

ABSTRACT
Selenoproteins are a unique class of proteins that contain the 21st amino acid, selenocysteine (Sec). Addition of Sec into a protein is achieved by recoding of the UGA stop codon. All 25 mammalian selenoprotein mRNAs possess a 3′ UTR stem-loop structure, the Selenocysteine Insertion Sequence (SECIS), which is required for Sec incorporation. It is widely believed that the SECIS is the major RNA element that controls Sec insertion, however recent findings in our lab suggest otherwise for Selenoprotein S (SelS). Here we report that the first 91 nucleotides of the SelS 3′ UTR contain a proximal stem loop (PSL) and a conserved sequence we have named the SelS Positive UGA Recoding (SPUR) element. We developed a SelS-V5/UGA surrogate assay for UGA recoding, which was validated by mass spectrometry to be an accurate measure of Sec incorporation in cells. Using this assay, we show that point mutations in the SPUR element greatly reduce recoding in the reporter; thus, the SPUR is required for readthrough of the UGA-Sec codon. In contrast, deletion of the PSL increased Sec incorporation. This effect was reversed when the PSL was replaced with other stem-loops or an unstructured sequence, suggesting that the PSL does not play an active role in Sec insertion. Additional studies revealed that the position of the SPUR relative to the UGA-Sec codon is important for optimal UGA recoding. Our identification of the SPUR element in the SelS 3′ UTR reveals a more complex regulation of Sec incorporation than previously realized.
KEYWORDS: Selenoproteins, Selenoprotein S, selenocysteine, mRNA translation, 3′ untranslated region
Introduction
Selenoproteins are a class of proteins that contain the 21st amino acid, Selenocysteine (Sec). In humans, there are 25 selenoproteins with diverse roles in many different cellular processes. As Sec is more reactive than cysteine at the physiological pH [1], many selenoproteins are involved in redox reactions and contain Sec in the active site. Translation of a selenoprotein mRNA poses an interesting challenge to cells because Sec is encoded by the UGA codon. Typically, UGA is used as a stop codon and signals termination of protein synthesis. This dual definition of the UGA codon requires that cells have the ability to distinguish between a UGA codon that is a stop signal and one that codes for Sec. In order to achieve this, cells utilize a host of cis- and trans-acting factors [2]. Within the 3′ untranslated region (3′ UTR) of all eukaryotic selenoprotein mRNAs, there is a stem-loop structure known as the Sec Insertion Sequence (SECIS) element that is required for UGA recoding [3]. The SECIS of each selenoprotein mRNA is unique, but all share a conserved core characterized by a quartet of non-Watson-Crick base pairs [4]. SECIS binding protein 2 (SBP2) binds to the conserved core of the SECIS element [5,6] and this interaction is crucial for UGA recoding. Mutations in SBP2 or in the core motif of the SECIS element result in clinical disease in humans [7–11]. The SECIS:SBP2 complex interacts with a host of other trans-acting factors which are involved in basal recoding including a specialized elongation factor (EF-Sec) [12,13] that recognizes and binds the unique Sec-tRNASec [14], and ribosomal protein L30, which acts at the ribosome to displace SBP2 from the SECIS [15]. Nucleolin [16] and eIF4a3 [17] have been shown to interact with the SECIS element to further regulate Sec incorporation into the growing polypeptide chain.
Originally, the efficiency of UGA recoding was thought to be controlled only by the interaction between SBP2 and the SECIS element of a selenoprotein mRNA. A growing interest in the field is the ability of other mRNA sequences to further regulate Sec insertion into selenoproteins. Previous studies have characterized a stem-loop structure, the Sec Redefinition Element (SRE) [18,19], in the coding region of Selenoprotein N (SelN) near the UGA-Sec codon. Single point mutations in the SelN SRE decreased UGA recoding in a dual luciferase reporter assay [20]. Furthermore, SelN protein and mRNA levels were greatly reduced in fibroblasts from patients carrying the same point mutations. In addition to sequences found in the coding region, our lab has previously shown that regions outside of the SECIS element in the 3′ UTR of Selenoprotein S (SelS) mRNA can affect UGA recoding [21], but specific elements were not defined.
SelS is an endoplasmic reticulum (ER) and plasma membrane protein that contains Sec as its penultimate amino acid residue. SelS was first identified as being differentially regulated in the diabetic mouse model, P. obesus [22]. Since its discovery, SelS expression has been shown to be inversely regulated by circulating levels of glucose and insulin [22,23]. Comparative genomic studies have revealed that, among selenoproteins, SelS is one of the most widely expressed across species [24]. SelS is upregulated under conditions that cause ER stress, such as increased pro-inflammatory cytokine levels and nutrient deprivation [23,25,26]. The unfolded protein response is activated when a large portion of proteins in the ER are not folded properly [27]. During this response, the translation of proteins is diminished [28,29], chaperone protein levels increase to facilitate proper folding [30,31], and misfolded proteins are degraded [32]. Unfolded proteins are removed from the ER in a process called ER-associated degradation (ERAD). Valosin-containing protein (VCP), which is an ATPase, and other proteins form a retrotranslocation channel in the ER membrane [33]. This channel moves unfolded proteins from the ER to the cytoplasm where they are ubiquitinated and degraded by the proteasome [32]. SelS has been shown to associate with the ERAD channel by binding with VCP and interacting with Selenoprotein K (SelK) [34] and Derlin-1 [35]. Overexpression of SelS has been reported to have protective effects against ER stress in multiple systems [36–39]. Conversely, knockdown of SelS decreases cell survival in conditions of increased ER stress [36,37,40], likely as a result of the toxic accumulation of unfolded proteins in the ER.
Recoding of the UGA codon as Sec is not always successful. Failure to insert Sec into SelS results in a truncated protein that is only two amino acids shorter than full-length SelS. Selenoproteins often require Sec in their active sites to efficiently perform their function. However, it has been reported that the truncated form of SelS is capable of interacting with VCP to form the ERAD channel and can function during ER stress [38,41]. In fact, the only unique function of Sec-containing SelS is an in vitro peroxidase activity [42–44], but the substrate for this activity in cells is unknown. The expression of full-length and truncated proteins is further complicated by the fact that there are two SelS mRNA variants [21], only one of which contains a SECIS element and can produce a Sec-containing protein. Unsuccessful recoding events and the presence of a mRNA variant that only encodes a truncated SelS protein suggests that production of full-length and truncated SelS may be tightly regulated.
We previously reported that the proximal 60 nucleotides of the SelS 3′ UTR were required for efficient UGA readthrough in an in vitro translation assay [21]. This result suggests that cis-acting sequences in the SelS 3′ UTR outside of the SECIS element may regulate the production of full-length and truncated SelS. In this study, we establish that our V5-surrogate assay for UGA recoding is a robust and accurate measure of Sec insertion in cells. Furthermore, we show that the first 91 nucleotides of the SelS 3′ UTR contain two elements: a proximal stem loop (PSL), that indirectly affects UGA recoding in a sequence independent manner, and a conserved sequence we have named the SelS Positive UGA Recoding (SPUR) element that is required for efficient Sec insertion in cells.
Results
V5-surrogate assay for Sec insertion
In order to study UGA recoding in the endogenous context of SelS, we previously developed a V5-reporter construct [21], shown in Fig. 1a. The V5-epitope tag was inserted into the human SelS cDNA between the UGA188 codon and UAA190, the natural stop codon, followed by the entire human SelS 3′ UTR (SelS-V5/UGA) (Fig. 1a, top). The V5-tag can be detected by Western blot when UGA188 is recoded, whereas termination at UGA188 will result in the truncated form of SelS which lacks V5. Originally, the SelS-V5/UGA construct was validated in an in vitro translation assay [21]. To test the assay in cells, the SelS-V5/UGA plasmid or a vector control were transfected into McArdle 7777 cells, a rat hepatoma cell line. McArdle 7777 cells are capable of expressing a wide range of selenoproteins and have been used to study selenoprotein synthesis [17,45]. After 24 hours, the transfected cells were harvested and lysates were run on SDS-PAGE for analysis by Western blot. As shown in Fig. 1b, the V5-tag is robustly expressed in cells transfected with SelS-V5/UGA but was not detected in vector only lysates. Antibodies against SelS and β-Tubulin were used to control for SelS expression and protein loading, respectively. Compared to the endogenous SelS levels in the vector-only control, SelS is overexpressed in the SelS-V5/UGA transfected cells. The β-Tubulin signal was consistent between samples, showing that the increased V5 and SelS signals are not due to differences in protein loading.
Figure 1.
