

ABSTRACT
The PDCD1 gene encodes PD-1, an important immune checkpoint protein and key immunotherapy target to treat cancer. PDCD1 is alternatively spliced to generate an exon 3-skipped isoform PD-1Δ3 that has been suggested to play an antagonistic role to PD-1, but the mechanism underlying alternative splicing of PDCD1 has never been explored. Here using a minigene system, we analysed the splicing pattern of PDCD1 in multiple cell lines and confirmed exon 3 skipping as the main alternative splicing event. Using deletion analysis of exon 3, we mapped two splicing enhancers in the exon: ESE3a and ESE3b. Using mutagenesis, RNA-affinity chromatography, mass spectrometry as well as depletion and overexpression of MATR3, we defined MATR3 as a splicing activator during PDCD1 exon 3 splicing that operates through binding to ESE3b. MATR3’s splicing-stimulatory activity is counteracted by an RNA secondary structure around ESE3b and an RNA helicase DDX5. Furthermore, we identified ASOs that efficiently promotes PDCD1 exon 3 skipping in both minigene and endogenous-gene contexts. Our data support further study of the ASOs as potential drug candidates to treat cancer.
KEYWORDS: PDCD1, PD-1, cancer, antisense oligonucleotide, MATR3, DDX5
Introduction
Alternative splicing (AS) is an important post-transcriptional regulatory mechanism of gene expression that allows generation of more than one mRNA species from a multi-exon gene [1]. AS serves as an important source for protein diversity [2]. Splicing pattern changes can lead to gain or loss of a critical domain in a protein, resulting in full or partial loss of function, gain of function, alteration in protein stability and/or alteration in subcellular localization. Therefore, protein isoforms generated by AS may have similar, distinct or even antagonistic functions depending on the extent of changes in the open reading frame, implicating the importance of AS in regulation of gene function. AS events in the immune system have been widely documented; some of them play critical roles in immune responses, for example, CD45 employs AS as a feedback mechanism to maintain T-cell homoeostasis [3]. However, there are still many AS events that have not been well understood, in spite of the genes being critical in regulating immune activities. One such gene is programmed cell death 1 (PDCD1) that encodes PD-1, also called CD279.
PD-1 is an inhibitory immune checkpoint protein of 268 amino acids, comprising several domains including an N-terminal immunoglobulin variable (IgV-like) domain that recognizes ligand signal, a short stalk that separates the IgV-like domain from the plasma membrane, a transmembrane domain, and a cytoplasmic tail that contains two tyrosine-based signalling motifs [4,5]. PD-1 is expressed on the surface of activated T cells, B cells, natural killer cells, microphages and dendritic cells. Upon engagement with its ligands PD-L1 or PD-L2, it dampens T cell activation and restrains T cell function through multiple mechanisms such as enhancement of apoptosis, alteration of metabolism, and induction of suppressive cytokines [6]. Therefore, activation of the PD-1 signalling not only prevents autoimmune diseases but also generates a tumour microenvironment (TME) favouring tumour growth. A key strategy that cancer cells take to evade antitumor immunity is to hijack the PD-1 pathway through, e.g. overexpression of PD-L1. PD-1 is also expressed in cancer cells, for instance, subpopulations of melanoma, promoting tumorigenesis through tumour cell-intrinsic mechanism [7]. Recently, PD-1 has been shown to express and function in neuronal cell types such as primary sensory neurons, where PD-L1 ligation inhibits nociceptive neuron activity [8], and in injured retinal ganglion cells, where it appears to contribute to retinal apoptosis [9].
The PDCD1 gene, which contains five exons, is alternatively spliced. Nielsen et al. reported the presence of five splicing isoforms including an exon 3-skipped PD-1 (PD-1Δ3) [10]. The PD-1Δ3 isoform, duo to loss of exon 3 that encodes the entire membrane-spanning domain, is presumably present in cells or secreted as a soluble form; therefore, its function is predicted to be different, or even antagonistic [10]. Wan et al. indeed identified soluble PD-1 (sPD-1) in human sera and synovial fluid, and found elevated sPD-1 levels in patients with rheumatoid arthritis [11]. Using quantitative RT-PCR (qRT-PCR), the authors revealed that PD-1Δ3 is the only sPD-1; no other isoforms were detected in peripheral blood and synovial T cells in any groups [11]. A recent study found a positive correlation between increased serum sPD-1 levels and prolonged survival of patients with a subtype of lung cancer after treatment with erlotinib [12]. These studies suggest that the soluble isoform probably has antagonistic effects on PD-1 by interfering with its signalling pathway.
Currently, development of antibodies to occlude the binding of PD-1 to its ligands has been a main theme in cancer immunotherapy. Antibodies against the extracellular IgV-like domain have been successfully generated to treat patients. For example, Nivolumab, a humanized monoclonal IgG4 anti-PD-1 antibody, is highly effective in patients with varied types of cancer [13]. While antibodies against PD-1 and PD-L1 generate exciting clinical efficacy, the responder rate is limited and efficacy still needs to be improved. An alternative way to target PD-1 is to employ antisense oligonucleotide (ASO) strategy at the RNA processing level by switching the splicing pattern of PDCD1 to that favouring exon 3 skipping. This approach not only inhibits PD-1 activity by reducing its expression but also generates an isoform that may further neutralize the PD-1 signalling. We have recently developed an antisense drug nusinersen (previously termed ASO10-27), which is 2′-O-methoxyethyl (MOE) modified with a phosphorothioate backbone, to treat a devastating genetic disease called spinal muscular atrophy (SMA) [14–17]. Nusinersen markedly improves motor function and lifespan of SMA patients and is the only approved drug to treat SMA [18]. SMA is caused by deficiency of the survival of motor neuron (SMN) protein that is encoded by two paralogous genes: SMN1, a gene that normally is the major source of SMN but mutated in SMA patients, and SMN2, an erroneously spliced gene that generates limited functional full-length SMN protein [19,20]. Nusinersen increases cellular SMN protein levels by correcting SMN2 splicing [21].
To develop an anti-cancer ASO drug targeting PDCD1, it is necessary to understand how its AS is modulated. Here, using a PDCD1 minigene, we first performed a deletion analysis of exon 3 to map splicing regulatory elements (SREs) and uncovered two exonic splicing enhancers (ESEs) in the exon; the stronger ESE is under combinatorial regulation involving at least two splicing factors MATR3 and DDX5 and a secondary RNA structure. Furthermore, using an ASO-tiling assay, we identified MOE-modified ASOs that efficiently promote PDCD1 exon 3 skipping at low doses in both minigene and endogenous-gene settings, highlighting their therapeutic potential for further development to treat cancer.
Results
PDCD1 exon 3 is a cassette exon harbouring two putative splicing enhancers
To study PDCD1 splicing, we constructed a PDCD1 minigene in pCI-neo vector. The minigene comprises the 76-nt coding region of exon 1, a 355-nt shortened intron 1, the entire genomic fragment from exon 2 to intron 4, and the first 59 nt of exon 5 (Figure 1a). We examined the splicing pattern of the minigene in various cell lines including human epithelial HeLa cells, human malignant melanoma A375 cells, human metastatic melanoma C8161 cells, human malignant glioma U87MG cells, human hepatoma HepG2 cells and human embryonic kidney 293 (HEK293) cells; splicing was analysed by fluorescence-labelled RT-PCR with minigene-specific primer sets [22]. In all the cell lines, two main isoforms, i.e. the full-length transcript and exon 3-skipped isoform were detected, though the percentage of exon 3 inclusion varies in different cell lines (Figure 1b). The two mRNA isoforms were also the predominant transcripts expressed by the endogenous PDCD1 gene in B lymphocyte RPMI 8226 cells (Supplementary Fig. S1). Therefore, the PDCD1 minigene recapitulates the splicing pattern of the endogenous gene. Our data confirm a previous report that exon 3 inclusion or skipping is the main PDCD1 AS event [11].
Figure 1.
