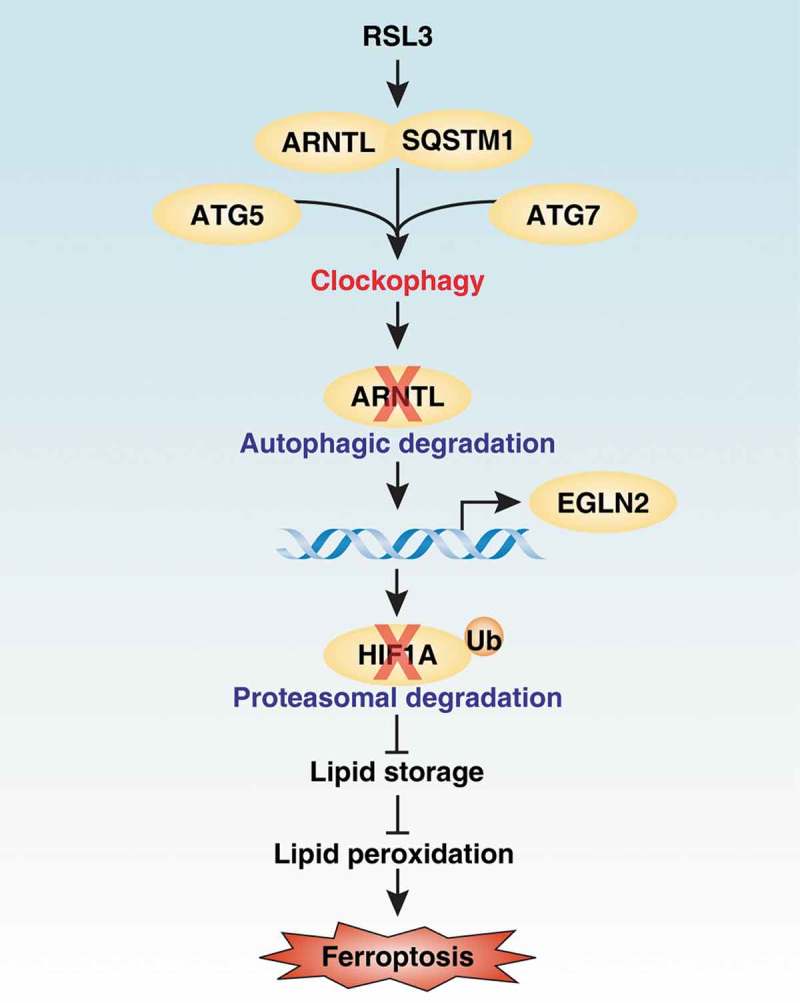
In many cases, autophagy is a homeostatic mechanism to remove unused proteins, damaged organelles, or invasive pathogens to promote cell survival. Alternatively, excessive or impaired autophagy can cause cell death through the degradation of anti-injury regulators or by causing metabolic crises. This type of autophagy-dependent cell death may occur in certain human diseases, such as neurological disease. Thus, it is important to define the process and function of autophagy in response to different triggers of cell death. Recently, we demonstrated that autophagy promotes ferroptotic cell death through the selective degradation of ARNTL/BMAL1 (aryl hydrocarbon receptor nuclear translocator-like), a key mammalian circadian clock regulator, via the cargo receptor SQSTM1/p62 (Figure 1) [1]. We termed this type of selective autophagy as “clockophagy,” and our findings increase the understanding of the mechanism of autophagy-dependent cell death.
Figure 1.