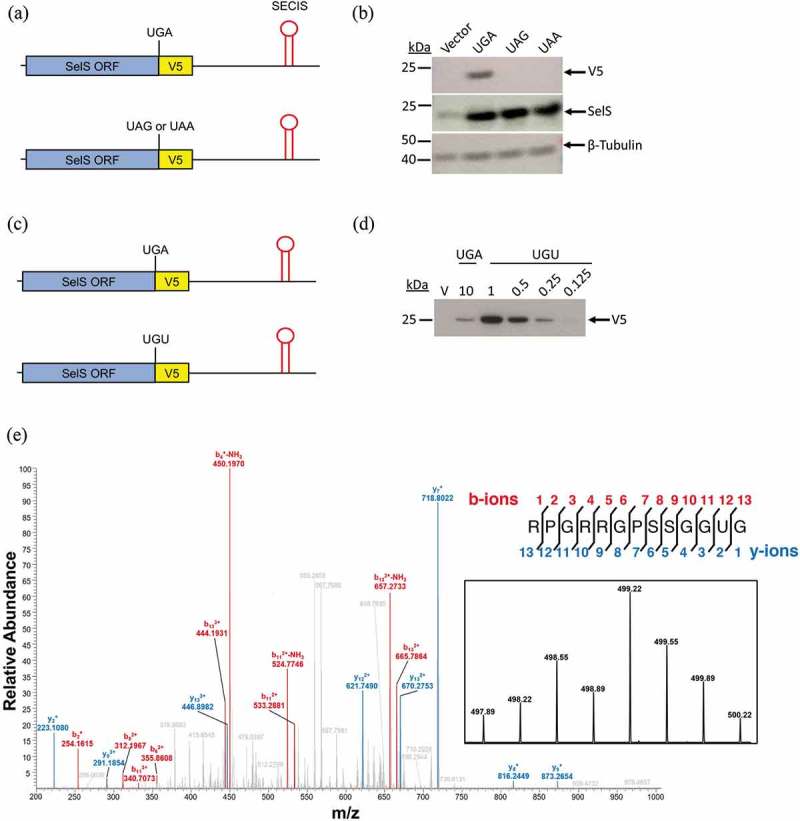
Validation of the SelS-V5 surrogate assay. a. Schematic representation of the SelS-V5/UGA construct. The V5-epitope tag was placed between the UGA-Sec and UAA-stop codons of the human SelS coding region followed by the entire SelS 3′ UTR. The UGA-Sec codon was mutated to UAG or UAA as indicated. b. Representative Western blot from three separate experiments of cells transfected with the SelS-V5/UGA and stop codon mutants as well as a vector control. Samples were immunoblotted with α-V5 antibody. The same blot was reprobed for SelS and β-Tubulin. c. The UGA codon in the SelS-V5/UGA construct was mutated to UGU. d. Western blot of 10 μg of lysate from McArdle 7777 cells transfected with SelS-V5/UGA or decreasing amounts (1 μg, 0.5 μg, 0.25 μg, and 0.125 μg) of lysate from SelS-V5/UGU transfected cells. e. SelS-V5/UGA was transfected into McArdle 7777 cells, immunoprecipitated with α-V5 beads, digested with chymotrypsin, and analysed by LC/MS2. Data represents analysis from three different transfections. The most abundant isotope of the Sec-containing peptide was analysed by MS2 fragmentation. Sequence: RPGRRGPSSGGUG, charge: +3, monoisotopic m/z: 499.2173 Da, [M + H] = 1495.6375. Fragment mass tolerance used for search = 0.6 Da, precursor mass tolerance 10 ppm. Fragments used for search – b; b-NH3 (red), y; y-NH3 (blue). Inset: Isotopic distribution of triply charged Sec-containing peptide.
We next wanted to validate that the V5-reporter assay was UGA codon-specific. One possibility is that the SelS 3′ UTR allows for readthrough of stop codons in general. However, when the UGA was mutated to another stop codon, UAA or UAG (Fig. 1a, bottom), V5 was not expressed (Fig. 1b). The UAA and UAG constructs both overexpressed SelS, suggesting that SelS is being translated, but termination occurs before the V5-tag can be synthesized (Fig. 1b).
In order to analyse the relative UGA recoding efficiency of the SelS-V5/UGA construct, the UGA188 codon was mutated to UGU, the codon for cysteine (Fig. 1c). Insertion of cysteine at UGU188 will result in all translated SelS protein containing the V5-tag. The SelS-V5/UGA lysate (10 μg) and increasing amounts of the SelS-V5/UGU lysate (0.125 μg, 0.25 μg, 0.5 μg, and 1.0 μg) were run on SDS-PAGE and analysed by Western blot (Fig. 1d). The V5 expression from increasing amounts of SelS-V5/UGU lysates was quantified (Fig. S1A) to generate a standard curve, which was linear (Fig. S1B). The SelS-V5/UGA lysate (10 μg) gave a V5 signal that was between what was observed for 0.125 and 0.25 μg of the SelS-V5/UGU lysate (Fig. S1A). Using the equation generated by the standard curve in Fig. S1B, the level of V5 expression from SelS-V5/UGA is approximately 2.0% compared to V5 expression from SelS-V5/UGU (100%). This recoding efficiency is 2.5-fold higher than what was previously reported for a luciferase-based recoding assay using the Glutathione Peroxidase 4 (GPx4) SECIS in the same cell-type [46].
Sec is inserted in the SelS-V5 surrogate assay
A limitation of UGA recoding reporter assays is that they only measure readthrough and cannot distinguish between Sec or another amino acid being inserted at the UGA codon. Metabolic labelling of cells with [75Se] in the form of selenous acid has been used to detect proteins that contain selenium but cannot determine whether other amino acids are incorporated. Therefore, we decided to use a mass spectrometric approach to identify the amino acid that is inserted at UGA in our V5-surrogate assay. V5-tagged SelS was immunoprecipitated from McArdle 7777 cells that were transfected with the SelS-V5/UGA construct. The immunoprecipitation efficiency was robust, with more than 90% depletion of the target (Fig. S2). The immunoprecipitated proteins were analysed by SDS-PAGE. The band corresponding to V5-tagged SelS was cut out of a Coomassie-stained gel, and the protein was digested in-gel with chymotrypsin. The digested sample was analysed by liquid chromatography-tandem mass spectrometry (LC/MS2) as described in the methods. The spectra generated by MS2 fragmentation were searched specifically against the human SelS protein sequence using Proteome Discoverer 2.2. The analysis revealed 26 unique peptides, covering 64% of the protein sequence. To detect the presence of Sec (U), the survey data were queried for the presence of a Cys residue (C) with an addition of 104.96 Da (C + 104.96). This modification accounts for the mass difference between U and C (41.9 Da), and the mass of the alkylation (57.02 Da). It also considers the isotopic distribution of selenium, which results in the most prominent peak being the M + 6 peak (5.99 Da). The C-terminal Sec-containing peptide, RPGRRGPSSGGUGF, was identified. The MS2 spectra of this peptide is shown in Fig. 1e. Also included is the MS1 profile of this peptide which has an isotope pattern consistent with the presence of selenium (Fig. 1e inset).
The digest was further analysed by parallel reaction monitoring (PRM) to search for the presence of the other 20 standard amino acid residues in place of Sec. The peptide sequences that were queried are shown in Table S1. No other amino acids were detected in the PRM analysis. Therefore, the SelS-V5 surrogate assay is a reliable method for measuring recoding of UGA as Sec in cells.
The proximal SelS 3′ UTR contains two distinct conserved sequences
We previously reported that deletion of the first 60 nucleotides of the SelS 3′ UTR decreased V5 expression in a cell-free translation assay [21]. To investigate whether this deletion had the same effect in cells, the Δ60 construct, which lacks the first proximal 60 nucleotides of the SelS 3′ UTR (Fig. 2a), was transfected into McArdle 7777 cells. V5 expression for the Δ60 construct was greatly reduced compared to the wild-type 3′ UTR (Fig. 2b).
Figure 2.
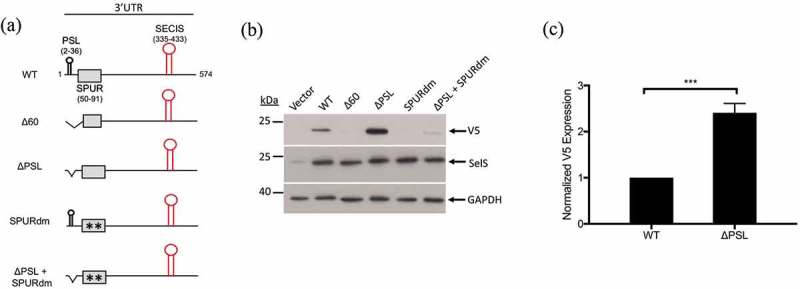
The proximal region of the SelS 3′ UTR contains elements that affect V5 expression. a. Schematic representation of the SelS-V5/UGA construct and the different 3′ UTR mutants (Δ60, ΔPSL, SPURdm, and ΔPSL+SPURdm). b. Representative Western blot of lysates from McArdle 7777 cells transfected with vector only or SelS-V5 constructs that contained different mutant 3′ UTRs. Lysates were resolved on SDS-PAGE, transferred to PVDF membrane, and immunoblotted for V5, followed by stripping and reprobing for SelS and GAPDH. c. Quantification of the relative V5 expression from the wild-type and ΔPSL constructs. V5 signals were normalized to SelS and GAPDH signals. Normalized V5 levels are expressed relative to wild-type. ***; p < 0.001. Data represents results from three independent experiments.
In our previous study, we proposed that a stem-loop structure in the proximal 60 nucleotides of the SelS 3′ UTR is responsible for this effect [21]. This proximal stem loop (PSL) spans nucleotides 3–36 of the SelS 3′ UTR and is conserved in sequence and predicted structure across species [21]. To test if the PSL alone is responsible for the decrease in UGA recoding, we deleted nucleotides 3–36 of the SelS 3′ UTR (ΔPSL) (Fig. 2a). Unexpectedly, the V5 signal from the ΔPSL construct increased by approximately 2.5-fold when compared to the wild-type 3′ UTR construct (Fig. 2b, c).
Since the PSL is not responsible for the effect of the 60-nucleotide deletion, we further analysed the proximal SelS 3′ UTR. Sequences of the SelS 3′ UTR from various species were collected from the NCBI database and analysed for conservation using the ClustalOmega server (https://www.ebi.ac.uk/Tools/msa/clustalo/) [47]. In addition to the PSL, we identified a non-conserved region (nucleotides 37–54), as well as a downstream sequence (nucleotides 55–91) that is 76% conserved, which we have named the SelS Positive UGA Recoding (SPUR) element (Fig. 3a). For comparison, nucleotides 93–335 (from the SPUR element to the beginning of the SECIS) are only 15% conserved. The consensus secondary structure of the proximal 3′ UTR from across species was determined using the prediction software Vienna RNA Websuite (http://rna.tbi.univie.ac.at/) [48]. Our analysis revealed a secondary structure for nucleotides 55–92 consisting of two small stem-loops denoted A and B (Fig. 3b, c). These secondary structures are strongly conserved across primates but begin to drop off as the diversity of species increases. The Δ60 deletion removes nucleotides 55–60 that are a part of SPUR element (Fig. 3a). Thus, we mutated two strongly conserved nucleotides in this region, C56→G and G61→C, referred to as SPURdm (Fig. 2a). This double point mutation greatly decreased V5 expression but did not affect the overexpression of SelS (Fig. 2b). There were no differences in loading based on GAPDH levels. Furthermore, deletion of the PSL in the context of the SPURdm was not able to rescue the V5 expression to wild-type levels (Fig. 2b).