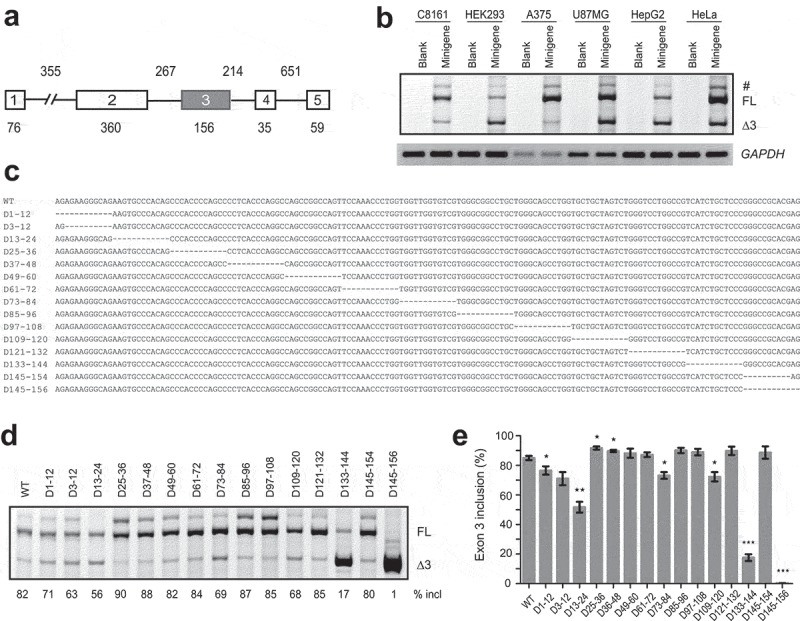
Splicing analysis of the PDCD1 minigene and mapping of SREs in exon 3 by a series of deletions. (a) Diagram of the PDCD1 minigene cloned in pCI-neo. Exons are shown with boxes and introns with lines. The exon and intron lengths are indicated. (b) The splicing pattern of the minigene was analysed in five different cell types: HEK293, HeLa, HepG2, U87MG, A375 and C8161. Thirty hours post-transfection, RNA was collected and analysed by Cy5-labelled RT-PCR. GAPDH was used as a loading control. Blank: empty vector; FL: full-length; Δ3: exon 3-skipped transcript. (c) Exon 3 sequence and positions of all deletions indicated by dashed lines. (d) Splicing analysis of WT and mutant minigenes shown in panel C. Each plasmid (500 ng) was transfected into A375 cells; RNA was collected 30 h later and evaluated by Cy5-labelled RT-PCR. (e) The histogram shows the quantitation of splicing from three experiments similar to the one shown in panel D. *P< 0.05, **P< 0.01, ***P< 0.001 versus WT (n = 3). #, an isoform with retention of the first 94 nt of intron 3.
We next explored key SREs in PDCD1 exon 3 via deletion analysis spanning the entire exon using the above minigene. Thirteen mutants, each having a 12-nt deletion, were created and named based on the coordinates of each deletion (Figure 1c). To circumvent the effects imposed by disruption of the 3′ splice site (3′ ss) or 5′ ss on exon 3 splicing, two extra mutants D3-12 and D145-154 were also generated. All 15 mutants were transfected into A375 cells for splicing analysis. While two mutants D25-36 and D37-48 displayed moderate improvement in exon 3 splicing, six mutants showed reduction in exon 3 inclusion (Figures 1d and e). The percentage of exon 3 inclusion for mutant D145-156 dropped sharply to 1%, reflecting disruption of the 5ʹ ss of intron 3 due to loss of the last dinucleotide AG in the exon. Exon 3 splicing also drastically deteriorated in mutants D133-144 and D13-24 with, respectively, 65% and 26% decrease in exon 3 inclusion. These data suggest the potential existence of a moderate ESE near the 5′ end of exon 3, here referred to as ESE3a, and a strong one near the 3′ end of the exon, here referred to as ESE3b.
Defining the key sequence of ESE3b and its cognate binding proteins
Strong ESEs usually represent ideal targets for ASO therapeutics. Therefore, we characterized the putative ESE3b via mutagenesis within the deletion region of D133-144. Six mutants with 3- or 6-nt deletions were generated and assayed in A375 cells. Compared to the wild-type (WT) minigene, deletion of TCA at positions 133–135 caused a 30% reduction in exon 3 inclusion and deletion of TCT at positions 135–138 gave a 27% decrease; combinatorial deletion of the six nucleotides resulted in a robust 58% decrease. No significant changes were observed for mutants with deletions at positions 139–144. This data demonstrate that the sequence UCAUCU at positions 133–138 in exon 3 is the core of ESE3b.
To further understand the hexamer (UCAUCU) in regulating PDCD1 exon 3 splicing, we conducted a comprehensive point-mutation analysis within the motif. The base of each of the six nucleotides was mutated to all other three bases, respectively, and a total of 18 mutants were obtained (Figure 2d). Splicing was analysed in A375 cells. We observed complex effects. Eight mutations (T133A, T133C, C134A, C134T, C134G, A135T, C135G, and T136A) caused no or little decrease in exon 3 inclusion, while 10 mutations (T133G, A135C, T136C, T136G, C137A, C137T, C137G, T138A, T138C, and T138G) inhibited exon 3 inclusion. The greatest decrease in exon 3 inclusion (by 39%) came from T138G. All mutations at positions 137 and 138 suppressed exon 3 splicing, suggesting that the last dinucleotide CT is key in the hexamer motif.
Figure 2.
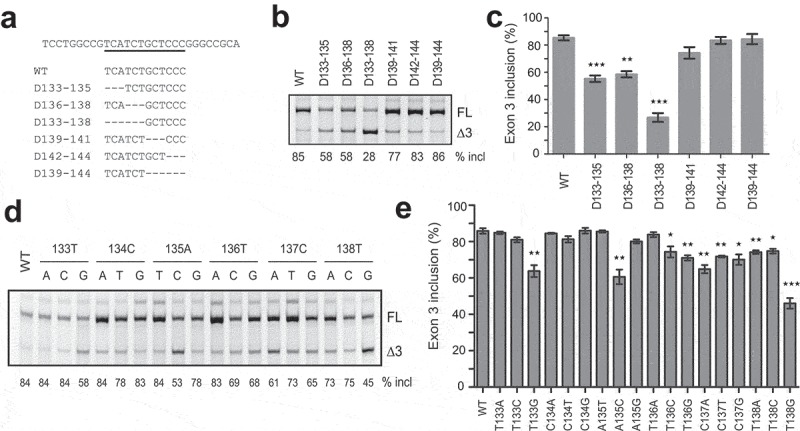
Identification of the key sequence of ESE3b via mutagenesis analysis. (a) Sequences around ESE3b in the WT minigene and deletion mutants. (b) Splicing analysis of mutant minigenes in panel A. Each plasmid (500 ng) was transfected into A375 cells. Total RNA was collected 30 h later and analysed by CY5-labelled RT-PCR. (c) Quantitation of splicing data for WT and mutant minigenes (n = 3). (d) Point-mutation analysis of the hexameric motif UCAUCU on PDCD1 exon 3 splicing in A375 cells. Each nucleotide was mutated into the other three nucleotide types. (e) Quantitation of splicing data for WT and mutant minigenes in panel D (n = 3). *P< 0.05, **P< 0.01, ***P< 0.001 versus WT.
We also tested several key minigene mutants in HeLa and HEK293 cells. The respective changes in the percentage of PDCD1 exon 3 inclusion caused by these mutations in HeLa cells were comparable to what we observed in A375 cells. Although splicing was much less efficient in HEK293 cells, the trend of change in exon 3 splicing caused by each mutation was highly similar among the three tested cell types, indicating that the mechanisms underlying the splicing changes induced by these mutations are conserved in these cell types (Supplementary Fig. S2).