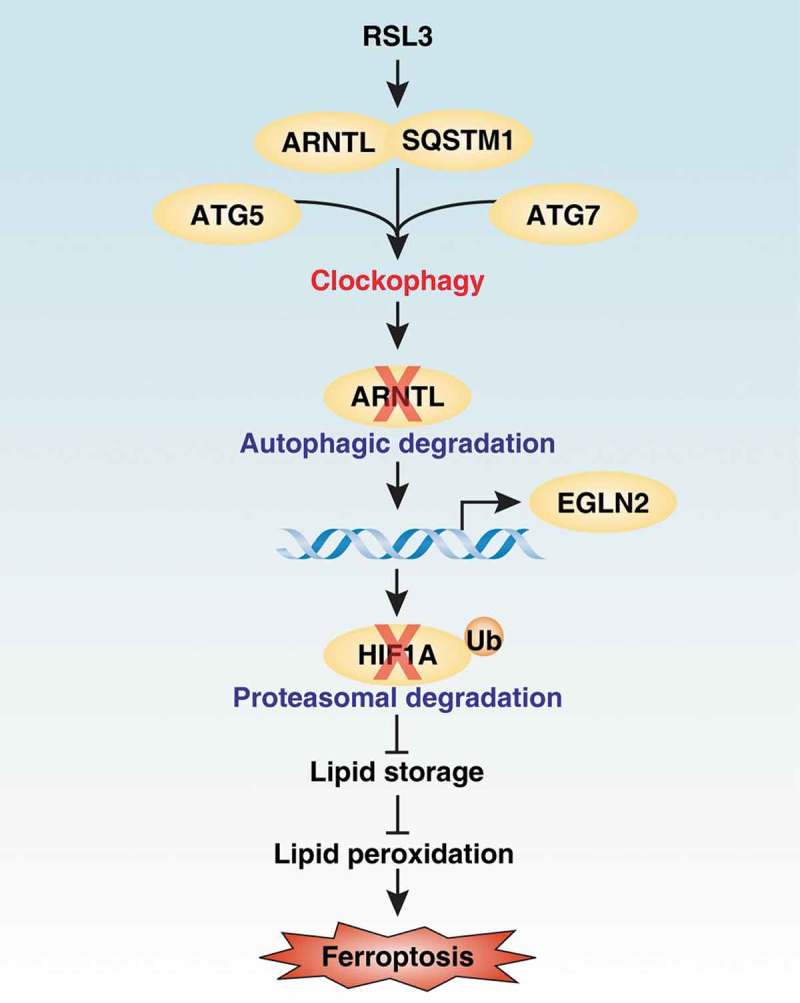
Effects of clockophagy on ferroptosis. Clockophagy promotes ferroptosis through the autophagic degradation of ARNTL via the cargo reseptor SQSTM1 and core autophagy components ATG5 and ATG7. The degradation of ARNTL can promote EGLN2 transcription, which leads to the proteasomal degradation of HIF1A. Consequently, the downregulation of HIF1A limits lipid storage and finally increases lipid peroxidation during ferroptosis.
Autophagy promotes ARNTL degradation during ferroptosis
Ferroptosis is a type of regulated cell death by oxidative stress, and is characterized by iron accumulation and lipid peroxidation. The major ferroptosis inducers can be divided into type 1 and type 2, based on their molecular target of either the antioxidant system or an enzyme: SLC7A11/system xc− and GPX4 (glutathione peroxidase 4), respectively. Circadian rhythms are endogenously generated rhythms and regulate a variety of physiological and pathological processes through the production and activation of a circadian clock gene or protein. Although a disrupted circadian clock is implicated in cell death, whether ferroptosis affects the expression of the circadian clock regulator is not clear. To address this question, we assayed the expression of ARNTL in both ferroptosis-resistant and -sensitive cancer cell lines. The protein expression of ARNTL is downregulated by type 2 activators (e.g., RSL3 and FIN56), but not by type 1 activators (e.g., erastin, sulfasalazine, and sorafenib) in ferroptosis-sensitive cancer cell lines (e.g., Calu-1, HT1080, and HL-60 cells). In contrast, the protein expression of ARNTL is not affected in ferroptosis-resistant cancer cell lines (e.g., THP1 cells). Thus, ARNTL exhibits a different protein expression change in response to various ferroptosis inducers.
Autophagy and the ubiquitin-proteasome system are the two major mechanisms for protein degradation. An autophagy inhibitor (e.g., spautin-1 or chloroquine), but not a proteasome inhibitor (e.g., MG-132), completely reverses RSL3-induced ARNTL protein degradation in Calu-1 and HT1080 cells. Autophagy-related (Atg) gene depletion analyses further demonstrate that both ATG5 and ATG7 (but not ATG9A) are required for RSL3-induced ARNTL protein degradation. A cargo receptor protein is important for selective autophagy by delivering the degraded components to the site of phagophore engulfment. Using affinity isolation assays coupled with mass spectrometry and RNAi, we demonstrated that the cargo receptor SQSTM1 (but not NBR1 [NBR1, autophagy cargo receptor], OPTN [optineurin], CALCOCO2/NDP52 [calcium binding and coiled-coil domain 2], or NCOA4 [nuclear receptor coactivator 4]) is required for autophagic ARNTL degradation. Protein structural analysis further shows that the ubiquitin-associated domain of SQSTM1 can be a major site for ARNTL binding. Thus, clockophagy requires ATG5, ATG7, and SQSTM1, but not ATG9A, during RSL3-induced ferroptosis.