Figure 3.
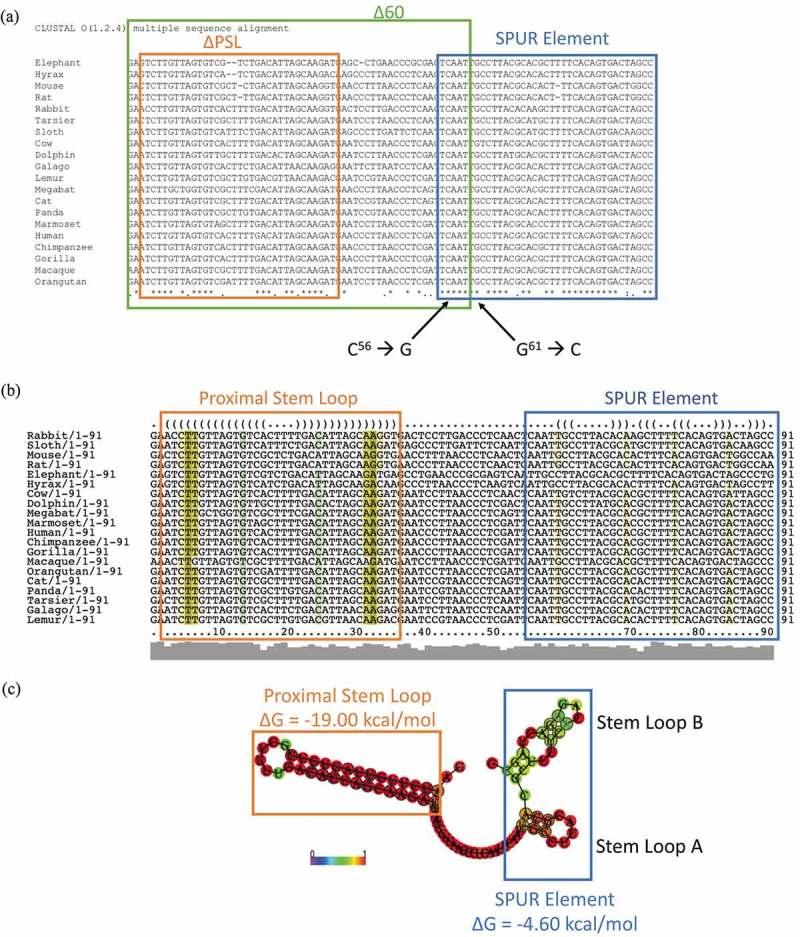
The proximal SelS 3′ UTR contains two conserved regions. a. The sequences of the first 91 nucleotides of SelS 3′ UTR from different mammals (Table S3) were analysed for nucleotide conservation by the ClustalOmega alignment program. Nucleotide positions with ‘*’ underneath have complete conservation across all species. Positions with ‘:’ have conservation between groups of strongly similar properties and positions marked with a ‘.’ have conservation between groups with weakly similar properties. The green box shows the first 60 nucleotides of the SelS 3′ UTR. The orange box shows the nucleotides that make up the PSL and the blue box encompasses the SPUR element. Nucleotides C56 and G61 were mutated to create the SPURdm. b. SelS 3′ UTR sequences were analysed for predicted structure using the RNAlifold program. The colour code indicates the number of base pair types found at each position: ochre-2, green-3, turquoise-4, blue-5, violet-6. Less saturated colours indicate that a base pair cannot be formed in some of the sequences. The orange box denotes the PSL and the blue box shows the SPUR element. c. The predicted consensus secondary structure of the first 91 nucleotides of the SelS 3′ UTR. Nucleotides shown in black circles indicate compensatory mutations within the sequences. The probability of a base pair is indicated on a scale from 0 (blue) to red (1) as shown in the colour bar. The PSL in orange has a highly conserved structure while the SPUR element (blue box) is made up of two smaller stem-loops. Free energies (ΔG) are shown for the PSL and the SPUR element.
Mutation of the PSL and SPUR element do not affect RNA levels or general translation
3′ UTRs have been reported to have a wide variety of functions, including the regulation of mRNA stability and translation [49]. Thus, the effect of mutations in the PSL and SPUR element on V5 expression could be due to mechanisms unrelated to UGA recoding. While the ΔPSL and the SPURdm did not affect SelS overexpression (Fig. 2b), we wanted to confirm that these mutations did not alter mRNA levels or general translation. To test if either the PSL or the SPUR element affect mRNA levels, the SelS-V5/UGA ΔPSL and SelS-V5/UGA SPURdm constructs (Fig. S3A) were transfected into McArdle 7777 cells along with a Renilla luciferase plasmid. Total RNA was extracted from each sample and cDNAs were generated. The relative transcript levels were measured by RT-qPCR using a forward primer in the nucleotide sequence corresponding to the V5-epitope tag and a reverse primer downstream in the SelS 3′ UTR. V5 RNA levels were normalized to Renilla and 18s RNA levels to control for transfection efficiency and loading, respectively. As shown in Fig. S3B, the deletion of the PSL or mutation of the SPUR element had no effect on transfected SelS-V5 mRNA levels when compared to the wild-type 3′ UTR. When the UGA codon in the WT, ΔPSL, and SPURdm SelS-V5 constructs was converted to a UGU-Cys codon, the mutations in the PSL and SPUR element no longer had an effect on V5 expression (Fig. S3C). Taken together, these results show that the differences observed in V5 expression with the ΔPSL and SPURdm mutants are due to changes in UGA recoding.
PSL and SPUR element mutations affect UGA recoding in HEK 293 cells
The experiments described above were all performed in the McArdle 7777 rat hepatoma line. To test whether these effects are cell-line or species specific, we transfected the SelS-V5/UGA constructs containing the ΔPSL and SPURdm mutations into the Human Embryonic Kidney cell line, HEK 293. V5 expression increased when the PSL was deleted while it decreased when the SPUR element was mutated (Fig. S4). Based on the above results, the SelS 3′ UTR contains two elements that affect UGA recoding in more than one cell type.
Point mutations in the SPUR element inhibit UGA recoding
We first analysed the SPUR element because it is required for efficient V5 expression. No homology was found between the SPUR sequence and the rest of the human genome. Furthermore, there were no robust hits when we searched multiple databases to identify RNA-binding proteins that have been shown to bind or crosslink to the region encompassing the SPUR element. Because these searches were uninformative, we employed a scanning point mutation approach to identify nucleotides that are required for function of the SPUR element. Nucleotides 55–92 of the SelS 3′ UTR are highly conserved across mammalian species and the SPURdm only takes into account a small portion of this conserved region (Fig 4a). Furthermore, this double mutation is predicted to alter the structure of the SPUR element by abolishing Stem Loop A (Fig. S5A). We generated eleven transversion point mutations (A ←→ T or C ←→ G) from nucleotides 46 to 104 (SPUR1-11) (Fig. 4b). SPUR1, 2, 10, and 11 flank the highly-conserved region whereas, SPUR3 through SPUR9 focus on some of the most highly conserved nucleotides within the SPUR element. As shown in Fig. S5A, SPUR1, 3, 5, 6, 7, and 9 do not affect the predicted structure while SPUR2, 4, and 8 do. These point mutation constructs were transfected into McArdle 7777 cells and analysed by Western blot (Fig. 4c). V5 expression was quantified and normalized to SelS and GAPDH (Fig. 4d). None of the point mutations had an effect on SelS overexpression. SPUR mutants 1, 10, and 11 had V5 signals similar to the wild-type 3′ UTR. Interestingly, SPUR5 did not show a statistically significant difference in V5 expression compared to the wild-type construct. In contrast, all other point mutations had a negative effect on V5 levels, reducing the signal by 60% or more (Fig. 4d). SPUR3 and 4, the two point mutations that make up the original SPUR double mutation, were deleterious to UGA recoding, each reducing V5 expression to less than 10% of wild-type levels. Thus, multiple nucleotides in the SPUR element are required for activity.
Figure 4.
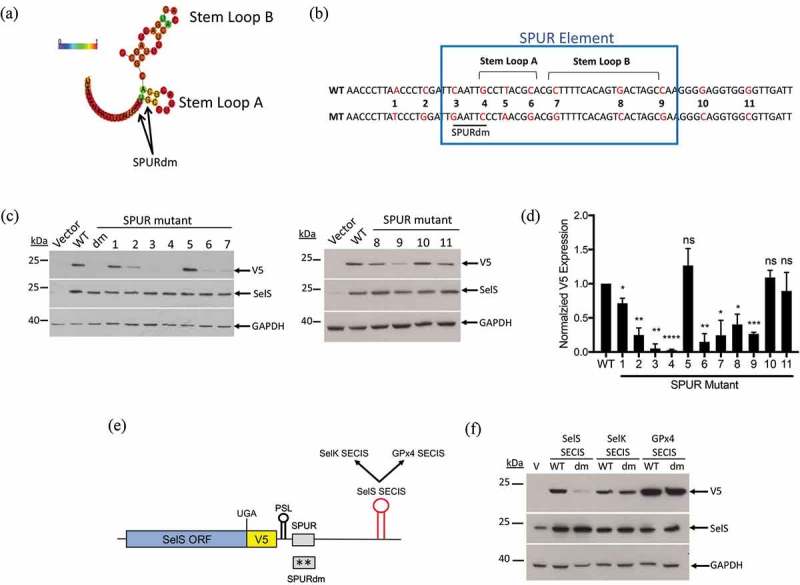
Requirements for SPUR element activity. a. Schematic showing the RNAlifold predicted structure of the human SPUR element. Nucleotide colours show probability of base pairing from 0 (blue) to 1 (red) as shown as described in the legend for Fig 3. The two nucleotides mutated in the SPURdm are indicated by arrows. b. Schematic of human SPUR element sequence. The blue box indicates the conserved nucleotides of the SPUR element. Stem Loops A and B are denoted by brackets. Red nucleotides in the sequence show the original nucleotide (WT) and what it was mutated to (MT). c. Representative Western blots showing the effect of single point mutations on V5 expression. Westerns were performed as described in Fig. 2b. d. Quantification of Western blots. V5 expression was quantified and normalized to SelS overexpression and GAPDH loading. Normalized V5 levels for each point mutant were compared to wild-type. Data represents results from two independent experiments analysed in duplicate (n = 4). *; p < 0.05, **; p < 0.01, ***; p < 0.001, ****; p < 0.0001, and ns – not significant). E. Schematic representation of the SelS-V5/UGA construct. The SelS SECIS element was replaced with the SECIS element from either SelK or GPx4. These SECIS elements were tested in both the wild-type SPUR and the SPURdm contexts. F. Representative Western blot from two independent experiments. WT = wild-type 3′ UTR, dm = SPURdm, and V = vector only. Western blots were performed as described in Fig 2(b).