SREs are often sites recognized by specific RNA-binding proteins. To find out if a RNA-binding protein may bind to the motif and exert its splicing-stimulatory activity, we performed RNA-affinity chromatography. A 16-mer biotinylated RNA sequence from position 129 to 144 in exon 3 and a negative control RNA that contains U138G mutation were obtained commercially and immobilized onto streptavidin beads (Figure 3a). After incubation of the two RNAs with HeLa cell nuclear extract under splicing conditions, the beads were extensively washed with buffer containing 100 mM KCl, and proteins that remained bound to the RNAs were analysed by SDS-PAGE and Coomassie-Blue staining (Figure 3b). Bands with differential appearance were excised from gels and analysed by mass spectrometry. Five RNA binding proteins with PSMs > 19 were caught: hnRNP UL1, Matrin 3 (MATR3), RBM14, hnRNP M and DHX15. MATR3 caught our attention as its optimal binding sequence (CAUCUU or AAUCUU) identified by a systematic method called RNAcomplete resembles the ESE3b region (UCAUCU) [23]. Using western blotting we confirmed that the binding of MATR3 to U138G RNA was much weaker than to WT RNA (Figure 3c). MATR3 is a 125 kDa nuclear matrix protein and has been recently reported as a broad AS regulator [24]. Downregulation of MATR3 indeed inhibited PDCD1 exon 3 splicing (Figure 3d), whereas other four proteins had little or no effect (Supplementary Fig. S3). Furthermore, we generated a construct in pCGT7 vector expressing T7 tagged MATR3 (T7-MATR3), which pronouncedly promoted PDCD1 exon 3 splicing (Figure 3e). We next created a mutant (Ins T) by inserting a T between positions 138 and 139 in exon 3 of the WT minigene to create the optimal motif CAUCUU. Another mutant Ins/T138G was also created by inserting a T into to the same spot in mutant T138G exon 3; the new RNA sequence CAUCGU is theoretically a better motif for MATR3 binding. Indeed both mutants displayed much better exon 3 splicing than their parental minigenes (Figure 3f). Our data suggest that MATR3 promotes PDCD1 exon 3 splicing through binding to ESE3b.
Figure 3.
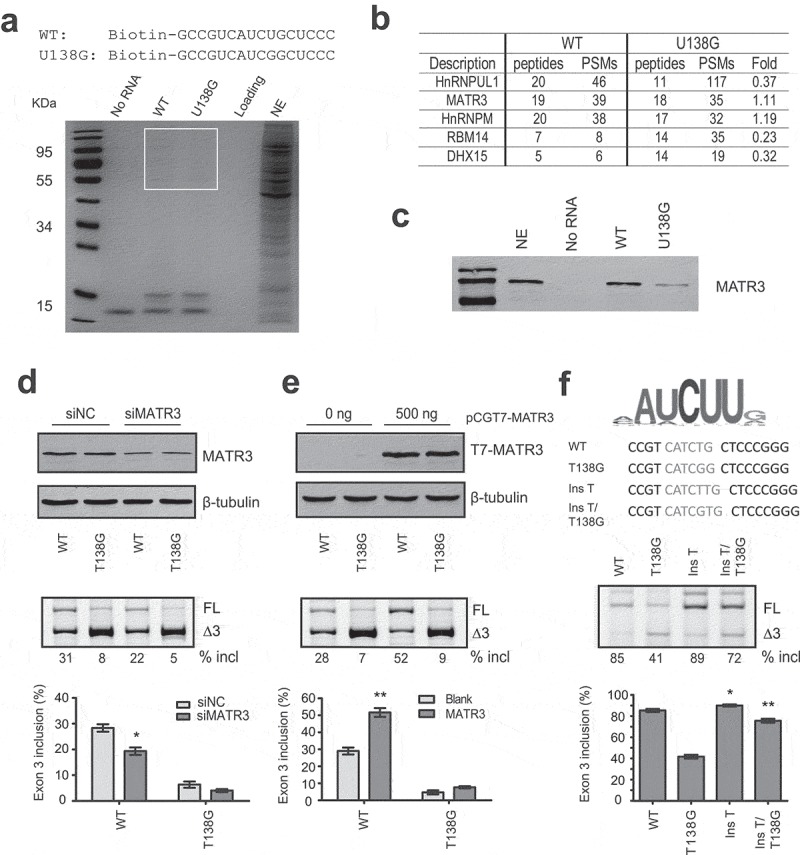
MATR3 binds to ESE3b and promotes PDCD1 exon 3 inclusion. (a) Sequences of WT and U138G biotinylated RNAs and Coomassie-Blue staining of protein samples obtained from RNA-affinity chromatography. Streptavidin beads covalently linked to the two RNAs were incubated with HeLa cell nuclear extract (NE) under splicing conditions, and the beads were then extensively washed with buffer containing 100 mM KCl. Captured proteins were eluted with Laemmli buffer, separated by SDS-PAGE and stained with Coomassie Blue. A reaction without RNA (No RNA) was used as a negative control. Sample without beads was shown as loading (the second lane from the right); 20 μl NE was loaded on the most right lane. Protein bands around ~10 kDa were streptavidin. Protein bands around 50–110 kDa were prominent on the gel and some show differential signal intensities between WT and U138G samples. Bands in boxed area were excised from the gel and subjected to mass spectrometry. (b) Mass spectrometry identified multiple proteins bound to the two RNA species with five RNA-binding proteins having PSMs over 19. (c) Western blot analysis of the eluted proteins with an anti-MATR3 antibody. 7.5% input of NE was loaded on the second left lane. (d) The effect of siRNA knockdown of MATR3 (siMATR3) on PDCD1 exon 3 splicing in the WT and T138G minigenes assayed in HEK293 cells (n = 3). (e) Overexpression of T7-MATR3 on WT and T138G PDCD1 exon 3 splicing assayed in HEK293 cells (n = 3). (f) Improvement of the MATR3 motif at ESE3b promotes exon 3 inclusion as assayed in A375 cells. For panels d-f, quantitation of exon 3 inclusion was shown below from three independent experiments. *P< 0.05, **P< 0.01 versus controls or parental plasmids.
RRM1 and RRM2 mediate splicing activation activity by MATR3
MATR3 is of 847 amino acids, comprising multiple domains including an N-terminal nuclear export signal, two zinc finger (ZF) domains, a PRI (PTB-RRM interaction) motif, two RNA recognition motifs (RRMs) and a C-terminal bi-partite nuclear localization signal (NLS). To identify which domains of MATR3 are required for promoting splicing of PDCD1 exon 3, a series of MATR3 deletion mutants were generated using the parental T7-MATR3-expressing plasmid (Figure 4a). All mutants retained the C-terminal NLS to ensure all proteins being localized in the nucleus. We examined the RNA-binding ability of two T7-tagged mutants (ΔRRMs and ΔC) by RNA-affinity chromatography (Supplementary Fig. S4). Indeed, T7-ΔRRMs that lacks two RRMs lost its ability to bind to the 16-mer WT RNA (see Figure 3a), whereas T7-ΔC, similar to WT MATR3, retained the binding ability.
Figure 4.
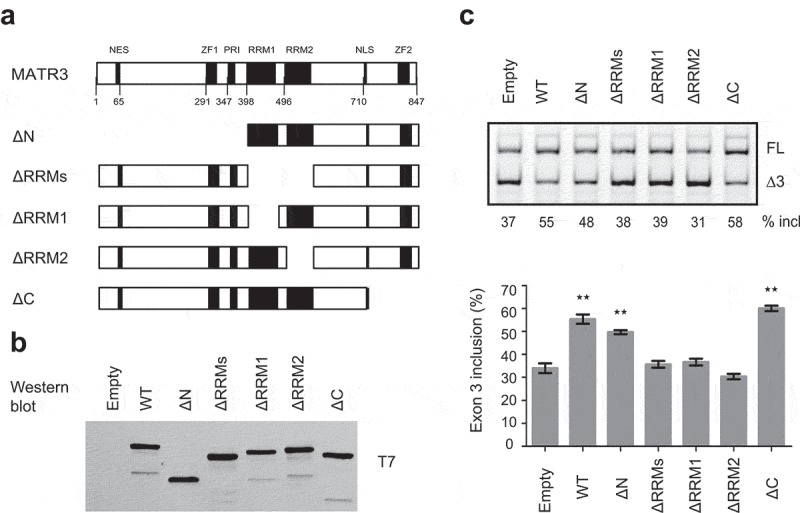
MATR3 promotes exon 3 inclusion through its RRM domain. (a) Diagram of the primary structure of MATR3 and its mutants. Mutant ΔN lacks the first 397 amino acids at the N terminus; mutant ΔRRMs lacks both RRMs (amino acids 398–571 being deleted), mutant ΔRRM1 lacks RRM1 (amino acids 398–473 being deleted), ΔRRM2 lacks RRM2 (amino acids 496–571 being deleted), and ΔC lacks 129 amino acids at the C terminus (from 719 to 847). (b) Western blotting using anti-T7 antibody shows proper expression of all mutants. (c) The effects of MATR3 mutants on PDCD1 minigene exon 3 splicing were analysed in HEK293 cells; Quantitation of exon 3 inclusion from three independent experiments is shown in the histogram below. **P< 0.01, versus T7-empty vector (n = 3).