Clockophagy promotes lipid peroxidation during ferroptosis
An early study reported that ferroptosis is distinct from autophagy and other types of regulated cell death, such as apoptosis and necroptosis. Increasingly, recent studies indicate that ferroptosis is a type of autophagy-dependent cell death that requires autophagy machinery to initiate oxidative injury. In particular, certain types of selective autophagy, such as NCOA4-mediated ferritinophagy and RAB7A-dependent lipophagy, promote ferroptotic cell death via the upregulation of iron-mediated oxidative stress or lipid peroxidation. We next investigated whether clockophagy also is involved in ferroptosis. The knockdown or knockout of ATG5, ATG7, or SQSTM1 in human cancer cells or mouse embryonic fibroblasts limits both type 1 and type 2 ferroptosis inducers from causing lipid peroxidation and subsequent cell death. Importantly, ARNTL depletion can overcome resistance to ferroptosis in THP1 cells, whereas ARNTL ovexpression can diminish sensitivity to ferroptosis in Calu-1 and HT1080 cells. The administration of antioxidants (e.g., ferrostatin-1 and liproxstatin-1) protects against ARNTL depletion-mediated cell death in response to RSL3. Furthermore, the transfection-enforced overexpression of ARNTL also reduces lipid peroxidation and restores cell viability in gpx4−/- cells. Together, these findings suggest that clockophagy promotes ferroptosis through the upregulation of lipid peroxidation.
Clockophagy promotes ferroptosis via the EGLN2-HIF1A pathway
The circadian transcription factor ARNTL governs gene expression dependent on time of day through the binding of E-box motifs in their promoters. Using bioinformatics, chromatin immunoprecipitation, and reporter gene analyses, we determined that EGLN2/PHD1 (egl-9 family hypoxia-inducible factor 2) is a novel and direct ARNTL target gene in RSL3-induced ferroptosis. Suppression of ARNTL expression increases EGLN2 expression leading to subsequent ferroptosis in Calu-1 and HT1080 cells. In contrast, the knockdown of EGLN2 by shRNAs blocks RSL3-induced ferroptotic cell death, indicating the EGLN1 is a positive regulator of ferroptosis. One major function of EGLN2 is to inhibit HIF1A (hypoxia inducible factor 1, alpha subunit) activation during hypoxia. We hypothesized that ARNTL-mediated EGLN2 downregulation would be required for ferroptosis resistance through the upregulation of HIF1A. Indeed, ARNTL can control HIF1A protein stability through a change of EGLN2 expression during ferroptosis. Using a combined pharmacological and genetic approach, we further demonstrated that the inhibition of HIF1A expression or activity restores the sensitivity to ferroptosis in EGLN2-knockdown or ARNTL-overexpressing HT1080 cells.
HIF1A generally functions as a pro-survival transcriptional regulator of cellular and systemic homeostatic response to hypoxia by upregulating gene expression involved in metabolism and immunity. Early studies showed that HIF1A can increase lipid storage and reduce fatty acid β-oxidation to promote tumor cell survival. We demonstrated that hypoxia pretreatment and the iron chelator desferrioxamine can increase HIF1A expression, which limits ferroptosis through increasing fatty acid uptake and lipid storage, thus minimizing peroxidation-mediated endomembrane damage. In particular, FABP3 (fatty acid binding protein 3, muscle and heart) and FABP7 (fatty acid binding protein 7, brain), two key HIF1A-target genes, may be responsible for this process.
In summary, our findings uncover a mechanism by which clockophagy drives ferroptosis, and identify a type of selective autophagy responsible for ARNTL degradation associated with an abnormal EGLN2-HIF1A pathway and subsequent lipid peroxidation. Our study also indicates that targeting clockophagy machinery may improve the anticancer activity of ferroptosis inducers in vitro and in vivo.
Funding Statement
M.Y. is supported by grants from the National Natural Science Foundation of China [81570154 and 81100359] and the Natural Science Foundation of Hunan Province [2016JJ3171]. R.K. is supported by a grant from the US National Institutes of Health [CA211070]. D.J.K. is supported by a grant from the US National Institutes of Health [GM131919]. D.T. is supported by grants from the American Cancer Society [Research Scholar Grant RSG-16-014-01-CDD].
Acknowledgments
We thank Dave Primm (Department of Surgery, University of Texas Southwestern Medical Center) for his critical reading of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Reference
- [1].Yang M, Liu J, Zhu S, et al. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci Adv. 2019;5:aaw2238. [DOI] [PMC free article] [PubMed] [Google Scholar]