As described above, the human SPUR element is predicted to form two small stem-loop structures, Stem Loop A and Stem Loop B (Fig. 4a). Interestingly, the SPUR4 and SPUR7 mutations are predicted to abolish the structure potential of Stem Loop A and Stem Loop B, respectively (Fig. S6A). To test if either stem-loop is responsible for the activity of the SPUR element, we generated compensatory mutations in SPUR4 and SPUR7, called AComp and BComp, which are predicted to reform Stem Loops A and B. However, compensatory mutations restoring the stem-loops did not rescue V5 expression (Fig. S6B). These results suggest that either predicted Stem Loops A and B do not form or that the structures are not important for SPUR activity.
The SPUR element does not function with other SECIS elements
SECIS elements vary in sequence and structure across selenoproteins. Therefore, we investigated whether the SPURdm would inhibit UGA recoding activity of other SECIS elements with similar structures. Since SelS contains a type II SECIS element with the AAR motif in the apical bulge, we selected two other type II SECIS elements, SelK and GPx4 [50,51] (Fig. S7). SelK contains Sec near the C-terminus, like SelS, whereas the Sec residue in GPx4 is in the N-terminal third of the protein. Chimeric 3′ UTRs were created in which the SelS SECIS was replaced with either the SelK or GPx4 SECIS elements (Fig. 4e). These were tested in both the wild-type SelS 3′ UTR and SPURdm contexts. As shown in Fig. 4f, the level of V5 expression from constructs with the wild-type 3′ UTR varied with the SECIS element. Compared to the SelS SECIS, V5 expression was lower with the SelK SECIS but higher with the GPx4 SECIS. This may be due to differences in recoding efficiencies when different SECIS elements are placed in the SelS 3′ UTR. As reported above, mutation of the SPUR element in the wild-type 3′ UTR inhibited V5 expression. However, the SPURdm mutation had little effect when the 3′ UTR contained either the SelK or GPx4 SECIS elements, instead of the SelS SECIS (Fig. 4f). These observations cannot be explained by differences in overexpression (SelS) or loading (GAPDH). These data suggest that the SPUR element cannot function with the SelK or GPx4 SECIS elements and that its activity may be specific to the SelS SECIS.
Deletion of the PSL promotes Sec insertion
Unlike the SPUR element, the SelS PSL inhibits UGA recoding since its deletion increased V5 expression in our surrogate assay. This activity is in contrast to the SRE, a stem-loop structure in the coding region of SelN, which is required for maximal UGA recoding [18]. Mutations in the SRE inhibited Sec insertion in an in vitro translation system and cells from patients with the same mutations had reduced SelN protein levels [20]. However, the SelN SRE also promoted readthrough of other stop codons in cells [18]. To exclude the possibility that deletion of the PSL increases basal readthrough by allowing the recruitment of near-cognate tRNAs at stop codons, we mutated the UGA codon to either UAG or UAA (Fig S8B). We then tested whether deletion of the SelS PSL would allow readthrough of the other stop codons. When the UGA codon in the SelS-V5/UGA ΔPSL construct was mutated to either the UAA or UAG stop codon (Fig. S8A), SelS was overexpressed, but there was no expression of the V5-tag (Fig. S8B).
We next wanted to determine whether deletion of the PSL promoted Sec incorporation in cells. V5-tagged SelS was immunoprecipitated from McArdle 7777 cells that were transfected with either the SelS-V5/UGA WT or the SelS-V5/UGA ΔPSL construct. The bands corresponding to V5-tagged SelS were processed for LC/MS2 analysis as described in the methods. The immunoprecipitated proteins from the wild-type and ΔPSL samples were positively identified as human SelS, with 25 and 34 unique peptides, covering 64% and 81% of the protein sequence respectively. Peptides from human SelS were not detected in immunoprecipitations from untransfected cell lysates.
A PRM analysis was performed on three independent immunoprecipitations to quantify the recoded SelS protein in the ΔPSL sample, relative to the wild-type sample. The experiment was set up to detect the Sec-containing peptide, RPGRRGPSSGGUGF, as well as two abundant peptides from different regions of the SelS sequence (Table S2). The internal peptides (Table S2) of the ΔPSL samples were increased by 2-fold compared to the wild-type sample (Fig. S8C and D). This was expected due to the increase of V5-containing protein seen with deletion of the PSL. The peak areas of the RPGRRGPSSGGUGF peptide were then compared between the wild-type and ΔPSL samples. The relative abundance of the Sec-containing peptide was 2-fold higher in the ΔPSL samples when compared to the wild-type samples (Fig. S8E) suggesting that the increase in V5-containing peptide is due to Sec insertion. The ratios of the Sec peptide to the average internal peptide abundance in the wild-type and ΔPSL samples were similar (Fig. S8F) suggesting that Sec is inserted into all V5-containing protein. To confirm this, the ΔPSL digest was analysed by PRM for the presence of the other 20 amino acid residues in place of Sec (Table S1). Based on the list of peptides queried, no amino acid substitutions were detected. Taken together, our results show that deletion of the PSL promotes Sec incorporation into the SelS-V5/UGA construct.
Other sequences can functionally replace the SelS PSL
The SelS PSL is a 34-nucleotide stem-loop with a 14-base pair stem and a 6-nucleotide loop. In order to identify the sequences or structures that were important for PSL function, we made mutations that removed the loop or introduced bulges in the stem (Fig. S9A). However, none of these mutations increased V5 expression compared to wild-type levels (Fig. S9B), suggesting that the exact sequence and/or structure may not be important for activity. We then investigated whether the SelS PSL could be replaced by other stem-loops from selenoprotein mRNAs. In addition to SelS, there are 7 other mammalian selenoproteins that contain C-terminal Sec residues [52]. Of these, six are predicted to possess a stem-loop near the beginning of their 3′ UTR based on RNA structure prediction software (Fig. S10). We tested the proximal stem loops from SelK and Selenoprotein O (SelO). The SelK PSL is smaller than the SelS PSL with an 8-base pair stem and 6-nucleotide loop, while the SelO PSL is larger and contains multiple bulges in its stem as well as an internal loop (Fig. 5a). The SelK and SelO PSLs were cloned into the SelS-V5/UGA ∆PSL construct. As reported above, V5 expression increased when the SelS PSL was deleted. Interestingly, insertion of either the SelK or SelO PSL into the ΔPSL construct reduced V5 expression back to wild-type 3′ UTR levels (Fig. 5b), demonstrating a function similar to the SelS PSL.
Figure 5.
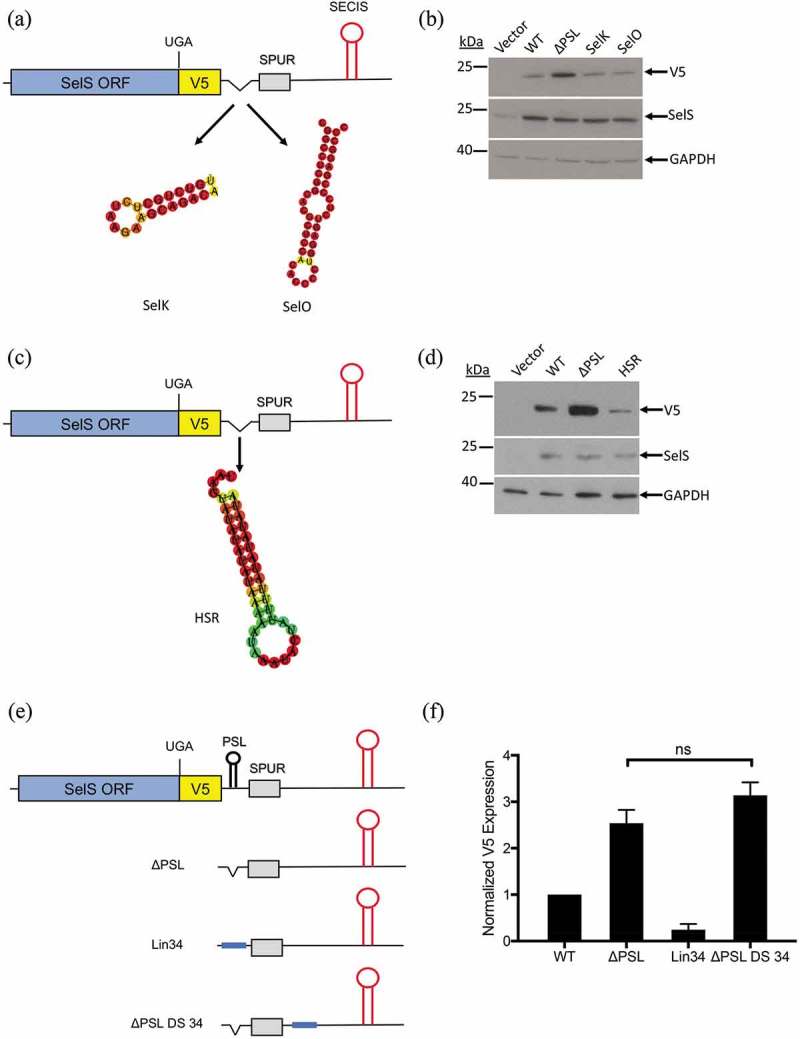
Other sequences can functionally replace the SelS PSL. a and c. Schematic representation of the SelS-V5/UGA ΔPSL construct. The SelS PSL was replaced with the proximal stem loop from either the SelK or SelO 3′ UTRs or the HSR found in the VEGF-A 3′ UTR. b and d. Western blot was performed as described in Fig 2(b). Representative of three independent experiments. e. Schematic representation of constructs used to test the linear sequence. The SelS PSL was replaced with an unstructured 34 nucleotide sequence (Lin34). The linear sequence was also placed downstream of the SPUR element in the ΔPSL context (ΔPSL DS 34). f. Quantification of Western blots. V5 expression was quantified and normalized to SelS overexpression and GAPDH loading. Data represents three independent experiments. Relative V5 levels are expressed relative to WT. No statistically significant changes were observed between ΔPSL and ΔPSL DS 34 (ns).