We next co-transfected each deletion plasmid (500 ng) with the plasmid expressing PDCD1 minigene into A375 cells, and both protein expression of the mutants and their effects on exon 3 splicing were analysed. Empty vector and the WT protein were used as controls. Interestingly, the mutant with deletion of the first 398 amino acids at the N terminus including the NES, ZF1 and PRI (mutant ΔN) still pronouncedly promoted exon 3 splicing, though its effect was slightly less than that by the WT protein. C-terminal deletion of 129 amino acids including ZF2 had no effect on exon 3 splicing. However, deletion of either RRM1 or RRM2 or both resulted in complete loss of the splicing-stimulatory effect by MATR3 (Figure 4c), Our data indicate that RRM1 and RRM2 are essential for the splicing activation by MATR3, whereas others including PRI and DNA-binding C2H2 ZF domains are not required.
An RNA secondary structure around ESE3b affects PDCD1 exon 3 splicing
Although MATR3 modulates PDCD1 exon 3 splicing, its effect is moderate, suggesting other factors may be involved. Predicted by UNAFold [25], a 32 nt-long sequence in exon 3 from position 117 to position 148 forms a potential stem-loop structure that contain a main loop and a small side loop (Figure 5). The structure here is referred to as SLE3. Mutation U138G, which not only compromises recognition of ESE3b by MATR3 but also strengthens the putative RNA secondary structure by adding two more pairs into the stem, robustly inhibited exon 3 splicing, whereas mutations T138A and T138C, which have little effect on SLE3 despite compromising MATR3 recognition, moderately inhibited exon 3 splicing (Figure 2d), supporting the notion that SLE3 suppresses PDCD1 exon 3 recognition. To further verify this notion, we analysed more mutations at position 131 or 137, or combinations of mutations at different positions in the context of the WT or T138G minigene (Figure 5a). We also generated two mutants CT>AG and CT>GG, which restore the 131–138 pair but destroy the 132–137 pair and thus moderately stabilize the RNA structure. WT and mutant minigenes were transfected into A375 cells and splicing was analysed by fluorescence-labelled RT-PCR (Figure 5b). We observed a clear correlation between the free energy of the RNA structure and the percentage of exon 3 inclusion during pre-mRNA splicing (Figure 5c).
Figure 5.
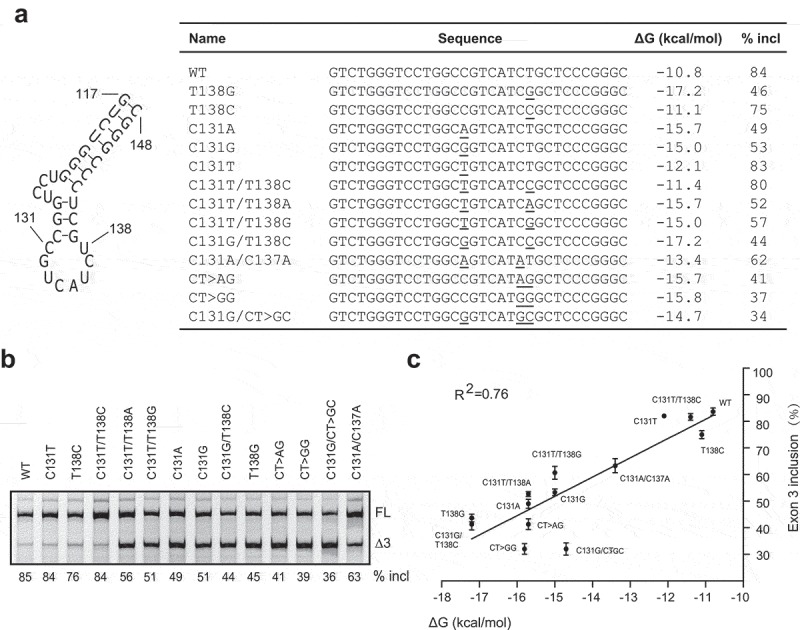
A putative RNA secondary structure located around ESE3b inhibits PDCD1 exon 3 splicing. (a) An RNA secondary structure is predicted by UNAFold in exon 3 from positions 117 to 148. For all mutants assayed, sequences at the structure region, predicted minimum free energies (ΔG, Kcal/mol) and percentages of exon 3 inclusion were listed. (b) Splicing of WT and mutant minigenes were analysed in 375 cells. The percentage of exon 3 inclusion for each minigene is shown below the gel. (c) The percentage of exon 3 inclusion of each mutation is presented as a scatter plot against the calculated minimum free energy. A strong correlation was observed by SPSS Pearson analysis (R2 = 0.76).
We also performed splicing analysis by mutating the small stem at positions 128–130 and 139–141 in exon 3 located between the side loop and main loop in the predicted RNA secondary structure. All possible 18 single-nucleotide mutations were generated and analysed in A375 cells (Supplementary Fig. S5). The alterations in the RNA structure around ESE3b for all mutants can be classified into three groups (Supplementary Fig. S6). Five mutants (G128A, C130G, T141A, T141C and T141G) in group 1 have a similar predicted RNA structure to that of the WT minigene. Four mutations (G128T, G128C, G129T and G129C) in group 2 destroy the middle small stem, resulting in the formation of a much bigger loop. A strong correlation between the percentage of exon 3 inclusion and the minimal free energy was observed for mutants in groups 1 and 2 (R2 = 0.61). Mutants in group 3 have major re-arrangements of the side loop, the middle stem and the main loop which should have complex effects on exon 3 splicing.
DDX5 represses PDCD1 exon 3 splicing
DDX5 is a nuclear RNA helicase. It has been shown to regulate AS through binding to GC-rich sequences, particularly at stem-loop regions [26]. The above secondary structure at the ESE3b region in PDCD1 exon 3 contains 72% GC. We hypothesized that DDX5 may bind to SLE3 and modulate exon 3 splicing owing to its vicinity to the 5ʹ splice site. To test this notion, we constructed a plasmid expressing T7-tagged DDX5 (T7-DDX5). The plasmid was co-transfected with WT, T138G or C131A/C137A minigene plasmids into A375 cells. The two mutants have a more stable stem-loop structure than the WT minigene at the ESE3b region (Figure 5a), and thus a comparison between them can tell whether the putative RNA secondary structure is involved. RNA samples were collected 30 h post-transfection for splicing analysis. Surprisingly, DDX5 inhibited exon 3 inclusion for all minigenes; and the effect was much stronger for the two mutants (43% and 49% decrease, respectively) than that for the WT minigene (25% decrease, Figure 6a). We next assessed the effect of DDX5 depletion on PDCD1 exon 3 splicing. HEK293 cells were chosen because the percentage of exon 3 inclusion for the WT PDCD1 minigene in these cells is ~22% and thus a splicing-stimulatory effect can be readily noted. Plasmids expressing WT, T138G and C131A/C137A minigenes were co-transfected with two siRNAs, respectively, and cells were collected 36 h post-transfection to analyse DDX5 levels as well as exon 3 splicing changes. Despite only ~50% reduction in DDX5 levels upon siRNA treatment, a robust increase of exon 3 inclusion was observed for both mutants (5.8-fold and 7.7-fold increase, respectively); in contrast, a 1.5-fold increase was observed for the WT minigene (Figure 6b). Both DDX5-overexpression and siRNA knockdown data suggest that DDX5 is a PDCD1 exon 3 splicing repressor. We next tested if the ATPase/helicase activity of DDX5 is required for its splicing repression. A previously described mutant R403L that lacks both ATPase and RNA unwinding activities was constructed in pCGT7 [27]. As shown in Figure 6a, the T7-tagged DDX5 mutant, though properly expressed, was completely inactive in suppressing PDCD1 exon 3 inclusion. The data suggest that the splicing inhibitory activity of DDX5 relies on its ATPase/helicase activity.
Figure 6.
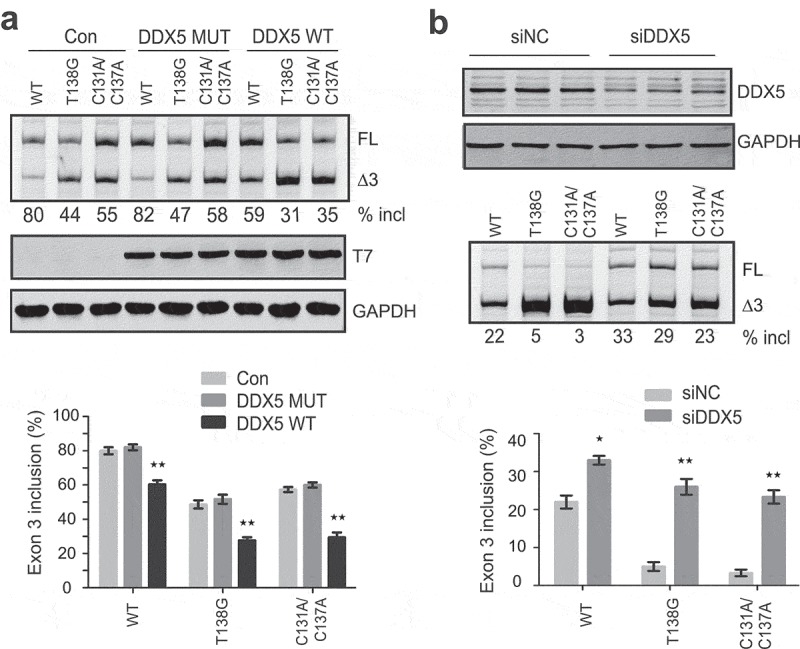
Effects of DDX5 overexpression and knockdown on PDCD1 exon 3 splicing. (a) Overexpression of T7-DDX5 (DDX5 WT) markedly inhibits PDCD1 exon 3 inclusion in 375 cells, whereas R403L mutation (DDX5 MUT) that abolishes ATPase and RNA unwinding activities lost the effect. Western blotting using an anti-T7 antibody confirmed proper expression of DDX5 WT and DDX MUT. The splicing inhibitory effect by DDX5 was more drastic for two mutants T138G and C131A/C137A (1.7- and 2.0- folds, respectively) with more stable RNA structures (see Figure 5) than that for the WT minigene. (b) Depletion of DDX5 with siRNA robustly promotes PDCD1 exon 3 splicing. 100 nM siRNA (siDDX5) or control (siNC) was co-transfected into HEK293 cells with each minigene plasmid (WT, T138G or C131A/C137A). DDX5 was detected with a polyclonal anti-DDX5 antibody with GAPDH as a loading control. The stimulatory effect of DDX5 depletion is more drastic for mutants T138G and C131A/C137A, consistent with the DDX5 overexpression data. The histograms on the bottom show the quantitation of exon 3 inclusion from three independent experiments. **P< 0.01 compared to no plasmid (n = 3); *P< 0.05 compared to si NC (n = 3).
A screen of ASOs that inhibit PDCD1 exon 3 inclusion
ASO-based splice switching technology has recently emerged as an attractive platform for new drug development. Using ASOs to promote PDCD1 exon 3 skipping and thus create a dysfunctional PD-1Δ3 protein isoform may represent a new approach to treat cancer. To identify ASOs that efficiently promote PDCD1 exon 3, we employed an ASO walk along the entire exon with the first and last ASOs covering a small part of flanking introns (Figure 7a). All 16 oligonucleotides, 15-nt long, were modified with a MOE ribose and phosphorothioate backbone. Two adjacent ASOs overlap by 5 nt. An oligonucleotide with the same chemistry but an unrelated sequence was used as a negative control. To test the effectiveness of these ASOs, we co-transfected each ASO individually with the PDCD1 minigene plasmid into A375 cells, and the splicing pattern of expressed RNAs was assayed by fluorescence-labelled RT-PCR. ASO3 and ASO4, both targeting the ESE3a region, drastically inhibited exon 3 splicing, while other ASOs and controls displayed no or negligible effects on exon 3 splicing (Figure 7b). The fact that ASOs targeting ESE3b had no effects on exon 3 splicing is not surprising as we showed above the presence of SLE3 in the region, which should prevent annealing of ASOs to their targets. To support the notion that the absence of effects of ASOs targeting ESE3b is due to SLE3, we performed an ASO study using PDCD1 minigene mutant G128C, which possesses a big loop in the RNA structure with ESE3b being well exposed in the single-strand RNA (ssRNA) region. Indeed, ASO14 and ASO14b that target ESE3b markedly repressed exon 3 inclusion in the mutant but not the WT minigene, whereas ASO13 that does not target the ESE3b sequence had no effects on exon 3 splicing in both the WT minigene and the mutant. (Supplementary Fig. S7).
Figure 7.
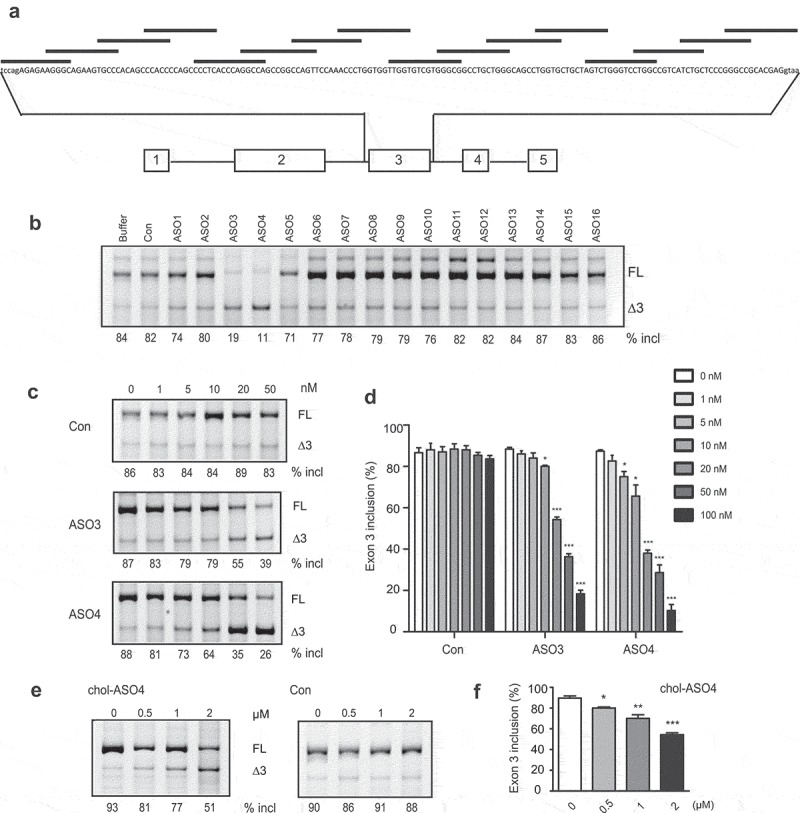
A screen of ASOs that inhibit PDCD1 exon 3 inclusion. (a) Schematic representation of ASOs used in the ASO walk along PDCD1 exon 3. (b) The effects of ASOs were examined with the PDCD1 minigene. Each ASO at 100 nM was co-transfected with the minigene into A375 cells. Buffer and a nonrelated ASO (con) were used as negative controls. (c) Both ASO3 and ASO4 show dose-dependent effects on PDCD1 exon 3 splicing. Each ASO at six different concentrations was co-transfected with the PDCD1 miniegene into A375 cells and spliced products were analysed by Cy5-labelled RT-PCR. (d) Quantitation of the effects of ASO3 and ASO4 at a series of concentrations on PDCD1 exon 3 splicing in the minigene context. (e) Cholesterol-conjugated ASO4 (cholASO4) at four indicated concentrations was examined with transcripts expressed from the endogenous PDCD1 in 8226 cells. Cells were collected 48 h post-ASO treatment and splicing were analysed by Cy5-labelled RT-PCR. (f) Quantitation of the effect of cholASO4 on the endogenous gene in 8226 cells. For panels d and f, n = 3, *P < 0.05, **P < 0.01, ***P < 0.001 versus 0 nM or 0 μM.