We next tested whether a non-selenoprotein stem-loop could take the place of the SelS PSL. The SelS PSL was replaced with a 36-nucleotide stem-loop structure of the Hypoxia Stability Region (HSR) found in the VEGF 3′ UTR [53] (Fig. 5c). When the HSR was inserted into the ΔPSL construct, V5 expression was reduced compared to the ΔPSL construct (Fig. 5d). Furthermore, this expression was lower than what was observed with the wild-type 3′ UTR.
Since stem-loops of different sequences and structures can functionally replace the SelS PSL, we then tested a linear sequence. We cloned a 34-nucleotide sequence into the ΔPSL construct (Fig. 5e, Lin34). This sequence is of similar GC content as the SelS PSL and is not predicted to form any structure. Furthermore, the addition of the linear sequence did not perturb the formation of Stem Loops A and B (Fig. S5B). As shown in Fig. 5f, the V5 signal from Lin34 was much lower than what was observed with the ΔPSL construct and, in fact, was only 25% of what we observed with the wild-type 3′ UTR. These results suggest that a linear sequence can functionally replace the SelS PSL.
One explanation for the above results is that deletion of PSL increased Sec insertion because the position of the SECIS element relative to the UGA-Sec codon has decreased, making the SECIS more efficient. If this is the case, inserting the 34-nucleotide linear sequence in another part of the ΔPSL 3′ UTR before the SECIS element should also decrease V5 expression. We added the linear sequence immediately downstream of the SPUR element into the non-conserved portion of the SelS 3′ UTR in the ΔPSL 3′ UTR context (Fig. 5e, ΔPSL DS 34). As shown in the graph in Fig. 5f, there was no significant difference in V5 expression between ΔPSL and ΔPSL DS 34. Therefore, deletion of the PSL does not increase UGA recoding due to the fact the SECIS element has been moved upstream.
The effect of the SPUR element is position dependent
Having excluded the SECIS element, we hypothesized that the effect of the ΔPSL mutation may be due to the fact that the position of the SPUR element has changed. In the SelS-V5/UGA construct, the SPUR element is 118 nucleotides downstream from the UGA-Sec codon because of the V5-tag, compared to 61 nucleotides in endogenous SelS. Deletion of the PSL would move the SPUR element 34 nucleotides closer to UGA-Sec. To test whether deleting the PSL would affect UGA recoding in the absence of the V5-tag, we generated a SelS construct that had a FLAG epitope tag at the N-terminus. The UGA codon was in its natural position relative to the SPUR element and constructs were made in the context of both the wild-type and ΔPSL 3′ UTRs (Fig. 6a, top two). These constructs were transfected into cells, and the FLAG-SelS was immunoprecipitated, run on SDS-PAGE, and the corresponding band was digested with endoproteinase GluC. Mass spectrometry was performed to detect the Sec-containing peptide, GGGACSWRPGRRGPSSGGUG (Fig. 6b). We found that deletion of the PSL in the FLAG-SelS/UGA188 context did not increase Sec insertion (Fig. 6c). These results were confirmed by metabolic labelling with [75Se] selenous acid of McArdle 7777 cells transfected with FLAG-SelS/UGA containing either the wild-type, ΔPSL, or SPURdm 3′ UTRs (Fig. S11A). Importantly, the SPURdm inhibited Sec incorporation which agrees with the V5-surrogate assay. Consistent with the mass spectrometry results, deletion of the PSL did not increase Sec incorporation (Fig. S11B).
Figure 6.
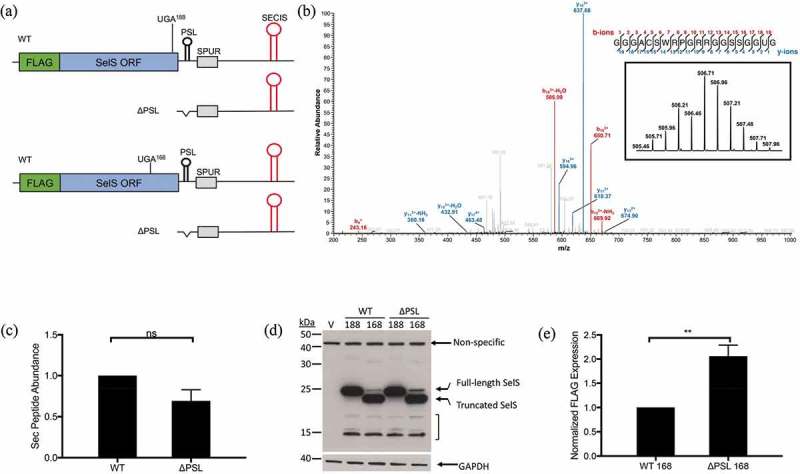
Relative position of SPUR element to UGA codon is important for activity. a. Schematic of FLAG-SelS/UGA188 (top two) and UGA168 (bottom two) constructs with either the wild-type or ΔPSL 3′ UTR. b. Representative analysis of Flag-SelS by mass spectrometry. FLAG-SelS was transfected into McArdle 7777 cells, immunoprecipitated using α-FLAG beads, digested with GluC, and analysed by mass spectrometry. Data represents analysis from two independent transfections. The most abundant isotope of the Sec-containing peptide was analysed by MS2 fragmentation. Sequence: GGGACSWRPGRRGPSSGGUG, charge: +4, monoisotopic m/z: 506.7096 Da, [M + H] – 2023.8167. Fragment mass tolerance used for search = 0.6 Da, precursor mass tolerance 10 ppm. Fragments used for search – b; b-NH3; b-H2O (red), y; y-NH3; y-H2O (blue). Inset: Isotopic distribution of triply charged Sec-containing peptide. c. Quantification of FLAG-SelS Sec-containing peptide by mass spectrometry. d. Representative Western blot. Vector only (V) FLAG-SelS/UGA188 (188) and UGA168 (168) with either the wild-type (WT) or ΔPSL 3′ UTR were transfected into McArdle 7777 cells. Westerns were probed with α-FLAG. Full-length and truncated forms of SelS are indicated. Possible SelS degradation bands are marked by a bracket. e. Quantification of Western blots. Expression of the full-length FLAG-SelS protein from the UGA168 lanes were quantified and normalized to GAPDH. Data represents three independent experiments (n = 3). Normalized full-length FLAG-SelS levels are expressed relative to WT. **; p < 0.01.
If the distance between the UGA-Sec and the SPUR element is important, we predict that deletion of the PSL in the FLAG-SelS context would increase Sec insertion if the UGA was moved upstream of its natural position. To test this, the UGA-Sec codon was moved upstream by 60 nucleotides to codon 168 and the natural UGA-Sec codon at position 188 was mutated to UGU-Cys (FLAG-SelS/UGA168; Fig. 6a, bottom two). This FLAG-SelS/UGA168 construct is analogous to the SelS-V5/UGA construct in that the UGA codon is 121 nucleotides upstream of the SPUR element. The UGA168 mutation was also made in the ΔPSL 3′ UTR context, which decreases the distance between the UGA-Sec and SPUR element to 87 nucleotides and is comparable to the SelS-V5/UGA ΔPSL construct. The FLAG-SelS/UGA188 and UGA168 constructs with either the wild-type or ΔPSL 3′ UTRs were transfected into McArdle 7777 cells and analysed by Western blot with the α-FLAG antibody. Because the there is only a two amino acid difference between full-length and truncated SelS when the UGA codon is at the natural position, there is no differentiation between the two forms (Fig. 6d, 188 WT and ΔPSL). As shown in Fig. 6d, most of the FLAG-SelS/UGA168 protein is truncated due to premature termination at UGA168 but full-length FLAG-SelS was also detected. As predicted, deletion of the PSL in the FLAG-SelS/UGA168 construct increased UGA recoding approximately 2-fold compared to the wild-type 3′ UTR (Fig. 6e). This is similar to the increase in Sec-insertion seen when the PSL is deleted in the SelS-V5/UGA context. These results suggest that position of the SPUR element relative to the UGA-Sec codon is important for its function.
Discussion
SelS is a widely expressed and highly conserved selenoprotein that has two known activities [24]. The most well-defined function is its role in the ERAD retrotranslocation channel, which removes unfolded proteins from the ER during ER stress [34,35]. SelS also has a peroxidase activity which has only been defined in vitro [42–44]. Interestingly, there is the potential to produce two different forms: a full-length, Sec-containing protein and a truncated protein resulting from premature termination at the UGA-Sec codon. The enzymatic functions of many selenoproteins require the Sec residue, and indeed, this is true of the peroxidase function of SelS [43]. Whether the Sec residue plays a role in the ERAD function of SelS has not been directly tested, but several lines of evidence suggest that the truncated protein is sufficient for this activity [38,41]. Given its important role in ERAD, it is not surprising that expression of SelS is increased at the transcriptional level by ER stress and proinflammatory cytokines [23,25,26]. However, whether the incorporation of Sec into SelS is regulated is unknown.