Robust decrease in exon 3 inclusion by ASO3 (by 65%) and ASO4 (by 73%) suggest the two ASOs have therapeutic potential. To further characterize the two ASOs, we conducted a dose-response study for each with six concentrations ranging from 1 to 100 nM. A marked inhibition of exon 3 splicing by ASO4 was observed at 10 nM (Figure 7c), which is lower than the minimal concentration required for ASO3 to efficiently work. We next asked if the effects of ASOs on splicing of minigene pre-mRNA are conserved for the endogenous gene. Immune cells, the main cell types that express PDCD1, are refractory to ASO transfection. To circumvent this, we attached a cholesterol molecule to the 3ʹ end of ASO4 so that it can be spontaneously taken up by suspended immune cells. Conjugation of cholesterol has been applied to siRNAs to achieve efficient cellular internalization in vitro and in vivo [28,29]. Cholesterol-conjugated ASO4, here referred to as cholASO4, was added into medium culturing RPMI 8226 cells at a series of concentrations; 48 h post-treatment, cells were collected for splicing analysis. We observed a clear dose-dependent deterioration in exon 3 splicing for transcripts expressed from the endogenous PDCD1 gene upon addition of cholASO4, and treatment with 2 μM decreased exon 3 inclusion from 90% to 54% (Figure 7f).
Discussion
PD-1 is a key inhibitory immune checkpoint protein and an important target for immune therapy against cancer. Understanding how its expression is regulated helps design approaches for PD-1 targeted therapies. Prior studies have shown that PDCD1 is alternatively spliced [10]; particularly, AS of exon 3 appears to create a protein isoform PD-1Δ3 that may antagonize PD-1 [11,12]. However, the mechanisms underlying the AS event have not been explored. In this study, using a minigene system we confirmed exon 3 skipping as the main AS event of PDCD1. Further deletion analysis identified two ESEs (ESE3a and ESE3b) in the exon. Using RNA-affinity chromatography, mass spectrometry, mutagenesis analysis of the PDCD1 minigene as well as overexpression and siRNA knockdown of MATR3, we uncovered a splicing activator, MATR3, which mediates the splicing enhancer activity of ESE3b by binding to a specific hexamer motif UCAUCU. Moreover, we found that a GC-rich RNA secondary structure present around the ESE3b region inhibits exon 3 splicing. DDX5, which has been reported to affect splicing via binding to GC-rich motifs and RNA secondary structures, markedly inhibits PDCD1 exon 3 splicing. Our data indicate that PDCD1 exon 3 splicing is under complex regulation, involving multiple factors. Finally, using an ASO walk in the minigene system, we identified two ASOs that efficiently promote PDCD1 exon 3 skipping. The strong splicing inhibition by the ASOs was also observed in an endogenous context in immune cells, suggesting their therapeutic potential.
Generally, SREs play important roles in determining the extent of inclusion for alternative spliced cassette exons with weak splice sites. Our data indicate auxiliary splicing signals in PDCD1 exon 3 significantly contribute to efficient recognition of the exon by the spliceosome. We focused on characterization of ESE3b since it appears to be a stronger enhancer than ESE3a in light of the initial deletion assay (Figure 1). Although multiple RNA-binding proteins were identified abundantly bound to the ESE3b region using RNA affinity chromatography coupled with mass spectrometry, only MATR3 was found to modulate PDCD1 exon 3 splicing. MATR3 is an RNA/DNA binding protein, mutations of which have been implicated in familial amyotrophic lateral sclerosis [30]. Recently, Coelho et al. identified 667 AS events in HeLa cells that are regulated by MATR3 [24], suggesting that the protein is a broad splicing regulator. Among the 667 AS events, nearly half were alternative cassette exons; MATR3 inhibits inclusion of 75% of them via PTB-dependent or -independent mechanisms while promotes inclusion of the rest. One mechanism proposed for MATR3 as an activator in the case of ABI2 exon 8 is to counteract PTB, a splicing suppressor that interacts with MATR3 via the RPI domain [24]. Surprisingly, we found that the stimulatory activity of MATR3 on PDCD1 exon 3 splicing is independent of PTB as deletion of PRI had no effect on its activity (Figure 4), indicating that MATR can adopt different mechanisms to activate splicing. In addition, zinc finger motifs are not required for MATR3’s activity in promoting PDCD1 exon 3 inclusion based on the MATR3 deletion analysis; this rules out the possibility that MATR3 functions at the epigenetic and transcriptional levels. As RRM1 and RRM2 are the only two domains essential for the splicing-stimulatory effect, MATR3 is likely to directly bind to the ESE3b motif through the two RRMs and serve as a steric blockage, which is similar to a mechanism proposed for HuR in its inhibition on SMN2 exon 7 splicing in our previous study [22]. Compared to the optimal MATR3 motif identified by a systemic RNAcompete analysis [23], the ESE3b motif is suboptimal, which explains why the effect of MATR3 on PDCD1 splicing is moderate. Indeed, two mutations that improve this MATR3 motif further enhanced PDCD1 exon 3 inclusion (Figure 3f).
Of note, the two MATR3 motif-improving mutations were cautiously designed to avoid significant changes in the predicted RNA secondary structure (SLE3) formed around ESE3b. It is well-established that RNA structure profoundly affects pre-mRNA splicing [31]. For example, an RNA structure termed TSL2 at the 5ʹ ss of SMN2 intron 7 impairs recognition of the spice site and thus inhibits exon 7 splicing [32]. Due to high GC content, SLE3 formed around ESE3b in PDCD1 exon 3 is highly stable. Our mutagenesis data demonstrates that the stability of SLE3 is negatively proportional to the percentage of exon 3 inclusion during PDCD1 pre-mRNA splicing, which is consistent with an early systematic splice-site analysis showing that high GC content near splice sites is often associated with RNA secondary structures that lead to alternatively spliced exons [33]. Presumably, SLE3, 8 nt away from the downstream exon-intron junction, blocks access of some key splicing factors that only recognize single-strand RNA and thus impairs recognition of the 5ʹ ss by the spliceosome.
ESE3b, the binding site for MATR3, is located in the main loop of the GC-rich SLE3. A recent genome-wide study using enhanced cross-linking and immunoprecipitation, called eCLIP, mapped DDX5 binding sites to GC-rich sequences near ssRNA or bulged RNA regions adjacent to dsRNA regions [26]. Therefore, MATR3 and DDX5 may compete to bind to ESE3b; in this case, the role of MATR3 is presumably to occlude DDX5 binding to ESE3b. This explains why RRM1 and RRM2 are sufficient to mimic the effect of the full-length protein. However, we could not rule out an active role of MATR3 in promoting PDCD1 exon 3 splicing. DDX5 is an ATP-dependent DEAD-box RNA helicase, functioning in various aspects of RNA metabolism including RNA biogenesis, splicing, transport and stability [34]. Lee et al. recently reported that DDX5 controls thousands of AS events in human K562 cells, much more than those regulated by hnRNP A1 [26]. One mechanism proposed for modulation of splicing by DDX5 is unwinding of the transient U1−5ʹ ss duplex [35]. However, the fact that only certain specific AS events are influenced by DDX5 suggests that its helicase activity is in general not essential or its effect is context-dependent. Here we show that DDX5 inhibits PDCD1 exon 3 splicing and the effect requires its ATPase/helicase activities. As mutants that carry a more stable SLE3 appeared more sensitive in response to DDX5 levels (Figure 6b), the splicing repression by DDX5 may involve the RNA stem-loop structure. Two prior studies have explored RNA structure-mediated splicing modulation by DDX5. DDX5 promotes MAPT exon 10 splicing via binding to a stem-loop structure downstream of the 5ʹ ss of the exon, which unwinds the structure and increases U1 snRNP annealing to the ss [36]; however, this stimulatory effect by DDX5 is dependent on a splicing factor RBM4. On the other hand, DDX5 inhibits inclusion of the alternative intron D exon (IDX) of the H-ras gene via interacting with a long stem-loop structure formed between IDX and a downstream ISS [37]. This inhibitory effect by DDX5 may involve a competition between DDX5 and hnRNP H in binding to the ISS [38]. Therefore, the consequence of DDX5 binding to an inhibitory RNA stem-loop structure largely depends on its interacting proteins, which is consistent with a recent report suggesting that splicing regulation operates in the context of a large assembly of splicing regulations (LASR) [39]. Interestingly, both MATR3 and DDX5 have been found to be components in LASR pelleted by Rbfox proteins in HEK293 cells as well as mouse brain [39], suggesting that the two proteins may often act together to modulate AS events.
A strong line of evidence that supports the presence of SLE3 is our ASO screening, in which all ESE3b-targeting ASOs were ineffective in inhibiting PDCD1 exon 3 splicing, suggesting inaccessibility of these ASOs to their complementary sequences owing to high stability of SLE3. Instead, both ESE3a-targeting ASOs, ASO3 and ASO4, displayed robust activity in suppressing PDCD1 exon 3 splicing in both minigene and endogenous-gene contexts. The mechanism of ESE3a promoting splicing is currently under investigation. However, the sequence from position 16 to position 40 targeted by ASO3 and ASO4 contains 64% Cs, suggesting that an RNA-binding protein favouring C-rich sequences may attach to the site and exerts its splicing-stimulatory activity. Further analysis of the action and interplay between all the cis-splicing signals, their cognate proteins and the secondary structure are required to better understand the regulatory landscape of the AS event.
Splice-switching ASOs have been lately proven to be an attractive new class of therapeutic modalities. Although major efforts for PD-1/PD-L1 targeted therapies focuses on developing antibodies, the fact that PDCD1 exon 3 is alternatively spliced presents an opportunity to manipulate PD-1 signalling through re-directing pre-mRNA splicing by ASOs, which may represent an alternate or even better approach to treat cancer, particularly considering that PD-1Δ3 still retains the ability to bind to PD-L1/PD-L2. Indeed, using a series of 15-mer ASOs in an antisense walk every 10 nt along a sequence covering the entire exon 3, we identified two ASOs that efficiently promote skipping of the exon. Particularly, treatment with cholASO4 at 2 μM resulted in a 40% reduction in PDCD1 exon 3 inclusion in cultured B cells. Note, ASO3 and ASO4 were hits from the initial walk and only 15-nt long. The ASO walk method, as we described previously [40], involves two steps: an initial coarse walk aiming to identify target regions, followed by a thorough microwalk in the promising regions aiming to optimize and identify lead ASOs. Therefore, there is much room for improvement. Further optimization using ASO microwalk of different lengths to fine-tune the targeting sequences is warranted to obtain a much more potent ASO. Although, several key questions remain to be addressed (for example, what percentage of total PD-1Δ3 is released from cells into blood and local tissues? And how it may counteract PD-1?), our data hold promise for development of a splice-switching ASO drug targeting PD-1 signalling.
Materials and methods
Plasmids
The PDCD1 minigene comprising of the 76-nt coding sequence of exon 1, a truncated 355-nt intron 1, the 360-nt exon 2, 267-nt intron 2, 156-nt exon 3, 214-nt intron 3, 35-nt exon 4, 651-nt intron 4 and a truncated 59-nt exon 5, followed by a consensus 5′ ss (CAGGTAAGTACTT) was amplified from human genomic DNA and constructed into PCI-neo vector using restriction enzymes NheI, XhoI and XbaI (New England Biolabs, Ipswich, MA, USA). Minigene mutants were created by site-specific mutagenesis with partially over-lapping primer pairs. Protein-expression plasmids were generated with vector pCGT7 using either the XbaI/KpnI (New England Biolabs) restriction sites (pCGT7-DDX5) or the SLIC method (pCGT7-MATR3) [41]. Protein deletion or point-mutation mutant were generated in their parental plasmids using the SLIC method or site-specific mutagenesis. All expressed proteins have an N-terminal T7 tag. Primer pairs used for cloning and qRT-PCR are listed in Supplementary Table 1.
Cell culture and transfections
A375, C8161, HEK293, HeLa, U87MG and HepG2 cells were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA, USA), RPMI 8226 cells were cultured in 1640 medium (Invitrogen), all supplemented with 10% (v/v) foetal bovine serum (FBS) and antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin) at 37 °C in a humidified 5% CO2 atmosphere. For splicing analysis of PDCD1 WT and mutant minigenes, 105 cells per well were seeded in 12-well plates in DMEM with 10% FBS; the next day, 500 ng of each minigene plasmid together with or without 500 ng of each protein-expression plasmid was delivered to cells using branched polyethylenimine reagent (Sigma-Aldrich, St. Louis, MO, USA). siRNA duplexes were purchased from Genepharma (Shanghai, China): si-MATR3 (sense: 5ʹ-AAAGACUUCCAUGGACUCUUA-3) for MATR3 knockdown, si-DDX5 (sense: 5ʹ-AACUCUAAUGUGGAGUGCGAC-3ʹ) for DDX5 knockdown, and siNC (sense: 5ʹ-UUCUCCGAACGUGUCACGUTT-3ʹ) as a negative control. All 2ʹ-O-methoxyethyl-modified oligonucleotides with a phosphodiester backbone including cholesterol-conjugated ASOs were purchased from Biosyntech (Suzhou, Jiangsu, China). siRNAs and ASOs were co-transfected with minigenes using Lipofectamine2000 (Life Technologies, Carlsbad, CA, USA). For transfection of cholesterol-conjugated ASOs, 5 × 105 RPMI 8226 cells were seeded in 1640 medium supplemented with reduced FBS (3%); the next day, the ASOs were added in medium for 48 h.
Fluorescence-labelled RT-PCR
Cells were harvested 30-h post-transfection, and total RNA was isolated with TRizol reagent (Vazyme, Nanjing, Jiangsu, China); 1 μg of each RNA sample was used per 20 μl reaction for first-strand cDNA synthesis with oligo (dT)18 and M-MLV reverse transcriptase (Vazyme). For minigenes, splicing products were amplified semi-quantitatively using 27 cycles (95ºC for 15 s, 58ºC for 15 s, and 72ºC for 35 s) with forward primer PDCD1-F (5ʹ- GCGGCCAGGATGGTTCTTA-3ʹ) and Cy5-labelled reverse primer Cy5-PDCD1-R (5ʹ-Cy5-AGAGAACACAGGCACGGCTGAG −3ʹ). For endogenous PDCD1, we used 33 cycles with Cy5-labelled PDCD1-F and Cy5-PDCD1-R to amplify cDNA. Cy5-labelled PCR products were separated on 6% native polyacrylamide gels, followed by fluorescence imaging with G:BOX Chem XL (Syngene, Cambridge, UK); signals were quantitated by image J software and exon 3 inclusion was expressed as a percentage of the total amount of spliced transcripts.
RNA pull-down
WT or U138G biotin-labelled RNA oligonucleotides were purchased from Genepharma. Briefly, following washing with washing buffer (20 mM Tris–HCl, pH 7.5, 100 mM KCl, 2 mM EDTA, 0.5 mM DTT, and 0.5 mM PMSF) for three times, 5μl streptavidin agarose beads (Sigma-Aldrich) were incubated 100 pmol with 5ʹ-biotinylated RNA for 2 h at 4°C under rotation. The beads were then rinsed with washing buffer to remove unbound RNAs. After changing washing buffer with interaction buffer (20 mM Tris–HCl, pH 7.5, 300 mM KCl, 0.2 mM EDTA, 0.5 mM DTT, and 0.5 mM PMSF), HeLa nuclear extract was added into the mixture and rotated at 4°C overnight. The beads were washed five times with interaction buffer and the proteins bound to the immobilized RNA were eluted by Laemmli buffer and heated at 90°C for 10 min.