In this study, we identified the SPUR element, which is the first example of a defined 3′ UTR sequence, outside of the SECIS, that regulates Sec incorporation in cells. The nucleotide sequence of the SPUR element is conserved in SelS from mammals, but it is not found in other genes and is not a known functional motif. Single point mutations across this element decreased UGA recoding, often by 60% or more. Intriguingly, mutation at nucleotide 65 (SPUR5) has little to no effect on UGA recoding, whereas nucleotides on either side are required. This observation raises the possibility that the SPUR element may be bipartite. Furthermore, while the SPUR element is predicted to form two small stem-loops, these structures do not appear to play a role in function based on our mutagenesis studies, suggesting that activity depends on the nucleotide sequence.
The 3′ UTR of selenoproteins mRNA transcripts all contain a SECIS element. Each of these SECIS elements contain a conserved motif required for SBP2 binding, but otherwise have variable sequences. Here, we have shown that the SPUR element mutations are deleterious to UGA recoding in the presence of the SelS SECIS, but not when the SelS SECIS is replaced with either the SelK or GPx4 SECIS elements. One possible explanation for this result is that the SPUR element has long-range interactions with specific nucleotides in the SelS SECIS. Indeed, there is a short sequence in the SPUR element that has the potential to base pair with the SelS SECIS as shown in Fig. S12. Experiments are currently in progress to test this hypothesis.
In contrast to the SPUR element, deletion of the PSL stimulated Sec incorporation in the V5-surrogate assay. We found that the SelS PSL could be functionally replaced with other stem-loops or even a linear sequence. These results suggest that removing the PSL indirectly affects Sec incorporation. We excluded the possibility that changing the position of the SECIS was responsible for the increase in UGA recoding observed with deletion of the PSL. Our experiments using the FLAG-SelS constructs support a model in which the efficiency of Sec incorporation is dependent on the distance between the SPUR element and the UGA-Sec codon. One could envision that the SPUR element may recruit the SelS SECIS and facilitate delivery of the Sec-tRNASec to the ribosome at the UGA-Sec codon (Fig. 7). This model may also explain why deletion of the PSL only increases recoding in the SelS-V5 construct and not in the FLAG-SelS construct. If ribosome proximity is important, then addition of the V5-tag between the UGA codon and the SPUR element can sterically hinder SECIS:ribosome interactions that are needed for Sec insertion. Deletion of the PSL in SelS-V5 could relieve this steric hindrance and allow the SelS SECIS to successfully meditate Sec insertion. It is tempting to speculate that the SPUR element plays an additional role in restricting Sec insertion to the C-terminus of SelS.
Figure 7.
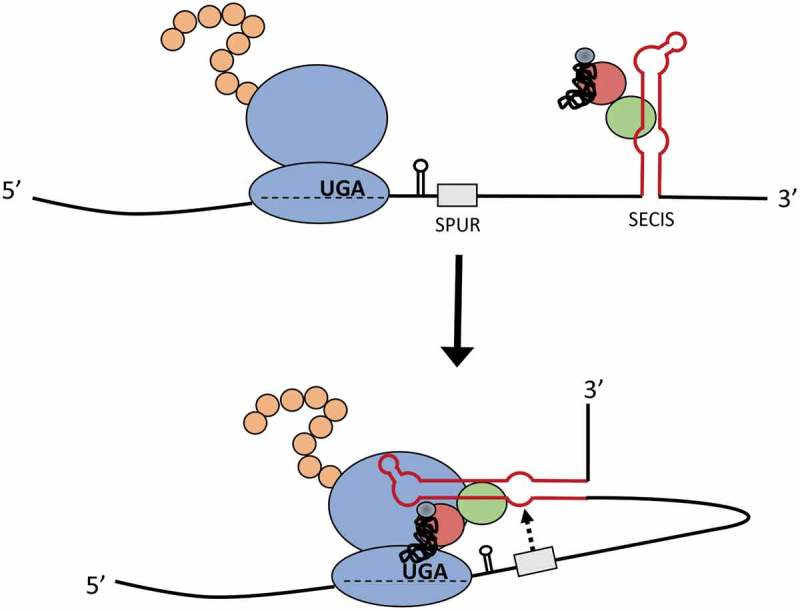
Model for the mechanism of action of the SPUR element. The SelS SECIS (red stem-loop) associates with SBP2 (green), EFSec (red), and the Sec tRNA (black). The interaction of the SPUR element (grey) with the SECIS brings the SECIS into position to interact with the ribosome at the UGA-Sec codon and facilitate Sec insertion.
Our results raise the intriguing question of why is the SelS PSL conserved if it does not play a role in UGA recoding. It is possible that the PSL is important for Sec incorporation in other tissues, at different developmental stages, or under specific stress conditions. Alternatively, the PSL may play a role in processes other than UGA recoding. We have shown that the PSL has no effect on mRNA stability or general translation but did not test if it has a role in splicing. We previously reported that there are two SelS mRNA variants produced through alternative splicing [21], only one of which contains a SECIS element. Both of these SelS transcripts are expressed in cell lines [21] and human tissues (Cockman EM unpublished), with the SECIS-containing transcript representing 85–90% of total SelS mRNA. The splice site for the SECIS-less mRNA is between nucleotides 13 and 14 of the 3′ UTR and is predicted to be sequestered in the 5ʹ stem of the PSL [21]. The structure of the PSL may be conserved across species due to evolutionary pressure to prevent splicing between nucleotide 13 and 14 and promote expression of the SECIS-containing SelS mRNA transcript.
Including SelS, there are 8 human selenoproteins that contain C-terminal Sec residues. Our analysis revealed that 6 of these 8 selenoproteins (including SelS and SelK) have the potential to form stem-loops in their 3′ UTR immediately downstream of the canonical stop codon (Fig. S10). We generated a SelK-V5/UGA construct in which the UGA-Sec codon is moved upstream from its natural position due to the V5-tag. Unlike our results with SelS, deletion of the SelK PSL had no effect on UGA recoding when compared to the wild-type 3′ UTR (Fig. S13). This result could be explained by the fact that SelK lacks a conserved sequence immediately downstream of the PSL that would be analogous to the SPUR element, based on our computational analysis. Thioredoxin reductase I (TR1) is another selenoprotein with a C-terminal Sec residue. Like SelK, we have not identified a SPUR-like element in the proximal region of the TR1 3′ UTR. Interestingly, Turanov et al reported that efficient Sec incorporation into TR1 only occurred when the UGA-Sec codon was in its natural C-terminal position [54]. This restriction of Sec incorporation appears to be defined by the TR1 SECIS element. Deletion of the TR1 PSL in the wild-type construct did not increase Sec insertion, but this was not tested in the context of the upstream UGA-Sec codons. Furthermore, our computational analyses do not exclude the existence of a SPUR-like element in more distal regions of the TR1 or SelK 3′ UTRs. The PSLs from TR1 and SelK do not encompass a splice site and their function is unknown.
In this study, we have developed important new tools for analysing Sec incorporation in mammalian cells. Standard approaches have used either modified reporter constructs which measure UGA recoding or metabolic labelling with [75Se] selenous acid. However, neither of these approaches specifically identify Sec as the incorporated amino acid nor do they exclude the possibility of the insertion of other amino acids. Using mass spectrometry, we have shown that Sec is incorporated into the SelS-V5 protein. Furthermore, none of the other 20 standard amino acids were detected in the peptide suggesting that only Sec is inserted at the UGA codon. To our knowledge, this study is the first report to demonstrate that a cell-based reporter assay for UGA recoding assay is a direct measure of Sec incorporation. In addition to studying the mechanism of Sec insertion, these same approaches could be used to study endogenous SelS.
Our findings have implications for future studies on elucidating the mechanism and regulation of Sec incorporation. Very few studies have investigated whether 3′ UTR sequences outside of the SECIS element contribute to the efficiency of Sec insertion. The SelS SECIS has been reported to have weak activity but this conclusion was based on the analysis of a minimal SECIS of ~100 nucleotides [55]. Our discovery of the SPUR element in SelS highlights the need to consider this possibility when analysing other selenoprotein mRNAs. Another consideration is whether modified nucleotides play a role in regulating Sec insertion. It has been shown in yeast that the replacement of uridine with pseudouridine at stop codons allows for the insertion of serine, threonine, or tyrosine [56]. Since yeast cannot synthesize selenoproteins, it remains to be investigated whether pseudouridine at the UGA codon would allow for the insertion of Sec in mammalian cells.
Under basal conditions, the SPUR element is required for efficient UGA recoding. It is tempting to speculate that the activity of the SPUR may be regulated which could affect the production of the Sec-containing and truncated SelS proteins in cells. Future studies on SelS will focus on understanding the mechanism by which the SPUR element effects UGA recoding and further elucidating how SelS is regulated post-transcriptionally.
Materials and methods
Sequence conservation and secondary structure prediction
Sequences used to perform conservation alignments and structure prediction were obtained using NCBI and Ensemble databases. Accession numbers for all sequences are listed in Table S3. Conservation alignments were performed using the ClustalOmega server (https://www.ebi.ac.uk/Tools/msa/clustalo/) [47]. Structure prediction and structure conservation was performed using the prediction software from RNAVienna websuites (http://rna.tbi.univie.ac.at/) [48].
DNA plasmids
The SelS/V5-UGA construct is previously described [21]. All mutagenic primers can be found in Table S4. Mutagenic PCR was performed using QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent). All other PCR was performed using Phusion Polymerase (New England Biolabs). Primers for insertion PCR can be found in Table S5.
To create GPx4 and SelK chimeric SECIS constructs, mutagenic primers were used to introduce a PacI site upstream and a NotI site downstream of the SelS SECIS in the SelS 3′ UTR (SelS PacI SECIS mutant and SelS NotI SECIS mutant). Primers corresponding to the SECIS elements of SelK (SelK SECIS PacI Fwd and SelK SECIS NotI Rev) or GPx4 (GPx4 SECIS PacI Fwd and GPx4 SECIS NotI Rev) were used to clone the SelK and GPx4 sequences with the PacI and NotI restriction sites. The SelK and GPx4 SECIS elements were digested and cloned into the PacI/NotI sites of the modified SelS 3′ UTR.