Western blotting
Protein samples separated by 10% SDS-PAGE were electroblotted onto PVDF membranes (Millipore, Bedford, MA, USA). The blots were then probed with primary mAbs or polyclonal antibodies (pAbs), followed by secondary IRDye@ 680RD goat anti-mouse or goat anti-rabbit antibody (LI-COR Biosciences, Lincoln, NE, USA). Anti-T7 mAb was a gift from Professor Adrian Krainer at Cold Spring Harbour Laboratory; anti-GAPDH and anti-β-tubulin mAbs were purchased from Santa Cruz Biotechnology (Dallas, TX, USA); anti-DDX5 and anti-Matrin3 pAbs were purchased from ProteinTech (Wuhan, Hubei, China). Protein signals were detected with an Odyssey Infrared Imaging System (LI-COR Biosciences).
Statistical analysis
Data from three independent experiments are presented as mean ± standard deviation. Statistical significance was analysed by Student’s t-test and one-way ANOVA with software SPSS 16.0.
Funding Statement
This work was supported by the National Natural Science Foundation of China (grant numbers 81530035, 81471298 and 81271423 to YH), Suzhou Clinical Research Centre of Neurological Disease (Szzx201503), Priority Academic Program Development of Jiangsu Higher Education Institutions, and Jiangsu Key Laboratory of Neuropsychiatric Diseases (BM2013003).
Acknowledgments
Y.H. gratefully acknowledges support from the National Natural Science Foundation of China (grants 81271423, 81471298, 81530035) and Priority Academic Program Development of Jiangsu Higher Education Institutions.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Wang ET, Sandberg R, Luo S, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Blencowe BJ. The relationship between alternative splicing and proteomic complexity. Trends Biochem Sci. 2017;42:407–408. [DOI] [PubMed] [Google Scholar]
- [3].Martinez NM, Lynch KW. Control of alternative splicing in immune responses: many regulators, many predictions, much still to learn. Immunol Rev. 2013;253:216–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. 2016;375:1767–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol. 2018;18:153–167. [DOI] [PubMed] [Google Scholar]
- [6].Sanmamed MF, Chen L. A paradigm shift in cancer immunotherapy: from enhancement to normalization. Cell. 2018;175:313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kleffel S, Posch C, Barthel SR, et al. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell. 2015;162:1242–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chen G, Kim YH, Li H, et al. PD-L1 inhibits acute and chronic pain by suppressing nociceptive neuron activity via PD-1. Nat Neurosci. 2017;20:917–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wang W, Chan A, Qin Y, et al. Programmed cell death-1 is expressed in large retinal ganglion cells and is upregulated after optic nerve crush. Exp Eye Res. 2015;140:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nielsen C, Ohm-Laursen L, Barington T, et al. Alternative splice variants of the human PD-1 gene. Cell Immunol. 2005;235:109–116. [DOI] [PubMed] [Google Scholar]
- [11].Wan B, Nie H, Liu A, et al. Aberrant regulation of synovial T cell activation by soluble costimulatory molecules in rheumatoid arthritis. J Immunol. 2006;177:8844–8850. [DOI] [PubMed] [Google Scholar]
- [12].Sorensen SF, Demuth C, Weber B, et al. Increase in soluble PD-1 is associated with prolonged survival in patients with advanced EGFR-mutated non-small cell lung cancer treated with erlotinib. Lung Cancer. 2016;100:77–84. [DOI] [PubMed] [Google Scholar]
- [13].Constantinidou A, Alifieris C, Trafalis DT. Targeting programmed cell death −1 (PD-1) and ligand (PD-L1): A new era in cancer active immunotherapy. Pharmacol Ther. 2018;194:84–106. [DOI] [PubMed] [Google Scholar]
- [14].Hua Y, Liu YH, Sahashi K, et al. Motor neuron cell-nonautonomous rescue of spinal muscular atrophy phenotypes in mild and severe transgenic mouse models. Genes Dev. 2015;29:288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hua Y, Sahashi K, Hung G, et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010;24:1634–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hua Y, Sahashi K, Rigo F, et al. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011;478:123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Passini MA, Bu J, Richards AM, et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med. 2011;3:72ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hoy SM. Nusinersen: First Global Approval. Drugs. 2017;77:473–479. [DOI] [PubMed] [Google Scholar]
- [19].Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. [DOI] [PubMed] [Google Scholar]
- [20].Lorson CL, Hahnen E, Androphy EJ, et al. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. 1999;96:6307–6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hua Y, Vickers TA, Okunola HL, et al. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82:834–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wu X, Wang SH, Sun J, et al. A-44G transition in SMN2 intron 6 protects patients with spinal muscular atrophy. Hum Mol Genet. 2017;26:2768–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ray D, Kazan H, Cook KB, et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013;499:172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Coelho MB, Attig J, Bellora N, et al. Nuclear matrix protein Matrin3 regulates alternative splicing and forms overlapping regulatory networks with PTB. Embo J. 2015;34:653–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Markham NR, Zuker M. UNAFold: software for nucleic acid folding and hybridization. Methods Mol Biol. 2008;453:3–31. [DOI] [PubMed] [Google Scholar]
- [26].Lee YJ, Wang Q, Rio DC. Coordinate regulation of alternative pre-mRNA splicing events by the human RNA chaperone proteins hnRNPA1 and DDX5. Genes Dev. 2018;32:1060–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lin C, Yang L, Yang JJ, et al. ATPase/helicase activities of p68 RNA helicase are required for pre-mRNA splicing but not for assembly of the spliceosome. Mol Cell Biol. 2005;25:7484–7493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ly S, Navaroli DM, Didiot MC, et al. Visualization of self-delivering hydrophobically modified siRNA cellular internalization. Nucleic Acids Res. 2017;45:15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Soutschek J, Akinc A, Bramlage B, et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432:173–178. [DOI] [PubMed] [Google Scholar]
- [30].Johnson JO, Pioro EP, Boehringer A, et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci. 2014;17:664–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lin CL, Taggart AJ, Fairbrother WG. RNA structure in splicing: an evolutionary perspective. RNA Biol. 2016;13:766–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Singh NN, Singh RN, Androphy EJ. Modulating role of RNA structure in alternative splicing of a critical exon in the spinal muscular atrophy genes. Nucleic Acids Res. 2007;35:371–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhang J, Kuo CC, Chen L. GC content around splice sites affects splicing through pre-mRNA secondary structures. BMC Genomics. 2011;12:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Xing Z, Ma WK, Tran EJ. The DDX5/Dbp2 subfamily of DEAD-box RNA helicases. Wiley Interdiscip Rev RNA. 2019;10:e1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Liu ZR. p68 RNA helicase is an essential human splicing factor that acts at the U1 snRNA-5ʹ splice site duplex. Mol Cell Biol. 2002;22:5443–5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kar A, Fushimi K, Zhou X, et al. RNA helicase p68 (DDX5) regulates tau exon 10 splicing by modulating a stem-loop structure at the 5ʹ splice site. Mol Cell Biol. 2011;31:1812–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Guil S, Gattoni R, Carrascal M, et al. Roles of hnRNP A1, SR proteins, and p68 helicase in c-H-ras alternative splicing regulation. Mol Cell Biol. 2003;23:2927–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Camats M, Guil S, Kokolo M, et al. P68 RNA helicase (DDX5) alters activity of cis- and trans-acting factors of the alternative splicing of H-Ras. PLoS One. 2008;3:e2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Damianov A, Ying Y, Lin CH, et al. Rbfox proteins regulate splicing as part of a large multiprotein complex LASR. Cell. 2016;165:606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hua Y, Vickers TA, Baker BF, et al. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 2007;5:e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Li MZ, Elledge SJ. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat Methods. 2007;4:251–256. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.