Cell culture
McArdle 7777 (rat hepatoma) and HEK 293 (human embryonic kidney) cells were obtained from American Type Culture Collection (ATCC). Cells were cultured in DMEM supplemented with 10% foetal bovine serum (Gibco) and 60 nM sodium selenite in 5% CO2 at 37°C.
V5-surrogate assay
McArdle 7777 cells and HEK 293 cells were plated at 3 × 105 cells/well in 2 mL of supplemented DMEM in a 6-well plate 24-hours before transfection. A total of 1 μg of DNA consisting of 600 ng pcDNA 3.1, 200 ng Firefly luciferase, and 200 ng of the appropriate SelS-V5 plasmid DNA were transfected using 8 μL of Lipofectamine reagent (ThermoScientific) following the manufacturer protocol. Cells were incubated with DNA:lipofectamine complexes for 24-hours (McArdle 7777 cells) or 48-hours (HEK 293 cells). After transfection, cells were pelleted and proteins extracted using NP40 lysis buffer (150 mM NaCl; 50 mM Tris Cl, pH 7.0; 1% NP40). Protein concentrations were measured using pre-diluted BSA standards (ThermoScientific) with Pierce 660 nm Protein Assay Reagent (ThermoScientific) and measuring absorbance at 660 nm using a spectrometer (SpectraMax190, Molecular Devices). To control for transfection efficiency, Firefly luciferase assays were performed by adding 100 μL of Luciferase Assay System Substrate (Promega) to 1 μg of protein lysate. Luciferase activity was measured in triplicate using a luminometer (Victor Nivo, Perkin Elmer).
Western blotting
Proteins were separated by SDS-PAGE and transferred to ImmunoBlot polyvinylidene fluoride (PVDF) membrane (Biorad). The primary antibodies used were α-SelS Prestige (Sigma, HPA010025), α-GAPDH (6C5) (Abcam, ab8245), α-V5 (Invitrogen, R960), α-FLAG (Sigma, A8592), and α-β-Tubulin (Sigma, T0198). The secondary antibodies used were α-mouse-HRP and α-rabbit-HRP (Jackson Immunochemicals, 150–035-003 and 111–0450144). Proteins were detected using Immobilon Western HRP substrate (Millipore) and imaged by exposure to Biomax MR film (Kodak) or the Amersham 600 Imager (GE). Analysis and quantification for Western blots were performed using ImageStudioLite (LI-COR Biosciences).
qRT-PCR
After transfection, cells were pelleted and RNA was extracted using Trizol (Invitrogen) according to the manufacturer's protocol. To remove contaminating plasmid DNA, RNA samples were digested with NaeI (New England Biolabs) followed by RQI DNase (Promega). RNA quantity was then measured using spectrophotometry at 260 and 280 nm and quality was assessed by agarose gel electrophoresis. RNA (2 μg) was used to make cDNA with the SuperScript VILO cDNA Synthesis kit (ThermoScientific). Primer sequences used for qRT-PCR can be found in Table S6. All reactions were performed in triplicate using 2X Fast SYBR Green Master Mix (Applied Biosystems) and set up in MicroAmp Fast Optical 96-well reaction plates with optical caps (Applied Biosystems). Reactions lacking cDNA template or reverse transcriptase were used as controls. Reactions were run on a StepOnePlus Real-Time PCR System (Applied Biosystems). Data was analysed using StepOne Software (Applied Biosystems).
Immunoprecipitation
McArdle 7777 cells were transfected with the SelS-V5/UGA WT, SelS-V5/UGA ΔPSL, Flag-SelS/UGA188 WT or Flag-SelS/UGA188 ΔPSL constructs using Lipofectamine reagent (Invitrogen). Cells were harvested 24-hours after transfection and lysed with NP40 lysis buffer containing 20% glycerol. Protein lysates (3 mg) were diluted 1:1 with radioimmunoprecipitation assay (RIPA) buffer (SelS-V5 lysates) or NP40 buffer w/glycerol (Flag-SelS lysates), and subjected to immunoprecipitation with 40 μL of α-V5 agarose beads (Abcam; ab1229; 50% slurry; 0.25 mg/mL bound goat polyclonal antibody) or α-Flag M2 affinity gel (Sigma; A2220; 50% slurry), overnight at 4°C. Lysates from untransfected McArdle 7777 cells were subjected to immunoprecipitation under the same conditions, as controls. The beads were centrifuged at 1500 g for 5 min at 4°C, and the unbound fraction was collected for analysis of immunoprecipitation efficiency. The beads were then washed twice with 1 mL (50 volumes) of RIPA buffer for SelS-V5 lysates or phosphate buffered saline with 0.1% tween-20 for Flag-SelS lysates. The immunoprecipitate was eluted off the beads by boiling with 1X Laemmli buffer and separated by SDS-PAGE (15%), followed by Coomassie staining (Gelcode blue safe protein dye; ThermoScientific) to visualize protein bands.
In-gel digestion and liquid chromatography-tandem mass spectrometry
For protein digestion, the bands corresponding to SelS-V5 or Flag-SelS (by size) were cut to minimize excess polyacrylamide, and divided into a number of smaller pieces. The gel pieces were washed with a solution of 50% ethanol/5% acetic acid, and dehydrated in acetonitrile. The bands were then reduced with DTT followed by alkylation with iodoacetamide prior to in-gel digestion. SelS-V5 bands were digested in-gel by adding 15 μL chymotrypsin (Sigma; 11,418,467,001; 25 ng/μL) in 50 mM ammonium bicarbonate, and incubating overnight at room temperature. Flag-SelS bands were digested in-gel by adding 15 μL endoproteinase GluC (Sigma; 11,047,817,001; 50 ng/μL) in 50 mM ammonium bicarbonate, and incubating overnight at room temperature. The resulting peptides were extracted from the polyacrylamide in two aliquots of 30 μL with a solution of 50% acetonitrile/5% formic acid. These extracts were combined and evaporated in a speedvac and then resuspended in 1% acetic acid (HPLC grade) in a final volume of 30 μL for LC/MS analysis.
The LC/MS system was a ThermoScientific Fusion Lumos mass spectrometry system. The HPLC column was a Dionex 15 cm x 75 μm id Acclaim Pepmap C18, 2 μm, 100 Å reversed-phase capillary chromatography column. Five μL of the extract were injected and the peptides were eluted from the column by an acetonitrile/0.1% formic acid gradient (2–70% over 2 h), at a flow rate of 0.25 μL/min, were introduced into the source of the mass spectrometer on-line. The microelectrospray ion source was operated at 2.5 kV. The digests were analysed using a data dependent survey analysis acquiring full scan mass spectra to determine peptide molecular weights and product ion spectra to determine amino acid sequence. The data dependent experiments were searched specifically against the sequence of SelS, where the Sec was replaced with a Cys residue, using SequestHT bundled in the Proteome Discoverer 2.2 program (ThermoScientific). These searches used an MS1 mass tolerance of 10 ppm, a MS2 mass tolerance of 0.6 Da, and considered oxidized Met, carbamidomethylation of Cys, and Cys + 104.96 Da modification (for selenocysteine identification) as a variable modification. Positive identification of Selenocysteine containing peptides required the presence of several sequence specific ions along with an MS1 profile consistent with the presence of selenium.
The digests were also analysed using Parallel Reaction Monitoring (PRM) in which specific m/z ratios were fragmented and analysed. Quantification of peptide abundances was performed using Xcalibur 4.0 software (ThermoScientific) to plot PRM chromatograms and integrating the peptide peak areas.
Metabolic labelling with 75Se
McArdle 7777 cells were transfected with Lipofectamine as described above. After 24 hours, media was changed to serum-free DMEM that was supplemented with 100 nM 75Se (specific activity, 6.29 μCi/μL; Research Reactor Centre, University of Missouri, Columbia, MO). 24 hours later, cells were washed with PBS and lysed with NP40 buffer as described above. Lysates were resolved by SDS-PAGE and imaged by PhosphorImager (GE Healthcare).
Statistical analysis
Where applicable, data have been represented as mean ± SD. Data were analysed by unpaired, two-tailed Student’s t test, using GraphPad Prism version 8 (GraphPad Software).
Funding Statement
This work was supported by the National Institutes of Health under Grant #R01DK107426 (D.M.D.) and Grant #R01GM077073 (P.R.C.). The Fusion Lumos instrument was purchased via an NIH shared instrument grant, 1S10OD023436-01.
Acknowledgments
We would like to thank Maggie Rybak and Hannah Singerline for technical assistance as well as the Proteomics Core at the Lerner Research Institute for assistance with mass spectrometry.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary material data for this article can be accessed here.
References
- [1].Byun BJ, Kang YK.. Conformational preferences and pKa value of Selenocysteine residue. Biopolymers. 2011;95:345–353. [DOI] [PubMed] [Google Scholar]
- [2].Seeher S, Mahdi Y, Schweizer U. Post-transcriptional control of selenoprotein biosynthesis. Curr Protein Pept Sci. 2012;13(4):337–346. [DOI] [PubMed] [Google Scholar]
- [3].Berry M, Banu L, Chen Y, et al. Recognition of UGA as a selenocysteine codon in type I deiodinase requires sequences in the 3ʹ untranslated region. Nature. 1991;353:273–276. [DOI] [PubMed] [Google Scholar]
- [4].Walczak R, Westhof E, Carbon P, et al. A novel RNA structural motif in the selenocysteine insertion element of eukaryotic selenoprotein mRNAs. Rna. 1996;2(4):367–379. [PMC free article] [PubMed] [Google Scholar]
- [5].Copeland P, Fletcher J, Carlson B, et al. A novel RNA binding protein, SBP2, is required for the translation of mammalian selenoprotein mRNAs. Embo J. 2000;19(306–314):306–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Fletcher JE, Copeland PR, Driscoll DM, et al. The selenocysteine incorporation machinery: interactions between the SECIS RNA and the SECIS-binding protein SBP2. RNA. 2001;7(10):1442–1453. [PMC free article] [PubMed] [Google Scholar]
- [7].Dumitrescu AM, Liao XH, Abdullah MS, et al. Mutations in SECISBP2 result in abnormal thyroid hormone metabolism. Nat Genet. 2005;37(11):1247–1252. [DOI] [PubMed] [Google Scholar]
- [8].Di Cosmo C, McLellan N, Liao XH, et al. Clinical and molecular characterization of a novel selenocysteine insertion sequence-binding protein 2 (SBP2) gene mutation (R128X). J Clin Endocrinol Metab. 2009;94(10):4003–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Azevedo MF, Barra GB, Naves LA, et al. Selenoprotein-related disease in a young girl caused by nonsense mutations in the SBP2 gene. J Clin Endocrinol Metab. 2010;95(8):4066–4071. [DOI] [PubMed] [Google Scholar]
- [10].Schoenmakers E, Agostini M, Mitchell C, et al. Mutations in the selenocysteine insertion sequence-binding protein 2 gene lead to a multisystem selenoprotein deficiency disorder in humans. J Clin Invest. 2010;120(12):4220–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Allamand V, Richard P, Lescure A, et al. A single homozygous point mutation in a 3ʹuntranslated region motif of selenoprotein N mRNA causes SEPN1-related myopathy. EMBO Rep. 2006;7(4):450–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tujebajeva RM, Copeland PR, Xu XM, et al. Decoding apparatus for eukaryotic selenocysteine insertion. EMBO Rep. 2000;1(2):158–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fagegaltier D, Hubert N, Yamada K, et al. Characterization of mSelB, a novel mammalian elongation factor for selenoprotein translation. Embo J. 2000;19(17):4796–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Xu X, Carlson B, Mix H, et al. Biosynthesis of selenocysteine on its tRNA in eukaryotes. PLoS Biol. 2007;5:e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chavette L, Brown BA, Driscoll DM. Ribosomal protein L30 is a component of the UGA-selenocysteine recoding machinery in eukaryotes. Nat Struct Mol Biol. 2005;12(15):408–416. [DOI] [PubMed] [Google Scholar]
- [16].Miniard A, Middleton L, Budiman M, et al. Nucleolin binds to a subset of selenoprotein mRNAs and regulates their expression. Nucleic Acids Res. 2010;38:4807–4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Budiman ME, Bubenik J, Miniard A, et al. Eukaryotic initiation factor 4a3 is a selenium-regulated RNA -binding protein that selectively inhibits selenocysteine incorporation. Mol Cell. 2009;35:479–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Howard M, Aggarwal G, Anderson A, et al. Recoding elements located adjacent to a subset of eukaryal selenocysteine-specifying UGA codons. EMBO. 2005;24(8):1596–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Howard M, Moyle M, Aggarwal G, et al. A recoding element that stimulates decoding of the UGA codons by Sec tRNA [Ser]Sec. RNA. 2007;13:912–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Maiti B, Arbogast S, Allamand V, et al. A mutation in the SEPN1 SRE reduces selenocysteine incorporation and leads to SEPN1-related myopathy. Hum Mutat. 2010;30(3):411–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bubenik J, Miniard A, Driscoll DM. Alternative transcripts and 3ʹUTR elements govern the incorporation of Selenocysteine into Selenoprotein S. PLOS One. 2013;8(4):e62102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Walder K, Kantham L, McMillan J, et al. Tanis: a link between type 2 diabetes and inflammation? Diabetes. 2002;51:1859–1866. [DOI] [PubMed] [Google Scholar]
- [23].Gao Y, Feng H, Walder K, et al. Regulation of the selenoproteins SelS by glucose deprivation and endoplasmic reticulum stress- SelS is a novel glucose-regulated protein. FEBS Lett. 2004;563(1–3):185–190. [DOI] [PubMed] [Google Scholar]
- [24].Shchedrina V, Everley R, Zhang Y, et al. Selenoprotein K binds multiprotein complexes and is involved in the regulation of endoplasmic reticulum homeostasis. J Biol Chem. 2011;286:42937–42948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Curran J, Jowett JBM, Elliott K, et al. Genetic variation in selenoprotein S influences inflammatory response. Nat Genet. 2005;37(11):1234–1241. [DOI] [PubMed] [Google Scholar]
- [26].Hannan N, Wanyonyi S, Konstantopolous N, et al. Activation of the selenoprotein SEPS1 gene expression by pro-inflammatory cytokines in HepG2 cells. Cytokine. 2006;33(5):246–251. [DOI] [PubMed] [Google Scholar]
- [27].Wu J, Kaufman R. From the acute ER stress to physiological roles of the unfolded protein response. Cell Death Differ. 2006;13:374–384. [DOI] [PubMed] [Google Scholar]
- [28].Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397(6716):271–274. [DOI] [PubMed] [Google Scholar]
- [29].Raven JF, Baltzis D, Wang S, et al. PKR and PKR-like endoplasmic reticulum kinase induce the proteasome-dependent degradation of cyclin D1 via a mechanism requiring eukaryotic initiation factor 2alpha phosphorylation. J Biol Chem. 2008;283(6):3097–3108. [DOI] [PubMed] [Google Scholar]
- [30].Wu J, Rutkowski DT, Dubois M, et al. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell. 2007;13(3):351–364. [DOI] [PubMed] [Google Scholar]
- [31].Yamamoto K, Sato T, Matsui T, et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell. 2007;13(3):365–376. [DOI] [PubMed] [Google Scholar]
- [32].Smith MH, Ploegh HL, Weissman JS. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science. 2011;334(6059):1086–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ye Y, Shibata Y, Kikkert M, et al. Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. PNAS. 2005;102(40):14132–14138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lee JH, Park KJ, Jang JK, et al. Selenoprotein S-dependent Selenoprotein K Binding to p97(VCP) Protein Is essential for endoplasmic reticulum-associated degradation. J Biol Chem. 2015;290(50):29941–29952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ye Y, Shibata Y, Yun C, et al. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nat Genet. 2004;429:841–847. [DOI] [PubMed] [Google Scholar]
- [36].Kim KH, Gao Y, Walder K, et al. SEPS1 protects RAW264.7 cells from pharmacological ER stress agent-induced apoptosis. Biochem Biophys Res Commun. 2007;354:127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fradejas NPM, Mora-Lee S, Tranque P, et al. SEPS1 gene is activated during astrocyte ischemia and shows prominent antiapoptotic effects. J Mol Neurosci. 2008;35:259–265. [DOI] [PubMed] [Google Scholar]
- [38].Kelly E, Greene C, Carroll T, et al. Selenoprotein S/SEPS1 modifies endoplasmic reticulum stress in Z variant alpha 1-antitrypsin deficiency. J Biol Chem. 2009;284:16891–16897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zheng HTZL, Huang CJ, Hua X, et al. Selenium inhibits high glucoseand high insulin induced adhesion molecule expression in vascular endothelial cells. Arch Med Res. 2008;39:373–379. [DOI] [PubMed] [Google Scholar]
- [40].Zhao Y, Li H, Men LL, et al. Effects of selenoprotein S on oxidative injury in human endothelial cells. J Transl Med. 2013;11:287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lee JH, Kwon JH, Jeon YH, et al. Pro178 and Pro183 of selenoprotein S are essential residues for interaction with p97(VCP) during endoplasmic reticulum-associated degradation. J Biol Chem. 2014;289(20):13758–13768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Christensen LC, Jensen NW, Vala A, et al. The human selenoprotein VCP-interacting membrane protein (VIMP) is non-globular and harbors reductase function in an intrinsically disordered region. J Biol Chem. 2012;287(31):26388–26399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Liu J, Rozovsky S. Contribution of selenocysteine to the peroxidase activity of selenoprotein S. Biochemistry. 2013;52(33):5514–5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Liu J, Li F, Rozovsky S. The intrinsically disordered membrane protein selenoprotein S is a reductase in vitro. Biochemistry. 2013;52(18):3051–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bubenik JL, Ladd AN, Gerber CA, et al. Known turnover and translation regulatory RNA-binding proteins interact with the 3ʹ UTR of SECIS-binding protein 2. RNA Biol. 2009;6(1):73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mehta A, Rebsch C, Kinzy S, et al. Efficiency of mammalian selenocysteine incorporation. J Biol Chem. 2004;279:37852–37859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Sievers F, Higgins DG. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol Biol. 2014;1079:105–116. [DOI] [PubMed] [Google Scholar]
- [48].Gruber AR, Lorenz R, Bernhart SH, et al. The Vienna RNA websuite. Nucleic Acids Res. 2008;36(Web Server issue):W70–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Mayya VK, Duchaine TF. Ciphers and executioners: how 3ʹ-untranslated regions determine the fate of messenger RNAs. Front Genet. 2019;10:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kryukov GV, Castellano S, Novoselov SV, et al. Characterization of mammalian selenoproteomes. Science. 2003;300(5624):1439–1443. [DOI] [PubMed] [Google Scholar]
- [51].Fagegaltier D, Lescure A, Walczak R, et al. Structural analysis of new local features in SECIS RNA hairpins. Nucleic Acids Res. 2000;2679–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Driscoll DM, Chavatte L. Finding needles in a haystack. In silico identification of eukaryotic selenoprotein genes. EMBO Rep. 2004;5(2):140–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ray PS, Jia J, Yao P, et al. A stress-responsive RNA switch regulates VEGFA expression. Nature. 2009;457(7231):915–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Turanov AA, Lobanov AV, Hatfield DL, et al. UGA codon position-dependent incorporation of selenocysteine into mammalian selenoproteins. Nucleic Acids Res. 2013;41(14):6952–6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Latreche L, Jean-Jean O, Driscoll DM. L C, Novel structural determinants in human SECIS elements modulate the translational recoding of UGA as selenocysteine. Nucleic Acids Res. 2009;37:5868–5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Karijolich J, Yu YT. Converting nonsense codons into sense codons by targeted pseudouridylation. Nature. 2011;474(7351):395–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.